

Abstract
There is substantial clinical interest in synthetic platelet analogs for potential application in transfusion medicine. To this end, our research is focused on self-assembled peptide–lipid nanoconstructs that can undergo injury site-selective adhesion and subsequently promote site-directed active platelet aggregation, thus mimicking platelet’s primary hemostatic actions. For injury site-selective adhesion, we have utilized a coagulation factor FVIII-derived VWF-binding peptide (VBP). FVIII binds to VWF’s D′–D3 domain while natural platelet GPIbα binds to VWF’s A1 domain. Therefore, we hypothesized that the VBP-decorated nanoconstructs will adhere to VWF without mutual competition with natural platelets. We further hypothesized that the adherent VBP-decorated constructs can enhance platelet aggregation when co-decorated with a fibrinogen-mimetic peptide (FMP). To test these hypotheses, we used glycocalicin to selectively block VWF’s A1 domain and, using fluorescence microscopy, studied the binding of fluorescently labeled VBP-decorated nanoconstructs versus platelets to ristocetin-treated VWF. Subsequently, we co-decorated the nanoconstructs with VBP and FMP and incubated them with human platelets to study construct-mediated enhancement of platelet aggregation. Decoration with VBP resulted in substantial construct adhesion to ristocetin-treated VWF even if the A1-domain was blocked by glycocalicin. In comparison, such A1-blocking resulted in significant reduction of platelet adhesion. Without A1-blocking, the VBP-decorated constructs and natural platelets could adhere to VWF concomitantly. Furthermore, the constructs co-decorated with VBP and FMP enhanced active platelet aggregation. The results indicate significant promise in utilizing the FVIII-derived VBP in developing synthetic platelet analogs that do not interfere with VWF-binding of natural platelets but allow site-directed enhancement of platelet aggregation when combined with FMP.
Introduction
von Willebrand factor (VWF) is a major protein that mediates physiological (hemostasis) as well as pathological (thrombosis) adhesion of platelets in vascular injury.1–3 VWF is secreted from the Weibel–Palade bodies of injured endothelial cells and alpha (α)-granules of activated platelets.4 Each monomeric subunit of VWF consists of several domains with specific bioactivity, e.g. the A1 domain mediates binding to the platelet glycoprotein GPIbα component, the A3 domain mediates binding to subendothelial collagen, the C1–C2 domain mediates binding to fibrinogen (Fg) and to integrin GPIIb–IIIa on activated platelets, the D′–D3 domain acts as a carrier for coagulation factor FVIII before thrombin-induced activation, and the A2 domain undergoes cleavage via a metalloprotease ADAMTS-13 enzyme for regulation of the VWF multimer size.5,6 The multi-domain VWF monomeric subunits can multimerize via disulphide bonds, and this multimeric VWF circulates as a globular protein.7,8 However, at a vascular injury site due to increasing hemodynamic shear, the globular VWF multimers can unravel and further self-associate to enhance the VWF availability for bioactive functions (Fig. 1).9
Fig. 1.
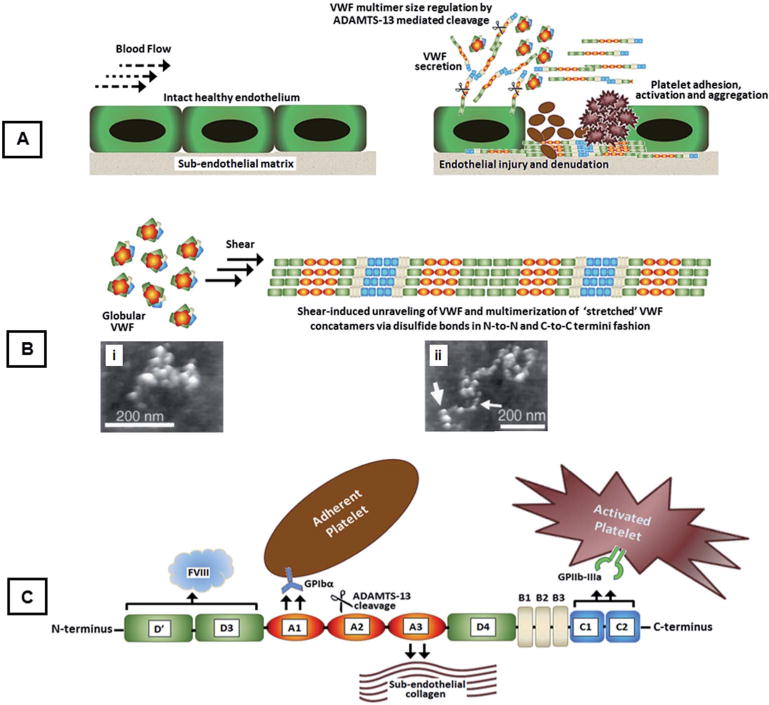
(A) Schematic of normal vascular endothelium and subsequent endothelial injury leading to VWF secretion, its shear-induced conformational change and multimerization on subendothelial collagen, and platelet adhesion, activation and aggregation on a VWF/collagen matrix; (B) schematic showing shear-induced conformational unraveling of VWF multimers leading to self-association along with atomic force microscopy (AFM) images of (i) globular and (ii) stretched VWF (adapted with permission from R. E. Marchant et al., Curr. Protein Pept. Sci., 2002, 3, 249–274); (C) a closer schematic look at the various domains of VWF with specific bioactive functions.
