

ABSTRACT
Although vaccinia virus (VACV) was once used as a vaccine to eradicate smallpox on a worldwide scale, the biological origins of VACV are uncertain, as are the historical relationships between the different strains once used as smallpox vaccines. Here, we sequenced additional VACV strains that either represent relatively pristine examples of old vaccines (e.g., Dryvax, Lister, and Tashkent) or have been subjected to additional laboratory passage (e.g., IHD-W and WR). These genome sequences were compared with those previously reported for other VACVs as well as other orthopoxviruses. These extant VACVs do not always cluster in simple phylogenetic trees that are aligned with the known historical relationships between these strains. Rather, the pattern of deletions suggests that all existing strains likely come from a complex stock of viruses that has been passaged, distributed, and randomly sampled over time, thus obscuring simple historical or geographic links. We examined surviving nonclonal vaccine stocks, like Dryvax, which continue to harbor larger and now rare variants, including one that we have designated “clone DPP25.” DPP25 encodes genes not found in most VACV strains, including an ankyrin-F-box protein, a homolog of the variola virus (Bangladesh) B18R gene which we show can be deleted without affecting virulence in mice. We propose a simple common mechanism by which recombination of a larger and hypothetical DPP25-like ancestral strain, combined with selection for retention of critically important genes near the terminal inverted repeat boundaries (vaccinia virus growth factor gene and an interferon alpha/beta receptor homolog), could produce all known VACV variants.
IMPORTANCE Smallpox was eradicated by using a combination of intensive disease surveillance and vaccination using vaccinia virus (VACV). Interestingly, little is known about the historical relationships between different strains of VACV and how these viruses may have evolved from a common ancestral strain. To understand these relationships, additional strains were sequenced and compared to existing strains of VACV as well as other orthopoxviruses by using whole-genome sequence alignments. Extant strains of VACV did not always cluster in simple phylogenetic trees based on known historical relationships between these strains. Based on these findings, it is possible that all existing strains of VACV are derived from a single complex stock of viruses that has been passaged, distributed, and sampled over time.
INTRODUCTION
The Orthopoxvirus genus encompasses many immunologically related poxviruses, which vary greatly in their capacity to infect different hosts. Of these, cowpox virus (CPXV) probably exhibits the greatest genetic diversity and a broad host range, while variola virus (VARV), which causes smallpox, exhibits relatively little genetic variation and naturally infects only humans. The CPXV group comprises at least 5 subtypes (1), and these encode all of the genes present in all other orthopoxviruses. This has led to the suggestion that gene loss has played an important role in Orthopoxvirus evolution and that ancestral CPXV-like strains have evolved into all of the modern orthopoxviruses through “reductive evolution” (2, 3).
The relationships between orthopoxviruses are important because in the late 18th century, Edward Jenner showed that humans could be vaccinated against a smallpox challenge by using material extracted from a cowpox lesion, a process far safer than the existing practice of variolation with VARV (4, 5). In the years that followed, and prior to the advent of modern in vitro culture methods, the various inocula used as vaccinating agents were distributed around the world while being repeatedly passaged in humans and amplified in animals. This generally involved virus culture on the skin of cows, rabbits, or sheep, but donkeys and chicken eggs were also sometimes used (6). During this era, it is now recognized that passage through different hosts would likely have attenuated these agents, and viruses causing less adverse side effects in humans were also likely to have been retained for use as future stocks (7), but how these interventions might have affected virus evolution is unknown. Curiously, the virus that was eventually used to eradicate smallpox was one now called vaccinia virus (VACV), another orthopoxvirus but one that is clearly different from any known strain of CPXV. The biological origin of VACV is uncertain, although it has been suggested that a horsepox-like virus was an ancestor, even though a surviving horsepox virus (HPXV) genome harbors many extra genes (8). This hypothesis is supported by Jenner's report that he obtained his later inocula from an infection in horses called “grease” (5).
During the period when VACV was being grown and distributed for use as a smallpox vaccine, it acquired many different names that reflect the country or health agency involved in its propagation (9). In many cases, these viruses became linked to geographical regions. The New York City Board of Health (NYCBH) strain was originally transported from England in 1856 and was later widely used in the United States and West Africa. A Russian strain, EM-63, was also (probably) derived from NYCBH by a circuitous route. Europe used many different strains, including Lister, Bern, Paris, Copenhagen, Tashkent, and Ankara, while the TianTan and Ikeda strains were used in China and Japan, respectively (9). Interestingly, viruses related to VACV continue to evolve in wild habitats, including South American cattle (10) and Southeast Asian water buffalo (11). This has led to speculation as to whether these are escaped human vaccine strains (12, 13). The historical records suggest that the different smallpox vaccines also varied greatly in virulence, a feature that is still poorly understood.
Some of these VACV strains have been sequenced entirely, and these data can provide valuable insights into the historical relationships between viruses and virus strains. Perhaps more importantly, the selection for less virulent but still effective vaccines over ∼200 years of virus propagation could provide unique insights into which Orthopoxvirus genes affect pathogenicity in humans. The most common differences that are detected by sequencing are single nucleotide polymorphisms (SNPs) and small insertions and deletions (indels), although these mutations accumulate in such great numbers that they provide few insights into the deeper elements of virus phylogenies. Comprehensive analyses of these genomic changes that lead to gene truncation and fragmentation can sometimes detect remnants of the evolutionary process still present in orthopoxvirus genomes (14). Large deletions and other kinds of genome rearrangements are also commonly detected (15, 16), especially around the terminal inverted repeats (TIRs) (17), and can significantly affect the lengths of VACV genomes. They can also alter the number of duplicated versus unique genes and can be associated with reduced infectivity and pathogenicity (16, 17). Because these larger rearrangements are less common and not readily obscured by sequence drift, they provide a more useful tool than SNPs for studying deeper evolutionary relationships between viruses. In some rare cases, one can also detect evidence of horizontal gene transfer between related orthopoxviruses, including two CPXV-like genes in VACV strain Lister (18) and a small region of sequence encoding HPXV-like SNPs in a Dryvax subclone (DPP17) (19). It was once common practice to periodically cocultivate smallpox vaccine strains with other viruses, including VARV (20) so as to “refresh” vaccine efficacy. Such activities could have produced recombinants between related orthopoxviruses.
Although a virus resembling HPXV is often assumed to be ancestral to VACV, the evolutionary path(s) from CPXV-like and HPXV-like viruses to VACV is unclear, as are the relationships between the many VACV strains. Using new and existing data obtained from whole-genome alignments between different VACVs, we describe here some probable routes by which the extant VACV strains can be related and how they might have evolved from a stock of virus containing a hypothetical HPXV-like ancestral strain. This approach has identified a gene, designated DVX_213, that seems to have been subject to widespread negative selection in VACV strains. However, the reasons for it being lost are not easily explained by DVX_213 being a virulence factor, as deleting it does not affect virulence, at least in mice.
MATERIALS AND METHODS
Viruses and cells.
Our VACV International Health Department-White (IHD-W) stock came originally from the collection of Sam Dales and is derived from the IHD-J (Japan) strain. Strain Tashkent was obtained from Geoff Smith, and strain Lister was purchased from the ATCC, along with Western Reserve (WR). Cowpox virus was obtained from M. Barry, and strain Copenhagen came originally from the collection of E. Paoletti. The Dryvax clones were described previously (19). Viruses were propagated on monkey kidney epithelial (BSC-40) cells in modified Eagle's medium supplemented with 5% fetal bovine serum, 1% nonessential amino acids, 1% l-glutamine, and 1% antibiotic at 37°C in a 5% CO2 atmosphere. To clone the viruses, BSC-40 cells were grown to 80% confluence in 24-well plates and then infected at a multiplicity of infection of ∼1 PFU/well in phosphate-buffered saline (PBS) for 1 h at 37°C. The inocula were replaced, and the viruses were cultured for 3 days and then harvested from wells containing only one plaque. Each virus was cloned twice more by limiting dilution, as described above. The clones that were eventually sequenced were designated DPP25 (Dryvax clone 25), TKT1 to TKT4, IHD-W1 to IHD-W3, and WR72.
Viral sequencing, data analysis, and annotation.
Each purified virus was cultivated on BSC-40 cells and purified by centrifugation through sucrose gradients (21). The DNA was extracted, and 500 ng of each virus DNA was sequenced by using a Roche 454 GS Junior system. Contigs were produced by using GS De Novo Assembler, and CLC Genomics Workbench 6 was used to complete the assembly of nearly full-length genomes. Roche 454 sequencers have difficulties in sequencing homopolymer runs. Any such conflicts between the reference sequences and our assemblies were generally assumed to be 454 errors, although PCR and Sanger sequencing were also used to verify some sequences. Bioinformatics analyses were performed by using Viral Genome Organizer (22, 23) and Viral Orthologous Clusters (24, 25) (http://www.virology.ca/). LAGAN (26) (http://lagan.stanford.edu) was used to produce multiple-genome alignments, and Base-by-Base (27) was used to check the alignments. To explore virus phylogenies, 98.8 kb of conserved DNA sequences (spanning the genes DVX_058 to DVX_155) was extracted from the multiple-genome alignment and analyzed with the Recombination Detection Program (RDP) (28), using 1,000 bootstrap replicates. JDotter was used to make dot alignments (29). The Genome Annotation Transfer Utility (GATU) (30) was used to transfer a reference annotation to our virus genome sequences, and Artemis (31) was used to visualize and edit the annotation. Table 1 lists the VACV genomes produced and/or cited in this study.
