

Abstract

Selective inhibitors of individual subfamilies of G protein-coupled receptor kinases (GRKs) would serve as useful chemical probes as well as leads for therapeutic applications ranging from heart failure to Parkinson’s disease. To identify such inhibitors, differential scanning fluorimetry was used to screen a collection of known protein kinase inhibitors that could increase the melting points of the two most ubiquitously expressed GRKs: GRK2 and GRK5. Enzymatic assays on 14 of the most stabilizing hits revealed that three exhibit nanomolar potency of inhibition for individual GRKs, some of which exhibiting orders of magnitude selectivity. Most of the identified compounds can be clustered into two chemical classes: indazole/dihydropyrimidine-containing compounds that are selective for GRK2 and pyrrolopyrimidine-containing compounds that potently inhibit GRK1 and GRK5 but with more modest selectivity. The two most potent inhibitors representing each class, GSK180736A and GSK2163632A, were cocrystallized with GRK2 and GRK1, and their atomic structures were determined to 2.6 and 1.85 Å spacings, respectively. GSK180736A, developed as a Rho-associated, coiled-coil-containing protein kinase inhibitor, binds to GRK2 in a manner analogous to that of paroxetine, whereas GSK2163632A, developed as an insulin-like growth factor 1 receptor inhibitor, occupies a novel region of the GRK active site cleft that could likely be exploited to achieve more selectivity. However, neither compound inhibits GRKs more potently than their initial targets. This data provides the foundation for future efforts to rationally design even more potent and selective GRK inhibitors.
G protein-coupled receptor (GPCR) kinases (GRKs) regulate cell signaling by phosphorylating the third intracellular loop and/or carboxyl terminal tail of active GPCRs, promoting the binding of arrestin and clathrin-mediated endocytosis.1 There are three vertebrate GRK subfamilies: GRK1 (which includes GRK1 and GRK7), GRK2 (GRK2 and GRK3), and GRK4 (GRK4, GRK5, and GRK6).2 The GRK1 and GRK4 subfamilies are more closely related to each other than to GRK2. GRK1 subfamily members are expressed primarily in rod and cone cells, whereas GRK2 and GRK4 subfamily members, except for GRK4, are broadly expressed. These enzymes play a beneficial adaptive role in cells by fine tuning signals through GPCRs and preventing damage from sustained signaling, and their activity may underlie the biased agonism observed at some pharmacologically relevant GPCRs.3 However, excess GRK activity is also highly correlated with disease. Overexpression of GRK2 and GRK5 have been characterized as biomarkers and causative factors in heart failure4 and cardiac hypertrophy,5,6 respectively. Cardiac-specific inhibition of GRK2 through viral-mediated delivery of the carboxyl-terminus of GRK2 (βARKct) effectively restores a normal phenotype in both cellular and animal models of heart failure,7,8 and GRK5 null mice are protected against hypertrophy.5 Thus, orally available and selective small molecule inhibitors of individual GRKs are expected to be of profound clinical importance not only for cardiovascular function but also in essential hypertension,9 Parkinson’s disease, and multiple myeloma.10,11 Compounds that directly or indirectly inhibit GRKs may also be useful in potentiating the activity of drugs that act as agonists at GPCRs.12,13
The development of protein kinase inhibitors is often hindered by a lack of selectivity or poor pharmacokinetic properties. Despite these hurdles, the FDA-approved drug paroxetine was recently shown to be an effective inhibitor of GRK2 with 50-fold selectivity over other GRK subfamilies,14 demonstrating that high selectivity, oral bioavailability, and good pharmacokinetic properties can be achieved in a single GRK inhibitor. Structural analysis demonstrated that the drug binds in the active site of GRK2, stabilizing the enzyme in a relatively closed, ADP bound-like conformation. However, paroxetine and its derivatives reported thus far still have much lower potency against GRKs than “off-target” serotonin transporters,15 emphasizing the need to identify alternative chemical scaffolds. Other selective small molecule inhibitors of GRK2 have been reported in the literature,16,17 but their mechanism of action is not understood. To date, there have been very few reports of GRK5-selective compounds (e.g., ref (18)), and how such molecules might bind to GRK5 has been assessed only via docking studies.
To rapidly identify alternate scaffolds with GRK subfamily selectivity, a collection of known kinase inhibitors assembled by the Structural Genomics Consortium at the University of Oxford was screened for compounds that preferentially increase the melting point (Tm) of either GRK2 or GRK5. Fourteen of these compounds were obtained from GlaxoSmithKline (GSK), of which 13 were confirmed to be GRK inhibitors in phosphorylation assays. Four compounds exhibited nanomolar IC50 values. The compounds can be clustered into three classes on the basis of structural similarity: an indazole class, a pyrrolopyrimidine class, and chemically unrelated compounds (other). The most potent, selective compounds from the indazole and pyrrolopyrimidine classes were cocrystallized with GRK2 and GRK1, respectively, in order to elucidate molecular determinants underlying their binding and selectivity.
Results and Discussion
Hit Identification
Label-free high-throughput screening methodologies such as differential scanning fluorimetry (DSF)19 forego the need to develop novel assay materials and/or reporters specific for individual protein targets. DSF has been successfully employed in prior screens,20 hit characterization and validation,21 selectivity profiling of kinase inhibitors,22 and the characterization of ligand interactions with GRKs.14,15 Thus, to identify scaffolds that selectively target either GRK2 or GRK5, DSF was used to screen a common collection of known protein kinase inhibitors (1002 compounds for GRK2 and 1096 for GRK5) including the GSK Published Kinase Inhibitor Set (PKIS). Compounds were screened at 10 μM concentration. Significant hits in these screens were defined by a ΔTm ≥ 2 °C relative to a DMSO control. Only eight compounds significantly stabilized GRK2, whereas 127 compounds stabilized GRK5 (Supporting Information Tables 1 and 2). All but one of the compounds that stabilized GRK2 also stabilized GRK5, although to a different extent. The pan-kinase inhibitor staurosporine, which served as a positive control in the screen, gave the second largest positive ΔTm for GRK2 (5.4 °C) and the largest for GRK5 (12 °C). Interestingly, five of the seven GRK2 inhibitors belong to a chemically related family of indazole/dihydropyrimidine-containing compounds that were developed by GSK as ROCK inhibitors.23−25 These compounds bear striking structural similarity to paroxetine (Figure 1a). A cluster of pyrrolopyrimidine-containing compounds that were developed as IGF-1R inhibitors26,27 was among the top hits for GRK5 (Figure 1b).
Figure 1.

Small molecule inhibitors of GRKs, ROCK, and IGF-1R. Compounds identified in a DSF screen were classified into three classes: (a) an indazole class of compounds, (b) a pyrrolopyrimidine class, and (c) other compounds. The previously identified inhibitor paroxetine is structurally related to the indazole compounds and is included in panel a for comparison. Shaded boxes illustrate where molecules are different from the reference GSK180736A or GSK2163632A compounds. (d) ROCK inhibitors CHEMBL22528230 and CHEMBL1082820,31 related to the indazole class. (e) IGF-1R inhibitors CHEMBL46455251 and CHEMBL507625,27 related to the pyrrolopyrimidine class.
