

ABSTRACT
The rabies virus (RABV) phosphoprotein P is a multifunctional protein: it plays an essential role in viral transcription and replication, and in addition, RABV P has been identified as an interferon antagonist. Here, a yeast two-hybrid screen revealed that RABV P interacts with the focal adhesion kinase (FAK). The binding involved the 106-to-131 domain, corresponding to the dimerization domain of P and the C-terminal domain of FAK containing the proline-rich domains PRR2 and PRR3. The P-FAK interaction was confirmed in infected cells by coimmunoprecipitation and colocalization of FAK with P in Negri bodies. By alanine scanning, we identified a single mutation in the P protein that abolishes this interaction. The mutant virus containing a substitution of Ala for Arg in position 109 in P (P.R109A), which did not interact with FAK, is affected at a posttranscriptional step involving protein synthesis and viral RNA replication. Furthermore, FAK depletion inhibited viral protein expression in infected cells. This provides the first evidence of an interaction of RABV with FAK that positively regulates infection.
IMPORTANCE Rabies virus exhibits a small genome that encodes a limited number of viral proteins. To maintain efficient virus replication, some of them are multifunctional, such as the phosphoprotein P. We and others have shown that P establishes complex networks of interactions with host cell components. These interactions have revealed much about the role of P and about host-pathogen interactions in infected cells. Here, we identified another cellular partner of P, the focal adhesion kinase (FAK). Our data shed light on the implication of FAK in RABV infection and provide evidence that P-FAK interaction has a proviral function.
INTRODUCTION
Rabies is a fatal human disease caused by viruses of the genus Lyssavirus belonging to the family Rhabdoviridae (order Mononegavirales). The best-characterized lyssavirus is rabies virus (RABV), which infects diverse mammalian species. Other lyssaviruses, European bat lyssaviruses 1 and 2 (EBLV-1 and -2), Australian bat lyssavirus (ABLV), Mokola virus (MOKV), and Duvenhage virus (DUVV), have been reported to cause lethal human rabies (1–3).
The single-stranded negative-sense RNA genome (∼12 kb) encodes five proteins, nucleoprotein (N), phosphoprotein (P), matrix protein (M), glycoprotein (G), and a polymerase (L), in the order 3′-N-P-M-G-L-5′. Viral transcription and replication take place within Negri bodies (NBs), which are inclusion bodies formed by viral infection and now considered viral factories (4). During transcription, a positive-strand leader RNA and five capped and polyadenylated mRNAs are synthesized. The replication process yields nucleocapsids containing full-length antigenome sense RNA, which in turn serve as templates for the synthesis of genome sense RNA.
Besides its crucial role in viral transcription and replication, the RABV P protein is a multifunctional interferon (IFN) antagonist protein. First, P protein prevents IFN induction by impairing IRF-3 phosphorylation and dimerization, leading to the inhibition of IFN production (5). Second, the P protein interacts with STAT1 and prevents JAK/STAT signaling through several mechanisms, including sequestration of STAT away from the nuclear compartment and inhibition of STAT-DNA binding (6–9). Finally, P interacts with the nuclear interferon stimulated gene (ISG), promyelocytic leukemia protein (PML), causing its mislocalization to the cytoplasm and leading to the disruption of PML nuclear bodies (11, 12). The fact that one PML isoform (PML IV) has a specific antiviral effect on RABV suggests that the P-PML interaction could counteract this antiviral response (13).
In addition to the dynein light chain (LC8) and STAT1, which were previously identified as cellular partners of the RABV P protein (6, 14), the focal adhesion kinase (FAK) was also isolated from a two-hybrid screen using RABV P as the bait. FAK is a cytoplasmic protein tyrosine kinase preferentially localized at cellular focal contacts. It plays a key role in cellular signaling pathways, which are important for transcriptional regulation, cell cycle progression, modulation of apoptosis, control of cell migration, and metastasis of transformed cells (15–18).
As viruses activate and hijack many signaling pathways to favor their own replication, we analyzed the role of FAK during RABV infection.
We have confirmed that P interacts with FAK in the context of viral infection. We have identified the binding domains of both proteins and precisely defined critical residues of P involved in the interaction. The role of FAK in the viral life cycle was investigated by generating a recombinant virus unable to interact with FAK or by depletion of FAK with small interfering RNA (siRNA). The results indicated that FAK, through its interaction with P, favors viral multiplication.
MATERIALS AND METHODS
Cells and viruses.
BSR cells, cloned from BHK 21 (baby hamster kidney), U373-MG (human glioblastoma astrocytoma), HEK (hamster embryonic kidney), and N2A (neuroblastoma) cells, were grown in Dulbbeco's modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS). The CVS strain of rabies virus was grown in BSR cells, and the N2C strain was grown in N2A cells.
Other strains of lyssaviruses (8743THA, 8918FRA, 9007FIN, 8619NIG, 8720ZIM, 86132SA, and 9810AUS) were provided by the National Reference Centre for Rabies, WHO Collaborating Centre for Reference and Research on Rabies, Paris, France, and were used to clone their P proteins in the pLex plasmid.
Antibodies and drugs.
The rabbit polyclonal anti-P antibody, the mouse monoclonal anti-P antibody, the mouse monoclonal anti-N antibody, and the rabbit polyclonal anti-M antibody were previously described (4, 19). The rabbit polyclonal anti-pY397FAK antibody (44-625G) was obtained from Invitrogen, and the rabbit polyclonal anti-FAK C-20 antibody was from Santa Cruz. The anti-α-tubulin antibody (DM1A) and Texas Red-phalloidin (S6501) were from Sigma. Secondary fluorescent antibodies were purchased from Molecular Probes.
Plasmids.
The yeast expression plasmids pLex10 and pGAD-GE were previously described (6). The vector pLex10 contains the yeast-selectable TRP1 gene and the lexA DNA-binding domain (DB) fused with the wild-type (WT) or mutant P protein. The plasmid pGAD contains the yeast-selectable LEU2 gene and the sequence encoding the GAL4 activation domain (AD) fused to the FAK or the N gene. P mutations were introduced in the WT P gene by two-step PCR-based site-directed mutagenesis using the forward primer 2HPCA1 (5′-GCCGAATTCATGAGCAAGATCTTT-3′) and the reverse primer 2HPCB1 (5′-CCGGTCGACTTAGCAGGATGTATA-3′) in combination with primers specific for the mutations. The pGAD-FAK construct was isolated from a yeast two-hybrid screen and contains the Rattus norvegicus C-terminal domain of the FAK. The plasmids pFAK-GFP and pFRNK-GFP, provided by J.-A. Girault (INSERM-Institut du Fer à Moulin), were described previously (20, 21). The pFAKΔFRNK-GFP plasmid was constructed by deletion of the FAK-related nonkinase (FRNK) sequence by using PCR with the forward primer 5′-GCCCTCGAGATATGGCAGCTGCT-3′ and the reverse primer 5′-GCCGGATCCATCCTCATCCGTTC-3′. The plasmid pRL-TK was described previously (22). The plasmids pTIT-N, pTIT-P, and pTIT-L were described previously (23) and were obtained from K. K. Conzelmann (Ludwig-Maximilians University of Munich). The plasmid rCVS-N2C was generously provided by M. Schnell (Thomas Jefferson University). The pcDNA3.1-P and pcDNA3.1-PR109A plasmids were constructed by two-step PCR-based site-directed mutagenesis using the forward primer 5′GCCGCTAGCATGAGCAAGATCTTT3′ and the reverse primer 5′-CCGGTCGACTTAGCAGGATGTATA-3′ in combination with primers specific for the mutations.
The pLex plasmids encoding the phosphoproteins P Nishigahara (P Ni) and P Ni-CE were constructed as described previously (21). The pLex plasmids encoding the P proteins of different lyssaviruses were prepared as follows. The cDNAs were obtained by reverse transcription (RT)-PCR from total RNA isolated from infected BSR cells using hexamer primers (Roche Boehringer). The sequences of the primers used for the PCR amplification are presented in Table 1, with the gene-specific sequence in boldface. The PCR products were cloned into pLex at EcoRI and SalI sites using the in-fusion cloning methodology (24).
TABLE 1.