TABLE 1.
Viruses cited in this work
| Virus identification | Virus | Source or strain | GenBank accession no. |
|---|---|---|---|
| CPX-GRI | Cowpox | Strain GRI-90 | X94355 |
| HPXV | Horsepox | MNR-76 | DQ792504.1 |
| RPXV | Rabbitpox | AY484669 | |
| CVA | Vaccinia | Chorioallantoic VACV Ankara | AM501482 |
| Cop | Vaccinia | Copenhagen | M35027 |
| DPP10 | Vaccinia | Dryvax | JN654977 |
| DPP13 | Vaccinia | Dryvax | JN654980 |
| DPP15 | Vaccinia | Dryvax | JN654981 |
| DPP17 | Vaccinia | Dryvax | JN654983 |
| DPP20 | Vaccinia | Dryvax | JN654985 |
| DPP21 | Vaccinia | Dryvax | JN654986 |
| DPP25 | Vaccinia | Dryvax | KJ125438 |
| DPP9 | Vaccinia | Dryvax | JN654976 |
| CL3 | Vaccinia | Dryvax (Acambis clone 3) | AY313848 |
| Duke | Vaccinia | Dryvax (clinical isolate) | DQ439815 |
| IHD-W | Vaccinia | IHD-W | KC201194 |
| LC16m0 | Vaccinia | Lister | AY678277 |
| LC16m8 | Vaccinia | Lister | AY678275 |
| VACV107 | Vaccinia | Lister | DQ121394 |
| VACV-LO | Vaccinia | Lister-LO | AY678276 |
| TP03 | Vaccinia | TianTan | KC207810 |
| TP05 | Vaccinia | TianTan | KC207811 |
| TT08 | Vaccinia | TianTan | JX489135 |
| TT09 | Vaccinia | TianTan | JX489136 |
| TT10 | Vaccinia | TianTan | JX489137 |
| TT11 | Vaccinia | TianTan | JX489138 |
| TT12 | Vaccinia | TianTan | JX489139 |
| WR | Vaccinia | Western Reserve | NC_006998 |
| GLV-1h68 | Vaccinia | Lister | EU410304 |
| 2k | Vaccinia | Dryvax (Acambis clone 2000) | AY313847 |
| 3737 | Vaccinia | Dryvax | DQ377945 |
Construction of mutant viruses.
Homologous recombination was used to delete the DVX_213 gene from VACV strain DPP25. PCR was used to amplify 350 bp of sequence flanking the left side of the DVX_213 locus, and the DNA was cloned into plasmid pDGloxPKODEL (32) by using HindIII and SalI sites. The PCR primers were 213 left F (5′-AATTAAGCTTGCTGTGTCAACGTCATTAT-3′) and 213 left R (5′-TAGTGTCGACCGCGTATCTTCATCCATT-3′). A PCR product encoding 410 bp of sequence to the right of DVX_213 was also amplified by using primers 213 right F (5′-TAATGCGGCCGCTCTGACTAATGCCGTCCT-3′) and 213 right R (5′-ATTATAGATCTCCTTAATTTCTTCTCCATCTCC-3′) and cloned into the targeting vector by using NotI and BglII sites. The vector (pDGloxP-DVX213KO) was then linearized with EcoRI and transfected into BSC-40 cells infected with VACV strain DPP25 at a multiplicity of infection (MOI) of 2. Mycophenolic acid-resistant (recombinant) viruses were isolated by using three rounds of liquid selection and four rounds of plaque picks. The resulting virus was called DPP25Δ213.
Animal studies.
Four-week-old female BALB/c mice were obtained from Charles River Laboratories. Each mouse was inoculated intranasally with 10 μl of PBS containing 107 PFU of purified virus or 10 μl of PBS alone. The body weights were recorded every day over a 1-month period, with any mouse being euthanized if it lost >30% of its body weight. All of these experiments were performed according to policies established by the Canadian Council on Animal Care and were approved by the University of Alberta Animal Care and Use Committee.
Nucleotide sequence accession numbers.
The data reported in this study have been deposited in the GenBank database under accession numbers KJ125438, KJ125439, KM044309, and KM044310.
RESULTS
DPP25 sequencing and annotation.
Dryvax is an old nonclonal smallpox vaccine that comprises a “swarm” of many different VACV substrains and which we had previously classified into four subgroups according to genome structure analysis (19). One of these subgroups was discovered previous to our investigations and is represented by a strain called Acambis clone 3 (CL3). It is of special interest because CL3 is reported to have been more virulent than other Dryvax clones (33). Using PCR primers that target sequences found only in CL3, we identified another related virus clone, which we have called DPP25 (Table 1). DPP25-like viruses comprise <1% of the viruses in a Dryvax stock and contain a 1.1-kbp segment harboring open reading frames (ORFs) DVX_214 to DVX_216, which are not found in any other VACV except strains CL3 (33) and HPXV (19). To gain a better understanding of the relationship between DPP25 and CL3, we sequenced DPP25 and deposited the data in the GenBank database under accession number KJ125438. This sequencing confirmed that DPP25 shares the same genome structure as CL3, although the two viruses can be distinguished by the many SNPs that differentiate all Dryvax clones. To facilitate a comparison of different VACV strains, we created a synthetic genome, called “DVX,” by merging the DPP25 sequence (which encodes the greatest amount of unique VACV sequence) with that of DPP15 (encoding the longest terminal inverted repeats among the Dryvax clones [19]). The relationship between DPP25/CL3 and other VACV strains is explored in greater detail below. The many different ways in which VACV genes are annotated make it difficult to understand the discussions that follow. Table 2 provides a mapping of the different gene names and numbers of different VACV strains, although we have limited the listing to the telomeric genes that are most germane to these analyses.
TABLE 2.
Alternative names of genes found within the telomeres of Dryvax strains that are also found in other strains of vaccinia virus
| Dryvax gene | Vaccinia virus strain gene name or ORF no. |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cop | CVA | Lister107 | TianTan (TP5) | IHDW1 | WR | TKT3 | RPXV | DPP13 | DPP15 | HPXV | |
| DVX left end | |||||||||||
| Chemokine-binding protein-DVX_001 | C23L/B29R | 001 | 001 | 001-2 | 001 | 001 | 001 | 001 | 001 | 001 | 002 |
| TNF-α receptor (CrmB)-DVX_002-3 | C22L/B28R | 002-3 | 002 | 003 | 002 | 002-5 | 002 | 002-3 | 002-3 | 003 | |
| Ankyrin-like-DVX_004-006 | C21L-C19L/B27R-B25R | 004 | 003ABCD | 003-5 | 006-8 | 003-5 | 004-6 | 004-6 | 004 | ||
| Ankyrin (CPXV-008)-DVX_007/8 | C18L, C17L/B24R, B23R | 005-7 | 004AB | 004-5 | 006-7 | 007-8 | 007-8 | 005abc | |||
| Unknown-DVX_009 | B22R | 008 | 005 | 006 | 008 | 002 | 009 | 009 | 006 | ||
| Unknown-DVX_010 | B21R/C15L | 009 | 006 | 007 | 009 | 003 | 010 | 010 | |||
| Surface glycoprotein fragment-DVX_011 | 008 | 011 | 011 | ||||||||
| Unknown-DVX_012 | C14L | 007 | 009 | 010 | 004 | 012 | 012 | ||||
| Serpin (SPI-1)-DVX_013 | C12L | 010 | 008 | 010 | 011 | 005 | 013 | 013 | |||
| Epidermal growth factor-DVX_014 | C11R | 011 | 009 | 011 | 012 | 009 | 006 | 006 | 014 | 014 | 016 |
| DVX right end | |||||||||||
| Ankyrin-DVX_211 | B18R | 194 | 205 | 200 | 199 | 192 | 178 | 194 | |||
| Interferon alpha/beta receptor-DVX_212 | B19R | 206 | 201 | 200 | 193 | 179 | 195 | ||||
| Ankyrin (Bangladesh B18R)-DVX_213 | B20R | 207 | 202 | 201-3 | 194 | 180 | 196 | ||||
| Kelch-like (EV-M-167)-DVX_214/6 | 203 | 204 | 195 | 197 | |||||||
| TIR paralog DVX_217/026 | 026 | ||||||||||
| TIR paralog DVX_218/025 | 025 | ||||||||||
| TIR paralog DVX_219/024 | 024 | ||||||||||
| TIR paralog DVX_220/023 | 023 | ||||||||||
| TIR paralog DVX_221/022 | 022 | ||||||||||
| TIR paralog DVX_222/021 | 021 | ||||||||||
| TIR paralog DVX_223/020 | 020 | ||||||||||
| TIR paralog DVX_224/019 | 214 | 019 | |||||||||
| TIR paralog DVX_225/018 | 215-7 | 207 | 018 | ||||||||
| TIR paralog DVX_226/017 | 208 | 017 | |||||||||
| TIR paralog DVX_227/015 | 218 | 208 | 209 | 015 | |||||||
| TIR paralog DVX_228/014 | 219 | 209 | 210 | 014 | |||||||
| TIR paralog DVX_229/013 | 220 | 210 | 204 | 205 | 196 | 013 | 013 | 198 | |||
| TIR paralog DVX_230/012 | 211 | 205 | 206 | 197 | 181 | 012 | 012 | 199 | |||
| TIR paralog DVX_231/011 | 212 | 011 | 011 | 200 | |||||||
| TIR paralog DVX_232/010 | B21R/C15L | 221 | 213 | 206 | 182 | 010 | 010 | 201 | |||
| TIR paralog DVX_233/009 | B22R | 222 | 197 | 214 | 207 | 198 | 183 | 009 | 009 | 202 | |
| TIR paralog DVX_234/008-7 | B23-B24R | 223-5 | 198 | 215-6 | 208-9 | 199-200 | 008-7 | 008-7 | 203abc | ||
| TIR paralog DVX_235/006-4 | B25-B27R | 226 | 199CD | 210-12 | 211-3 | 201-3 | 006-4 | 006-4 | 204 | ||
| TIR paralog DVX_236/003-2 | B28R | 227-8 | 200 | 217 | 213 | 214-7 | 204 | 003-2 | 003-2 | 205 | |
| TIR paralog DVX_237/001 | B29R | 229 | 201 | 218-9 | 214 | 218 | 205 | 184 | 001 | 001 | 206 |
IHD-W sequencing and annotation.