The primary mechanism of platelet adhesion at a vascular injury site is the tethering of platelet GPIbα to VWF’s exposed A1 domains. Therefore, simulating this functional aspect is a critical component of our research to design an artificial platelet analog. The clinical interest for artificial platelet analogs stems from the issues that natural platelet-based products pose, e.g. shortage in supply, very short shelf-life (3–5 days) due to high risk of pathologic contamination, storage lesions and a variety of biological side effects.10 An effective approach to design artificial platelet analogs is to decorate the surface of biocompatible intravenously-administrable particles with motifs that render platelet-mimetic hemostatic functions. To this end, we have focused on mimicking platelet’s key primary hemostatic actions of injury site-selective adhesion and site-selective amplification of platelet aggregation and have combined them on a single synthetic platform. For this, we have utilized self-assembly of lipid–peptide bioconjugates to form unilamellar liposomal constructs (~150 nm in diameter) that are heteromultivalently decorated with VWF-binding peptides (VBP), collagen-binding peptides (CBP) and active platelet glycoprotein GPIIb–IIIa binding fibrinogen-mimetic peptides (FMP). Our design rationale is that the VBP and CBP will promote injury site-selective adhesion of the constructs via VWF- and collagen-binding, while the FMP will promote site-directed aggregation of active platelets onto the adhered constructs to amplify primary hemostasis. We have recently demonstrated the platelet-mimetic abilities of our constructs at the cellular scale.11–13
Building on these studies, we now focus on establishing a molecular scale mechanistic model of the platelet-mimetic functions exhibited by the nanoconstructs. As the first component of this mechanistic investigation, here we report on our analysis of how surface decoration with VBP enables the binding of the constructs on VWF. In this context, we rationalized that an artificial platelet design for VWF-binding should not interfere with the binding of available natural platelets to the same VWF. Hence, the mechanisms of nanoparticle binding to VWF should be different from that of natural platelets binding to VWF. To achieve this exclusivity at the molecular scale, we have focused on the FVIII-binding D′–D3 domain of VWF and have utilized an FVIII-derived VBP that has moderately high affinity (IC50 ~ 9 μM) to this domain.14,15 It is also reported that although each VWF monomeric subunit contains one D′–D3 domain that can theoretically bind to one FVIII molecule, physiologically VWF contains ‘bound FVIII in ~1 : 50 (FVIII : VWF) ratio.16 Hence we rationalized that the physiological VWF will have sufficient ‘unoccupied’ D′–D3 domains that can enable adhesion of VBP-decorated nanoparticles under flow, while available natural platelets can still, simultaneously, bind to the A1 domain of VWF via GPIbα without mutual interference. Also, since the VBP we used does not include the thrombin-binding Arg1689 region but rather residues 2303–2332 of the C2 domain of FVIII,14,15 we further rationalized that the binding of VBP to VWF will not be thrombin-cleavable. Furthermore, we hypothesized that co-decoration of the VBP-decorated nanoconstructs with FMP motifs will potentially enable the constructs to recruit and amplify the aggregation of locally activated platelets and thereby cumulatively enhance primary hemostasis, as shown in Fig. 2. Here we report on our experimental studies of our rationale and hypotheses at cellular and molecular scales in vitro.
Fig. 2.
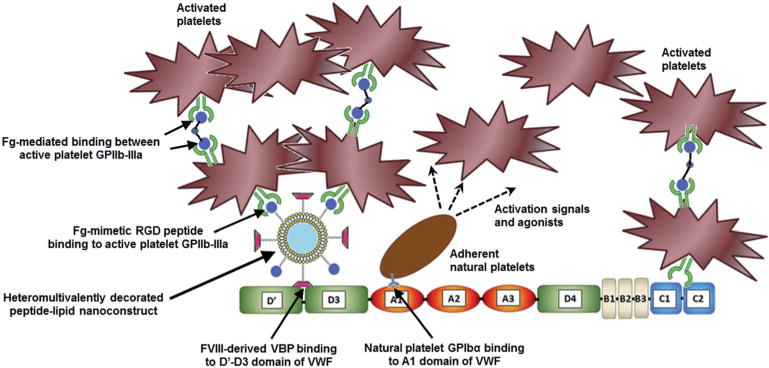
A schematic of the envisioned mechanism of action of the VBP–FMP-co-decorated liposomal constructs interacting with VWF and platelets to enhance the primary hemostatic processes of platelet recruitment and aggregation. In the schematic, ‘Fg’ stands for fibrinogen. The VWF-binding peptide (VBP) peptide is the sequence TRYLRIHPQSWVHQI and the fibrinogen-mimetic peptide (FMP) containing the arginine–glycine–aspartic acid (RGD) sequence is cyclo-CNPRGDY(OEt)RC.