Sequences of primers used for PCR amplification of cDNA corresponding to the P proteins of lyssaviruses
| Lyssavirus isolate | Primersa |
|---|---|
| 8743THA | 5′-GACTGGCTGGAATTCATGAGCAAGATCTTTGTCAATC-3′ |
| 5′-TTGGCTGCAGGTCGACTCAGCGGGATGTATAACGGT-3′ | |
| SHBRV | 5′-GACTGGCTGGAATTCATGAGCAAGATCTTTGTCAATC-3′ |
| 5′-TTGGCTGCAGGTCGACTCAGGGGAACGCATAACGC-3′ | |
| 9810AUS (ABLV) | 5′-GACTGGCTGGAATTCATGAGCAAGATCTTTGTCAATC-3′ |
| 5′-TTGGCTGCAGGTCGACTTAACATGACATGTAACGGTTC-3′ | |
| 8619NIG (LBV) | 5′-GACTGGCTGGAATTCATGAGCAAGGGATTAATACACC-3′ |
| 5′-TTGGCTGCAGGTCGACGTAATTGTGATCTATCTCCTCAA-3′ | |
| 8720ZIM (MOKV) | 5′-GACTGGCTGGAATTCATGAGCAAAGATTTGGTGCATC-3′ |
| 5′-TTGGCTGCAGGTCGACTTACTCTGCCTCCTGGAGCC-3′ | |
| 8918FRA (EBLV-1) | 5′-GACTGGCTGGAATTCATGAGCAAGATCTTTGTCAATC-3′ |
| 5′-TTGGCTGCAGGTCGACTCAATACGCAAGATACTGTTGA-3′ | |
| 9007FIN (EBLV-2) | 5′-GACTGGCTGGAATTCATGAGCAAGATCTTTGTCAACC-3′ |
| 5′-TTGGCTGCAGGTCGACTCAATATGCCAGATACCGATTG-3′ | |
| 86132A (DUVV) | 5′-GACTGGCTGGAATTCATGAGCAAGGATTTTTATCAATC-3′ |
| 5′-TTGGCTGCAGGTCGACTCAATAGGTCAGGTATTTGTCA-3′ |
The P sequences are in boldface, and restriction sites are underlined.
Yeast two-hybrid assays.
The P sequence of RABV (strain CVS) was fused to the DNA-binding domain of LexA and used as bait to screen the nerve growth factor-induced PC12 (rat adrenal pheochromocytoma cell line) cDNA library in which each DNA was fused to the sequence encoding the GAL4 activation domain, as described previously (6). Briefly, the yeast L40 strain containing the two LexA-responsive reporter genes HIS3 and lacZ was first transformed with the bait plasmid pLex-P using a lithium acetate protocol. pLex-P-expressing L40 cells selected and grown in Trp-deficient medium were then transformed with plasmid DNA from the PC12 cDNA library. Double transformants were grown on plates containing medium lacking Trp and Leu (Trp− Leu−) to select for the presence of both the bait and library plasmids and deprived of His (Trp− Leu− His−) to select for protein-protein interaction. Positive clones were then assayed for β-galactosidase activity. These clones conferred on L40 the abilities to grow in the absence of histidine and to produce β-galactosidase activity in the presence of the LexA BD-P hybrid, but not with LexA BD alone or with LexA BD-lamin.
The β-galactosidase activities of histidine-positive clones were tested by 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) overlay as follows: an X-Gal mixture containing 0.5% agar, 0.1% SDS, 6% dimethylformamide, and 0.04% X-Gal was overlaid on fresh transformants grown on Trp− Leu− plates, and blue clones were detected after 60 min to 18 h at 30°C.
Minireplicon assay system and luciferase assay.
The minireplicon assay system was slightly modified from that of Le Mercier et al. (25). N2A cells were grown in 12-well plate (3 × 105 cells per well) in DMEM supplemented with 5% FCS and incubated for 24 h at 37°C. Cells were transfected with 0.75 μg pDI-luc, 1.2 μg pcDNA1-N, 1.2 μg pcDNA3.1-P or pcDNA3.1-PR109A, 0.3 μg pcDM8-L, 1 μg pT7 encoding the T7 RNA polymerase, and 0.3 μg pRL-TK using Lipofectamine 2000 as described by the manufacturer (Invitrogen). The N, P, and L proteins and the RNA minigenome formed a functional RNP template resulting in luciferase gene transcription, and thus, the transcriptional activity of the reconstituted RNP was related to the amount of luciferase. Forty-eight hours after transfection, the measurement of firefly and Renilla luciferase activities was performed using the dual-luciferase assay (Promega) according to the manufacturer's protocol. Relative expression levels were calculated by dividing the firefly luciferase values by those of Renilla luciferase.
Construction and recovery of rN2C virus carrying the P mutant P.R109A.
The full-length recombinant N2C (prN2C) infectious clone was described previously (26). The amplified fragment carrying the R109A mutation in the P protein (P.R109A) was inserted into the original full-length genomic plasmid using two unique restriction sites, AvrII in the sequence of the N gene and SpeI in the C-terminal region of the P gene. The fragment carrying the R109A mutation was constructed by two-step PCR amplification of the full-length genomic plasmid using the forward primer A1-AvrII (5′GCCTTGTATCACCCTAGGGAA3′) and the reverse primer B1-SpeI (5′CGGTCGACTAGTAATTGGAAT3′), in combination with the primers carrying the mutation P.R109A (restriction sites are underlined). The PCR product was digested with AvrII and SpeI and was introduced into the full-length genomic plasmid to obtain the resulting plasmid, prN2C-P.R109A.
Recombinant viruses were recovered as described previously, with the following modifications (26, 27). Briefly, N2A cells (106 cells) were transfected using Lipofectamine 2000 (Invitrogen) with 0.85 μg of full-length rN2C R109A cDNA, in addition to 0.4 μg pTIT-N, 0.2 μg pTIT-P, 0.2 μg pTIT-L, and 0.15 μg pTIT-G, which encode the N, P, L, and G proteins of rabies virus strain SAD-L16. These plasmids were cotransfected with 0.25 μg of a plasmid expressing the T7 RNA polymerase. Six days posttransfection, the supernatant was passaged on fresh N2A cells, and the recombinant viruses were amplified for 3 days and then detected by immunofluorescence staining with anti-P and anti-N antibodies.
Cell infection and transfection.
Cells were grown on glass coverslips in 6-well plates and infected with rabies virus at different multiplicities of infection (MOI) and at various times postinfection (p.i.). Cells grown to 80% confluence were transfected with different quantities of plasmid using Lipofectamine 2000 as described by the manufacturer (Invitrogen).
Detection of viral mRNA by qRT-PCR.
Quantification of viral mRNA was performed by quantitative RT (qRT)-PCR). Total RNA was extracted from cells using the Nucleospin RNA II kit (Macherey-Nagel) according to the manufacturer's instructions. First-strand cDNA was synthesized using a reverse primer specific for the P gene, 2HPCB1 (5′-CCGGTCGACTTAGCAGGATGTATA-3′). Quantitative PCR (qPCR) was performed using P gene-specific primers (qPCRsP, 5′-CAACCTTGGTGAGATGGTTAGGGT-3′, and qPCRasP, 5′-GTTGACCGTGACATAGGATAC-3′). Primers specific for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were used to quantify the cellular RNA content (qPCRsGAPDH, 5′-GATGGGTGTGAACCACGAGAAA-3′, and qPCRasGAPDH, 5′-ATCACGCCACAGCTTTCCAG-3′). The reaction was performed using the SYBR green Quantifast PCR kit (Qiagen) containing 2 μl of reverse transcription product, 12.5 μl of SYBR green mix, and 125 pmol of each primer. The qPCR was performed with a standard protocol using the Mx3000P system, and data were analyzed with the MxPro software (Stratagene). The relative P mRNA quantities for both N2C-WT and N2C R109A viruses were normalized to the amount of GAPDH, and the ratio of P mRNA N2C-WT to P mRNA N2C R109A was determined using the 2−ΔΔCT method described previously (28).
Northern blot analysis.
RNA was isolated from cells with the RNA Now kit (Ozyme). Total RNA was separated on a 1.5% agarose gel under denaturing conditions and blotted onto nylon membranes (Roche Molecular Biochemicals). Hybridizations were performed with digoxigenin (DIG)-labeled oligonucleotides recognizing the RABV N or GAPDH sequence. Bound probe was detected by incubation with anti-DIG antibody conjugated to alkaline phosphatase, followed by addition of CDP Star substrate (Roche Molecular Biochemicals).
siRNA transfection.
A pool of 4 siRNAs targeting FAK (5′-GCGAUUAUAUGUUAGAGAU-3′, 5′-GGGCAUCAUUCAGAAGAUA-3′, 5′-UAGUACAGCUCUUGCAUAU-3′, and 5′-GGACAUUAUUGGCCACUGU-3′) was purchased from ThermoScientific (On-Targetplus human FAK siRNA-Smart pool and On-Targetplus Nontargeting pool). Cells were seeded at 0.5 × 105/ml/well in 24-well plates the day before. Transient transfections were performed using Dharmafect reagent according to the manufacturer's instructions (Dharmacon, GE Healthcare). A final siRNA concentration of 25 nM was used. Cells were infected (MOI = 3) 48 h posttransfection. Western blot analysis (WB) was performed on cell lysates collected at various time points postinfection. At an siRNA concentration of 25 nM, cell viability measured by trypan blue exclusion was >95%.
Coimmunoprecipitation.