As we were exploring the links between VACV strains, we discovered that the sequence for strain IHD-W (GenBank accession number KC201194) lacks the right TIR region. (For simplicity, we refer to this sequence as IHD-W0, to differentiate it from IHD-W clones in general.) To address this problem, we isolated three different IHD-W clones from a laboratory stock and sequenced them. All three clones were nearly identical. Consequently, we annotated only one of them (IHD-W1) and deposited the data in GenBank under accession number KJ125439. A feature of IHD-W strains is that they form comet-like plaques due to a K151E substitution mutation in the DVX_168 (Cop A34R) gene (34). Indeed, our IHD-W clones also exhibited this phenotype and encode the K151E mutation, confirming that the virus is likely of IHD-W origin.
The IHD-W1 strain still appears to come from another swarm of viruses, as do the Dryvax and TianTan clones that we have previously characterized. Previous studies have found that most of the small indels in VACV are associated with repeats, and this theme was seen again here (19, 35). For example, Sanger resequencing confirmed that, relative to the previously reported IHD-W0 strain sequence, our IHD-W clones encode a frameshift in the DVX_176 (A40R) gene (36). This is caused by an A inserted into an A5 patch (Fig. 1A). We also noted an AT insertion in an (AT)3 patch in the DVX_124 (D8L) gene (37, 38) and an AC insertion in an (AC)2 patch in the DVX_192 (A56R) gene (Fig. 1B and C). The A56R gene encodes the VACV hemagglutinin (HA) and serves a variety of functions (39–41). Interestingly, the IHD-W strain was reported to be HA negative due to a 2-nucleotide (AC) insertion into A56R (42) and to form syncytia in infected cells (43). We observed this mutation in our IHD-W strains, although it is not reported in the published sequence (Fig. 1C). Whether these mutations have any collective influence on virus growth remains to be investigated.
FIG 1.

Frameshift mutations in IHD-W strains. Whole-genome sequencing detected three different frameshift mutations not seen in the original IHD-W sequence (IHD-W0). Relative to strain DPP25, these insertions (*) created mutations within the A40R, D8L, and A56R genes. These insertions comprised an extra A added to an A5 track in A40R (A), an extra dinucleotide (AT) added to an (AT)3 repeat in D8L (sequencing also revealed a C-to-T mutation in the original IHD-W0 clone, indicated by #) (B), and an extra dinucleotide(AC) added to an (AC)2 repeat in A56R (C).
Tashkent sequencing and annotation.
Tashkent is an old VACV strain that caused such a high frequency of adverse effects that its use as a smallpox vaccine was discontinued. We sequenced four clones isolated from a stock provided by G. Smith and assembled and annotated two of them (TKT3 and TKT4 [GenBank accession numbers KM044309 and KM044310, respectively]). Both viruses are similar but not identical. Compared to TKT3, the TKT4 viral genome encodes 8 small indels and 35 SNPs and lacks two 427-bp fragments (containing sequences homologous to DVX_005) within its TIRs.
When one uses DPP25 as a reference, a number of deletions are detected in these strains, which are discussed in detail below. Another interesting feature of the Tashkent clones is that they carry several intact genes that likely affect virulence. For example, only a limited number of VACV strains (e.g., Lister, some TianTan, and a few Dryvax clones) harbor a functional copy of A53R (CrmC, a soluble tumor necrosis factor [TNF] receptor), whereas this gene is intact in TKT3 and TKT4. We noted that Tashkent also harbors a complete C4L gene (an NF-κB inhibitor [44]), which is absent in CVA strains and all known Dryvax strains.
Relationship between Dryvax and IHD-W clones.
The VACV IHD-W strain is derived from VACV strain IHD-J, which was cloned in Japan by Y. Ichihashi and first described in 1971 (43; H. Shida and S. Dales, personal communication). In turn, strain IHD-J comes from a stock called “International Health Department,” which was derived by passage of the VACV New York City Board of Health (NYCBH) strain through 51 rounds of intracerebral infection in mice followed by four passages on egg chorioallantoic membranes. Thus, this new IHD-W1 sequence provided a useful tool for testing how VACV sequences might drift over the course of continued passage in different laboratories, since the IHD strains and the Dryvax strains share an ancestor, the NYCBH strain.
As an initial test of the methods available for comparing virus genomes, we used dot matrix analysis (45) to identify any major sequence differences between the IHD and Dryvax strains. When we compared IHD-W1 to DPP25, the only notable difference was a 2-kbp insert near the right TIR, noted above, and found only in VACV strains DPP25 and CL3 (Fig. 2A, arrow). This sequence is also missing in DPP13, a much more common type of clone isolated from the same Dryvax stock as DPP25 (Fig. 2B, arrow), and if one compares DPP13 with IHD-W1, one can see that the two genomes are essentially identical (Fig. 2C). We also compared our IHD-W1 sequence with the one previously reported for IHD-W0. Aside from a computational artifact caused by the missing portion of the right TIR sequence, we also detected a 4.9-kb deletion spanning the ATI gene in IHD-W0 (Fig. 2D, arrow). This seems to be a unique feature of this particular clone, as further PCR testing confirmed that the ATI gene predicted from sequencing is present in our IHD-W stock. The several critical genetic differences between our IHD-W clones and the one sequenced previously raised some concerns that these strains might have been mixed up at some point in their history. However, we also used ∼100 kbp of sequences derived from the conserved regions bounded by DVX_058 to DVX_155 (Cop F9L to A24R) and assembled a number of phylogenetic trees (see below). These methods clearly clustered all four IHD-W clones into one very short branch in all three trees (see Fig. 4). The VACV ATI gene seems to be a “hot spot” for mutation, with different deletions also being detected in VACV strains as diverse as Copenhagen (46), buffalo poxvirus (11), and Belo Horizonte virus (47). It seems that some IHD-W clones encode an ATI mutation as well.
FIG 2.

Whole-genome alignments comparing the Dryvax, IHD-W, and Tashkent strains. Dot plots were used to detect major differences in VACV genome structures. (A) DPP25 versus IHD-W1. The two genomes are nearly identical apart from a 2-kbp deletion in IHD-W1 (arrow). (B) DPP25 versus DPP13. DPP13 encodes the same 2-kbp deletion seen in IHD-W1 (arrow). (C) IHD-W1 versus DPP13. The two genomes are structurally identical. (D) IHD-W1 versus IHD-W0 (note that the reported IHD-W0 sequence is missing the right TIR). The IHD-W0 clone bears a 4.9-kbp deletion in the ATI gene (arrow). (E) DPP25 versus TKT3. The TKT3 strain encodes a 6.1-kbp deletion (DVX_007-013) also detected in strain WR (1), a 3.7-kbp deletion (DVX_016-025) also detected in strain Copenhagen (2), and two TKT3-specific deletions (3.5 kbp and 1.3 kbp [3 and 4, respectively]). (F) TKT3 versus TKT4. TKT4 bears two 427-bp deletions in the very short TIRs (arrow).
FIG 4.

Phylogenetic relationships between VACVs. A multiple alignment was compiled by using 98.8 kbp of sequences encoding the core region of the VACV genome (DVX_058 [F9L] to DVX_155 [A24R]). (A) Least-squares method. (B) Neighbor-joining method. (C) Maximum likelihood method. All three approaches clustered individual VACVs into groupings reflecting the vaccine origin and generally assigned HPXV as a distinct outgroup. The RDP program (28) was used to produce the alignments and trees, with 1,000 bootstrap replicates.
These initial comparisons showed that VACV genomes can retain a common global structure over the course of extended passage in different laboratories. However, one can detect idiosyncratic patterns of mutation (e.g., repeat-induced frameshifts and ATI mutations), and determination of the relatedness between strains thus requires a variety of methods of analysis. Bearing this point in mind, we then examined the relationships between DPP25 and other less-well-connected and/or -related VACV strains.
Relationship between Dryvax and Tashkent clones.
Four large deletions are detected in the Tashkent clones relative to DPP25 (Fig. 2E, deletions 1 to 4). The first deletion spans DVX_007 to DVX_013 (6.1 kbp), and the same deletion is seen in strain WR. The second deletion spans DVX_016 to DVX_025 (3.7 kbp) and is identical to the one found in Copenhagen. The third deletion spans DVX_213 to DVX_216 (3.5 kbp) and overlaps both sides of a 2-kbp deletion detected in strain WR (see Fig. 6). Tashkent retains the joint connecting DVX_216 to DVX_013, suggesting that the 2-kbp deletion characteristic of the DPP13/IHDW1/WR group has undergone some expansion. In addition, all of the Tashkent clones share a fourth unique 1.3-kbp deletion at their right TIRs (DVX_231-232). As noted above, relative to TK3, TKT4 also lacks a 427-bp fragment (homologous to DVX_005) within its TIRs (Fig. 2F, arrow).
