

Abstract
The transfer of the gamma phosphate from ATP to sphingosine (Sph) to generate a small signaling molecule, sphingosine 1-phosphate (S1P), is catalyzed by sphingosine kinases (SphK), which exist as two isoforms, SphK1 and SphK2. SphK is a key regulator of S1P and the S1P:Sph/ceramide ratio. Increases in S1P levels have been linked to diseases including sickle cell disease, cancer, and fibrosis. Therefore, SphKs are potential targets for drug discovery. However, the current chemical biology toolkit needed to validate these enzymes as drug targets is inadequate. With this review, we survey in vivo active SphK inhibitors and highlight the need for developing more potent and selective inhibitors.
Sphingosine 1-phosphate (S1P) is a simple polar lipid that circulates at low micromolar concentrations in rodents, humans, and, presumably, other mammals. S1P exerts its effects via five cell surface G protein coupled receptors (S1P1–5) and less well-characterized intracellular targets.1 S1P signaling was validated as a drug target through the study of a sphingosine analogue, FTY720 (reviewed by Brinkmann2). Administration of FTY720 to mice and rats evokes both lymphopenia and first-dose bradycardia.3 The efficacy of FTY720 (and its excellent pharmacokinetics) in a variety of transplantation and autoimmune disease models prompted further study that culminated in its development as a medicine (fingolimod, Gilenya) for remitting relapsing multiple sclerosis.
FTY720 is a prodrug; its first metabolite, phospho-FTY720 (formed by sphingosine kinase (SphK)), is an S1P analogue that is an agonist at the S1P1, 3, 4, and 5 receptors.4,5 Subsequent studies with FTY720 analogues and genetically modified mice revealed that agonists of the S1P1 receptor drive lymphopenia and, in primates, bradycardia, thus implicating endogenous S1P in control of lymphocyte trafficking and heart rate.6 The insights gained through the study of FTY720, and its clinical success, have encouraged investigations to validate additional S1P signaling pathway members as drug targets, including individual S1P receptors, S1P lyase (cleaves S1P to hexadecenal and phospho-ethanolamine), and the S1P synthetic enzyme, SphK (Figure 1). Our goal with this review is to describe the current state of affairs regarding sphingosine kinase as a potential drug target, as revealed by chemical biology tools that are sphingosine kinase inhibitors.
Figure 1.

Sphingosine’s metabolic fates.
Biochemically, S1P is formed by the transfer of phosphate from ATP to the 1-hydroxyl group in sphingosine (Sph), which is catalyzed by SphK. Two isoforms of SphK exist in mammals: SphK1 and SphK2. SphK1 is the smaller protein (384 vs 618 amino acids), and the amino acid sequence of the two enzymes is 80% similar and 45% overall identical.7 Variants (minor changes at the amino termini) of SphK1 and SphK2 arise from alternate splicing of their respective genes, but the biologic relevance, if any, of these are unknown. The essential functions of SphK1 and SphK2 are redundant in the sense that mice lacking either enzyme are viable, fertile, and without obvious phenotype8−10 (humans lacking either enzyme have not been reported to date), and the recombinant enzymes have similar KM and Vmax constants. Salient differences include subcellular localization (SphK2 has a nuclear localization sequence that is lacking in SphK1) and substrate specificity (unlike SphK1, SphK2 phosphorylates a variety of sphingosine analogues, for example, the aforementioned FTY720).
SphK1 is the more extensively studied regarding inhibitor development.11 In 2013, a high-resolution X-ray crystal structure of human SphK1 was reported.12 The structure revealed a two-domain architecture similar to that of nicotinamide adenine dinucleotide kinases and diacylglycerol kinases, albeit with substantial differences in the non-ATP substrate binding domain. Although a structure of SphK2 has not been announced, the SphK1 structure enables the construction of a homology model that might prove to be useful in guiding inhibitor design and development.
Metabolism and Intracellular Action of S1P
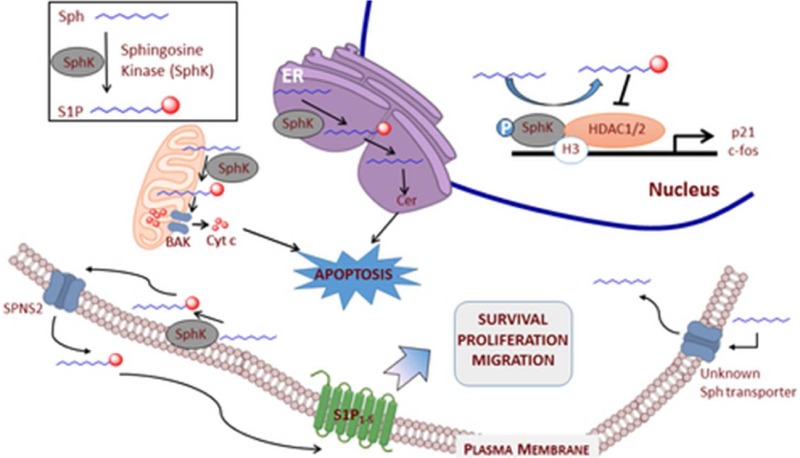
Genetic evidence from mice indicates that sphingosine, via SphK1 and SphK2, is the sole source of S1P. S1P can be cleaved by S1P lyase, revert to sphingosine via the action of phosphatases, or can be extruded from the cell by the transporter, SPNS2.13 All three routes are available in most cells examined, but the relative fluxes of S1P through these three pathways is largely unknown. However, the propensity of cells to secrete S1P while avidly taking up sphingoid bases (sphingosine, FTY720) contributes to the high extracellular concentration of S1P and low extracellular sphingosine (this ratio is reversed inside cells).