Biochemical Evaluation
Fourteen compounds (Figure 1a–c), including all GRK2 hits except staurosporine, and the most stabilizing pyrrolopyrimidine-containing compounds were obtained from GSK and retested using DSF on GRKs representing each vertebrate subfamily (GRK1, 2, and 5) (Table 1). Of these compounds, only GW806742X failed to reconfirm. The ability of these compounds to shift the Tm of the related AGC family kinase member protein kinase A (PKA) was also assessed (Table 1). In no case was ΔTm greater for PKA than for one of the tested GRKs. In vitro phosphorylation assays were conducted with each GRK using tubulin and 5 μM ATP as substrates to determine IC50 values (Table 2). The most potent inhibitors, GSK2163632A, GSK180736A, and GSK2110236A, were capable of inhibiting GRK1, GRK2, and GRK5 with log IC50 values of −6.9, −6.6, and −5.5, respectively. ΔTm values were strongly correlated with the potency of inhibition against GRK2 (Figure 2a) and were less strongly correlated in the case of GRK5 (Figure 2b). There was no correlation for GRK1 (Figure 2c). As expected from the DSF data, the GW806742X compound failed to inhibit any of the kinases and was not further considered.
Table 1. Small Molecule Thermostabilization of GRKs and PKA.
| GRK1 | GRK2 | GRK5 | PKA | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| rank/ΔTm (°C)a | Tm (°C) | ΔTm (°C) | Tm (°C) | ΔTm (°C) | Tm (°C) | ΔTm (°C) | Tm (°C) | ΔTm (°C) | ||
| paroxetineb | 20.4 | 0.5 | 45.3 | 7.8 | 26.8 | 0.0 | 50.1 | 4.7 | ||
| indazole class | GSK180736A | GRK2: 3/5.3 | 25.3 | 5.4 | 49.9 | 12.4 | 28.7 | 1.9 | 49.2 | 3.8 |
| GSK299115A | GRK2: 4/4.8 | 23.4 | 3.5 | 48.3 | 10.7 | 28.1 | 1.3 | 49.5 | 4.1 | |
| GSK466317A | GRK2: 8/2.9 | 24.6 | 4.7 | 45.0 | 7.4 | 28.0 | 1.3 | 50.6 | 5.2 | |
| GSK317354A | GRK2: 6/3.7 | 23.2 | 3.3 | 49.0 | 11.4 | 27.3 | 0.6 | 45.9 | 0.5 | |
| GSK270822A | GRK2: 5/3.8 | 24.6 | 4.7 | 47.8 | 10.3 | 29.4 | 2.6 | 51.9 | 6.5 | |
| pyrrolopyrimidine class | GSK2163632A | GRK5: 4/9.2 | 26.3 | 6.4 | 41.4 | 3.8 | 33.0 | 6.3 | 48.5 | 3.1 |
| GSK2110236A | GRK5: 7/8.3 | 18.5 | –1.4 | 40.6 | 3.0 | 32.6 | 5.8 | 46.6 | 1.2 | |
| GSK2220400A | GRK5: 13/6.9 | 28.6 | 8.7 | 38.8 | 1.2 | 30.5 | 3.8 | 45.4 | 0.0 | |
| GSK1713088A | GRK5: 11/7.4 | 39.1 | 19.2 | 44.2 | 6.7 | 40.3 | 13.6 | 45.8 | 0.4 | |
| GSK1326255A | GRK2: 6/3.3 | 25.1 | 5.2 | 42.4 | 4.9 | 29.3 | 2.5 | 48.5 | 3.1 | |
| other | GW693881A | GRK5: 12/6.9 | 24.8 | 4.9 | 40.0 | 2.5 | 27.7 | 0.9 | 47.4 | 2.0 |
| GW416981X | GRK5: 15/6.5 | 24.8 | 4.9 | 40.4 | 2.8 | 28.7 | 2.0 | 47.7 | 2.3 | |
| GSK1007102B | GRK2: 1/5.4 | 26.7 | 6.8 | 49.3 | 11.8 | 28.7 | 2.0 | 52.2 | 6.8 | |
| GW806742X | GRK5: 16/6.5 | 20.3 | 0.4 | 36.4 | –1.1 | 27.1 | 0.4 | 45.6 | 0.2 | |
Rank denotes the rank ordering of the compounds in the primary screen. ΔTm values are relative to the intrinsic melting point of each kinase, determined independently for primary and confirmation screens.
Paroxetine was not included in the primary screen but is included due to its structural similarity to the GSK180736A/indazole cluster of compounds and as a biochemical benchmark.
Table 2. Potency and Selectivity of Inhibitors among the GRKs and PKA.
| GRK1 |
GRK2 |
GRK5 |
PKA | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| log IC50 | foldb | log IC50 | fold | log IC50 | fold | log IC50 | Sseld | |||
| paroxetinea | –3.4 | –5.9 | –3.9 | >−3.3c | 0.085 | GRK2 | ||||
| indazole class | GSK180736A | >−3 | NA | –6.6 | 5.0 | –4.0 | 1.3 | >−3.3 | 0.022 | GRK2 |
| GSK299115A | >−3 | NA | –5.5 | 0.40 | –4.1 | 1.6 | –4.2 | 0.360 | GRK2 | |
| GSK466317A | –3.0 | 0 | –4.5 | 0.040 | –4.4 | 3.2 | –4.9 | 1.001 | NON | |
| GSK317354A | >−3 | NA | –5.6 | 0.50 | –3.2 | 0.20 | –3.7 | 0.109 | GRK2 | |
| GSK270822A | –3.1 | 1 | –4.9 | 0.10 | –4.2 | 2.0 | –4.3 | 0.883 | NON | |
| pyrrolopyrimidine class | GSK2163632A | –6.9 | 3162 | –4.7 | 0.063 | –5.5 | 40 | >−3.3 | 0.199 | GRK1 |
| GSK2110236A | –6.2 | 631 | –4.7 | 0.063 | –5.5 | 40 | >−3.3 | 0.561 | NON | |
| GSK2220400A | –5.0 | 40 | –3.7 | 0.0063 | –5.2 | 20 | >−3.3 | 0.770 | NON | |
| GSK1713088A | –4.9 | 32 | –5.2 | 0.20 | –5.5 | 40 | >−3.3 | 0.968 | NON | |
| GSK1326255A | –3.3 | 1 | –5.1 | 0.16 | –5.6 | 50 | >−3.3 | 0.586 | NON | |
| other | GW693881A | >−3 | NA | –3.7 | 0.0063 | –4 | 1.3 | >−3.3 | 1.005 | NON |
| GW416981X | –3.0 | 0 | >−3 | NA | –4.6 | 5.0 | >−3.3 | 0.330 | GRK5 | |
| GSK1007102B | –4.2 | 6 | –6.0 | 1.3 | –5.5 | 40 | –6.3 | 0.917 | NON | |
| GW806742X | >−3 | NA | >−3 | NA | >-3 | NA | >−3.3 | 1.386 | NON | |
Paroxetine was not in the primary screen but is included as a benchmark to calculate fold changes in potency.