FIG 6.

A 2-kbp TIR deletion shared by DPP13, IHD-W, and WR strains. Shown are the left and right boundaries of the deletion, aligned against the longer sequences encoded by DPP25 and CL3 strains. For clarity, the connecting sequences in DPP25 and CL3 have been omitted (…). The conservation of this deletion in so many different strains suggests a common ancestry.
Relationship between DPP25 and horsepox virus.
An important aspect of poxvirus evolutionary modeling concerns the hypothesis that as viruses spread into new biological niches, the process of speciation appears to be associated with gene loss (3). If this is true, then the simplest evolutionary scheme would involve a DPP25-like virus evolving from an even larger virus. Horsepox virus (HPXV) is the largest known example of what is still clearly a vaccinia virus, if one defines this assignment based upon a relationship supported by phylogenetic trees, and perhaps retains some resemblance to a hypothetical common ancestor. By using a dot matrix plot, it can be seen that HPXV and DPP25 share the same gene content and gene order from DVX_014 (vaccinia virus growth factor) to DVX_213 as well as from ORFs DVX_214 to DVX_216 (containing fragments of a Kelch-like protein) (Table 3). However, DPP25 also encodes duplicated segments of DNA bearing the genes DVX_010 to DVX_013 in both the right and left TIRs, whereas this sequence is found only in the right end of HPXV (Fig. 3A, deletion 3). Compared to HPXV, DPP25 also bears a 10.7-kbp deletion near the left TIR boundary and a 5.5-kbp deletion near the right TIR boundary (Fig. 3A, deletions 1 and 2, respectively). (The 5.5- and 10.7-kbp deletions differentiate HPXV from all other vaccinia virus strains and are discussed in greater detail below.) Collectively, these data suggest that DPP25/CL3 shares a unique sequence with HPXV, located near the right TIR boundary, but that the overall genome structures have been impacted by events that have changed the location of the TIR boundaries, inverted and duplicated sequences now located in the TIRs, and deleted two large segments of DNA.
TABLE 3.
Right TIR boundaries of vaccinia virusesh
| Strain | % retained gene contenta |
Genes comprising TIRs | |||
|---|---|---|---|---|---|
| DVX_211 (Cop B18) | DVX_212 (Cop B19) | DVX_213 | DVX_214-6b | ||
| Lister | 71 (N) | 0 | 0 | 0 | CPX-GRI 208,c CPX-GRI 209,c CPX-GRI 210-214 |
| DPP15 | 100 | 72 (N) | 0 | 0 | DVX_026-001 |
| TKT3 | 100 | 100 | 14 (N) | 15 (C) | DVX_013, DVX_012, DVX_009-001 |
| Cop | 100 | 100 | 16 (N) | 0 | DVX_011-001 |
| CVA | 100 | 100 | 28 (N) | 0 | DVX_019-006, DVX_002-001d |
| GLV-1h68 | 100 | 100 | 28 (N) | 0 | DVX_019-013, DVX_008, DVX_003-001e |
| DPP13 | 100 | 100 | 69 (N) | 28 (C) | DVX_013-001 |
| IHDW1 | 100 | 100 | 69 (N) | 28 (C) | DVX_013-001 |
| WR | 100 | 100 | 69 (N) | 28 (C) | DVX_013,f DVX_012,f DVX_018-014, DVX_006-001 |
| TP5 | 100 | 100 | 76 (N) | 0 | DVX_015-001 |
| TT12 | 100 | 100 | 76 (N) | 0 | DVX_015, DVX_014, DVX_010-001g |
| RPXV | 100 | 100 | 100 | 3 (N) | DVX_012-001 |
| DPP25 | 100 | 100 | 100 | 100 | DVX_013-001 |
N and C indicate that the value reported is the percentage of the gene's N or C terminus that is retained, respectively.
Resembles CPX-GRI B19R but is fragmented into three pieces (DVX_214-6) in DPP25. B19 belongs to the BTB/Kelch gene family.
The Lister homologs of CPX-GRI 208 and 209 are not located in TIR.
CVA encodes another 1.8-kbp deletion (DVX_003-005) in the TIRs.
GLV-1h68 encodes additional 2.6-kbp (DVX_009-012) and 1.7-kbp (DVX_004-007) deletions in the TIRs.
The WR homologs of DVX_013 and DVX_012 are not located within the TIR but are still present downstream of DVX_216.
TT12 encodes another 3-kbp deletion (DVX_011-013) in its TIRs.
Note that the conjunction site between DVX_216 and DVX_013 is still conserved in TKT3 virus, although TKT3 has a shorter TIR (only DVX_006 due to multiple deletions in the terminal regions).
FIG 3.

Whole-genome alignments comparing orthopoxvirus strains. Dot plots were used to detect major differences in Orthopoxvirus genome structures. (A) DPP25 versus HPXV. DPP25 bears a 10.7-kbp deletion relative to HPXV (deletion 1), which has been partially replaced by a fragment of DNA bearing DVX_010 to DVX_013 and found in the HPXV right telomere (deletion 3). Two 5.5-kbp fragments have also been lost from both DPP25 TIRs (deletion 2). (B) DPP25 versus WR. WR bears 6.1-kbp (deletion 1) and 2-kbp (deletion 2) deletions relative to DPP25 as well an inversion of sequences that occupy gap 3 (DVX_018-014 in WR was replaced with DVX_011-007 in DPP25) and comprising segment 4 (showing a translocation of DVX_013-012 upstream of DVX_018-014). (C) IHD-W1 versus WR. The 2-kbp deletion detected in panel C (deletion 2) is not seen, suggesting that IHD-W1 and WR share the same 2-kbp deletion relative to DPP25. (D) DPP25 versus Copenhagen. Copenhagen bears 3.7-kbp (deletion 1), 4.1-kbp (deletion 2), and 5.7-kbp (deletion 3) deletions relative to DPP25. (E) DPP25 versus RPXV. The two structures are nearly identical except for a 3.3-kbp deletion in RPXV (deletion 1) next to the right TIR of RPXV. (F) DPP25 versus TP5. A 500-bp patch of 59-bp repeats in TP5 replaces 1.2 kbp of sequence bearing DVX_004-6 in TP5 (deletion 1). There are also deletions of 2.4 kbp from DPP25 (DVX_214-6) and 2.3 kbp from TP5 (DVX_015-014), creating gap 2. (G) DPP25 versus CVA. CVA encodes 1.8-kbp deletions within TIRs (DVX_003-005) (deletion 1). There are also deletions of 3.4 kbp (DVX_213-6) from DPP25 and 3.6 kbp (DVX_019-014) from CVA, creating gap 2. (H) DPP25 versus Lister. A gap deletes 9.3 kbp (DVX_211-216 and DVX_013-010) from Lister and substitutes 1.7 kbp of sequence (List195-6 or CPX-GRI 208-9). (I) CPX-GRI versus HPXV. HPXV bears three deletions relative to CPX-GRI. These deletions span 1.4 kbp, 1.6 kbp, and 3.6 kbp (deletions 1 to 3, respectively).
Relationship between DPP25 and other extant VACV strains.
Dot matrix comparisons of DPP25 with other VACV strains detect an array of both simple and more complex sequence rearrangements. For example, the genomes of rabbitpox virus (RPXV) and VACV strain Copenhagen are colinear with DPP25. However, RPXV is differentiated from DPP25 by a single 3.3-kbp deletion (Fig. 3E, deletion 1), whereas Copenhagen exhibits three deletions of 3.7, 4.1, and 5.7 kbp (Fig. 3D, deletions 1 to 3, respectively). The rightmost of these deletions again spans ORFs DVX_214-216 and DVX_013 in RPXV and then further extends into DVX_213 and DVX_012 in Copenhagen.
In contrast, it is more difficult to relate DPP25 to viruses such as TianTan, CVA, and Lister (Fig. 3F to H). We found one region in the right-hand termini bearing two different sequences in each dot plot. When we compared DPP25 to CVA, we observed a 3.6-kbp fragment (DVX_019-014) being replaced by a 3.4-kbp fragment (DVX_213-216) in DPP25. Similarly, when we compared DPP25 to Lister, we observed an expansion of a 1.7-kbp fragment into a 9.3-kbp region in DPP25. Finally, comparison of DPP25 with TianTan (TP5) showed a 2.3-kbp substitution of DVX_014-014 in TP5 with a 2.4-kbp segment in DPP25 (DVX_213-216). Although there has been a complex pattern of genome rearrangements in the region near the right TIR boundary, a common theme is that one detects variably sized deletions spanning DVX_211 to DVX_214-216 and into DVX_012.
Although WR is the accepted VACV reference strain, in hindsight, the choice might have been unfortunate due to the complexity of the rearrangements that it appears to have suffered relative to other VACV strains. Compared to DPP25, dot plot analysis detected a 6.1-kbp (DVX_007-013) deletion in the WR left TIR and a 2-kbp deletion in right end of the WR genome. In addition, WR encodes a translocation that inserted 3.0 kbp of sequence comprising DVX_018-014 after DVX_013-012 (Fig. 3B, deletion 4). WR is the only VACV bearing this translocation, which complicates comparisons with other strains.
VACV phylogenetic trees.