While the GPCRs S1P1–5 are well-characterized and universally accepted as mediating S1P signaling, there is evidence, albeit less well developed currently, that S1P has additional intracellular targets. Among these potential targets is ceramide synthase 2 (CerS2), which controls levels of ceramide as a negative feedback loop to regulate levels of ceramide to induce apoptosis.14 When synthesized in the nucleus by SphK2, S1P is reported to inhibit histone deacetylase 1 and 2 (HDAC 1/2) with the effect of modulating transcription of, for example, cyclin dependent inhibitor p21 and the transcriptional regulator c-fos (Figure 2).15 If the interaction of SphK2 with transcription complexes is generally true, then SphK2 inhibitors might exert genomic effects by shifting the balance of some histone modifications. In the cytosol, S1P activates NF-κB by stimulating the E3 ubiquitin ligase of tumor necrosis factor receptor-associated factor 2 (TRAF2) to affect inflammatory, antiapoptotic, and immune processes.16 In the mitochondria, S1P potentiates respiration by interacting with prohibitin 2, a protein that controls mitochondrial assembly and function.17 Furthermore, interaction of S1P with the effector protein BAK contributes to mitochondrial membrane permeabilization and release of cytochrome c to mediate apoptosis.18 Secretion of S1P was first reported to be mediated by several ABC (ATP binding cassette) transporters (ABCA1, ABCC1, and ABCG2).19,20 More recently, a member of a different transporter class, spinster homologue 2 (SPNS2),13 was found to mediate release of S1P (and phospho-FTY720) from a variety of cell types.
Figure 2.

Intracellular S1P signaling affects transcription and mitochondrial integrity to induce apoptosis, which is distinct from inside–out signaling via S1P receptors.
Physiologically, the main source of S1P is erythrocytes, which are rich in SphK1 while lacking S1P lyase and SPNS2.21 Thus, whole blood S1P concentration ([S1P]) is low micromolar and plasma [S1P] is high nanomolar, whereas tissue S1P levels are very low (indeed, so low as to make quantification problematic, particularly in view of blood contamination).22,23 The steep gradient of S1P is thought to be a controlling element regarding lymphocyte egress from secondary lymphoid tissue. Exogenous isotope- or mass-labeled S1P is rapidly (minutes) cleared from the bloodstream.24−26 As might be expected, SphK1 null mice have about 50% of normal S1P blood levels.9,27 Unexpectedly, SphK2 null mice have S1P levels that are 2–3-fold higher than those in wild-type mice.23,28,29 The latter curious phenomenon may be a result of impaired transport of S1P from blood to tissue mediated by SphK2,23 but, if verified with SphK2 inhibitors and extended to nonrodent species, the results indicate that SphK1 and SphK2 inhibitors can be used to lower or raise S1P blood levels, respectively. On a practical note, the change in blood S1P levels in response to SphK inhibitors provides a convenient pharmacodynamic measure of target engagement.
Possible Indications for SphK Inhibitors
In addition to the aforementioned effects on heart rate and lymphocyte trafficking, plasma S1P is also implicated in vascular barrier function.30,31 Thus, agents that raise circulating S1P might mitigate the vascular leak characteristic of, for example, ischemia–reperfusion injury. As would be expected of a molecule that signals through a set of widely expressed G protein coupled receptors, S1P stimulates a variety of signaling cascades. The propensity of S1P to oppose apoptosis pathways and to promote cell migration coupled with the exaggerated expression of SphK1 in some human tumor specimens32−34 and a negative correlation between SphK1 mRNA levels and survival35 has prompted repeated suggestion that SphK1 inhibitors could be useful for an oncology indication. Recently, S1P was found to be elevated in the blood of sickle cell disease patients, and infusion of a SphK1 inhibitor (PF543) reduced sickling in a mouse model of the disease.36 Although there are far more Sphk1 inhibitors described than SphK2 inhibitors, one SphK2 inhibitor (ABC294640) has been deployed in a variety of animal models (see below).