Experimental
Materials
Phosphate Buffered Saline (PBS), glass coverslips and microscope slides, 3.8% w/v sodium citrate, paraformaldehyde (PFA), and bovine serum albumin (BSA) were obtained from Thermo Fisher Scientific (Waltham, MA). FVIII free and physiologic human von Willebrand factor (VWF) and alpha (α) thrombin were obtained from Hematologic Technologies (Essex Junction, VT). Glycocalicin was purchased from USCN Life Science (Wuhan, China). Calcein was purchased from Invitrogen (Carlsbad, CA) and ristocetin from Helena Laboratories (Beaumont, TX). For liposomal construct fabrication, cholesterol was purchased from Sigma Aldrich (Saint Louis, MO). The lipids distearyl phosphatidyl choline (DSPC), poly(ethylene glycol)-modified DSPE (DSPE–PEG2000), carboxypoly(ethylene glycol)-modified DSPE (DSPE–PEG2000–COOH), and rhodamine B–dihexadecanoyl-sn-glycero-3-phosphoethanolamine (DHPE–rhodamine) were purchased from Avanti Polar Lipids (Alabaster, AL). Adenosine diphosphate (ADP) was purchased from Bio/Data Corporation (Horsham, PA). The peptides TRYLRIHPQSWVHQI (VBP) and cyclo-CNPRGDY(OEt)RC (FMP) were synthesized and characterized as reported in our previous publications.11,12
Preparation of surfaces, VBP-decorated constructs and platelets for binding experiments
The platelet-adhesive role of VWF is physiologically and pathologically facilitated by shear-induced ‘conformational unraveling’ of globular VWF under a blood flow environment to expose the GPIbα-binding A1 domain (Fig. 1). It has been reported that such platelet-adhesive conformational changes in VWF can also be achieved under static incubation conditions by treating VWF with the antibiotic ristocetin.17,18 Hence in our experiments, we have used ristocetin-treated VWF adsorbed on a glass coverslip surface as our ‘test’ substrate, versus untreated VWF as control. Additionally, we have used BSA-coated coverslips as a second control surface, since albumin is known to have no specific adhesive activity towards platelets or VBP.19–21 For blocking ristocetin-treated VWF’s exposed A1 domain, we used glycocalicin, which is the carbohydrate-rich extramembranous portion of platelet GPIbα.22,23 Since it is the GPIbα that undergoes binding to VWF’s A1 domain, we rationalized that pre-treatment with soluble excess glycocalicin will effectively inhibit this specific binding to the A1 domain while still allowing bioactivity of the other domains of VWF. Our rationale is strengthened by reports of using glycocalicin as a viable substitute for recombinant GPIbα in the VWF:ristocetin cofactor assay.24 Exposure of platelets and VBP-decorated constructs to VWF without glycocalicin pre-treatment was used as the control condition. For all platelet-based experiments, platelets were isolated using serial centrifugation from human whole blood collected via venipuncture from aspirin-refraining healthy donors using institution-approved protocols. The platelets were stained with calcein (λex = 495 nm, λem = 515 nm, green fluorescence) to enable imaging of their binding to experimental surfaces. For all construct-based experiments, VBP-decorated as well as VBP–FMP-co-decorated lipid–peptide nanoconstructs were formed using reverse-phase evaporation and extrusion, resulting in ~150 nm diameter (confirmed by dynamic light scattering) vesicles bearing 5 mol% VBP (for single decoration) or 2.5 mol% of VBP and FMP each (for co-decoration). To enable imaging of constructs, DHPE–rhodamine (λex = 540 nm, λem = 625 nm, red fluorescence) was incorporated in the construct membrane at 1 mol% during fabrication.
Binding studies with platelets on VWF surface
FVIII-free VWF (10 μg ml−1 in 1× PBS pH 7.4) was adsorbed on glass coverslips by incubating overnight at 4 °C. The VWF-adsorbed surfaces were exposed to incubation with calcein-stained (green) platelet suspension (2 × 106 platelets per μl in 1% BSA/1× PBS, pH 7.4) in the presence of ristocetin (1 mg ml−1 in 1× PBS, pH 7.4) for 1 h at room temperature. In control experiments, similar VWF-adsorbed surfaces were first exposed to incubation with soluble glycocalicin (1 μg ml−1 in 1× PBS, pH 7.4) in the presence of ristocetin (1 mg ml−1 in 1× PBS, pH 7.4) for 1 h at room temperature; the surfaces were then gently washed with PBS and subsequently incubated with the platelet suspension. In additional control experiments, the VWF-adsorbed surfaces were not subjected to ristocetin treatment but exposed to incubation with the platelet suspension with or without prior exposure to glycocalicin. In another control experiment, BSA-coated coverslip surfaces were exposed to ristocetin followed by incubation with the platelet suspension. In all these experiments, the platelets were not deliberately activated with ADP, since the tethering interactions between platelet GPIbα and VWF A1 domain can occur for ‘resting’ platelets marginating to a vascular injury site.25,26 The coverslips were mounted onto glass microscope slides, imaged with a Zeiss Axio Observer.D1 inverted fluorescence microscope fitted with a photometrics chilled CCD camera, and the degree of platelet binding was quantified by surface averaged intensity analysis for calcein (green) fluorescence.
Binding studies with VBP-decorated constructs on VWF surface
The VBP-decorated constructs, at a concentration of 2 × 106 particles per μl in 1× PBS, pH 7.4, were incubated with FVIII-free VWF-adsorbed surfaces with and without ristocetin treatment. Also, in comparison studies, the construct incubation with the ristocetin-treated VWF surface was carried out after exposing the surface to glycocalicin pre-incubation. In control experiments, VBP-decorated constructs were incubated with BSA-coated surfaces, and undecorated constructs were incubated with VWF-adsorbed surfaces in the presence of ristocetin. The VBP we used is derived from the C2 domain (residues 2303–2332) of FVIII.14,15 Physiologically, VWF-bound FVIII is cleaved by thrombin at the Arg372 and Arg740 positions in the heavy chain A1–A2 domains27,28 and at the Arg1689 position of the light chain (C2-containing) domain of FVIII29,30 to release the A3–C1–C2 based activated FVIIIa fragment. Based on these reports, we rationalized that the VBP-mediated binding of constructs to VWF should remain unaffected by thrombin. To test this, VBP-decorated rhodamine-labeled (red) constructs were allowed to adhere to ristocetin-treated VWF-adsorbed surfaces, and then the surfaces were exposed to incubation with thrombin (0.067 μg ml−1 in 1× PBS, pH 7.4) for 1 h at room temperature. Fluorescence from VWF-bound constructs was imaged prior to and after thrombin exposure and quantified by intensity analysis. In a separate group of experiments, all the above studies with VBP-decorated constructs were carried out using surfaces adsorbed with physiological VWF instead of FVIII-free VWF to test our hypothesis component that the physiological VWF is capable of binding to the VBP-decorated constructs. As before, the construct-binding was imaged by fluorescence microscopy and quantified by intensity analysis.