We next examined the phylogenetic relationships between these viruses. Different Dryvax genomes encode a widely distributed scattering of polymorphic sites, showing that no one virus can properly represent an entire stock (19). Depending upon the method used, these analyses can also produce different trees, and this can also depend upon the choice of genome alignments (48). To minimize these problems, we used three different methods (least squares, neighbor joining, or maximum likelihood), as many of the clones as possible from each group, and a common genomic alignment spanning ∼100 kbp of conserved sequence from DVX_058 to DVX_155 (Cop F9L to A24R). To incorporate an additional example of another Lister strain (LIVP), we computationally deleted sequences from a recombinant derivative, GLV-1h68 (49). The virus designated WR72 was a clone obtained from our stock of VACV strain WR and is of ATCC origin.
The different methods correctly assigned the viruses into related groups (e.g., Dryvax, TianTan, WR, Ankara, Lister, and IHD-W) and produced very similar phylogenetic trees (Fig. 4). In two of the three assemblies, HPXV mapped separately from all other VACVs, while in the third assembly, it clustered near the first divergence point separating Dryvax clones from other VACVs (although topologically, this is no different from the other two plots if this is an unrooted tree). Broadly speaking, this analysis also identified two VACV subgroups mapping downstream of the HPXV-VACV divergence point. One is an “American cluster,” encompassing only Dryvax clones, another comprises mostly European and Asian strains. This makes sense since Dryvax came from a seed stock supplied to the New York City Health Department in 1856 and has likely been passaged in isolation for >100 years. The exceptions to this division are the WR and IHD-W strains. Like Dryvax, they were also derived from the New York City Board of Health stocks, and why they might not cluster within the Dryvax quasispecies is discussed below. This analysis also clearly identifies the impact of laboratory cloning, which produces very short branch lengths (e.g., TP3/5, IHD-W, and LC16 clones) compared to unpurified stocks that have been passaged in animals (Dryvax and Lister) or eggs (TianTan). By the same token, the branches leading to the Dryvax clones are among the shortest in these trees, suggesting that Dryvax stocks may still retain some features characteristic of the oldest (and/or least-passaged) VACV strains.
This analysis supports the hypothesis that a stock of virus sharing features characteristic of both HPXV and VACV lies at the root of these trees. Moreover, the relatively short length of the Dryvax branches, the nearness to the tree's roots, and the detection of HPXV-like sequences in one particular clone (8) suggested that Dryvax may still retain some of the features of this hypothetical common ancestor. To gain further insights into these relationships, we turned to a third method, which uses the TIRs and the TIR boundaries to provide additional insights into VACV evolution.
Three features of the genome structure common to all VACVs.
The VACV telomeres comprise the most divergent and distinct features of the different viruses. We performed a BLAST search using ∼20 kbp of sequence from the left end of HPXV (HPXV_003 to HPXV_020a). This search showed that all other VACVs have lost 10.7 kbp of HPXV-related sequences from within this region (Fig. 5A). The deletion has a left end that now comprises the left boundary of the HPXV TIR (and that of CPXV as well) and is marked by DVX_009 (Cop B22R), while the right edge is marked by the growth factor gene (DVX_014 [Cop C11R]). DNA containing DVX_010 to DVX_013 replaces sequences in this gap and is present in both VACV TIRs, while HPXV/CPXV retains these sequences only at the right end of the genome. It has been suggested that the VACV telomeres were created by a transposition of these sequences from the right to the left end of an HPXV-like genome (8). However, since the HPXV TIR still harbors homologs of the genes DVX_001 to DVX_009, it is more likely that the entire region (from DVX_013 to DVX_001 and corresponding to HPXV_196 to the end of the HPXV genome) transposed from the right to the left end of this hypothetical common ancestor. This would have created a new joint site connecting the SPI-1 (DVX_013) and growth factor (DVX_014) genes.
FIG 5.

Left and right VACV genome boundaries. NCBI Mega BLAST searches were used to produce the primary alignments, which were then verified by dot plot analyses (Fig. 2 and 3). (A) Alignment of 20 kbp of HPXV sequence (ORFs 003 to 020a) versus the indicated VACV strains. All VACV strains encode a 10.7-kb deletion, relative to HPXV, where the right boundary abuts the position of the VACV growth factor (VGF) gene (DVX_014 [C11R]). DVX_014 is typically located adjacent to the SPI-1 (serpin-1) gene due to a translocation of sequences (DVX_010-013) found in the right end of the HPXV genome. (B) An alignment of 24 kbp of HPXV sequences (ORFs 194 to 206) versus the indicated VACV strains. A 5.5-kbp deletion is shared by nearly all VACVs, although WR encodes sequences translocated from the left end of an HPXV-like genome.
We repeated the BLAST analysis using 24 kbp from the right end of the HPXV genome and encompassing HPXV_194 to HPXV_206 (Fig. 5B). This analysis detected a 5.5-kbp deletion within HPXV_200 that is shared by many VACVs, although further evolution of the deletion is seen in some strains. For example, TianTan strain TP5 contains the 5.5-kbp deletion, but other TianTan strains contain further expansions and minor rearrangements. These observations suggest that all extant VACV strains are derived from a virus bearing a 5.5-kbp deletion in the right TIR, relative to HPXV. Figure 5B also illustrates a unique feature of Dryvax strains CL3 and DPP25 in that they retain sequences (1.1 kbp) syntenic with HPXV_197 (DVX_214 to DVX_216) and immediately to the left of the 5.5-kbp deletion. All other extant VACVs have lost various amounts of DNA in this region.
These analyses suggest that VACV strains are defined by three unique genetic features, relative to larger orthopoxviruses like HPXV. These features comprise the 10.7- and 5.5-kbp deletions and the translocation of DNA bearing DVX_010 to DVX_013, to create a new joint site connecting the SPI-1 and growth factor genes. If one accepts the hypothesis that larger viruses are probably representative of older strains, a virus with a DPP25- or CL3-like genome might be a representative of this ancestral VACV strain. Although we have not explored the question exhaustively, the origins of HPXV may also be linked back to a CPXV-like virus, assuming that a CPX-GRI-like precursor strain suffered three large deletions.
The right TIR boundary is a defining VACV genetic feature.
To a first approximation, all VACVs harbor much the same gene content and gene order, starting with DVX_001 (Cop C23L) and ending near the TIR boundary with DVX_211 (Cop B18R) (Fig. 2 and 3 and Table 3), although there are exceptions in the form of idiosyncratic deletions and inversions. In contrast, the length of each VACV TIR differs due to variations in the setting of the right TIR boundary. This not only determines the overall length of the TIRs but also encompasses two additional genes (DVX_212 and DVX_213) and a gene fragment (DVX_214 to DVX_216) that are commonly affected by rearrangements in this region. As we show below, the location of the right TIR boundary is a feature that differentiates and defines different families of VACV. To simplify this comparison, we used DVX notation to unify the gene names and summarize the TIR boundaries in Table 3.
One way to illustrate this point is to compare the right TIR boundaries of the DPP13, IHD-W, and WR strains. All three viruses are reported to be derived from an NYCBH stock, and yet simple phylogenies do not readily support this history (Fig. 4). However, all three viruses share the same common 2-kb deletion compared to DPP25 (Table 3 and Fig. 6), with the result that the right TIR boundaries in WR, IHD-W0, and DPP13 all terminate at 69% of the length of DVX_213 (Table 3). DPP13 and IHD-W1 also share other identical genome features (Fig. 2C), further supporting a common origin in a DPP13-like clone. WR has clearly also been subjected to extensive additional mutation, as relative to DPP25 and other VACV strains, it encodes another 6.1-kbp deletion of the DVX_013-007 genes from the TIRs, and the gene order between DVX_018 and DVX_014 has been rearranged (Fig. 3). Presumably, this occurred after the isolation and subsequent propagation of the WR strains.
By this analysis, one can see how the quasispecies that still comprises Dryvax stocks (19), and which was presumably also a feature of the original NYCBH stock, might give rise to specific viral lineages. For example, viruses resembling DPP13 comprise ∼40% of the viruses in Dryvax and encode a right TIR boundary that terminates at 69% of the length of DVX_213. A DPP13-like strain was likely the ancestor of not only the WR and IHDW strains but also VACV strain Duke (19). Alternatively, DPP15-like viruses comprise ∼50% of Dryvax clones and encode TIR boundaries that terminate at 72% of DVX_212 (Table 3). These viruses share a right TIR with Acambis 2k-like clones (19). In contrast to the diversity present in Dryvax/NYCBH-derived strains, all of the Chinese TianTan strains sequenced to date bear a distinctive right TIR boundary that terminates at 76% of DVX_213, on the N-terminal side of DVX_015 (Table 3).
This kind of analysis also provides insights into other less-well-established VACV relationships. For example, Lister strains have the fewest genes immediately adjacent to the right TIR boundary, which is located at 71% of DVX_211 (Table 3). Lister is a complex virus and harbors two genes, List195 (18) and List196 (50), that are otherwise found only in VACV USSR and Evans strains (18). However, GLV-1h68, a recombinant clone isolated from an old Lister stock called LIVP, lacks List195 and List196 (49), suggesting that this stock may still contain a mixture of viruses. Regardless of the specific relationships, GLV-1h68 shares its right TIR boundary with MVA's parent strain CVA (both terminate at 28% of DVX_213 [Table 3]), which implies that Lister and CVA share an origin.
Biological function of DVX_213.