Cells (106 cells) were harvested by scraping into cold phosphate-buffered saline (PBS) and lysed on ice in 500 μl of buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, and an anti-protease cocktail. The cytoplasmic fraction was incubated overnight at 4°C with the rabbit polyclonal anti-FAK antibody. Immune complexes were precipitated by incubation with protein A-Sepharose for 1 h at 4°C, washed three times, and denatured in Laemmli buffer. Immunoprecipitated proteins were analyzed by Western immunoblotting using the mouse monoclonal anti-P antibodies.
Western blot analysis.
Cells were washed and resuspended in PBS, lysed in hot Laemmli sample buffer, and boiled for 5 min. Proteins were analyzed by electrophoresis on SDS-PAGE and transferred onto a nitrocellulose membrane. The membrane was blocked with 10% skim milk in PBS for 2 h and incubated overnight at 4°C with the corresponding antibodies. The blots were then washed extensively in PBS-Tween and incubated for 1 h with the appropriate peroxidase-coupled secondary antibodies (Amersham). Bands were developed using ECL substrate (Amersham). Alternatively, the blots were incubated with Fluor 800-conjugated IgG or Fluor 680-conjugated IgG secondary antibody (Cell Signaling) for 1 h at room temperature. After washing, the membranes were scanned with the Odyssey infrared imaging system (Li-Cor, Lincoln, NE) at a wavelength of 700 or 800 nm. Protein spot levels were determined by using Image J quantification software or Image Studio software (Li-Cor).
35S-labeling experiments and immunoprecipitation.
At 16 h postinfection, the culture medium was replaced with cysteine- and methionine-free medium, and the culture was incubated for 1 h. The cells were then labeled for 15 min with 2 ml of prewarmed medium containing 20 μCi of [35S]methionine and [35S]cysteine (0.8 MBq) (Express protein labeling mix; PerkinElmer). The cells were washed in cold PBS and lysed in 500 μl of buffer, as described above. The cytoplasmic fractions were incubated for 2 h at 4°C with anti-P or anti-N antibody. Immune complexes were precipitated by incubation with protein A-Sepharose for 1 h at 4°C, washed three times, denatured in Laemmli buffer, and analyzed by SDS-PAGE. Quantification of radioactivity was performed with a phosphorimager.
Immunofluorescence staining and confocal microscopy.
Cells were fixed for 10 min with 4% paraformaldehyde (PFA) and permeabilized for 5 min with 0.1% Triton X-100 in PBS. The cells were incubated with the indicated primary antibodies for 1 h at room temperature, washed, and incubated for 1 h with Alexa Fluor-conjugated secondary antibodies. Following washing, the cells were fixed and mounted with Immu-Mount (Thermo Scientific) containing DAPI (4′,6-diamidino-2-phenylindole). Images were captured using a Leica SP2 confocal microscope (63× oil immersion objective) or a Zeiss Axio Observer fluorescence microscope (63× oil immersion objective).
RESULTS
Identification of FAK as a RABV P protein binding partner.
The yeast two-hybrid system was used to search for proteins interacting with RABV (strain CVS) P protein. A nerve growth factor-induced PC12 cell cDNA library was first screened in a two-hybrid assay with the full-length P protein (residues 1 to 297), as described previously, for the identification of LC8 and STAT1 (6, 14). As most of the positive clones encoded LC8, which strongly interacts with P (14), we introduced two amino acid substitutions (D143A and Q147A) into P that abolish binding to LC8 (29). Using the mutated P as the bait, we selected and sequenced 25 positive clones, 5 of which encoded different portions of the C-terminal domain of FAK (GenBank accession number AF020777).
FAK is expressed in most tissues, and its sequence is highly conserved across species. The protein is a 125-kDa non-receptor protein tyrosine kinase located primarily at focal adhesions (FAs) and serves as a key component in the transmission of signals between the extracellular matrix and the cytoplasm. The complete protein is 1,052 amino acids long and contains a central kinase domain (residues 422 to 676) flanked by large N- and C-terminal domains (Fig. 1A). The N-terminal domain, referred to as the FERM domain (residues 36 to 258), regulates FAK activity, whereas the C-terminal domain comprises the focal-adhesion-targeting (FAT) domain (residues 914 to 1052) and an unstructured proline-rich region between the catalytic and FAT domains. FAT is responsible for localizing FAK to FAs and contains binding sites for other proteins, such as paxillin, talin, and vinculin (30). In addition to FAK, FRNK is also a product of the gene but is autonomously expressed under the control of an alternative, intronic promoter. FRNK (residues 676 to 1052) is composed of the FAT domain and proline-rich domains (PRR) important for adaptor protein binding.
FIG 1.
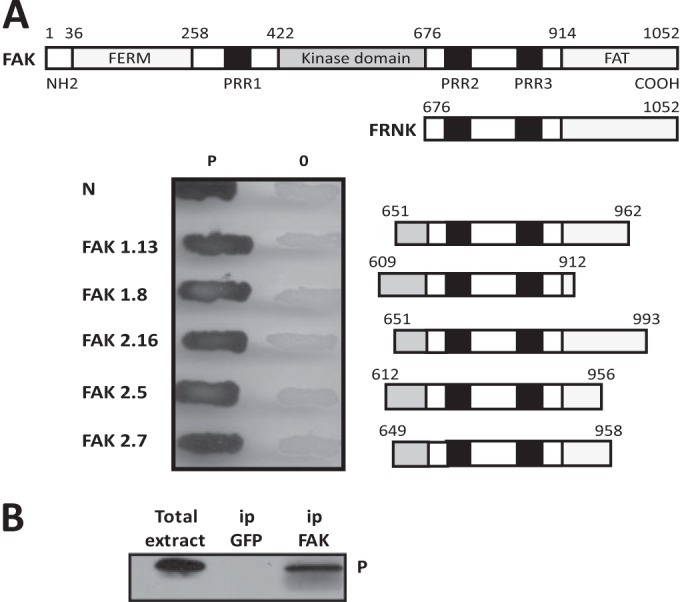
The RABV P protein binds FAK. (A) Mapping of the P protein binding domain within FAK by two-hybrid screen. The major domains of FAK (the FERM, kinase, and FAT domains) and the PRR are shown. FRNK is transcribed from a second promoter within the FAK gene and comprises the FAT domain and 2 PRR (top). Several pGAD clones containing different C-terminal fragments of FAK fused to the Gal4 AD were isolated during the two-hybrid screen. Yeast cells (strain L40) were cotransformed with these pGAD plasmids and the pLex plasmid encoding P fused to the DB or empty pLex plasmid (0). The P-FAK interaction is indicated by the appearance of blue colonies in the presence of X-Gal (bottom). P-N interaction was also analyzed. (B) Detection of P-FAK interaction in infected cells. HEK293T cells were transfected with a plasmid expressing FAK. At 24 h posttransfection, the cells were infected with wild-type CVS at an MOI of 3 PFU/cell. At 24 h after infection, cells were harvested and lysed. The cell lysate was incubated with a rabbit polyclonal anti-FAK or anti-GFP antibody. Immune complexes were precipitated (ip) by incubation with protein A-Sepharose and analyzed by Western immunoblotting with mouse monoclonal anti-P antibody. In the left portion of the blot, total cellular extracts were also analyzed.
The interaction between the full-length P protein and five of the positive clones encoding different fragments of the C-terminal domain was confirmed by two-hybrid assay (Fig. 1A).
To demonstrate that P protein indeed associates with full-length FAK in vivo, HEK293T cells were transfected with a plasmid expressing FAK before infection by RABV (CVS) at an MOI of 3. At 24 h postinfection, the cell extracts were immunoprecipitated using an anti-FAK antibody or green fluorescent protein (GFP) antibody as a control. The retained proteins were then analyzed by Western blotting with an antibody directed against P. As shown in Fig. 1B, the P protein was specifically coprecipitated with FAK, but not with the GFP antibody (Fig. 1B).
Identification of P-FAK interacting domains.
The domain of FAK interacting (GE Healthcare) with P was deduced from the sequences of the positive cDNA clones encoding variants of the C-terminal part of FAK and containing the unstructured proline-rich region flanked by different portions of the linker domain between the kinase domain and the FAT domain (Fig. 1A). The fact that all these clones shared a common part extending from residues 649 to 916 indicated that this linker region, which comprised the proline-rich domains PRR2 and PRR3, was sufficient for binding to P.
RABV P protein contains three functional domains separated by two intrinsically disordered regions: the N-terminal domain (PNTD, residues 1 to 52), interacting with the soluble N protein (called N°) and L protein (31–33); the dimerization domain (Pdim, residues 91 to 131) (34); and the C-terminal domain (PCTD, residues 186 to 297), involved in binding to the N RNA (35–38), as well as cellular proteins STAT1 and PML (6, 11).