Studies involving simultaneous binding of constructs and platelets on VWF
For these experiments, the VWF-adsorbed surfaces were treated with ristocetin and then exposed to incubation with calcein-stained (green) platelets and rhodamine-labeled (red) VBP-decorated constructs, simultaneously. For control conditions, the VWF-adsorbed ristocetin-treated surfaces were incubated with platelets and unmodified (no VBP decoration) constructs, simultaneously. A further component of our hypothesis was to study whether co-decoration of VBP-decorated constructs with fibrinogen-mimetic peptides (FMPs) enables the VWF-adhered constructs to promote amplified aggregation of active platelets via platelet GPIIb–IIIa interaction with the FMPs (shown in Fig. 2). We have previously demonstrated that FMP-decorated biotinylated constructs pre-adhered on avidin-coated surfaces can promote enhanced aggregation of active platelets, while without FMP-decoration or without platelet activation such aggregation was minimal.12 Building on these prior studies, constructs co-decorated with VBP and FMP peptides (VBP–FMP-liposomes) were incubated simultaneously with platelets on VWF-adsorbed ristocetin-treated surfaces. We rationalized that if the constructs undergo VBP-mediated adhesion on the VWF surfaces and, in effect, promote FMP-mediated enhanced aggregation of active platelets on them, then this will be exhibited by the significant overlap between construct fluorescence (red) and platelet fluorescence (green). Comparison with experimental results from incubation of platelets with constructs that bear VBP-decoration only but not FMP co-decoration was regarded as a control. Also for comparison purposes, in one group of these experiments, the platelets were not activated by any external addition of agonists, while in another group the platelets were pre-activated by agonist (ADP) treatment. The rationale for such experimental design was that, for the ‘predominantly unactivated platelet’ group, some of the platelets will effectively bind naturally to VWF’s A1 domain, and this binding will activate these platelets resulting in secretion of agonists (e.g. ADP) to activate more platelets locally. These locally activated platelets can then undergo direct binding to VWF’s C1–C2 domain as well as undergo FMP-mediated enhanced aggregation onto the VWF-adhered constructs. In comparison, for the ‘significantly pre-activated platelet’ group, the pre-existing population of activated platelets can possibly undergo amplified aggregation promoted by the VBP–FMP-constructs. Aggregation of pre-activated platelets on VWF in the presence of ristocetin but with unmodified constructs (no VBP and FMP decoration) was also included for comparison in this group. As before, the binding of constructs and platelets was imaged and the overall platelet recruitment/aggregation (green fluorescence) was quantified by fluorescence intensity analysis.
Statistical analysis
All fluorescence data of construct binding and platelet adhesion were quantified as surface-averaged fluorescence intensity. Student’s t-test was used to analyze the difference between two means. All other statistical analyses between multiple groups were performed using a one-way ANOVA with Tukey method. In all analyses, significance was considered to be p < 0.05.
Results
Binding of platelets on VWF surfaces
Fig. 3 shows representative fluorescence images as well as the quantitative data for binding of calcein-stained (green) platelets to FVIII-free VWF in the presence or absence of ristocetin treatment and additional presence or absence of glycocalicin pre-incubation. As evident from the results, ristocetin treatment led to a significant increase in platelet adhesion to VWF compared to conditions without ristocetin. However, this adhesion was significantly reduced when glycocalicin pre-incubation was used on the ristocetin-treated VWF-surface prior to platelet incubation. In fact, this reduced adhesion was found to be statistically similar to that of platelet adhesion to VWF without ristocetin or platelet adhesion to the control BSA surface. These results suggest that incubation with glycocalicin was effective in specifically blocking the A1-domain of VWF to cause significant reduction of platelet adhesion.
Fig. 3.
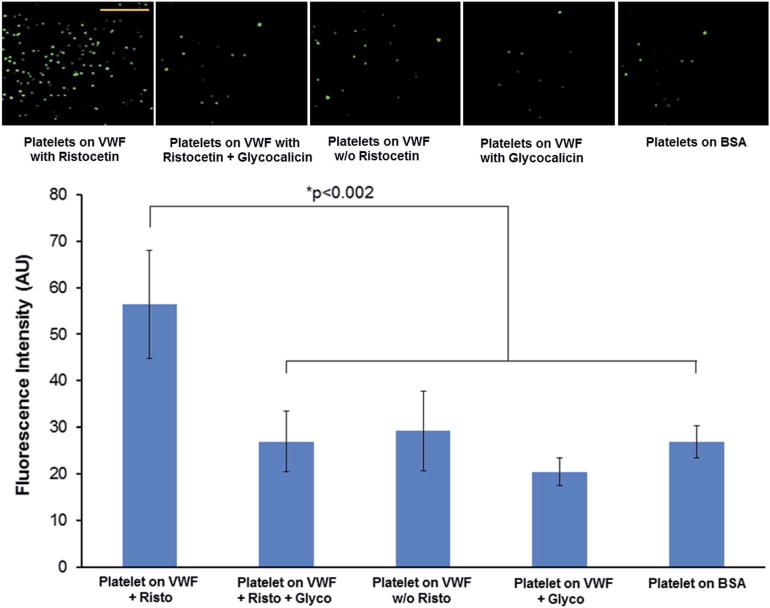
Representative fluorescence microscopy images (scale bar 100 μm) and quantitative fluorescence intensity data of interaction of calceinstained (green) platelets to glass coverslip-adsorbed FVIII-free VWF in the presence or absence of ristocetin (Risto) treatment with additional presence or absence of glycocalicin (Glyco) pre-incubation. Platelets were found to significantly bind to Risto treated VWF compared to binding in the absence of Risto; the platelet-binding to Risto-treated VWF was significantly reduced by pre-incubation with Glyco (p < 0.002) and this reduction was comparable to low platelet-binding on the negative control BSA surface.
Binding of VBP-decorated liposomal constructs on VWF surfaces
Fig. 4 shows representative fluorescence micrographs as well as the quantitative data for the binding of VBP-decorated constructs to FVIII-free VWF in the presence or absence of ristocetin treatment and additional presence or absence of glycocalicin preincubation. As evident from the results, the presence or absence of glycocalicin pre-incubation had no drastic effects on the binding of VBP-decorated constructs (red) to VWF. The binding of the constructs was only slightly lowered (statistically not significant) if the VWF-adsorbed surfaces were not first treated with ristocetin. In contrast, the binding of the VBP-decorated constructs was significantly reduced on the negative control BSA surface, and this reduced level was similar to that of undecorated constructs on the VWF-adsorbed surface. Comparison of results in Fig. 4 and 3 demonstrates that glycocalicin can significantly reduce platelet-adhesion to ristocetin-treated VWF but does not affect the binding of VBP-decorated constructs to VWF under the same conditions. This suggests that the VBP possibly interacts with a VWF domain that is different from the platelet GPIbα-binding A1 domain.