A notable feature of the right TIR boundary is that nearly all VACVs (except for RPXV, DPP25, and CL3) have mutated or lost DVX_213. This is an ankyrin-F-box-containing protein of unknown function and a homolog of the variola virus (Bangladesh) B18R gene. Moreover, in a previous study, a similar DVX_213-encoding strain, called CL3, was shown to be the most virulent clone among six different Dryvax-derived isolates (51). We wondered if DVX_213 might be a virulence factor and tested it by inserting a yellow fluorescent protein (YFP)-GTP cassette into the DVX_213 locus in DPP25. The DPP25 and DPPΔ213 strains were then tested for virulence in BALB/c mice by using an intranasal route of infection (Fig. 7). Although a sufficient dose of virus (107 PFU) was used to cause significant morbidity relative to the sham-treated controls, we saw no difference in the degree of illness between the mice inoculated with the DPP25 virus and those inoculated with the DPPΔ213 virus. Thus, at least in mice, we can find no evidence that DVX_213 is a virulence factor and potentially a trait subject to negative selection over the course of VACV propagation and use.
FIG 7.

Effect of DVX_213 on pathogenicity in mice. Sucrose gradients were used to purify the parent strain (DPP25) and a recombinant virus bearing a deletion of the DVX_213 gene (DPP25Δ213). These viruses were used to inoculate BALB/c mice by the intranasal route using 107 PFU per mouse and three (control and DVX25) or five (DPP25Δ213) mice per group. Control mice were sham treated with 10 μl of PBS. The body weights were recorded each day over 1 month, and no animals met the requirement (>70% weight loss) for euthanization. There was no statistical difference between the impacts of the two viruses on animal health.
DISCUSSION
In this study, we have used a global comparison of whole-genome sequences to explore the relationships between extant VACV strains. This study confirmed the well-established observations that the central part of the VACV genome is highly conserved between strains and that most of the major diversity is found in the telomeres. More specifically, we observed three distinct features conserved in all VACV strains (relative to HPXV and other larger orthopoxviruses) and showed that the right TIR boundary can serve as a specific marker for differentiating VACV strains.
Integrating these data into a model for VACV evolution is most easily done if one proposes that an early intermediate in VACV evolution resembled DPP25/CL3. DPP25-like viruses now comprise only a small fraction of the four types of virus still found in Dryvax stocks (19). As defined by the right TIR boundaries, DPP15-like and DPP13-like viruses comprise the majority of these viruses, while the two minor types (resembling DPP25 and DPP17) make up <1% of the viruses in this stock (19). Theoretically, all it would take to generate DPP13 from DPP25 would be to delete a 2-kbp fragment encompassing DVX_213 to DVX_216 from DPP25. DPP15 could be produced by a further rearrangement of the right TIR of a DPP25- or DPP13-like virus. We can imagine that a similar process would produce other VACV strains. For example, an RPXV-like virus could be produced by deleting a 3.3-kbp fragment containing DVX_214 to DVX_216 and DVX_013/229 from the right TIR boundary of DPP25, producing a TIR encompassing DVX_012 to DVX_001 (Table 3), and a related process can be used to explain the origin of strain Copenhagen. Of course, one does find viruses with extended TIRs. For example, DPP15 has the longest TIRs, encompassing DVX_001 to DVX_026. As we noted previously, the formation of this virus could be explained by translocating the DPP25 left telomere (extending up to DVX_026) to the virus right end by illegitimate recombination and, in doing so, connecting it to DVX_212 (19).
Based on these observations, we suggest that a simple illegitimate recombination model can explain the origins of most VACV strain variants. This model is shown in Fig. 8, where we show two identical viruses (viruses A and B) in which genes 1, 2, and 3 are duplicated in the telomeres. A large deletion could be produced by recombination between genes 3 and 5 (Fig. 8, scheme 1). If this were a reciprocal event, it would produce viruses A′ and B′, where virus A′ lacks gene 4 and virus B′, bearing an unstable gene duplication, would rapidly revert to the parent form. (Of course, such deletions do not have to involve two viruses, but the principles of the scheme for producing all three types of genome rearrangements are most easily illustrated as shown.). If one inverts the relative orientation of the genomes, the same kind of error-prone process can produce large deletions linked to shortened telomeres (Fig. 8, scheme 2) and deletions associated with extended telomeres (Fig. 8, scheme 3). Importantly, the process illustrated in Fig. 8 (scheme 3) could explain how a DPP25-like virus could be derived from an HPXV-like ancestor. This would involve deleting HPXV_007 to HPXV_015b (corresponding to genes 4 and 5 in the model) and translocating HPXV_196 to HPXV_199 (DVX_013 to DVX_010; gene 200 in the model) from the right to the left end of the genome, making the TIR longer. An actual example of such a process is illustrated by an analysis of myxoma virus variants. One strain was described where two genes present in the right TIR were translocated onto the left TIR with the creation of a small deletion (52), exactly as predicted by this model.
FIG 8.

Illegitimate recombination model for VACV genome rearrangements. For clarity, we show all four schemes involving two different (red and black) parental viruses, although a simple deletion (schemes 1 and 2) could involve just a single virus. We also show these events as being reciprocal, although no evidence supports or refutes this mechanism. The numbers represent genes, some of which are duplicated and present in reverse order in the TIRs. In scheme 1, illegitimate recombination (IR) between nonhomologous sites creates a virus lacking gene 4 (virus A′) and one bearing two copies of the gene. In the absence of selection for the extra copy, virus B′ would delete a copy of gene 4 (through homologous recombination [HR]) and revert to the wild-type state (virus B″). Scheme 2 shows shortening of the telomeres through gene deletion. Illegitimate recombination creates a virus bearing a large deletion and a shorter right TIR (virus A′) as well as a virus bearing a copy of gene 200 flanked by duplicated copies of gene 3 (virus B′). As in scheme 1, virus B′ would likely revert back to a wild-type state. Note that the two viruses are aligned in opposite orientations to each other in this and subsequent schemes (arrows). Scheme 3 shows a method for producing a virus with a large deletion and an extended telomere. Recombination between genes 199 and 5 produces virus A′ (missing gene 200) and a translocation of genes 4 and 5 from the left to the right end of the genome. Virus B′ inherits a longer TIR that now includes gene 200. Scheme 4 shows VACV strain WR. VACV strain WR is proposed to have been generated by two events. First, gene 3 was deleted by a process like that shown in scheme 1, and illegitimate recombination between genes 2 and 6 then produced a virus (virus A″) exhibiting an inversion involving gene 3.
The origin of VACV strain WR can be explained by using a slight variation on these schemes (Fig. 8, scheme 4). The WR genome carries DVX_216 followed by DVX_013 and DVX_012 connecting to DVX_018. To produce this arrangement, gene 3 (which corresponds to the genes DVX_013 to DVX_007) is first deleted from the left end but not the right. We then postulate that recombination near virus A′ gene 3 and virus B′ gene 5 occurs. This process would create the appearance of an inversion involving genes 3 to 5 (from DVX_013-12 to DVX_018-014).
The schemes outlined above would tend to erode single-copy sequences lying adjacent to the TIR boundaries, raising questions about what selective pressures might constrain this process. It has been noted that the VACV growth factor gene (DVX_014) is rarely affected by deletions, suggesting that it marks a constrained boundary in the left TIR (48). At the right end of the VACV genome, one sees that DVX_214-216 has been truncated or deleted in all extant VACVs and that while some VACV genomes retain DVX_214, most TIR boundaries end within DVX_213 (Table 3 and Fig. 9A). This suggests that these three open reading frames are dispensable. What seems to be critically important is that nearly all viruses, with a few exceptions (e.g., strain Lister), retain DVX_212 (Cop B19R), encoding an interferon alpha/beta receptor homolog (53). Interferons promote very effective antiviral responses (54), and it is not surprising that most VACVs retain an inhibitory capacity (54). Thus, the left and right TIR boundaries (i.e., the conserved core) are likely defined by the VACV growth factor gene and the interferon alpha/beta receptor homolog.
FIG 9.
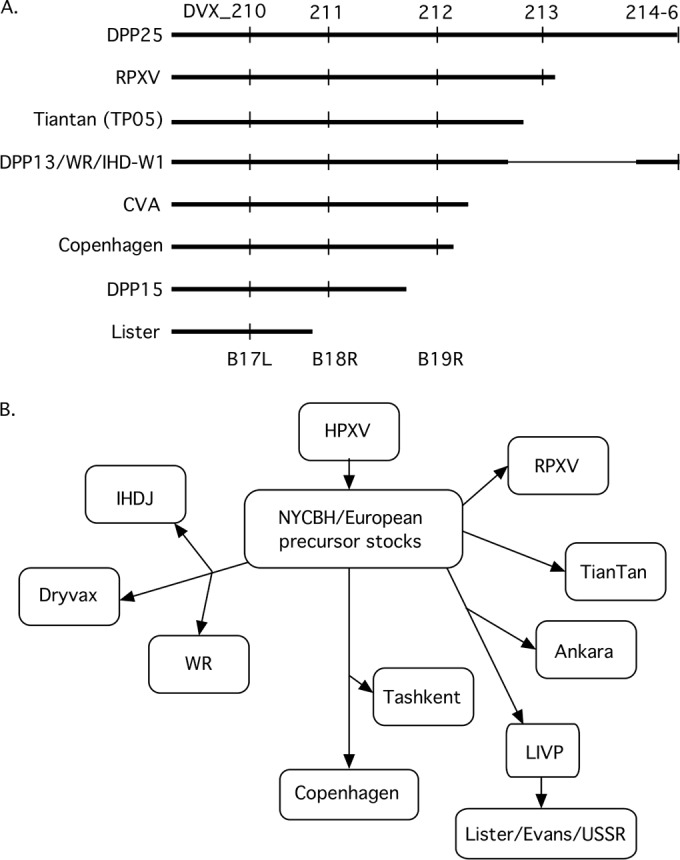
Model for VACV evolution. (A) Graphical depiction of the right genomic boundaries of VACVs. (B) Postulated historical relationships between different VACV strains. The analysis is based upon a combination of known historical relationships, conserved deletions, and right TIR boundaries. For example, Ankara and GLV-1h68 (a LIVP clone) share the same TIR boundary (where 28% of the N terminus of DVX_213 connects to DVX_019), suggesting that they might be derived from a common older stock. Tashkent also shares a 3.7-kbp deletion with Copenhagen along with three additional large deletions, one of them identical to a deletion in WR. However, since WR is derived from NYCBH, this leads us to assume that this particular deletion was also present in the old NYCBH/European precursor stocks before the NYCBH strain was transported from Europe in 1865 and isolated from Tashkent. This might account for its survival in WR and Tashkent, even though these two viruses share no known historical relationship. In contrast, one can find no shared, and yet still distinctive, features linking RPXV or TianTan to other particular VACV strains (see the text for additional discussion).