To identify the FAK binding domain on P, deletion mutants of the P protein fused to LexA were tested for the ability to bind the FAK fragment of one positive clone (2.7). As shown in Fig. 2, truncation of the first 172 N-terminal residues (PΔN172) abolished the association of P with FAK, whereas truncation of the 125 C-terminal residues (amino acids 172 to 297; PΔC125) had no effect on the association, indicating that the binding domain was located in the amino-terminal part of P. As expected, PΔN172 still interacted with N (33–35). As PΔN52 still interacted with FAK, the PNTD (first 52 residues) of P was not implicated in the interaction with FAK. Internal truncations between residues 52 and 172 and further deletions in the dimerization domain (residues 91 to 131) impaired the interaction, indicating that residues between 106 and 131 were necessary for FAK binding (Fig. 2). Sequence alignment of this domain of the lyssavirus P proteins indicated conserved hydrophobic residues and several amino acids having similarity (Fig. 3A). Site-directed mutagenesis performed on these residues indicated that substitution of alanine for one of the residues, R106, R109, R113, F114, W118, or I125, abolished the interaction between RABV P and FAK (Fig. 3B, right lane). As expected, none of the mutations affected binding to N (Fig. 3B, left lane).
FIG 2.
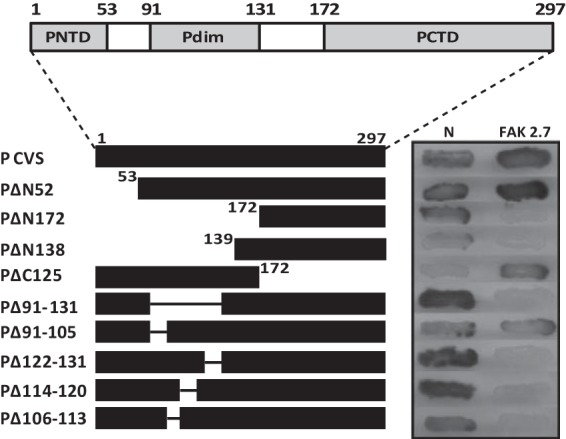
Mapping of the FAK binding domain within the P protein by two-hybrid screen. (Top) RABV P contains three modular domains: PNTD (residues 1 to 52), Pdim (residues 91 to 131) (34), and PCTD (residues 186 to 297). (Bottom) The interactions between deletion mutants of the P protein fused to the LexA DB and the 649-to-958 fragment of FAK (clone 2.7) fused to the Gal4 AD were assayed as for Fig. 1. In parallel, interaction with RABV N was also analyzed.
FIG 3.
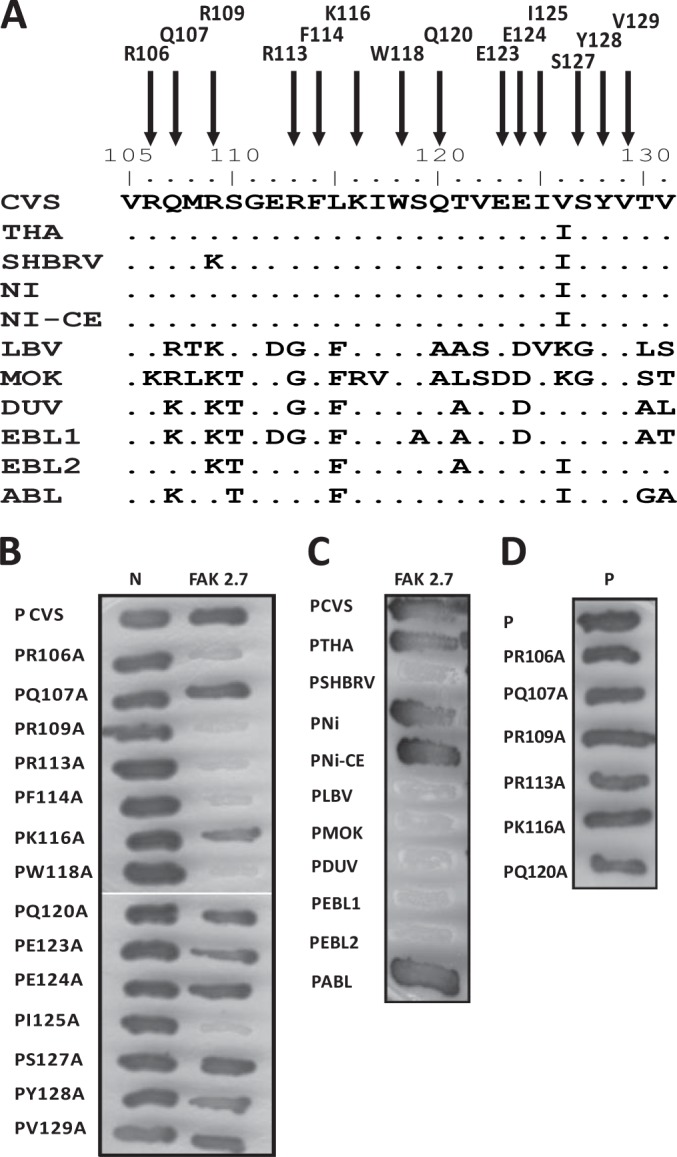
Identification of P residues essential for FAK interaction and analysis of the interaction of FAK with the P proteins of lyssaviruses by two-hybrid screen. (A) Sequence alignment of the FAK interaction domain (residues 106 to 131) of lyssavirus P proteins. Shown are the amino acid sequences of the P proteins of the RABV strains CVS, SHBRV, THA, Ni, and Ni-CE and the lyssaviruses LBV, MOKV, DUVV, EBLV-1, EBLV-2, and ABLV. (B) Binding of mutant P proteins to FAK and RABV N. The interactions between mutant P proteins (in which amino acids have been replaced by an alanine) and FAK (residues 649 to 958) or RABV N were assayed as for Fig. 1A. (C) Binding of FAK to P proteins of lyssaviruses. The interactions were performed as for Fig. 1A. (D) Binding of mutant P proteins to P. The interactions were performed as for Fig. 1A.
Interactions between FAK and other lyssavirus P proteins in the yeast two-hybrid system.
To investigate whether the interaction was conserved among other members of the genus Lyssavirus, we selected several viral species representative of lyssavirus diversity, including 8743THA (THA), silver-haired bat rabies virus (SHBRV), Nishigahara (Ni) and its attenuated Ni derivative strain Ni-CE, the closely related Australian bat lyssavirus (ABLV), Duvenhage virus (DUVV), EBLV-1 and EBLV-2, and lyssaviruses more distantly related to RABV, Lagos bat virus (LBV) and Mokola virus (MOKV). The interaction of the FAK clone 2.7 with the full-length P proteins of the different viruses was investigated with the yeast two-hybrid system. The P proteins of THA, Ni, Ni-CE, and ABLV displayed interaction with FAK, whereas the P proteins of SHBRV, LBV, MOKV, DUVV, EBLV-1, and EBLV-2 failed to interact with FAK (Fig. 3C). Interestingly, the substitution in position 109 of lysine for arginine was completely correlated with the lack of interaction, especially in the case of the P protein of SHBRV, for which the only residue modified in the binding domain is a lysine in position 109 compared to THA, Ni, and Ni-CE (Fig. 3A). This result indicated that R109 was one of the crucial residues for the interaction.
FAK accumulates in Negri bodies during viral infection.
As P interacts with FAK in the context of viral infection, we analyzed the localization of FAK in infected cells. U373-MG cells were mock transfected or transfected with plasmid encoding FAK-GFP and then infected at an MOI of 3 for 24 h. The cells were analyzed by confocal microscopy using the appropriate antibodies (Fig. 4). The endogenous FAK, as well as the overexpressed FAK-GFP, localized in NBs (Fig. 4A and B). When infected cells were previously transfected with a plasmid encoding the C-terminal domain of FAK corresponding to FRNK-GFP, the NBs contained the FRNK protein. In contrast, the FAK mutant with the FRNK domain deleted (FAKΔFRNK), which did not interact with P, did not localize in the NBs (Fig. 4B). These results indicate that FAK accumulates in NBs and that this recruitment is mediated by the interaction of P with FAK.
FIG 4.

Localization of FAK in infected cells. (A) Cells were mock infected (top row) or infected with RABV (strain CVS) at an MOI of 3 for 24 h (bottom row). The cells were analyzed by confocal microscopy after staining with anti-P antibody, rabbit anti-FAK antibody, or Texas red-phalloidin. DAPI (blue) was used to stain the nuclei (Merge). Colocalization is apparent as yellow coloration in the merged images. Scale bars, 15 μm. (B) Cells were transfected with plasmids encoding FAK-GFP, FRNK-GFP, or FAKΔFRNK-GFP and then infected at an MOI of 3 for 24 h. The cells were analyzed by confocal microscopy after staining with anti-P antibody. DAPI (blue) was used to stain the nuclei (Merge). Colocalization is apparent as yellow coloration in the merged images. Scale bars, 15 μm.
Recovery and characterization of recombinant RABV carrying the R109A mutation in the P protein.