Fig. 4.
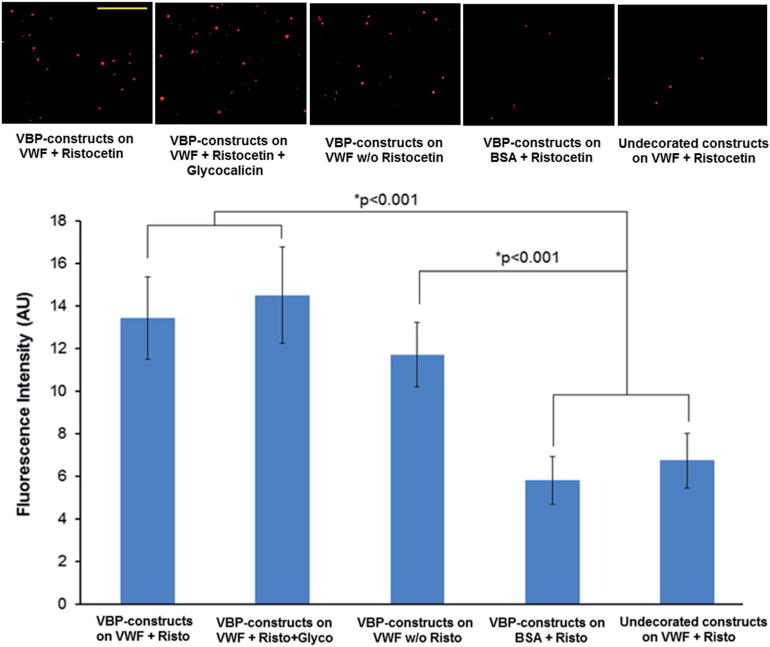
Representative fluorescence microscopy images (scale bar 100 μm) and quantitative fluorescence intensity data of interaction of rhodamine-labeled (red) VBP-decorated constructs to glass coverslip-adsorbed FVIII-free VWF in the presence or absence of ristocetin (Risto) treatment with additional presence or absence of glycocalicin (Glyco) pre-incubation. The constructs were found to substantially bind to VWF even when Glyco pre-incubation (VWF A1 blocking) was used, and this binding was significantly reduced (p < 0.001) only when the VBP-decorated constructs were exposed to BSA or undecorated constructs were exposed to the VWF surface. The constructs showed only slightly lower binding to VWF without Risto treatment.
From additional experiments, Fig. 5A shows the comparison of fluorescence intensity results of VWF surface-adhered VBP-decorated constructs prior to and after exposure to thrombin. As evident from the results, the fluorescence intensity before and after thrombin-exposure remains statistically unchanged, suggesting that VBP binding (hence construct binding) to the VWF was not cleaved by thrombin. This is important to ensure that the construct adhesion is stable under the locally relevant presence of thrombin. From further experiments, Fig. 5B shows the fluorescence intensity results for VBP-decorated constructs binding to FVIII-free VWF compared to physiologic VWF adsorbed on glass coverslips in the presence of ristocetin. As evident from the results, there was no statistical difference in construct binding between the two VWF scenarios, suggesting that VBP-decorated constructs will remain capable of effectively binding to physiological VWF in vivo.
Fig. 5.
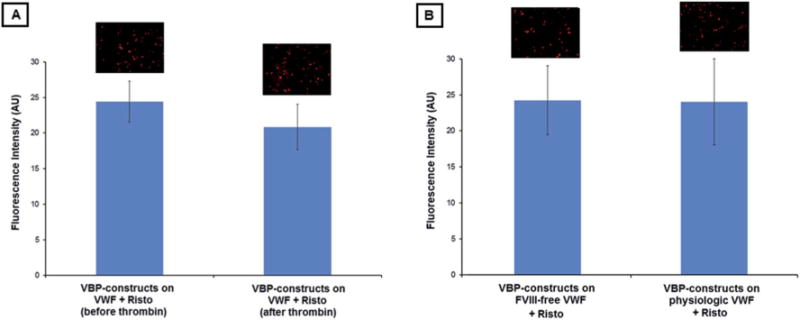
(A) Representative fluorescence microscopy images and quantitative fluorescence intensity data of VBP-decorated liposomal constructs bound to Risto-treated VWF before and after thrombin exposure. (B) Representative fluorescence microscopy images and quantitative fluorescence intensity data of VBP-decorated liposomal constructs bound to Risto-treated FVIII-free VWF versus physiological VWF. No statistical difference was observed in either case.