Increase in potency relative to paroxetine.
Compound solubility limited the assay to effectively measure only IC50 values lower than 1 mM (GRK assays) or 0.5 mM (PKA assays); thus, potencies weaker than this are reported as log IC50 ≥ −3 or ≥ −3.3, respectively.
Selectivity is determined by having a Ssel of <0.5; thus, all values ≥0.5 are considered to be nonselective.
Figure 2.

GRK2 and 5 inhibitor potency correlates with ΔTm and buried surface area. Compounds with undetermined IC50 values in Table 2 are omitted. (a) ΔTm of GRK2 is strongly correlated (Pearson’s r = −0.833, P = 0.0004) with potency. (b) ΔTm of GRK5 is also significantly correlated (r = −0.6309, P = 0.0156). (c) ΔTm of GRK1 does not, however, correlate (r = −0.091). (d) Buried surface area of small molecules crystallized in complex with GRK2 is significantly correlated with their potency (r = −0.787, P = 0.0316). In each panel, paroxetine is denoted by a square.
Importantly, some of these inhibitors were highly selective: GSK180736A was ≥400-fold selective for GRK2 over both GRK1 and GRK5, GSK2163632A was 160-fold selective for GRK1 over GRK2, and GSK2163632A and GSK2110236A were 6-fold selective for GRK5 over GRK2. Next, whether PKA could be inhibited by these compounds was assessed. Three indazole class compounds (GSK299115A, GSK466317A, and GSK270822A) had <100 μM IC50 values against PKA, whereas GSK1007102B exhibited a nanomolar IC50 value. Remarkably, neither the most potent and selective indazole (GSK180736A) nor pyrrolopyrimidine (GSK2163632A) compound were able to effectively inhibit PKA at the concentrations tested. However, it should be noted that the assays used to evaluate the GRKs and PKA differ both in substrate (protein vs peptide) and concentration of enzyme and ATP. Estimation of the Ki values by application of the Cheng–Prussoff equation, however, suggests that these compounds are still >100-fold more potent against GRKs than PKA, whose IC50 values were typically too high to be measured. To quantify the selectivity of the compounds more objectively, Ssel values28 were calculated for each compound based on their ability to inhibit GRK1, 2, and 5 and PKA (Table 2). Consequently, the compounds can be characterized as GRK1, GRK2, GRK5, or nonselective (Ssel ≥ 0.5) (Table 2). Although most of the pyrrolopyrimidine inhibitors were nonselective as judged by Ssel, they are probably more aptly described as GRK1 or GRK5 selective because each compound in this class could inhibit either GRK1 or GRK5 more potently than GRK2 and also had no measurable effect on PKA activity.
Structure of the GRK2·GSK180736A–Gβγ Complex
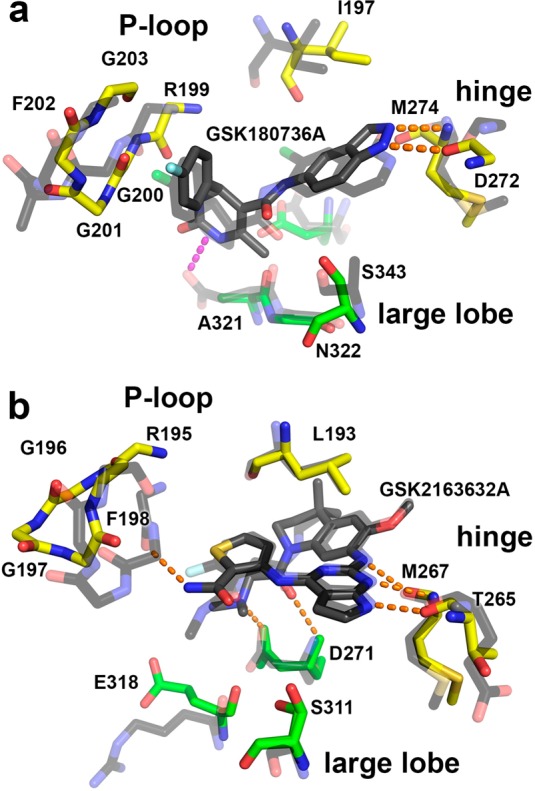
In the 2.56 Å resolution structure of the GRK2·GSK180736A–Gβγ complex (Table 3), GSK180736A binds in the active site of the enzyme in a manner similar to that of paroxetine (PDB entry 3V5W) (Figure 3a,b) and is ordered (average B = 65.7 Å2) approximately as well as the rest of the small lobe (average B = 58.9 Å2). The source compound is probably racemic, but the electron density map clearly favors the S-enantiomer. GSK180736A has an interaction surface of 290 A2, slightly more than that of paroxetine (280 A2) and the benzolactam paroxetine derivative CCG206868 (270 A2). GSK180736A thus confirms the trend that more buried surface area leads to more potent inhibition, at least in GRK2 (Figure 2d). Its indazole ring occupies the adenine subsite in the same manner as the benzodioxole ring of paroxetine, where it forms two conventional hydrogen bonds with backbone atoms in the hinge of the kinase domain, and its fluorophenyl group occupies the polyphosphate subsite in an analogous way to that of paroxetine and CCG206868. The amide linker joining the indazole and dihydropyrimidine forms a hydrogen bond with Ser334, a signature residue characteristic of GRKs among the AGC kinase family.29 The dihydropyrimidine ring of GSK180736A exhibits a large degree of mobility based on its electron density and temperature factors, but it is positioned such that it could form a hydrogen bond with backbone carbonyl of Arg199 in the P-loop and van der Waals interactions with residues in the large lobe (Figure 3a). The AST is not as well ordered in the GSK180736A complex relative to that of paroxetine (PDB entry 3V5W), with only sparse density observed for residues 476–479. This is likely due to structural differences between the central rings of these two inhibitors (Figure 1), which are positioned such that they can make direct contacts with the AST loop.