To study the role of the P-FAK interaction during the viral cycle, a recombinant virus harboring a P protein unable to interact with FAK was generated by using the reverse genetics system. Since the FAK binding domain overlapped with the dimerization domain, it was important that the mutation of P residues impaired FAK interaction without altering the P-P interaction. As shown in Fig. 3D, the mutations R106A, R109A, and R113A did not affect P dimerization (Table 2). Given that a mutation of arginine in position 109 was naturally found in the P proteins of several lyssaviruses, we decided to recover a recombinant mutant carrying the R109A mutation in the P protein. We used the recombinant CVS-N2C strain (rN2C), which is closely related to the CVS-11 strain with no mutation in the P gene. The R109A mutation was introduced into the P gene of the infectious clone of rN2C to generate the rN2C-P.R109A mutant. The recombinant viruses rN2C-WT and rN2C-P.R109A were then recovered in mouse neuroblastoma N2A cells (Fig. 5A, left), as described previously (26). The rN2c-WT virus was confirmed to be phenotypically close to CVS-N2C in terms of protein expression and growth kinetics (not shown). Importantly, after rescue of the recombinant rN2C-P.R109A, RT-PCR/sequencing analysis of the P gene confirmed that the rescued virus retained the R109A mutation and did not reveal any compensatory mutation in the P gene.
TABLE 2.
Summary of the interactions of the P mutants with FAK, P CVS, and N
| P mutant | Interaction witha: |
||
|---|---|---|---|
| FAK | P CVS | N | |
| P CVS | + | + | + |
| PR106A | − | + | + |
| PQ107A | + | + | + |
| PR109A | − | + | + |
| PR113A | − | + | + |
| PF114A | − | − | + |
| PK116A | + | + | + |
| PW118A | − | − | + |
| PQ120A | + | + | + |
| PE123A | + | NT | + |
| PE124A | + | NT | + |
| PI125A | − | − | + |
| PS127A | + | NT | + |
| PY128A | + | − | + |
| PV129A | + | + | + |
+, positive interaction; −, negative interaction; NT, not tested.
FIG 5.

The P.R109A protein of the recombinant rN2C-P.R109A virus does not interact with FAK. (A) N2A cells were infected with either the rN2C-WT or rN2C-P.R109A virus at an MOI of 2 for 24 h. (Left) Cell extracts were analyzed by Western blotting with anti-P, anti-N, anti-M, and anti-tubulin antibodies. (Right) N2A cells were transfected with plasmid encoding FAK and then infected with either the rN2C-WT or rN2C-P.R109A virus. The cell lysates were immunoprecipitated with a rabbit polyclonal anti-FAK antibody, and immune complexes were analyzed by WB with a mouse anti-P antibody. (B) BSR cells were transfected with plasmid encoding FAK-GFP and then infected with either the rN2C-WT or rN2C-P.R109A virus. Epifluorescence images were obtained from cells stained with anti-P antibody. DAPI (blue) was used to stain the nuclei (Merge). Colocalization is apparent as yellow coloration in the merged images. Scale bars, 15 μm.
Cells were transfected with a plasmid encoding FAK-GFP and then infected with either the WT or mutant P.R109A virus. Coimmunoprecipitation experiments confirmed that the WT P protein interacted with FAK whereas the P mutant did not (Fig. 5A, right). In parallel, immunofluorescence experiments were performed under the same conditions described above. FAK-GFP accumulated in the NBs of cells infected by the WT virus but not in those formed by the mutant virus (Fig. 5B). This confirmed that the interaction of P with FAK resulted in the recruitment of P in NBs.
To determine the effects of this mutation on viral growth, N2A cells were infected with the WT or mutant virus at different MOI, and the virus released in the culture medium at different times postinfection was titrated. Single-step (MOI = 1 or 3) or multiple-step (MOI = 0.3) growth curves indicated that production of the mutant virus was 5- to 10-fold lower than that of the WT (shown for an MOI of 1 in Fig. 6A). In parallel, the amounts of viral proteins in infected cell extracts were analyzed by Western blotting. The P protein expression of the mutant virus was reduced compared to the WT virus (Fig. 6B). The same observation was made for the N protein (not shown). To investigate whether this reduction was due to the inhibition of viral protein synthesis, we performed labeling experiments with [35S]methionine/[35S]cysteine. Cells infected with either the WT or mutant P.R109A at an MOI of 3 were labeled for 15 min at 12 h p.i. and 24 h p.i. Cell extracts were then immunoprecipitated with anti-P antibody, and the immune complexes were analyzed by SDS-PAGE (Fig. 6C). The P protein interacts strongly with the N protein, and therefore, both viral proteins were present in the immune complexes. Viral protein synthesis was reduced in cells infected with the mutant compared to the WT, and this reduction was more pronounced at 24 h p.i. than at 12 h p.i. (by a factor of 5 compared to a factor of 3, respectively) (Fig. 6C). To test whether the inhibition of viral translation could reflect an inhibition of viral transcription, we performed Northern blot analysis on RNA isolated from N2A cells infected by both viruses at 12 h p.i. and 24 h p.i. Primary transcription was also analyzed in the presence of the protein synthesis inhibitor cycloheximide (CLX). As genome replication requires the ongoing synthesis of the N protein, treatment of cells with CLX results exclusively in primary transcription obtained from the incoming virion genomes and proteins. Whereas the primary transcription products could not be detected in cells infected by WT or mutant virus, the total amount of N mRNA produced by the mutant virus appeared to be reduced (Fig. 7A). As Northern blotting is not the most sensitive method, we used quantitative RT-PCR to detect the P transcripts found in infected cells. In the absence of CLX, the amount of P mRNA produced by the mutant was estimated to be 5 times smaller than the amount produced by the WT (Fig. 7B). The mRNA levels resulting from primary transcription (in the presence of CLX) were very similar in the two infections. These results revealed that secondary transcription of the viral mutant was impaired, whereas its viral primary transcription was not, suggesting that steps preceding transcription, such as viral entry or uncoating, were not altered but, rather, that a posttranscriptional step (such as protein synthesis and/or replication) was affected.
FIG 6.

Characterization of the recombinant rN2C-P.R109A virus. (A) Growth kinetics of rN2C-WT and rN2C-P.R109A viruses. N2A cells were infected at an MOI of 1, and samples were harvested for titration at the indicated times p.i. Viral titers represent averages of titers from at least 3 independent experiments. The error bars indicate standard deviations. (B) Viral protein expression in noninfected N2A cells (NI) or in N2A cells infected (MOI = 1) with rN2C-WT and rN2C-P.R109A viruses at different times p.i. The cell extracts were analyzed by Western blotting using anti-P and anti-tubulin (Tub) antibodies. The levels of P proteins were quantified by Western blot scanning and normalized with respect to the amount of tubulin. The P/tubulin ratio was set to 1 for cell extracts collected 12 h p.i. with rN2C-WT virus (lane 2 from left). As expected, the isoforms of P (P2 and P3) were also detected by the anti-P antibody (47). The blot shown is representative of 3 assays. (C) Viral protein synthesis in noninfected N2A cells (NI) or N2A cells infected (MOI = 1) with rN2C-WT and rN2C-P.R109A viruses. (Left) At 12 or 24 h p.i., the cells were pulse-labeled with [35S]methionine and [35S]cysteine for 15 min. The lysates were immunoprecipitated with anti-P antibodies. The immunoprecipitates were analyzed by SDS-PAGE, followed by autoradiography. (Right) Quantification was performed with a phosphorimager.
FIG 7.