Simultaneous binding of peptide-decorated constructs and platelets on VWF surfaces
Fig. 6 A1-to-D3 show representative fluorescent images from these studies along with schematic depictions of envisioned interactive mechanisms of the red liposomal constructs and green platelets on VWF. Fig. 6E shows quantitative fluorescence intensity data of platelet fluorescence (calcein green fluorescence) as a measure of overall platelet recruitment and aggregation on the VWF surface when co-incubated with the various test and control liposomal constructs. The co-localization of red constructs and green platelets is shown in pseudocolor yellow (in Fig. 6 A3, B3, C3 and D3). The control conditions included constructs without any peptide decoration (unmodified). As evident from the results, without VBP-decoration, the liposomal constructs could not substantially bind to the VWF surface (minimum red fluorescence in A1), but the platelets could themselves naturally bind to the VWF surface (green fluorescence in A2). This resulted in minimum co-localization of red and green fluorescence (yellow in A3). Incubation of the VBP-decorated constructs (red) and platelets (green) on ristocetin-treated VWF-adsorbed surfaces resulted in their concomitant binding on the surface without mutual interference (B1 and B2) with minimal co-localization (yellow in B3). When the constructs were co-decorated with VBP and FMP motifs, their incubation with platelets on the VWF-adsorbed surface demonstrated a slight increase of the overall platelet recruitment and aggregation on the surfaces (increased platelet fluorescence shown by the third bar in 6E), and the corresponding images showed slight enhancement of yellow overlap (C3) indicating increased co-localization of red and green fluorescence (C1 and C2). These results were obtained with platelets isolated from freshly drawn whole blood via serial centrifugation but without deliberate pre-activation by ADP. We have previously shown by flow cytometry analyses that such freshly prepared platelet suspensions still have ~20–25% of the platelets activated, possibly due to blood draw and storage.31 Therefore, we rationalize that the slight enhancement in platelet recruitment/aggregation is a cumulative result of these low percentages of pre-active platelets binding directly to the C-domain of VWF as well as to the FMP ligands co-decorated on the surface of the VWF-adhered VBP-decorated constructs (schematic shown in the image panel), plus, the binding of a small number of platelets that may get locally activated due to action of agonists secreted by the VWF-adherent platelets themselves. In comparison, when the platelets were deliberately pre-activated by ADP treatment prior to incubation with peptide-modified constructs on ristocetin-treated VWF-adsorbed surfaces, the overall platelet fluorescence (fifth bar in 6E) and the corresponding co-localization (yellow in D3) of constructs (red, D1) and platelets (green, D2) were found to be significantly enhanced. This enhancement was also statistically higher than when such ADP-activated platelets were incubated with unmodified (no VBP and FMP decoration) constructs (fourth bar in 6E). These results suggest that in the presence of pre-activated platelets, the VBP-decorated constructs do not interfere with platelet binding to VWF but rather amplify recruitment/aggregation of the active platelets as a cumulative effect of the platelets directly binding to VWF’s C-domains as well as significantly binding to the FMP ligands co-decorated on the construct surface (schematic shown in fluorescence image panels of Fig. 6). Therefore, the ‘primary hemostasis’ processes of platelet adhesion and aggregation can be efficiently mimicked and amplified by our platelet-inspired nanoconstruct design, possibly by the mechanism depicted in Fig. 2.
Fig. 6.
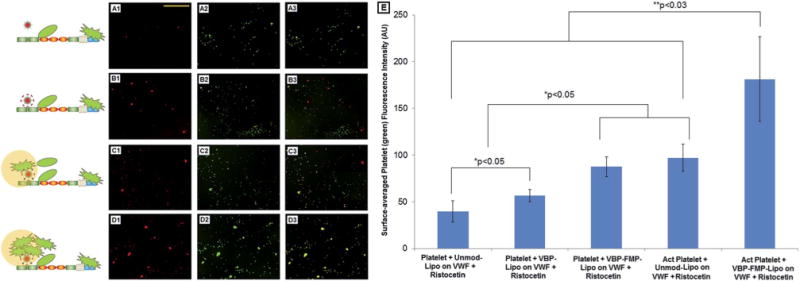
(A1–D3) Representative fluorescence microscopy images (along with envisioned mechanistic schema) of peptide-decorated rhodamine-labeled (red) constructs and calcein-stained (green) platelets incubated simultaneously on Risto-treated VWF adsorbed on glass coverslips. (E) Quantitative overall fluorescence intensity data of platelets (green) adhered and aggregated on the VWF-adsorbed coverslips. A1, B1, C1 and D1 represent construct binding; A2, B2, C2 and D2 represent platelet binding; A3, B3, C3 and D3 represent merged results to exhibit co-localization in yellow. The conditions tested were undecorated (Unmod-Lipo), VBP-decorated (VBP-Lipo) and VBP–FMP-co-decorated (VBP–FMP-Lipo) liposomal nanoconstructs incubated with predominantly inactive platelets (Platelet) and ADP-activated platelets (Act Platelet). Undecorated constructs showed minimal VWF-binding and platelet co-localization, VBP-decorated constructs showed concomitant VWF-binding with platelets but minimal platelet co-localization, and VBP–FMP-co-decorated constructs showed substantial VWF-binding as well as platelet co-localization, especially if platelets were already in a pre-activated state.
Discussion
Hemostasis is a complex multi-step process involving platelet margination, adhesion, activation and aggregation (primary hemostasis), coagulation processes on an adhered active platelet membrane (secondary hemostasis), and subsequent spatiotemporal regulation of clot retraction. Design of platelet-inspired synthetic hemostats should aim at adapting various functional components of these natural phenomena. To this end, several nano- and microscale design approaches are investigating (i) surface-modification of synthetic particles with platelet aggregation-promoting fibrinogen and fibrinogen-derived peptides, (ii) collagen or VWF-adhesion promoting recombinant glycoprotein moieties, (iii) encapsulation of platelet agonists and coagulation promoters within particles, and (iv) fabrication of particles with platelet-mimetic physico-mechanical properties that allow platelet-mimetic margination.10 While these approaches have been mutually independent, for an optimized design of platelet-inspired synthetic hemostat, several of these components may potentially need to be integrated. In this aspect, a crucial component is the integration of the ‘adhesion-promoting’ and ‘aggregation-promoting’ components on a single particle. Past strategies to achieve this by co-decorating a particle surface with adhesion- and aggregation-promoting recombinant protein moieties have indicated difficulties stemming from mutual steric interference between the moieties due to their large size.11,32 In our research, we have been able to resolve this issue by co-decorating a particle surface heteromultivalently with adhesion- and aggregation-promoting small molecular weight peptides that do not have mutual steric interference.11–13 Compared to particles that bear only adhesion-promoting or only aggregation-promoting moieties, our ‘functionally integrated’ design that combines adhesion- and aggregation-promoting functionalities has indicated a statistically enhanced capability of hemostasis in a mouse tail transection model.13 Building on these studies, we are presently focused on establishing a molecular scale mechanistic model for the hemostatic action of our platelet-inspired constructs. As a first step towards this in the current study, we have investigated whether our VWF-binding peptide (VBP) is capable of promoting construct adhesion on VWF without interfering with the natural platelet interaction to VWF’s A1 domain via platelet GPIbα. Our results indicate that even when VWF’s A1 domain is significantly ‘blocked’ by treatment with glycocalicin (resulting in significant reduction of natural platelet adhesion), the VBP-decorated constructs remain capable of binding to VWF, possibly via a different VWF domain. Furthermore, in the absence of glycocalicin-based ‘blocking’ of the A1 domain, the VBP-decorated constructs and platelets remain capable of simultaneously binding to VWF without mutual interference. In addition, when the VBP-decorated constructs were co-decorated with active platelet GPIIb–IIIa-binding FMPs, the constructs were capable of cumulatively increasing the recruitment and aggregation of active platelets on the VWF-adsorbed surfaces.