Table 3. Crystal Refinement Statistics.
| protein complex | GRK2·GSK180736A–Gβγ | GRK1·GSK2163632A |
|---|---|---|
| X-ray source | APS 21ID-F | APS 21ID-F |
| wavelength (Å) | 0.9787 | 0.9787 |
| Dmin (Å) | 25.00–2.59 (2.63–2.59)a | 25.00–1.85 (1.88–1.85) |
| space group | C2221 | P22121 |
| cell constants (Å) | a = 61.4, b = 241.4, c = 212.5 | a = 45.5, b = 66.6, c = 205.5 |
| unique reflections | 51 212 (1843) | 54 333 (2671) |
| Rsym (%) | 20.1 (100) | 10.3 (100) |
| completeness (%) | 100 (100) | 99.9 (99.9) |
| ⟨I⟩\⟨σI⟩ | 18.7 (1.8) | 90 (1.8) |
| redundancy | 26.2 (25.6) | 21.8 (19.5) |
| refinement resolution (Å) | 25.00–2.59 (2.63–2.59) | 25.00–1.85 (1.89–1.85) |
| total reflections used | 47 772 (2677) | 51 503 (3672) |
| RMSD bond lengths (Å) | 0.004 | 0.002 |
| RMSD bond angles (deg) | 0.740 | 1.833 |
| Est. coordinate error (Å) | 0.482 | 0.137 |
| Ramachandran plot | ||
| most favored, outliers (%) | 91.5, 0.6 | 98.4, 0.0 |
| Rwork | 25.1 (39.5) | 21.4 (35.4) |
| Rfree | 27.6 (40.6) | 25.3 (28.5) |
| protein atoms | 8225 | 4025 |
| water molecules | 69 | 218 |
| inhibitor atoms | 27 | 39 |
| average B-factor (Å2) | 58.5 | 48.8 |
| protein | 58.9 | 49.0 |
| inhibitor | 65.7 | 35.9 |
| MolProbity score | 2.18 (94th percentile) | 0.83 (100th percentile) |
| MolProbity Cβ deviations | 0 | 1 |
| MolProbity bad backbone bonds | 0 | 0 |
| MolProbity bad backbone angles | 2 | 3 |
| PDB entry | 4PNK | 4PNI |
Values in parentheses correspond to the highest resolution shell of data.
Figure 3.

Comparison of cocrystal structures of GSK inhibitors with those of paroxetine in complex with GRK1 and GRK2. (a) GRK2·GSK180736A (stick model with dark gray carbons) is well-ordered in the active site. Three σ |Fo| – |Fc| omit map density is shown as a light gray mesh in panels a and c. (b) GSK180736A binds in a manner that is nearly superimposable with paroxetine (black carbons, PDB entry 3V5W). (c) Crystal structure of the GRK1·GSK2163632A complex. (d) Occupation of the AST subsite by GSK2163632A alters the conformation of the AST loop (magenta) compared to that in the GRK1·paroxetine complex (PDB entry 4L9I). Orange dashed lines indicate hydrogen bonds or salt bridges.
There is a high level of sequence conservation within the GRK2 and ROCK1 active sites, as both are members of the AGC kinase family and contain nearly identical residues at signature positions.29 Comparison with cocrystal structures of the indazole derivatives, including that of CHEMBL225282 (PDB entry 3V8S),30 and of the isoquinoline derivatives, CHEMBL1222571 (3NCZ)25 and CHEMBL1082820 (3NDM)31 (Figure 1d), in complex with ROCK1 reveal that they form analogous interactions with the hinge of the kinase domain. Their terminal phenyl groups all interact with the P-loop in an analogous way to that of GSK180736A (Figures 3b and 4a). It is thus reasonable to expect that the closely related GSK466317A, GSK317354A, and GSK299115A compounds identified in the DSF screen would bind in an analogous way to both ROCK1 and GRK2.
Figure 4.
Comparison of GRK·inhibitor complexes with ROCK1 and IR bound to related compounds. (a) Superposition of the GRK2·GSK180736A structure (yellow carbons for residues in small lobe and green carbons for large lobe, GRK2 numbering) with that of ROCK1·CHEMBL1082820 (transparent black carbons) reveals similar hydrogen bonds (orange dashed lines) formed with the hinge in the adenine subsite and similar docking of the halogen-substituted phenyl in the polyphosphate subsite. Numbering corresponds to human GRK2. (b) Superposition of the GRK1·GSK2163632A structure with that of the IR kinase·CHEMBL464552 complex (transparent black carbons) shows excellent alignment with the exception of the P-loop. Numbering corresponds to bovine GRK1.
Structure of the GRK1·GSK2163632A Complex
A crystal structure of GRK5 in complex with one of the screening targets would greatly facilitate rational design efforts. However, to date, no crystal structures of GRK5 have been reported. On the other hand, GRK1 is known to crystallize more readily than GRK5, is more closely related in sequence with GRK5 than GRK2, and has been used previously to assess the interactions of small molecule GRK inhibitors. Therefore, to investigate how pyrrolopyrimidine inhibitors inhibit GRKs, the 1.85 Å resolution crystal structure of the GRK1·GSK2163632A complex was determined (Table 3). The inhibitor binds in the active site such that two of the cyclic amines of its pyrrolopyrimidine form hydrogen bonds with the backbone oxygen and nitrogen of Met267 and Thr265, respectively, in the hinge of the kinase domain (Figure 3c). One of the exocyclic amines of this group forms a third longer hydrogen bond to the backbone oxygen of Met267. The dimethylaminoacetyl arm of the inhibitor forms two more specific interactions with residues just after the hinge: a hydrogen bond with the backbone amide and a salt bridge with the side chain of Asp271. On the other end of the molecule, the thiophenecarboxamide occupies the ribose subsite of the active site where it forms a π-hydrogen bond with the aromatic ring of Phe198 in the P-loop, which adopts an unusual “tucked under” conformation and thereby fills the polyphosphate subsite. Thus, the mode of binding is much different than that of paroxetine and GSK180736A (Figure 3d). The inhibitor also forms extensive van der Waals interactions with residues 474–478 of the AST, which adopt a conformation not previously observed in any GRK1 structure.15,32−34 The inhibitor buries 410 Å2 of solvent-accessible surface area. Because GSK2163632A and its analogues were initially developed as IGF-1R inhibitors, it is not surprising that the conformation of the molecule and its interactions in the active site are analogous to those made by the closely related CHEMBL464552 compound (GSK1838705A, Figures 1e and 4b) with the insulin receptor (IR) kinase domain (PDB entry 3EKK).27 The GRK1 and IR complexes exhibit remarkably similar kinase domain conformations (1.4 Å RMSD for Cα atoms in the kinase domain) despite their low 23% sequence identity. In terms of variable active site residues that directly interact with the inhibitor, they differ only conservatively: GRK1-Ile266, -Leu321, and -Ser331 vs IR-Leu1078, -Met1139, and -Gly1149, respectively. However, the IR kinase domain does not exhibit the same P-loop conformation, perhaps due to differences in the chemical moiety analogous to the thiophenecarboxamide arm in CHEMBL464552 (Figures 1 and 4b). Not being an AGC kinase, the IR kinase domain also lacks an AST element.
GSK2163632A stabilizes one of the most open states of the GRK1 kinase domain yet observed. The complex is most similar to the GRK1·ATP structure (PDB 3C4W) (1.05 Å RMSD for 479 atomic pairs) as determined by the PDBeFold server.35 If the small lobe of the kinase domain (residues 185–268 and 494–511) is used to align these structures, then the large lobe in the GSK2163632A complex is more open by 7°.36 Thus, the selectivity of the pyrrolopyrimidine compounds for the GRK1 or GRK5 is most likely a combination of the ability of the kinase domain to adopt this unusual conformation as well as its ability to interact productively with the unique residues found in the AST.