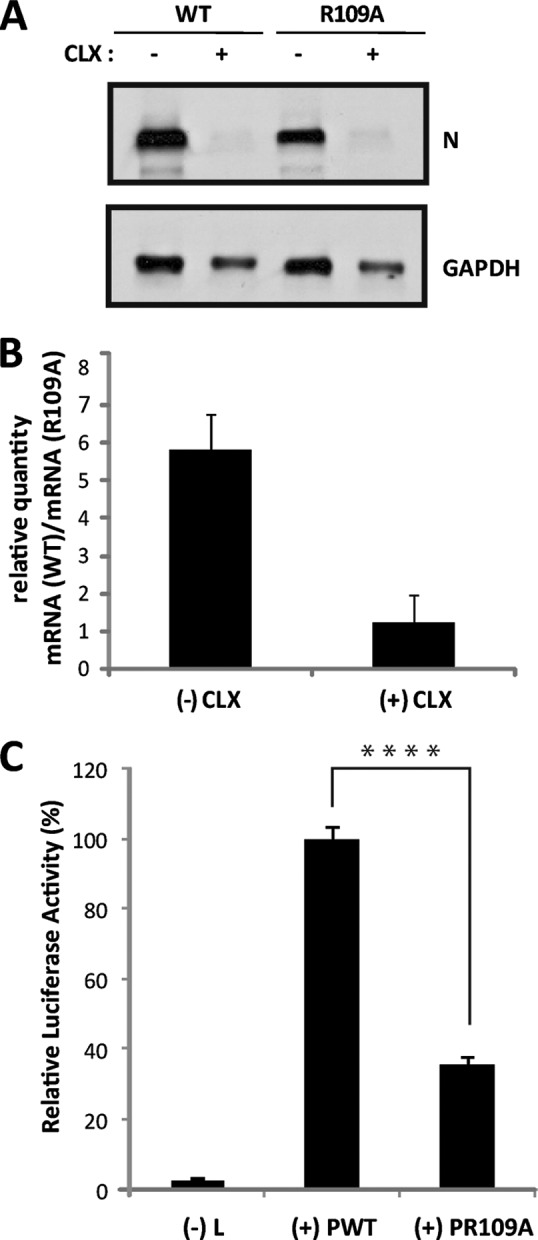
Effect of the substitution R109A on viral RNA synthesis in the context of viral infection or in a minireplicon system. (A and B) Viral RNA synthesis in infected cells. (A) N2A cells were infected (MOI = 1) with rN2C-WT and rN2C-P.R109A viruses in the absence (−CLX) or the presence (+CLX) of cycloheximide. After 16 h of infection, total RNA was extracted from the cells, and samples (10 μg of RNA per lane) were analyzed by Northern blotting for the presence of rabies virus N mRNA and GAPDH mRNA, as described in Materials and Methods. (B) After 16 h of infection under the same conditions as for panel A, total RNA extracted from cells was used for P mRNA quantification by RT-qPCR, as described in Materials and Methods. Mean values and standard deviations from two independent experiments are shown. (C) Luciferase expression from a RABV minireplicon system. N2A cells were transfected with plasmids encoding L, N, P, or P.R129A protein and T7 polymerase and with the plasmid (pRL-TK) encoding Renilla luciferase in the presence of the RABV minireplicon system plasmid for the expression of firefly luciferase. The L plasmid was missing in one experiment, which served as a negative control [(−) L]. After 36 h, cell extracts were analyzed for luciferase activity. The data represent separate assays for firefly luciferase normalized to expression of Renilla luciferase and are expressed as percentages of the control. The error bars indicate the standard deviations (three independent experiments). A significant difference between WT P (PWT) and P.R109A was determined using a Student t test (****, P < 0.0001).
To determine more specifically the effect of the lack of P-FAK interaction on viral RNA synthesis, we used the luciferase reporter system in a minireplicon system allowing reconstitution of a functional rabies virus RNP. The luciferase gene reporter is framed by 3′ leader and 5′ trailer sequences and is under the control of rabies virus transcription signals (25). Therefore, the production of luciferase activity is the result of the encapsidation of the minigenome followed by its transcription by the L-P complex and its replication in the presence of the N protein. Upon cell transfection with the corresponding expression vector, the P.R109A protein was expressed at a level comparable to that of wild-type P (not shown). Quantification of luciferase activity showed that the substitution P.R109A resulted in a reduction of viral transcription and replication to about 65% (Fig. 7C), demonstrating that the binding of P to FAK is required for efficient viral RNA synthesis.
All these data are consistent and established a strong correlation between the P-FAK interaction and a positive effect on RABV replication.
FAK depletion leads to the reduction of viral protein expression.
We then analyzed the effect of downregulation of FAK on viral infection by RNA interference (RNAi)-mediated silencing. U373-MG cells were transfected with a pool of 4 siRNAs targeting FAK, as well as FRNK (25 nM), or with nontargeting control (siCONT) for 48 h; then, the cells were infected (MOI = 3). Cells were harvested at different times postinfection, and the lysates were analyzed by Western blotting to determine the expression of FAK, viral protein, and tubulin as a control (Fig. 8). FAK-targeting siRNA, but not control siRNA, clearly depleted FAK in both infected and noninfected cells. FAK depletion had no major effect on cell viability (>95%) as measured by trypan blue exclusion (not shown). The knockdown of FAK expression resulted in a significant decrease in the amount of the viral P protein (by 35%) at 24 h p.i. Although this reduction was moderate, it indicated that specific inhibition of FAK synthesis led to a reduction of viral protein expression and confirmed that the P-FAK interaction positively regulated RABV infection.
FIG 8.
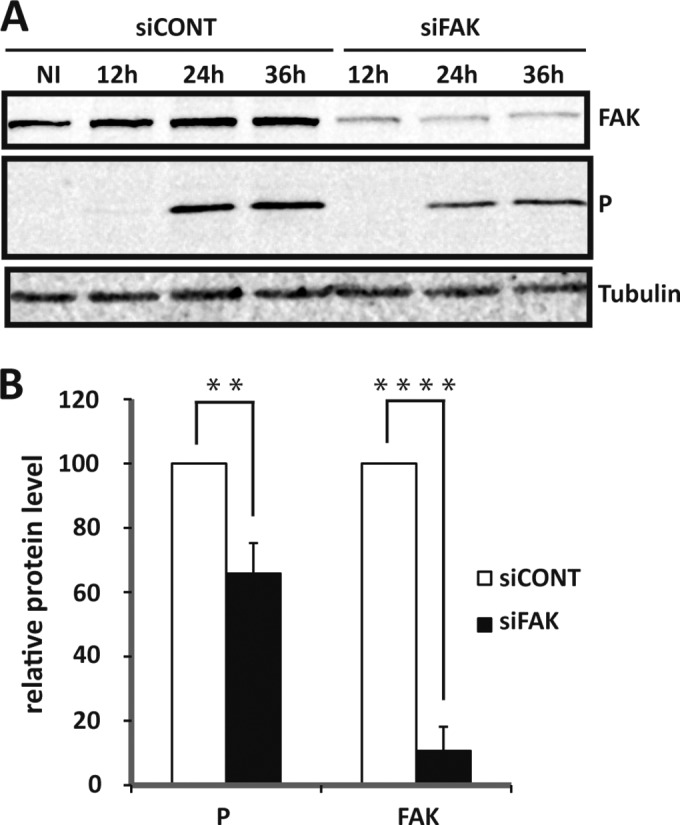
Effect of specific depletion of FAK by siRNA interference on RABV replication. (A) U373-MG cells were transfected with nontargeting (siCONT) or FAK-targeting (siFAK) siRNA and not infected (NI) or infected (MOI = 3) with RABV 48 h posttransfection. Cell extracts were analyzed by Western blotting at various times (12 h, 24 h, and 36 h p.i.) using anti-FAK, anti-P, and anti-tubulin antibodies. (B) Western blots from 3 independent experiments were quantified using immunoblot scanning and normalized with respect to the amount of tubulin. The amounts of P and FAK were measured at 24 h p.i., and an arbitrary level of 100 was applied to the amount of protein obtained in cells transfected with siCONT for comparison. Significant differences between siCONT and siFAK conditions were determined using a Student t test (**, P < 0.005; ****, P < 0.0001). The error bars indicate the standard deviations.
DISCUSSION
In this study, using the yeast two-hybrid system, we have identified FAK as a cellular partner of the RABV P protein. The binding involves the 106-to-131 domain corresponding to the dimerization domain of P and the C-terminal domain of FAK that comprises the linker region between the kinase and FAT domains and containing the proline-rich domains PRR2 and PRR3 (16). As described in the literature, these unstructured PRR motifs have a scaffolding function, recruiting SH3 domain-containing proteins into a multiprotein complex. It has also been shown that WW domains bind PRR, but with a lower affinity. Although the P protein does not comprise any SH3 domain or WW domain, other regions in the C-terminal linker domain of FAK could be involved in the interaction with P.
The P-FAK interaction was confirmed by coimmunoprecipitation and colocalization in infected cells. Indeed, endogenous FAK or overexpressed FAK-GFP was found to accumulate in NBs of infected cells. This recruitment was mediated by the interaction of P with FAK, since a FAK-GFP mutant unable to bind P protein was not retained in NBs.
The data on the P-FAK interaction in the genus Lyssavirus suggest that the interaction is not conserved. However, as the P binding domain on FAK (the linker region between the kinase and FAT domains) is the most variable region (39), we cannot exclude the possibility that bat lyssavirus P protein might interact with bat FAK. Whether this interaction is conserved remains unclear, as we have not investigated the point.
Alanine scanning mutagenesis of the dimerization domain of P (residues 106 to 131) allowed the identification of the crucial residues (R106, R109, R113, F114, W118, and I125) involved in the interaction with FAK. The structure of the dimerization domain of P indicated that the residues R106, R109, and R113 are not involved in dimer formation (40). Indeed, substitutions of Ala for these residues inhibit P-FAK interaction but do not lead to loss of dimerization.
As Arg in position 109 is not present in the P sequence of lyssaviruses that did not interact with FAK, the P.R109A mutant virus was constructed to investigate the role of FAK during viral infection. As expected, FAK-GFP did not accumulate in the NBs formed by the mutant virus. This mutant was affected at a posttranscriptional step involving protein synthesis and/or replication, since primary transcription was not affected. This indicates that viral entry is not impaired. Although we cannot exclude the possibility that the substitution R109A also affects the interaction of P with another cellular partner, the fact that depletion of FAK by siRNA resulted in a decrease in viral protein expression indicates that FAK favored RABV replication and that this effect is potentially mediated by P-FAK interaction, for which Arg in position 109 is a critical residue. From our data, we do not know whether the P-FAK interaction could promote virus replication either by enhancing a positive effect of FAK or by inhibiting an antiviral effect of FAK. It is likely that this interaction does not have an essential function in the viral life cycle but rather a regulatory role in some processes involved in pathogenicity or apoptosis.