At the molecular level, the VBP itself has only moderately high affinity to VWF, as indicated by its IC50 value of ~9 μM for inhibiting FVIII binding to VWF.14,15 However, decoration of multiple copies of this peptide on a nano- or microparticle surface is expected to significantly enhance the overall affinity of the particles to VWF. Such overall affinity enhancement via multi-copy decoration of ligands on nanoparticles has been reported for a variety of surface-engineered nanoparticle designs,33–35 we rationalize that optimization of VBP decoration density on our nanoconstructs will render a similar enhancement of VWF-binding of our constructs. In the current studies we have utilized only one fixed molar composition of VBP in surface-decorating the nanoconstructs, since the focus of this study was to investigate whether VBP-decoration allows construct binding to VWF without interfering with natural platelets. In future studies, the VBP-decoration density will be varied and correlated with overall construct binding affinity to VWF utilizing established surface plasmon resonance (SPR) techniques.36 It is also interesting to note that the binding of the VBP-decorated constructs to VWF adsorbed on glass coverslips showed no statistical difference between ristocetin-treated versus ristocetin-untreated conditions, whereas a significant difference in platelet-binding was noted between those two conditions. This is indicative of the possibility that even without ristocetin treatment, the incubation and adsorption of VWF onto glass slides renders some conformational exposure of VWF that may allow interaction with VBP-binding regions but not sufficient conformational changes to allow substantial exposure of the platelet GPIbα binding A1 domain. Such possibility can be further rationalized from the fact that when VBP-liposomes or unmodified liposomes are exposed to soluble VWF without ristocetin and allowed to flow over collagen-coated surfaces at low shear (<10 dyn cm−2), only minimal binding of the VBP-liposomes on the collagen-coated surface is observed (cf. ESI†). Also, the adhesion of the VBP-decorated constructs on VWF surfaces was comparable between FVIII-free VWF and physiologic VWF and was unaffected by thrombin. Since the VBP is derived from residues 2303–2332 of the C2 domain of FVIII that does not contain the thrombin-binding Arg1689 site and since physiologically FVIII binds to VWF’s D′–D3 domain, we rationalize that the VBP-decorated constructs bind to VWF’s D′–D3 domain without interfering with the platelet-binding A1 domain. Future studies will be directed at validating this molecular model rationale by utilizing D′–D3-domain specific and A1-domain specific antibodies to VWF. Altogether, our results indicate substantial promise of utilizing the VBP-peptide to promote VWF adhesion of platelet-inspired nanoconstructs towards efficient design of synthetic platelet analogs. The VBP-decorated design may also be potentially used to develop vehicles that can actively target vascular pathology sites rich in endothelium- and platelet-secreted VWF for drug delivery.37
Supplementary Material
Acknowledgments
The research was supported by NIH Grant Number 1R01HL121212-01 (Sen Gupta). Modery-Pawlowski is supported by NSF GRFP Grant Number DGE-0951783.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c3nr06400j
References
- 1.Denis C, Lenting PJ. Int J Hematol. 2012;95:353–361. doi: 10.1007/s12185-012-1041-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Meyer SF, Deckmyn H, Vanhoorelbeke K. Blood. 2009;113:5049–5057. doi: 10.1182/blood-2008-10-165621. [DOI] [PubMed] [Google Scholar]
- 3.Ruggeri ZM, Mendolicchio GL. Circ Res. 2007;100:1673–1685. doi: 10.1161/01.RES.0000267878.97021.ab. [DOI] [PubMed] [Google Scholar]
- 4.Weibel ER, Palade GE. J Cell Biol. 1964;23:101–112. doi: 10.1083/jcb.23.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luo GP, Ni B, Yang X, Wu YZ. Acta Haematol. 2012;128:158–169. doi: 10.1159/000339426. [DOI] [PubMed] [Google Scholar]
- 6.Lopez JA, Dong JF. Semin Hematol. 2004;41:15–23. doi: 10.1053/j.seminhematol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 7.Siediecki CA, Lestini BJ, Kottke-Marchant K, Eppell SJ, Wilson DL, Marchant RE. Blood. 1996;88:2939–2950. [PubMed] [Google Scholar]