There is one last conclusion that can be drawn from this analysis, concerning the origin(s) of VACV. The history of smallpox vaccination does not provide any obvious clues as to whether all modern VACV strains are derived from a single stock of widely shared vaccine or were independently collected by different individuals. However, if one examines the phylogenetic trees shown in Fig. 4, it is apparent that viruses that should be linked by historical connections (e.g., WR, Dryvax, and IHD-W) map all over the place. Moreover, viruses that have no known historical connections still share a number of common and distinctive deletions (e.g., strains Tashkent and WR share the same 6.1-kbp deletion relative to DPP25). The most obvious way to explain these features is if all extant VACV strains are derived from one common stock (“X”), a large pool of viruses encapsulating much of the diversity still seen in modern strains (Fig. 9B). This population lies at the root of the phylogenetic trees mapped in Fig. 4, and each of the branch points reflects an attempt to computationally illustrate the consequences of sampling different populations of viruses out of a larger pool of viruses. Although WR, Dryvax, and IHD-W all share an origin within the NYCBH stock, the pool that became Dryvax probably represents only a subset of strains within stock X (and NYCBH), and this has partially obscured the links to WR and IHD-W. Similarly, although Lister and its related strains have no immediate history linking them to NYCBH, they could have been originally sampled from the same common pool of viruses as WR and IHD-W, thus creating the apparent relationships. This situation is perhaps not surprising given the way in which VACV strains were shared in the 19th century. Until stocks started being passaged in animals, serial passage through individuals (55) and the sharing of scabs (55) would have created population bottlenecks, sampling randomly and reducing the diversity at each stage. Each subset of viruses would then have evolved in isolation, creating new variants that acquired an idiosyncratic assemblage of SNPs. However, regardless of the drift occurring after “speciation,” all of the viruses would still retain a common array of deletions, deletions presumably once scattered among the viruses in stock X. Thus, the structure of extant VACV strains suggests a common origin in a single stock of now widely shared virus.
ACKNOWLEDGMENTS
We thank W. Magee, R. Noyce, and other members of our laboratory for their helpful advice. We also thank J. Cao and S. Tyler at the National Microbiology Laboratory, Public Health Agency of Canada, for providing vaccinia virus DNA and the sequences of their WR clones.
This work was supported by operating grants from the Canadian Institutes for Health Research and the Natural Sciences Engineering Research Council and by infrastructure awards from the Canada Foundation for Innovation. L.Q. was a recipient of a Faculty of Medicine and Dentistry 75th Anniversary award.
REFERENCES
- 1.Carroll DS, Emerson GL, Li Y, Sammons S, Olson V, Frace M, Nakazawa Y, Czerny CP, Tryland M, Kolodziejek J, Nowotny N, Olsen-Rasmussen M, Khristova M, Govil D, Karem K, Damon IK, Meyer H. 2011. Chasing Jenner's vaccine: revisiting cowpox virus classification. PLoS One 6:e23086. doi: 10.1371/journal.pone.0023086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shchelkunov SN, Safronov PF, Totmenin AV, Petrov NA, Ryazankina OI, Gutorov VV, Kotwal GJ. 1998. The genomic sequence analysis of the left and right species-specific terminal region of a cowpox virus strain reveals unique sequences and a cluster of intact ORFs for immunomodulatory and host range proteins. Virology 243:432–460. doi: 10.1006/viro.1998.9039. [DOI] [PubMed] [Google Scholar]
- 3.Hendrickson RC, Wang C, Hatcher EL, Lefkowitz EJ. 2010. Orthopoxvirus genome evolution: the role of gene loss. Viruses 2:1933–1967. doi: 10.3390/v2091933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baxby D. 1979. Edward Jenner, William Woodville, and the origins of vaccinia virus. J Hist Med Allied Sci 34:134–162. [DOI] [PubMed] [Google Scholar]
- 5.Baxby D. 1977. The origins of vaccinia virus. J Infect Dis 136:453–455. doi: 10.1093/infdis/136.3.453. [DOI] [PubMed] [Google Scholar]
- 6.Geddes AM. 2006. The history of smallpox. Clin Dermatol 24:152–157. doi: 10.1016/j.clindermatol.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 7.Parrino J, Graham BS. 2006. Smallpox vaccines: past, present, and future. J Allergy Clin Immunol 118:1320–1326. doi: 10.1016/j.jaci.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tulman ER, Delhon G, Afonso CL, Lu Z, Zsak L, Sandybaev NT, Kerembekova UZ, Zaitsev VL, Kutish GF, Rock DL. 2006. Genome of horsepox virus. J Virol 80:9244–9258. doi: 10.1128/JVI.00945-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobs BL, Langland JO, Kibler KV, Denzler KL, White SD, Holechek SA, Wong S, Huynh T, Baskin CR. 2009. Vaccinia virus vaccines: past, present and future. Antiviral Res 84:1–13. doi: 10.1016/j.antiviral.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagasse-Sugahara TK, Kisielius JJ, Ueda-Ito M, Curti SP, Figueiredo CA, Cruz AS, Silva MM, Ramos CH, Silva MC, Sakurai T, Salles-Gomes LF. 2004. Human vaccinia-like virus outbreaks in Sao Paulo and Goias States, Brazil: virus detection, isolation and identification. Rev Inst Med Trop Sao Paulo 46:315–322. doi: 10.1590/S0036-46652004000600004. [DOI] [PubMed] [Google Scholar]
- 11.Bhanuprakash V, Venkatesan G, Balamurugan V, Hosamani M, Yogisharadhya R, Gandhale P, Reddy KV, Damle AS, Kher HN, Chandel BS, Chauhan HC, Singh RK. 2010. Zoonotic infections of buffalopox in India. Zoonoses Public Health 57:e149–155. doi: 10.1111/j.1863-2378.2009.01314.x. [DOI] [PubMed] [Google Scholar]
- 12.Singh RK, Hosamani M, Balamurugan V, Bhanuprakash V, Rasool TJ, Yadav MP. 2007. Buffalopox: an emerging and re-emerging zoonosis. Anim Health Res Rev 8:105–114. doi: 10.1017/S1466252307001259. [DOI] [PubMed] [Google Scholar]
- 13.de Assis FL, Vinhote WM, Barbosa JD, de Oliveira CHS, de Oliveira CMG, Campos KF, Silva NS, de Souza Trindade G, Kroon EG, Abrahao JS, Jonatas. 2013. Reemergence of vaccinia virus during zoonotic outbreak, Para State, Brazil. Emerg Infect Dis 19:2017–2020. doi: 10.3201/eid1912.130589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hatcher EL, Hendrickson RC, Lefkowitz EJ. 2014. Identification of nucleotide-level changes impacting gene content and genome evolution in orthopoxviruses. J Virol 88:13651–13668. doi: 10.1128/JVI.02015-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Archard LC, Mackett M, Barnes DE, Dumbell KR. 1984. The genome structure of cowpox virus white pock variants. J Gen Virol 65(Part 5):875–886. [DOI] [PubMed] [Google Scholar]
- 16.Paez E, Dallo S, Esteban M. 1985. Generation of a dominant 8-MDa deletion at the left terminus of vaccinia virus DNA. Proc Natl Acad Sci U S A 82:3365–3369. doi: 10.1073/pnas.82.10.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moyer RW, Graves RL, Rothe CT. 1980. The white pock (mu) mutants of rabbit poxvirus. III. Terminal DNA sequence duplication and transposition in rabbit poxvirus. Cell 22:545–553. [DOI] [PubMed] [Google Scholar]
- 18.Garcel A, Crance JM, Drillien R, Garin D, Favier AL. 2007. Genomic sequence of a clonal isolate of the vaccinia virus Lister strain employed for smallpox vaccination in France and its comparison to other orthopoxviruses. J Gen Virol 88:1906–1916. doi: 10.1099/vir.0.82708-0. [DOI] [PubMed] [Google Scholar]
- 19.Qin L, Upton C, Hazes B, Evans DH. 2011. Genomic analysis of the vaccinia virus strain variants found in Dryvax vaccine. J Virol 85:13049–13060. doi: 10.1128/JVI.05779-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fenner F, Henderson A, Arita I, Jezek Z, Ladnyi ID. 1988. Smallpox and its eradication. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 21.Lee HJ, Essani K, Smith GL. 2001. The genome sequence of Yaba-like disease virus, a yatapoxvirus. Virology 281:170–192. doi: 10.1006/viro.2000.0761. [DOI] [PubMed] [Google Scholar]