These two structures and the activity-based data in Table 2 also allow for an assessment of structure–activity relationships (Supporting Information).
Conclusions
From the initial hits in the primary DSF screen, there was a nearly perfect reconfirmation rate (13 out of 14 tested), although the rank order was not the same (Table 1). The high hit rates observed in this study are consistent with increases in the number of inhibitory compounds identified when using full-length kinases as the receptor37 instead of isolated kinase domains38 as well as the fact that the compounds in the set were already optimized for homologous active sites. The Tm shifts strongly correlated with the potencies of inhibition for GRK2 and GRK5 (Figure 2a,b), consistent with a strong correlation between ΔTm and IC50 values reported for other kinases.37,39 Given the high structural similarity among the GRK subfamilies (∼45% identity in the kinase domain), it is not immediately clear why fewer compounds showed temperature shifts for GRK2 compared to GRK5 (8 vs 127 compounds from the primary screen). It may simply reflect that it is harder to identify stabilizing interactions in proteins with intrinsically higher stability. It is notable that ΔTm is less correlated with the IC50 value for GRK1 and GRK5 (Figure 2b,c) than for GRK2 (Figure 2a). This may indicate that some hits are stabilizing GRK1 and GRK5 in ways other than binding in the active site or that when the total range of Tm shifts between strong and weak inhibitors is lower, there is greater error in rank ordering of the compounds and thus a weaker correlation with IC50 value.
Analysis of inhibition of other kinases by the compounds from the indazole class, which are GRK2-selective, reveal that they are fairly selective for AGC family kinase members (cross-screening data for the GSK PKIS compounds against 224 kinases is available in the ChEMBL database). Other inhibited kinases include myotonic dystrophy kinase-related Cdc42-binding kinase (MRCK), mitogen- and stress-activated protein kinase (MSK), ribosomal S6 kinase (RSK), janus kinase (JAK), and Aurora A kinase. Among the indazole compounds, GSK466317A is the most promiscuous, as it inhibits more than 10 other AGC kinases by more than 50% at 1 μM, which implies that the presence of a dihydropyridone ring and trifluoromethyl group leads to a loss of selectivity. The compounds from the pyrrolopyrimidine class had a much broader inhibition pattern, exhibiting potent inhibition against PKC-activated protein kinase D (PRKD), ROCK, RSK, testis-specific serine/threonine kinase (TSSK), CDC-like kinase 2 (CLK2), polo-like kinase 1 (PLK1), TTK protein kinase (TTK), anaplastic lymphoma kinase (ALK), epidermal growth factor receptor (EGFR), transforming growth factor beta type 2 receptor (IIR), insulin receptor tyrosine kinase (INSR), and tousled-like kinase (LTK). This data implies that these diverse groups of kinases must be capable of presenting similarly complementary chemical environments for the pyrrolopyrimidine scaffold and will represent significant off-targets that must be selected against in future rounds of design. The four compounds grouped to the “other” class (GW693881A, GW416981X, GSK1007102B, and GW806742X, Figure 1c) exhibited strong inhibition of many of the 221 kinases evaluated, suggesting that they are pan-kinase inhibitors.
Another advantage of having screened previously reported kinase inhibitors is the existence of experimental data on the selectivity, potency, and structure of these compounds directed against other kinases. The indazole-containing compounds, typified by GSK180736A, confirm a structural linkage between the active sites of GRK2 and ROCK1,29 along with a few other AGC kinase subfamilies based on cross-screening data (Figure 4a). GSK180736A was initially developed as a ROCK1-selective inhibitor with 200-fold higher potency for ROCK1 over other kinases such as RSK1 and p70S6K.24 GRK2 is inhibited 18-fold less potently than ROCK1, although the assay conditions differ. Interestingly, the trifluoromethyl substitution of GSK317354A and the naphthyl substitution of GSK270822A increase potency of ROCK inhibition by 10- and 5-fold, respectively, whereas they dramatically reduced GRK inhibition (Table 2).23 The chemical differences among these compounds most directly impact their interactions with the P-loop, suggesting that optimization of interactions with the P-loop is a route by which selectivity for GRK2 can be gained. This hypothesis is further strengthened when considering inhibition of PKA (Table 2). Namely, the presence of the fluorophenyl moiety of GSK180736A abrogates inhibition of PKA, which is, however, measurable for the closely related GSK466317A, GSK317354A, GSK270822A, and GSK299115A compounds. Superposition of the structures of GRK2 and ROCK in complex with similar inhibitors (GSK180736A and CHEMBL108280) provides a structural explanation for how the P-loop can mediate compound selectivity (Figure 4a). For example, one residue that differs in identity in the P-loop between these enzymes (GRK2-Gly201/ROCK1-Ala86) shows a 2.8 Å movement in the Cα atom positioning in the structure of GRK2·GSK180736A as compared with ROCK1·CHEMBL108280. Thus, maintaining a fluorophenyl moiety in future generations of inhibitors may be key for maintaining GRK2 selectivity over PKA.
The central ring of GSK180736A provides a hydrogen bond to the P-loop, increases the Tm of GRK2 by an additional 4 °C, and likely helps to induce closure of the GRK2 kinase domain by an additional 2° relative to paroxetine. Other potent ROCK inhibitors (Figure 1d) either interact with the side chain of Asp202 (analogous to GRK2-Ala321) or lack a central ring all together, as is the case with CHEMBL225282 (Figure 4b). These data suggest that optimization of the central ring system of GSK180736A in a manner that renders it incompatible with ROCK signature residues may greatly increase the selectivity toward GRK2.
The pyrrolopyrimidine compounds, typified by GSK2163632A, exhibit very strong (>500-fold) selectivity for IGF-1R over GRKs, although once again these values were measured under different assay conditions. However, GSK2163632A in complex with GRK1 is nearly superimposable with the closely related CHEMBL464552 in complex with the insulin receptor (IR) kinase domain (Figure 4b). Occupation of the AST subsite by these drugs may lower their potency toward GRKs because the drug must compete with or remodel the AST loop. Even so, it is clear from the GRK1·GSK2163632A structure that compounds capable of selective interactions with the AST of individual GRKs are a viable avenue for development of more selectivity among GRK subfamilies.