- 8.Savage B, Sixma JJ, Ruggeri ZM. Proc Natl Acad Sci U S A. 2002;99:425–430. doi: 10.1073/pnas.012459599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan H, Deng N, Zhang S, Cao Y, Wang Q, Liu X, Zhang Q. J Hematol Oncol. 2012;5:65–75. doi: 10.1186/1756-8722-5-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Modery-Pawlowski CL, Tian LL, Pan V, McCrae KR, Mitragotri S, Sen Gupta A. Biomaterials. 2013;34:526–541. doi: 10.1016/j.biomaterials.2012.09.074. [DOI] [PubMed] [Google Scholar]
- 11.Ravikumar M, Modery CL, Wong TL, Dzuricky MJ, Sen Gupta A. Bioconjugate Chem. 2012;23:1266–1275. doi: 10.1021/bc300086d. [DOI] [PubMed] [Google Scholar]
- 12.Ravikumar M, Modery CL, Wong TL, Sen Gupta A. Biomacromolecules. 2012;13:1495–1502. doi: 10.1021/bm300192t. [DOI] [PubMed] [Google Scholar]
- 13.Modery-Pawlowski CL, Tian LL, Ravikumar M, Wong TL, Sen Gupta A. Biomaterials. 2013;34:3031–3041. doi: 10.1016/j.biomaterials.2012.12.045. [DOI] [PubMed] [Google Scholar]
- 14.Nogami K, Shima M, Nakai H, Tanaka I, Suzuki H, Morichika S, Shibata M, Saenko EL, Scandella D, Giddings JC, Yoshioka A. Br J Haematol. 1999;107:196–203. doi: 10.1046/j.1365-2141.1999.01673.x. [DOI] [PubMed] [Google Scholar]
- 15.Nogami K, Shima M, Giddings JC, Takeyama M, Tanaka I, Yoshioka A. Int J Hematol. 2007;85:317–322. doi: 10.1532/IJH97.06192. [DOI] [PubMed] [Google Scholar]
- 16.Bendetowicz AV, Morris JA, Wise RJ, Gilbert GE, Kaufman RJ. Blood. 1998;92:529–538. [PubMed] [Google Scholar]
- 17.Dong JF, Berndt MC, Schade A, McIntire LV, Andrews RK, Lopez JA. Blood. 2001;97:162–168. doi: 10.1182/blood.v97.1.162. [DOI] [PubMed] [Google Scholar]
- 18.Sadler JE. Blood. 2010;116:155–156. doi: 10.1182/blood-2010-04-276394. [DOI] [PubMed] [Google Scholar]
- 19.Keogh JR, Velander FF, Eaton JW. J Biomed Mater Res. 1992;26:441–456. doi: 10.1002/jbm.820260403. [DOI] [PubMed] [Google Scholar]
- 20.Balakrishnan S, Latour RA. Langmuir. 2012;28:2745–2752. doi: 10.1021/la204777x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang C, Jina J, Zhaoa J, Jianga W, Yin J. Colloids Surf, B. 2013;102:45–52. doi: 10.1016/j.colsurfb.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 22.Clemetson KJ, Naim HY, Lüscher EF. Proc Natl Acad Sci U S A. 1981;78:2712–2716. doi: 10.1073/pnas.78.5.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okamura I, Lombart C, Jamieson GA. J Biol Chem. 1976;251:5950–5955. [PubMed] [Google Scholar]
- 24.Vanhoorelbeke K, Pareyn I, Schlammadinger A, Vauterin S, Hoylaerts MF, Arnout J, Deckmyn H. Thromb Haemostasis. 2005;93:165–171. doi: 10.1160/TH04-04-0402. [DOI] [PubMed] [Google Scholar]
- 25.Reininger AJ, Heijnen HFG, Schumann H, Specht HM, Schramm W, Ruggeri ZM. Blood. 2006;107:3537–3545. doi: 10.1182/blood-2005-02-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varga-Szabo D, Pleines I, Nieswandt B. Arterioscler, Thromb, Vasc Biol. 2008;28:403–412. doi: 10.1161/ATVBAHA.107.150474. [DOI] [PubMed] [Google Scholar]
- 27.Nogami K, Zhou Q, Wakabayashi H, Fay PJ. Blood. 2005;105:4362–4368. doi: 10.1182/blood-2004-10-3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wakabayashi H, Griffiths AE, Fay PJ. J Biol Chem. 2010;285:25176–25184. doi: 10.1074/jbc.M110.106906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nogami K, Shima M, Hosokawa K, Nagata M, Koide T, Saenko EL, Tanaka I, Shibata M, Yoshioka A. J Biol Chem. 2000;275:25774–25780. doi: 10.1074/jbc.M002007200. [DOI] [PubMed] [Google Scholar]
- 30.Myles T, Yun TH, Leung LLK. Blood. 2002;100:2820–2826. doi: 10.1182/blood-2002-03-0843. [DOI] [PubMed] [Google Scholar]
- 31.SenGupta A, Huang G, Lestini BJ, Sagnella S, Kottke-Marchant K, Marchant RE. Thromb Haemostasis. 2005;93:106–114. doi: 10.1160/TH04-06-0340. [DOI] [PubMed] [Google Scholar]
- 32.Okamura Y, Handa M, Suzuki H, Ikeda Y, Takeoka S. J Artif Organs. 2006;9:251–258. doi: 10.1007/s10047-006-0345-0. [DOI] [PubMed] [Google Scholar]
- 33.Hong S, Leroueil PR, Majoros IJ, Orr BG, Baker JR, Jr, Holl MMB. Chem Biol. 2007;14:107–115. doi: 10.1016/j.chembiol.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 34.Tassa C, Duffner JL, Lewis TA, Weissleder R, Schreiber SL, Koehler AN, Shaw SY. Bioconjugate Chem. 2010;21:14–19. doi: 10.1021/bc900438a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reulen SWA, Dankers PYT, Bomans PHH, Meijer EW, Merkx M. J Am Chem Soc. 2009;131:7304–7312. doi: 10.1021/ja807723p. [DOI] [PubMed] [Google Scholar]
- 36.Canovi M, Lucchetti J, Stravalaci M, Re F, Moscatelli D, Bigini P, Salmona M, Gobbi M. Sensors. 2012;12:16420–16432. doi: 10.3390/s121216420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sen Gupta A. Nanomedicine. 2011;7:763–779. doi: 10.1016/j.nano.2011.04.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.