Interestingly, FAK is related to another tyrosine kinase, proline-rich tyrosine kinase 2 (PYK2), in sequence (46% identical and 65% similar at the protein level). Thus, the FAK and PYK2 proteins share a four-domain organization and are both involved in the regulation of similar cellular signaling pathways (reviewed in references 18 and 30). Therefore, PYK2 expression has been shown to compensate for some functions, although not all, of FAK in cells derived from FAK knockout mice (41). In addition, it has been reported that FAK is subjected to alternative splicing of several coding or noncoding exons that are likely to have important biological functions (39). These data could explain, due to some compensatory mechanism, why the effect of FAK depletion on viral protein expression is not stronger.
FAK is a protein tyrosine kinase, located primarily at FAs, that serves as a key component in the transmission of signals between the extracellular matrix and the cytoplasm and that has important cellular functions through regulation of the cytoskeleton. FAK regulates processes such as cellular survey, proliferation, and migration. Viruses activate intracellular signaling pathways, thereby facilitating viral entry, viral replication, and release of new viruses. Therefore, we have investigated some of these signaling pathways following activation of FAK during viral infection by WT and P.R109A viruses. Following activation by autophosphorylation at Tyr397, FAK recruits and activates members of the Src kinase family. The phosphorylation of FAK by Src (or other Src family kinases) at tyrosines 576 and 577 results in full activation of FAK and ultimately leads to activation of downstream targets, such as phosphoinositol 3-kinase (PI3-K)/Akt. We did not observe any difference in phosphorylation of FAK Tyr397 or FAK Tyr576/577 in cells infected by both viruses (not shown). Along this line, activation of Akt signaling by phosphorylation at Ser473 was similarly induced by both viral infections (not shown).
As FAK also plays an integral role in the regulation of the actin cytoskeleton, we have analyzed the effect of viral infection on actin networks. Both WT and mutant viruses induce alteration of the actin network (with a decrease in F-actin staining), as previously reported by Ceccaldi et al. (42). Several recent studies provided evidence that FAK can also regulate certain cellular functions in a kinase-independent manner. Among emerging functions of FAK, recent data implicate FAK as a regulator of mitochondrial antiviral signaling protein (MAVS), although its precise function in regulating RIG-I-like receptor (RLR) signaling remains unclear (43). Indeed, Bozym et al. have shown that FAK interacts with MAVS at the mitochondrial membrane and favors MAVS-mediated signaling by a kinase-independent mechanism resulting in the activation of antiviral signaling, such as IFN production (43). We hypothesized that P-FAK interaction could negatively regulate the host antiviral response and as a consequence support efficient replication of RABV. Unfortunately, we did not see any effect of the defect in P-FAK interaction on IFN production or the NF-κB pathway, and in addition, FAK deficiency did not result in reduced activation of IFN signaling (not shown).
Activation of FAK by viral infection has been reported for some viruses, and different roles of FAK have been proposed, although the mechanisms have not been elucidated. For influenza virus, FAK has been shown to regulate two independent processes, one mediating viral entry and the other involved in viral RNA replication (44).
In the case of hepatitis B virus, the HBx protein activates FAK, and this activation is important for multiple HBx functions, such as cellular transformation and cancer progression (45). Herpes simplex virus 1 (HSV-1) and HSV-2 induce rapid phosphorylation of FAK in several human target cells, and this activation has been shown to facilitate viral entry and nuclear transport of viral capsids (46).
Our data indicate that FAK has a positive effect on RABV growth by stimulating viral RNA replication and/or viral mRNA translation. Interestingly, the positive role of FAK in viral RNA synthesis is correlated with its interaction with P and association with the NBs, which are sites of viral transcription and replication, suggesting a possible interaction with the polymerase complex. Although FAK does not seem to be involved in the activation of IFN production in the context of RABV infection, we cannot exclude the possibility that FAK has an antiviral effect, such as a role in innate immunity, that might be counteracted by P alone or through its sequestration in NBs, resulting indirectly in a proviral role of the P-FAK interaction.
ACKNOWLEDGMENTS
We are very grateful to Yves Gaudin for his support during this project and for reading the manuscript. We acknowledge Frauke Beilstein for careful review of the manuscript. We thank Sébastien Nisole and Ahmet Civas for helpful discussions. We acknowledge Jean-Antoine Girault for the plasmid encoding FAK-GFP; Naoto Ito for the plasmids expressing P Ni and P Ni-CE; Bernard Dietzschold for providing the full-length cDNA clone of SHBRV strain SBO; and K. Conzelmann for the pTIT-P, pTIT-N, and pTIT-L plasmids of SAD. We thank Matthias Schnell for his support and for the plasmid rN2C.
This work was supported by a grant from Fondation pour la Recherche Médicale (FRM DEQ20120323711). Confocal microscopy was performed on the “Plate-forme Imagerie et Biologie Cellulaire” of the CNRS campus, supported by the Institut Fédératif de Recherche 87 “La plante et son environnement” and the program ASTRE of the Conseil Général de l'Essonne.
REFERENCES
- 1.Calisher CH, Ellison JA. 2012. The other rabies viruses: the emergence and importance of lyssaviruses from bats and other vertebrates. Travel Med Infect Dis 10:69–79. doi: 10.1016/j.tmaid.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morimoto K, Patel M, Corisdeo S, Hooper DC, Fu ZF, Rupprecht CE, Koprowski H, Dietzschold B. 1996. Characterization of a unique variant of bat rabies virus responsible for newly emerging human cases in North America. Proc Natl Acad Sci U S A 93:5653–5658. doi: 10.1073/pnas.93.11.5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le Mercier P, Jacob Y, Tordo N. 1997. The complete Mokola virus genome sequence: structure of the RNA dependent RNA polymerase. J Gen Virol 78:1571–1576. [DOI] [PubMed] [Google Scholar]
- 4.Lahaye X, Vidy A, Pomier C, Obiang L, Harper F, Gaudin Y, Blondel D. 2009. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: evidence that NBs are sites of viral transcription and replication. J Virol 83:7948–7958. doi: 10.1128/JVI.00554-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brzozka K, Finke S, Conzelmann KK. 2005. Identification of the rabies virus alpha/beta interferon antagonist: phosphoprotein P interferes with phosphorylation of interferon regulatory factor 3. J Virol 79:7673–7681. doi: 10.1128/JVI.79.12.7673-7681.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vidy A, Chelbi-Alix M, Blondel D. 2005. Rabies virus P protein interacts with STAT1 and inhibits interferon signal transduction pathways. J Virol 79:14411–14420. doi: 10.1128/JVI.79.22.14411-14420.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brzozka K, Finke S, Conzelmann KK. 2006. Inhibition of interferon signaling by rabies virus phosphoprotein P: activation-dependent binding of STAT1 and STAT2. J Virol 80:2675–2683. doi: 10.1128/JVI.80.6.2675-2683.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vidy A, El Bougrini J, Chelbi-Alix MK, Blondel D. 2007. The nucleocytoplasmic rabies virus P protein counteracts interferon signaling by inhibiting both nuclear accumulation and DNA binding of STAT1. J Virol 81:4255–4263. doi: 10.1128/JVI.01930-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moseley GW, Lahaye X, Roth DM, Oksayan S, Filmer RP, Rowe CL, Blondel D, Jans DA. 2009. Dual modes of rabies P-protein association with microtubules: a novel strategy to suppress the antiviral response. J Cell Sci 122:3652–3662. doi: 10.1242/jcs.045542. [DOI] [PubMed] [Google Scholar]
- 10.Reference deleted.