Methods
Protein Purification
Bovine GRK11–535, bovine and human GRK2S670A, and bovine GRK5 were purified via a common procedure consisting of Ni-NTA affinity, Source15S, and tandem S200 size-exclusion chromatography as previously described.14,15,40 A soluble mutant (C68S) of the Gβγ complex was purified by sequential Ni-NTA affinity, MonoQ, and tandem S200 size-exclusion chromatography.15 A bacterial expression plasmid (pET15b) encoding the catalytic subunit of PKA was obtained from Dr. Susan Taylor41 via AddGene (plasmid 14921). Rosetta(DE3)pLysS cells were transformed and grown at 37 °C while shaking at 150 rpm to an optical density of approximately 0.8. Cells were induced by adding IPTG to a final concentration of 200 μM. Expression was then carried out at 20 °C for approximately 20 h. Cells were harvested by centrifugation and lysed via sonication in a buffer containing 20 mM HEPES, pH 7.0, 50 mM NaCl, 5 mM β-mercaptoethanol, and 0.5 mM EDTA. Samples were centrifuged at 40 000g for 1 h to pellet cell debris, and the soluble fraction was loaded on a Ni-NTA column. The column was washed with lysis buffer with 10 mM imidazole, pH 8, added prior to elution with lysis buffer with 200 mM imidazole, pH 8. Eluted protein was concentrated and loaded onto tandem S200 size-exclusion columns to achieve the desired purity.
Primary Inhibitor Screen
Compounds from the PKIS were screened at 10 μM against purified human GRK2S670A and bovine GRK5 at 2 μM as previously reported.22
Kinetic Assays
GRK kinetic assays were conducted in a buffer containing 20 mM HEPES, pH 7.0, 2 mM MgCl2, 0.025% (w/v) n-dodecyl-β-d-maltoside with 50 nM bovine GRK, 5 μM ATP, and 500 nM tubulin in 5 min reactions. The low salt concentration and DDM were used to maximize GRK activity and to disrupt small molecule aggregates from forming, respectively. Reactions were quenched with SDS loading buffer, separated via SDS-PAGE, dried, and exposed with a phosphorimaging screen prior to quantification via Typhoon imager, as previously reported.14,15 The PKA inhibition assays were performed with the ADP-Glo system using 0.1 μg of PKA (438 nM final), 1 μg of CREBtide substrate, and 100 μM ATP for 30 min. After the initial reaction, ADP-Glo reagent was added to the reaction and allowed to incubate for an additional 40 min. Lastly, the kinase detection reagent was added and allowed to incubate for 30 min, and the luminescence was measured with a Pherastar imaging system. All data was analyzed, and inhibition curves were fit via GraphPad Prism.
DSF
DSF was conducted using a ThermoFluor plate reader (Johnson & Johnson) in a buffer containing 20 mM HEPES, pH 7.0, 5 mM MgCl2, 2 mM DTT, and 1 mM CHAPS with 0.2 mg mL–1 final concentration of GRK or PKA and 100 μM 1-anilinonaphthalene-8-sulfonic acid.14
GRK2·GSK180736A–Gβγ Structure Determination
Human GRK2 and Gβγ were mixed in a 1:1 ratio and concentrated to a final total protein concentration of approximately 10 mg mL–1 in the presence of 500 μM GSK180736A (from a 10 mM stock in DMSO) and 2 mM MgCl2. Crystals were obtained via hanging drop vapor diffusion using 0.8 μL of protein solution mixed with 0.8 μL of well solution (1 mL), which consisted of 1.2 M NaCl, 100 mM MES, pH 6.75, and 15% (w/v) PEG3350. Crystals appeared in approximately 1 week and continued to grow in size for several weeks. During harvesting, the crystals were cryoprotected via the addition of 25% (v/v) ethylene glycol to the harvested crystals prior to flash freezing in liquid N2. Diffraction data was collected at the Advanced Photon Source (APS) on LS-CAT beamline ID-F at a wavelength of 0.9787 Å. Data was collected from four sweeps collected from a single crystal. Indexing, integration, and scaling were performed with HKL2000.42 Initial phasing was performed via molecular replacement solution using PDB entry 3V5W as a search model in PHASER.14,43,44 Refinement was performed with the REFMAC5 module of CCP4 alternating with model building in Coot.44−46 The final model was verified with MolProbity.47
GRK1·GSK2163632A Crystal Structure Determination
GSK2163632A (10 mM stock in DMSO) and MgCl2 were added to a concentrated protein stock to achieve final concentrations of 10 mg mL–1 GRK1535, 500 μM inhibitor, and 2 mM MgCl2. Crystals were obtained via hanging drop vapor diffusion using a mixture of 0.8 μL of protein and 0.8 μL of well solution (1 mL), which consisted of 1.0 M NaCl, 100 mM MES, pH 6.0, and 12% (w/v) PEG3350. Crystals appeared in approximately 2 weeks and continued to grow in size for at least a week. During harvesting, the crystals were cryoprotected by addition of 25% (v/v) ethylene glycol to the drops prior to flash freezing in liquid nitrogen. Diffraction data was collected at the Advanced Photon Source (APS) on LS-CAT beamline ID-F at a wavelength of 1.09785 Å. Diffraction data was collected from three total sweeps on two crystals. Indexing, integration, and scaling were performed with HKL2000.42 A molecular replacement solution was achieved with the PHASER module of CCP4 using PDB entry 3C50(33) as a search model. Refinement validation was performed as described above.
Structural Analysis
Buried surface area calculations were performed with the AREAIMOL extension of CCP4.48,49 Structural similarity calculations were performed via the PDBeFold server,35 and domain rotations calculations were performed on isolated kinase domains via the DynDom server.36,50
Acknowledgments
We thank D. Drewry, B. Hardy, and W. J. Zuercher at GlaxoSmithKline for providing samples of the 14 compounds from the Published Kinase Inhibitor Set shown in Figure 1. This work was supported by National Institutes of Health grants [HL071818 and HL086865] to J.J.G.T. and an American Heart Association grant [N014938] to K.T.H. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE-AC02-06CH11357, and use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and Michigan Technology Tri-Corridor Grant [085P1000817]. J.M.E., M.S., and S.K. are supported by the Structural Genomics Consortium, a registered charity (no. 1097737) that receives funds from AbbVie, Bayer, Boehringer Ingelheim, the Canada Foundation for Innovation, the Canadian Institutes for Health Research, Genome Canada, GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and the Wellcome Trust [092809/Z/10/Z].