- 22.Upton C, Hogg D, Perrin D, Boone M, Harris NL. 2000. Viral genome organizer: a system for analyzing complete viral genomes. Virus Res 70:55–64. doi: 10.1016/S0168-1702(00)00210-0. [DOI] [PubMed] [Google Scholar]
- 23.Lefkowitz EJ, Upton C, Changayil SS, Buck C, Traktman P, Buller RM. 2005. Poxvirus Bioinformatics Resource Center: a comprehensive Poxviridae informational and analytical resource. Nucleic Acids Res 33:D311–D316. doi: 10.1093/nar/gki110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ehlers A, Osborne J, Slack S, Roper RL, Upton C. 2002. Poxvirus orthologous clusters (POCs). Bioinformatics 18:1544–1545. doi: 10.1093/bioinformatics/18.11.1544. [DOI] [PubMed] [Google Scholar]
- 25.Upton C, Slack S, Hunter AL, Ehlers A, Roper RL. 2003. Poxvirus orthologous clusters: toward defining the minimum essential poxvirus genome. J Virol 77:7590–7600. doi: 10.1128/JVI.77.13.7590-7600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brudno M, Do CB, Cooper GM, Kim MF, Davydov E, Green ED, Sidow A, Batzoglou S. 2003. LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res 13:721–731. doi: 10.1101/gr.926603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brodie R, Smith AJ, Roper RL, Tcherepanov V, Upton C. 2004. Base-By-Base: single nucleotide-level analysis of whole viral genome alignments. BMC Bioinformatics 5:96. doi: 10.1186/1471-2105-5-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. 2010. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brodie R, Roper RL, Upton C. 2004. JDotter: a Java interface to multiple dotplots generated by dotter. Bioinformatics 20:279–281. doi: 10.1093/bioinformatics/btg406. [DOI] [PubMed] [Google Scholar]
- 30.Tcherepanov V, Ehlers A, Upton C. 2006. Genome Annotation Transfer Utility (GATU): rapid annotation of viral genomes using a closely related reference genome. BMC Genomics 7:150. doi: 10.1186/1471-2164-7-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 32.Rintoul JL, Wang J, Gammon DB, van Buuren NJ, Garson K, Jardine K, Barry M, Evans DH, Bell JC. 2011. A selectable and excisable marker system for the rapid creation of recombinant poxviruses. PLoS One 6:e24643. doi: 10.1371/journal.pone.0024643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osborne JD, Da Silva M, Frace AM, Sammons SA, Olsen-Rasmussen M, Upton C, Buller RM, Chen N, Feng Z, Roper RL, Liu J, Pougatcheva S, Chen W, Wohlhueter RM, Esposito JJ. 2007. Genomic differences of vaccinia virus clones from Dryvax smallpox vaccine: the Dryvax-like ACAM2000 and the mouse neurovirulent clone-3. Vaccine 25:8807–8832. doi: 10.1016/j.vaccine.2007.10.040. [DOI] [PubMed] [Google Scholar]
- 34.Blasco R, Sisler JR, Moss B. 1993. Dissociation of progeny vaccinia virus from the cell membrane is regulated by a viral envelope glycoprotein: effect of a point mutation in the lectin homology domain of the A34R gene. J Virol 67:3319–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coulson D, Upton C. 2011. Characterization of indels in poxvirus genomes. Virus Genes 42:171–177. doi: 10.1007/s11262-010-0560-x. [DOI] [PubMed] [Google Scholar]
- 36.Wilcock D, Duncan SA, Traktman P, Zhang WH, Smith GL. 1999. The vaccinia virus A4OR gene product is a nonstructural, type II membrane glycoprotein that is expressed at the cell surface. J Gen Virol 80(Part 8):2137–2148. [DOI] [PubMed] [Google Scholar]
- 37.Matho MH, Maybeno M, Benhnia MR, Becker D, Meng X, Xiang Y, Crotty S, Peters B, Zajonc DM. 2012. Structural and biochemical characterization of the vaccinia virus envelope protein D8 and its recognition by the antibody LA5. J Virol 86:8050–8058. doi: 10.1128/JVI.00836-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodriguez JR, Rodriguez D, Esteban M. 1992. Insertional inactivation of the vaccinia virus 32-kilodalton gene is associated with attenuation in mice and reduction of viral gene expression in polarized epithelial cells. J Virol 66:183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turner PC, Moyer RW. 2006. The cowpox virus fusion regulator proteins SPI-3 and hemagglutinin interact in infected and uninfected cells. Virology 347:88–99. doi: 10.1016/j.virol.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 40.Turner PC, Moyer RW. 2008. The vaccinia virus fusion inhibitor proteins SPI-3 (K2) and HA (A56) expressed by infected cells reduce the entry of superinfecting virus. Virology 380:226–233. doi: 10.1016/j.virol.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeHaven BC, Girgis NM, Xiao Y, Hudson PN, Olson VA, Damon IK, Isaacs SN. 2010. Poxvirus complement control proteins are expressed on the cell surface through an intermolecular disulfide bridge with the viral A56 protein. J Virol 84:11245–11254. doi: 10.1128/JVI.00372-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown CK, Bloom DC, Moyer RW. 1991. The nature of naturally occurring mutations in the hemagglutinin gene of vaccinia virus and the sequence of immediately adjacent genes. Virus Genes 5:235–242. doi: 10.1007/BF00568973. [DOI] [PubMed] [Google Scholar]
- 43.Ichihashi Y, Dales S. 1971. Biogenesis of poxviruses: interrelationship between hemagglutinin production and polykaryocytosis. Virology 46:533–543. doi: 10.1016/0042-6822(71)90057-2. [DOI] [PubMed] [Google Scholar]
- 44.Ember SW, Ren H, Ferguson BJ, Smith GL. 2012. Vaccinia virus protein C4 inhibits NF-kappaB activation and promotes virus virulence. J Gen Virol 93:2098–2108. doi: 10.1099/vir.0.045070-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang Y, Zhang L. 2004. Rapid and sensitive dot-matrix methods for genome analysis. Bioinformatics 20:460–466. doi: 10.1093/bioinformatics/btg429. [DOI] [PubMed] [Google Scholar]
- 46.Goebel SJ, Johnson GP, Perkus ME, Davis SW, Winslow JP, Paoletti E. 1990. The complete DNA sequence of vaccinia virus. Virology 179:247–266, 517–263. [DOI] [PubMed] [Google Scholar]
- 47.Trindade GS, da Fonseca FG, Marques JT, Diniz S, Leite JA, De Bodt S, Van der Peer Y, Bonjardim CA, Ferreira PC, Kroon EG. 2004. Belo Horizonte virus: a vaccinia-like virus lacking the A-type inclusion body gene isolated from infected mice. J Gen Virol 85:2015–2021. doi: 10.1099/vir.0.79840-0. [DOI] [PubMed] [Google Scholar]
- 48.Qin L, Liang M, Evans DH. 2013. Genomic analysis of vaccinia virus strain TianTan provides new insights into the evolution and evolutionary relationships between orthopoxviruses. Virology 442:59–66. doi: 10.1016/j.virol.2013.03.025. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Q, Liang C, Yu YA, Chen N, Dandekar T, Szalay AA. 2009. The highly attenuated oncolytic recombinant vaccinia virus GLV-1h68: comparative genomic features and the contribution of F14.5L inactivation. Mol Genet Genomics 282:417–435. doi: 10.1007/s00438-009-0475-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gubser C, Bergamaschi D, Hollinshead M, Lu X, van Kuppeveld FJ, Smith GL. 2007. A new inhibitor of apoptosis from vaccinia virus and eukaryotes. PLoS Pathog 3:e17. doi: 10.1371/journal.ppat.0030017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weltzin R, Liu J, Pugachev KV, Myers GA, Coughlin B, Blum PS, Nichols R, Johnson C, Cruz J, Kennedy JS, Ennis FA, Monath TP. 2003. Clonal vaccinia virus grown in cell culture as a new smallpox vaccine. Nat Med 9:1125–1130. doi: 10.1038/nm916. [DOI] [PubMed] [Google Scholar]
- 52.Kerr PJ, Rogers MB, Fitch A, Depasse JV, Cattadori IM, Twaddle AC, Hudson PJ, Tscharke DC, Read AF, Holmes EC, Ghedin E. 2013. Genome scale evolution of myxoma virus reveals host-pathogen adaptation and rapid geographic spread. J Virol 87:12900–12915. doi: 10.1128/JVI.02060-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alcami A, Symons JA, Smith GL. 2000. The vaccinia virus soluble alpha/beta interferon (IFN) receptor binds to the cell surface and protects cells from the antiviral effects of IFN. J Virol 74:11230–11239. doi: 10.1128/JVI.74.23.11230-11239.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith GL, Benfield CT, Maluquer de Motes C, Mazzon M, Ember SW, Ferguson BJ, Sumner RP. 2013. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J Gen Virol 94:2367–2392. doi: 10.1099/vir.0.055921-0. [DOI] [PubMed] [Google Scholar]
- 55.Fenner F. 1988. Smallpox and its eradication. World Health Organization, Geneva, Switzerland. [Google Scholar]