- 11.Blondel D, Regad T, Poisson N, Pavie B, Harper F, Pandolfi PP, De The H, Chelbi-Alix MK. 2002. Rabies virus P and small P products interact directly with PML and reorganize PML nuclear bodies. Oncogene 21:7957–7970. doi: 10.1038/sj.onc.1205931. [DOI] [PubMed] [Google Scholar]
- 12.Chelbi-Alix MK, Vidy A, El Bougrini J, Blondel D. 2006. Rabies viral mechanisms to escape the IFN system: the viral protein P interferes with IRF-3, Stat1, and PML nuclear bodies. J Interferon Cytokine Res 26:271–280. doi: 10.1089/jir.2006.26.271. [DOI] [PubMed] [Google Scholar]
- 13.Blondel D, Kheddache S, Lahaye X, Dianoux L, Chelbi-Alix MK. 2010. Resistance to rabies virus infection conferred by the PMLIV isoform. J Virol 84:10719–10726. doi: 10.1128/JVI.01286-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raux H, Flamand A, Blondel D. 2000. Interaction of the rabies virus P protein with the LC8 dynein light chain. J Virol 74:10212–10216. doi: 10.1128/JVI.74.21.10212-10216.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ilic D, Damsky CH, Yamamoto T. 1997. Focal adhesion kinase: at the crossroads of signal transduction. J Cell Sci 110:401–407. [DOI] [PubMed] [Google Scholar]
- 16.Mitra SK, Hanson DA, Schlaepfer DD. 2005. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 17.Oktay MH, Oktay K, Hamele-Bena D, Buyuk A, Koss LG. 2003. Focal adhesion kinase as a marker of malignant phenotype in breast and cervical carcinomas. Hum Pathol 34:240–245. doi: 10.1053/hupa.2003.40. [DOI] [PubMed] [Google Scholar]
- 18.Parsons JT. 2003. Focal adhesion kinase: the first ten years. J Cell Sci 116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- 19.Raux H, Iseni F, Lafay F, Blondel D. 1997. Mapping of monoclonal antibody epitopes of the rabies virus P protein. J Gen Virol 78:119–124. [DOI] [PubMed] [Google Scholar]
- 20.Contestabile A, Bonanomi D, Burgaya F, Girault JA, Valtorta F. 2003. Localization of focal adhesion kinase isoforms in cells of the central nervous system. Int J Dev Neurosci 21:83–93. doi: 10.1016/S0736-5748(02)00126-0. [DOI] [PubMed] [Google Scholar]
- 21.Ito N, Moseley GW, Blondel D, Shimizu K, Rowe CL, Ito Y, Masatani T, Nakagawa K, Jans DA, Sugiyama M. 2010. Role of interferon antagonist activity of rabies virus phosphoprotein in viral pathogenicity. J Virol 84:6699–6710. doi: 10.1128/JVI.00011-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcin D, Curran J, Kolakofsky D. 2000. Sendai virus C proteins must interact directly with cellular components to interfere with interferon action. J Virol 74:8823–8830. doi: 10.1128/JVI.74.19.8823-8830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Conzelmann KK. 1998. Nonsegmented negative-strand RNA viruses: genetics and manipulation of viral genomes. Annu Rev Genet 32:123–162. doi: 10.1146/annurev.genet.32.1.123. [DOI] [PubMed] [Google Scholar]
- 24.Berrow NS, Alderton D, Sainsbury S, Nettleship J, Assenberg R, Rahman N, Stuart DI, Owens RJ. 2007. A versatile ligation-independent cloning method suitable for high-throughput expression screening applications. Nucleic Acids Res 35:e45. doi: 10.1093/nar/gkm047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le Mercier P, Jacob Y, Tanner K, Tordo N. 2002. A novel expression cassette of lyssavirus shows that the distantly related Mokola virus can rescue a defective rabies virus genome. J Virol 76:2024–2027. doi: 10.1128/JVI.76.4.2024-2027.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wirblich C, Schnell MJ. 2011. Rabies virus (RV) glycoprotein expression levels are not critical for pathogenicity of RV. J Virol 85:697–704. doi: 10.1128/JVI.01309-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wirblich C, Tan GS, Papaneri A, Godlewski PJ, Orenstein JM, Harty RN, Schnell MJ. 2008. PPEY motif within the rabies virus (RV) matrix protein is essential for efficient virion release and RV pathogenicity. J Virol 82:9730–9738. doi: 10.1128/JVI.00889-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 29.Poisson N, Real E, Gaudin Y, Vaney MC, King S, Jacob Y, Tordo N, Blondel D. 2001. Molecular basis for the interaction between rabies virus phosphoprotein P and the dynein light chain LC8: dissociation of dynein-binding properties and transcriptional functionality of P. J Gen Virol 82:2691–2696. [DOI] [PubMed] [Google Scholar]
- 30.Schaller MD. 2010. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J Cell Sci 123:1007–1013. doi: 10.1242/jcs.045112. [DOI] [PubMed] [Google Scholar]
- 31.Chenik M, Schnell M, Conzelmann KK, Blondel D. 1998. Mapping the interacting domains between the rabies virus polymerase and phosphoprotein. J Virol 72:1925–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Castel G, Chteoui M, Caignard G, Prehaud C, Mehouas S, Real E, Jallet C, Jacob Y, Ruigrok RW, Tordo N. 2009. Peptides that mimick the amino terminal end of the rabies virus phosphoprotein have antiviral activity. J Virol 83:10808–10820. doi: 10.1128/JVI.00977-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mavrakis M, Mehouas S, Real E, Iseni F, Blondel D, Tordo N, Ruigrok RW. 2006. Rabies virus chaperone: identification of the phosphoprotein peptide that keeps nucleoprotein soluble and free from non-specific RNA. Virology 349:422–429. doi: 10.1016/j.virol.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 34.Gerard FC, Ribeiro Ede A Jr, Leyrat C, Ivanov I, Blondel D, Longhi S, Ruigrok RW, Jamin M. 2009. Modular organization of rabies virus phosphoprotein. J Mol Biol 388:978–996. doi: 10.1016/j.jmb.2009.03.061. [DOI] [PubMed] [Google Scholar]
- 35.Mavrakis M, McCarthy AA, Roche S, Blondel D, Ruigrok RW. 2004. Structure and function of the C-terminal domain of the polymerase cofactor of rabies virus. J Mol Biol 343:819–831. doi: 10.1016/j.jmb.2004.08.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacob Y, Real E, Tordo N. 2001. Functional interaction map of lyssavirus phosphoprotein: identification of the minimal transcription domains. J Virol 75:9613–9622. doi: 10.1128/JVI.75.20.9613-9622.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Assenberg R, Delmas O, Morin B, Graham SC, De Lamballerie X, Laubert C, Coutard B, Grimes JM, Neyts J, Owens RJ, Brandt BW, Gorbalenya A, Tucker P, Stuart DI, Canard B, Bourhy H. 2010. Genomics and structure/function studies of Rhabdoviridae proteins involved in replication and transcription. Antiviral Res 87:149–161. doi: 10.1016/j.antiviral.2010.02.322. [DOI] [PubMed] [Google Scholar]
- 38.Delmas O, Assenberg R, Grimes JM, Bourhy H. 2010. The structure of the nucleoprotein binding domain of lyssavirus phosphoprotein reveals a structural relationship between the N-RNA binding domains of Rhabdoviridae and Paramyxoviridae. RNA Biol 7:322–327. doi: 10.4161/rna.7.3.11931. [DOI] [PubMed] [Google Scholar]
- 39.Corsi JM, Rouer E, Girault JA, Enslen H. 2006. Organization and post-transcriptional processing of focal adhesion kinase gene. BMC Genomics 7:198. doi: 10.1186/1471-2164-7-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ivanov I, Crepin T, Jamin M, Ruigrok RW. 2010. Structure of the dimerization domain of the rabies virus phosphoprotein. J Virol 84:3707–3710. doi: 10.1128/JVI.02557-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sieg DJ, Ilic D, Jones KC, Damsky CH, Hunter T, Schlaepfer DD. 1998. Pyk2 and Src-family protein-tyrosine kinases compensate for the loss of FAK in fibronectin-stimulated signaling events but Pyk2 does not fully function to enhance FAK- cell migration. EMBO J 17:5933–5947. doi: 10.1093/emboj/17.20.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ceccaldi PE, Valtorta F, Braud S, Hellio R, Tsiang H. 1997. Alteration of the actin-based cytoskeleton by rabies virus. J Gen Virol 78:2831–2835. [DOI] [PubMed] [Google Scholar]
- 43.Bozym RA, Delorme-Axford E, Harris K, Morosky S, Ikizler M, Dermody TS, Sarkar SN, Coyne CB. 2012. Focal adhesion kinase is a component of antiviral RIG-I-like receptor signaling. Cell Host Microbe 11:153–166. doi: 10.1016/j.chom.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elbahesh H, Cline T, Baranovich T, Govorkova EA, Schultz-Cherry S, Russell CJ. 2014. Novel roles of focal adhesion kinase in cytoplasmic entry and replication of influenza A viruses. J Virol 88:6714–6728. doi: 10.1128/JVI.00530-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bouchard MJ, Wang L, Schneider RJ. 2006. Activation of focal adhesion kinase by hepatitis B virus HBx protein: multiple functions in viral replication. J Virol 80:4406–4414. doi: 10.1128/JVI.80.9.4406-4414.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheshenko N, Trepanier JB, Gonzalez PA, Eugenin EA, Jacobs WR Jr, Herold BC. 2014. Herpes simplex virus type 2 glycoprotein H interacts with integrin alphavbeta3 to facilitate viral entry and calcium signaling in human genital tract epithelial cells. J Virol 88:10026–10038. doi: 10.1128/JVI.00725-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chenik M, Chebli K, Blondel D. 1995. Translation initiation at alternate in-frame AUG codons in the rabies virus phosphoprotein mRNA is mediated by a ribosomal leaky scanning mechanism. J Virol 69:707–712. [DOI] [PMC free article] [PubMed] [Google Scholar]