Supporting Information Available
Description of structure–activity relationships; results from the primary screen. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The atomic model and structure factors for the GRK2·GSK180736A–Gβγ and GRK1·GSK2163632A complexes have been deposited with the Protein Data Bank as entries 4PNK and 4PNI, respectively.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Gurevich E. V.; Tesmer J. J.; Mushegian A.; Gurevich V. V. (2012) G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol. Ther. 133, 40–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mushegian A.; Gurevich V. V.; Gurevich E. V. (2012) The origin and evolution of G protein-coupled receptor kinases. PLoS One 7, e33806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter E.; Ahn S.; Shukla A. K.; Lefkowitz R. J. (2012) Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 52, 179–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhart A. D.; Ozaki T.; Tevaearai H.; Rockman H. A.; Koch W. J. (2002) Vascular-targeted overexpression of G protein-coupled receptor kinase-2 in transgenic mice attenuates β-adrenergic receptor signaling and increases resting blood pressure. Mol. Pharmacol. 61, 749–758. [DOI] [PubMed] [Google Scholar]
- Gold J. I.; Gao E.; Shang X.; Premont R. T.; Koch W. J. (2012) Determining the absolute requirement of G protein-coupled receptor kinase 5 for pathological cardiac hypertrophy: short communication. Circ. Res. 111, 1048–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorriento D.; Santulli G.; Fusco A.; Anastasio A.; Trimarco B.; Iaccarino G. (2010) Intracardiac injection of AdGRK5-NT reduces left ventricular hypertrophy by inhibiting NF-κB-dependent hypertrophic gene expression. Hypertension 56, 696–704. [DOI] [PubMed] [Google Scholar]
- Rockman H. A.; Chien K. R.; Choi D. J.; Iaccarino G.; Hunter J. J.; Ross J.; Lefkowitz R. J.; Koch W. J. (1998) Expression of a β-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc. Natl. Acad. Sci. U.S.A. 95, 7000–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raake P. W.; Schlegel P.; Ksienzyk J.; Reinkober J.; Barthelmes J.; Schinkel S.; Pleger S.; Mier W.; Haberkorn U.; Koch W. J.; Katus H. A.; Most P.; Muller O. J. (2013) AAV6.βARKct cardiac gene therapy ameliorates cardiac function and normalizes the catecholaminergic axis in a clinically relevant large animal heart failure model. Eur. Heart J. 34, 1437–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felder R. A.; Sanada H.; Xu J.; Yu P. Y.; Wang Z.; Watanabe H.; Asico L. D.; Wang W.; Zheng S.; Yamaguchi I.; Williams S. M.; Gainer J.; Brown N. J.; Hazen-Martin D.; Wong L. J.; Robillard J. E.; Carey R. M.; Eisner G. M.; Jose P. A. (2002) G protein-coupled receptor kinase 4 gene variants in human essential hypertension. Proc. Natl. Acad. Sci. U.S.A. 99, 3872–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manago F.; Espinoza S.; Salahpour A.; Sotnikova T. D.; Caron M. G.; Premont R. T.; Gainetdinov R. R. (2012) The role of GRK6 in animal models of Parkinson’s disease and l-DOPA treatment. Sci. Rep. 2, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedemann R. E.; Zhu Y. X.; Schmidt J.; Yin H.; Shi C. X.; Que Q.; Basu G.; Azorsa D.; Perkins L. M.; Braggio E.; Fonseca R.; Bergsagel P. L.; Mousses S.; Stewart A. K. (2010) Kinome-wide RNAi studies in human multiple myeloma identify vulnerable kinase targets, including a lymphoid-restricted kinase, GRK6. Blood 115, 1594–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz K.; Lohse M. J.; Quitterer U. (2003) Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature 426, 574–579. [DOI] [PubMed] [Google Scholar]
- Gainetdinov R. R.; Bohn L. M.; Walker J. K.; Laporte S. A.; Macrae A. D.; Caron M. G.; Lefkowitz R. J.; Premont R. T. (1999) Muscarinic supersensitivity and impaired receptor desensitization in G protein-coupled receptor kinase 5-deficient mice. Neuron 24, 1029–1036. [DOI] [PubMed] [Google Scholar]
- Thal D. M.; Homan K. T.; Chen J.; Wu E. K.; Hinkle P. M.; Huang Z. M.; Chuprun J. K.; Song J.; Gao E.; Cheung J. Y.; Sklar L. A.; Koch W. J.; Tesmer J. J. (2012) Paroxetine is a direct inhibitor of G protein-coupled receptor kinase 2 and increases myocardial contractility. ACS Chem. Biol. 7, 1830–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homan K. T.; Wu E.; Wilson M. W.; Singh P.; Larsen S. D.; Tesmer J. J. (2014) Structural and functional analysis of G protein-coupled receptor kinase inhibition by paroxetine and a rationally designed analog. Mol. Pharmacol. 85, 237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino M.; Furugori T.; Mori T.; Moriyama S.; Fukuzawa A.; Shibano T. (2002) Rational design and evaluation of new lead compound structures for selective betaARK1 inhibitors. J. Med. Chem. 45, 2150–2159. [DOI] [PubMed] [Google Scholar]
- Kassack M. U.; Hogger P.; Gschwend D. A.; Kameyama K.; Haga T.; Graul R. C.; Sadee W. (2000) Molecular modeling of G-protein coupled receptor kinase 2: docking and biochemical evaluation of inhibitors. AAPS PharmSci. 2, E2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S. Y.; Lee B. H.; Jung H.; Yun C. S.; Ha J. D.; Kim H. R.; Chae C. H.; Lee J. H.; Seo H. W.; Oh K. S. (2013) Design and synthesis of novel 3-(benzo[d]oxazol-2-yl)-5-(1-(piperidin-4-yl)-1H-pyrazol-4-yl)pyridin-2-amine derivatives as selective G-protein-coupled receptor kinase-2 and -5 inhibitors. Bioorg. Med. Chem. Lett. 23, 6711–6716. [DOI] [PubMed] [Google Scholar]
- Niesen F. H.; Berglund H.; Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221. [DOI] [PubMed] [Google Scholar]
- DeSantis K.; Reed A.; Rahhal R.; Reinking J. (2012) Use of differential scanning fluorimetry as a high-throughput assay to identify nuclear receptor ligands. Nucl. Recept. Signaling 10, e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genick C. C.; Barlier D.; Monna D.; Brunner R.; Be C.; Scheufler C.; Ottl J. (2014) Applications of biophysics in high-throughput screening hit validation. J. Biomol. Screening 19, 707–714. [DOI] [PubMed] [Google Scholar]
- Fedorov O.; Niesen F. H.; Knapp S. (2012) Kinase inhibitor selectivity profiling using differential scanning fluorimetry. Methods Mol. Biol. 795, 109–118. [DOI] [PubMed] [Google Scholar]
- Goodman K. B.; Cui H.; Dowdell S. E.; Gaitanopoulos D. E.; Ivy R. L.; Sehon C. A.; Stavenger R. A.; Wang G. Z.; Viet A. Q.; Xu W.; Ye G.; Semus S. F.; Evans C.; Fries H. E.; Jolivette L. J.; Kirkpatrick R. B.; Dul E.; Khandekar S. S.; Yi T.; Jung D. K.; Wright L. L.; Smith G. K.; Behm D. J.; Bentley R.; Doe C. P.; Hu E.; Lee D. (2007) Development of dihydropyridone indazole amides as selective Rho-kinase inhibitors. J. Med. Chem. 50, 6–9. [DOI] [PubMed] [Google Scholar]
- Sehon C. A.; Wang G. Z.; Viet A. Q.; Goodman K. B.; Dowdell S. E.; Elkins P. A.; Semus S. F.; Evans C.; Jolivette L. J.; Kirkpatrick R. B.; Dul E.; Khandekar S. S.; Yi T.; Wright L. L.; Smith G. K.; Behm D. J.; Bentley R.; Doe C. P.; Hu E.; Lee D. (2008) Potent, selective and orally bioavailable dihydropyrimidine inhibitors of Rho kinase (ROCK1) as potential therapeutic agents for cardiovascular diseases. J. Med. Chem. 51, 6631–6634. [DOI] [PubMed] [Google Scholar]
- Ginn J. D.; Bosanac T.; Chen R.; Cywin C.; Hickey E.; Kashem M.; Kerr S.; Kugler S.; Li X.; Prokopowicz A. III; Schlyer S.; Smith J. D.; Turner M. R.; Wu F.; Young E. R. (2010) Substituted 2H-isoquinolin-1-ones as potent Rho-kinase inhibitors: part 2, optimization for blood pressure reduction in spontaneously hypertensive rats. Bioorg. Med. Chem. Lett. 20, 5153–5156. [DOI] [PubMed] [Google Scholar]
- Chamberlain S. D.; Redman A. M.; Patnaik S.; Brickhouse K.; Chew Y. C.; Deanda F.; Gerding R.; Lei H.; Moorthy G.; Patrick M.; Stevens K. L.; Wilson J. W.; Brad Shotwell J. (2009) Optimization of a series of 4,6-bis-anilino-1H-pyrrolo[2,3-d]pyrimidine inhibitors of IGF-1R: elimination of an acid-mediated decomposition pathway. Bioorg. Med. Chem. Lett. 19, 373–377. [DOI] [PubMed] [Google Scholar]
- Chamberlain S. D.; Redman A. M.; Wilson J. W.; Deanda F.; Shotwell J. B.; Gerding R.; Lei H.; Yang B.; Stevens K. L.; Hassell A. M.; Shewchuk L. M.; Leesnitzer M. A.; Smith J. L.; Sabbatini P.; Atkins C.; Groy A.; Rowand J. L.; Kumar R.; Mook R. A. Jr.; Moorthy G.; Patnaik S. (2009) Optimization of 4,6-bis-anilino-1H-pyrrolo[2,3-d]pyrimidine IGF-1R tyrosine kinase inhibitors towards JNK selectivity. Bioorg. Med. Chem. Lett. 19, 360–364. [DOI] [PubMed] [Google Scholar]
- Uitdehaag J. C.; Zaman G. J. (2011) A theoretical entropy score as a single value to express inhibitor selectivity. BMC Bioinf. 12, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homan K. T.; Tesmer J. J. (2014) Molecular basis for small molecule inhibition of G protein-coupled receptor kinases. ACS Chem. Biol. 10.1021/cb5003976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R.; Martin M. P.; Liu Y.; Wang B.; Patel R. A.; Zhu J. Y.; Sun N.; Pireddu R.; Lawrence N. J.; Li J.; Haura E. B.; Sung S. S.; Guida W. C.; Schonbrunn E.; Sebti S. M. (2012) Fragment-based and structure-guided discovery and optimization of Rho kinase inhibitors. J. Med. Chem. 55, 2474–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosanac T.; Hickey E. R.; Ginn J.; Kashem M.; Kerr S.; Kugler S.; Li X.; Olague A.; Schlyer S.; Young E. R. (2010) Substituted 2H-isoquinolin-1-ones as potent Rho-kinase inhibitors: part 3, aryl substituted pyrrolidines. Bioorg. Med. Chem. Lett. 20, 3746–3749. [DOI] [PubMed] [Google Scholar]
- Huang C. C.; Orban T.; Jastrzebska B.; Palczewski K.; Tesmer J. J. (2011) Activation of G protein-coupled receptor kinase 1 involves interactions between its N-terminal region and its kinase domain. Biochemistry 50, 1940–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P.; Wang B.; Maeda T.; Palczewski K.; Tesmer J. J. (2008) Structures of rhodopsin kinase in different ligand states reveal key elements involved in G protein-coupled receptor kinase activation. J. Biol. Chem. 283, 14053–14062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesmer J. J. G.; Nance M. R.; Singh P.; Lee H. (2012) Structure of a monomeric variant of rhodopsin kinase at 2.5Å resolution. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 68, 622–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2256–2268. [DOI] [PubMed] [Google Scholar]
- Hayward S.; Kitao A.; Berendsen H. J. (1997) Model-free methods of analyzing domain motions in proteins from simulation: a comparison of normal mode analysis and molecular dynamics simulation of lysozyme. Proteins 27, 425–437. [DOI] [PubMed] [Google Scholar]
- Rudolf A. F.; Skovgaard T.; Knapp S.; Jensen L. J.; Berthelsen J. (2014) A comparison of protein kinases inhibitor screening methods using both enzymatic activity and binding affinity determination. PLoS One 9, e98800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastassiadis T.; Deacon S. W.; Devarajan K.; Ma H.; Peterson J. R. (2011) Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov O.; Huber K.; Eisenreich A.; Filippakopoulos P.; King O.; Bullock A. N.; Szklarczyk D.; Jensen L. J.; Fabbro D.; Trappe J.; Rauch U.; Bracher F.; Knapp S. (2011) Specific CLK inhibitors from a novel chemotype for regulation of alternative splicing. Chem. Biol. 18, 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodowski D. T.; Tesmer V. M.; Benovic J. L.; Tesmer J. J. (2006) The structure of G protein-coupled receptor kinase (GRK)-6 defines a second lineage of GRKs. J. Biol. Chem. 281, 16785–16793. [DOI] [PubMed] [Google Scholar]
- Narayana N.; Cox S.; Shaltiel S.; Taylor S. S.; Xuong N. (1997) Crystal structure of a polyhistidine-tagged recombinant catalytic subunit of cAMP-dependent protein kinase complexed with the peptide inhibitor PKI(5–24) and adenosine. Biochemistry 36, 4438–4448. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B.; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saff E. B.; Kuijlaars A. B. J. (1997) Distributing many points on a sphere. Math. Intell. 19, 5–11. [Google Scholar]
- Lee B.; Richards F. M. (1971) The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 55, 379–400. [DOI] [PubMed] [Google Scholar]
- Hayward S.; Berendsen H. J. (1998) Systematic analysis of domain motions in proteins from conformational change: new results on citrate synthase and T4 lysozyme. Proteins 30, 144–154. [PubMed] [Google Scholar]
- Sabbatini P.; Korenchuk S.; Rowand J. L.; Groy A.; Liu Q.; Leperi D.; Atkins C.; Dumble M.; Yang J.; Anderson K.; Kruger R. G.; Gontarek R. R.; Maksimchuk K. R.; Suravajjala S.; Lapierre R. R.; Shotwell J. B.; Wilson J. W.; Chamberlain S. D.; Rabindran S. K.; Kumar R. (2009) GSK1838705A inhibits the insulin-like growth factor-1 receptor and anaplastic lymphoma kinase and shows antitumor activity in experimental models of human cancers. Mol. Cancer Ther. 8, 2811–2820. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.