First-Generation SphK Inhibitors
An ideal chemical probe inhibitor of SphK has single-digit nanomolar potency, is highly selective (100-fold or greater) for one isoform, and is sufficiently metabolically stable and nontoxic to be deployed in a rodent model with once or twice daily administration. Testing in the appropriate null mouse and a less active enantiomer, if available, are highly desirable as negative controls. Furthermore, the ability of the compound to change the pharmacodynamic marker (blood S1P) in a dose-dependent manner is expected. Engagement of the target in the tissue of interest, as revealed by changes in sphingosine or S1P levels, is essential. To date, no SphK inhibitor for either isoform that meets all of these criteria has been reported, although some later-generation SphK inhibitors approach this ideal. A SphK inhibitor clinical candidate would need to meet additional criteria, primarily safety and efficacy in appropriate animal models, and, depending on indication, oral availability. However, there is insufficient evidence available currently that provides guidance as to the degree of isoform selectivity that is desirable for a SphK inhibitor drug.
The earliest SphK inhibitors were low-potency, not selective, and inadequately characterized (Figure 3). The first inhibitors announced are simple analogues of Sph. Reduction of the double bond in Sph generated d,l-threo-dihydrosphingosine (DHS, safingol), which has an estimated Ki of ∼3–6 μM for SphK1.37 While DHS acts as a competitive inhibitor of Sph, it is a substrate for SphK2 and enters the sphingolipid metabolic pathway.7 Unfortunately, DHS is also a protein kinase Cα inhibitor and has other off-target effects.38
Figure 3.

First-generation sphingosine kinase inhibitors.
Dimethylation of Sph affords N,N-dimethylsphingosine (DMS), and this compound displays properties similar to DHS with a Ki of ∼5 μM at SphK1.39 A related structure is SK1-I, which is also a water-soluble analogue of sphingosine. It is a low-potency SphK1 inhibitor (Ki of 10 μM) with minimal effects against of a panel of kinases (5 μM).40 SK1-I decreased S1P levels in vitro, which was followed by an increase in ceramide levels. This compound was cytotoxic to human leukemia Jurkat-T and glioblastoma cells, and administration in vivo had a significant effect in reducing tumor volumes in xenographs.40,41 A number of nonlipid inhibitors were discovered through a screening campaign conducted by French et al.42,43 SKI-II is the most well-characterized among this group of early compounds; it is a nonselective SphK inhibitor and has an inhibition constant of 17 μM.44 SKI-II is competitive with Sph and has been documented to inhibit proliferation of various cancer cell lines.43 Oral administration of SKI-II in mice revealed in vivo activity and afforded sufficient compound exposure to inhibit tumor growth with a 100 mg/kg dosing regimen. SKI-II also has a favorable half-life of ∼15 h in mice. In a dextran sulfate sodium (DSS) mouse model of ulcerative colitis, SKI-II treatment decreased disease progression with concomitant decrease in colonic levels of inflammatory cytokines TNFα, interleukin (IL)-1β, interferon gamma (IFN)-γ, and IL-6 and reduction of S1P levels.45 Recent studies suggest several possible mechanisms of action for SKI-II. For example, treatment of several cell lines with SKI-II activated the proteasome, which induced degradation of SphK1.46 In this case, its activity is linked to proteasomal activation to increase destruction of SphK1. Further studies also indicate a significant reduction of SphK1 half-life as a consequence of lysosomal degradation that involves cathepsin B.47 More recently, SKI-II is also shown to inhibit the last enzyme in the de novo synthesis of ceramide, dihydroceramide desaturase (Ki = 0.3 μM), to regulate dihydroceramides and ceramide levels and the downstream metabolite, S1P.48 Thus, it appears that the in vivo effects observed with SKI-II may be a consequence of multiple pathways. Further complicating studies with SKI-II is a recent report stating that it is about 2-fold selective for SphK2 (Ki = 7.9 μM) over SphK1 (Ki = 16 μM) and thus is essentially a dual inhibitor. That SKI-II is a SphK1 inhibitor and binds at the Sph binding pocket rather than the ATP binding domain has been unequivocally demonstrated by a co-crystal structure with SphK1 at 2.3 Å resolution (Figure 4).12 SKI-II occupies the alkyl portion of the Sph binding domain: the chlorophenyl ring points toward the interior of the lipid binding pocket and is situated closer to the hydrophilic aminodiol region of Sph. In addition to a hydrogen bond between Thr196 and NH of the aminothiazole group, what is notable about this inhibitor is the presence of a hydrogen bond between the phenol hydroxyl group and Asp178 of helix α7, which, in tandem with α8, locks the gate to prevent Sph from entering. This co-crystal structure has been used for structure-based drug design of SphK inhibitors (vide infra).
Figure 4.
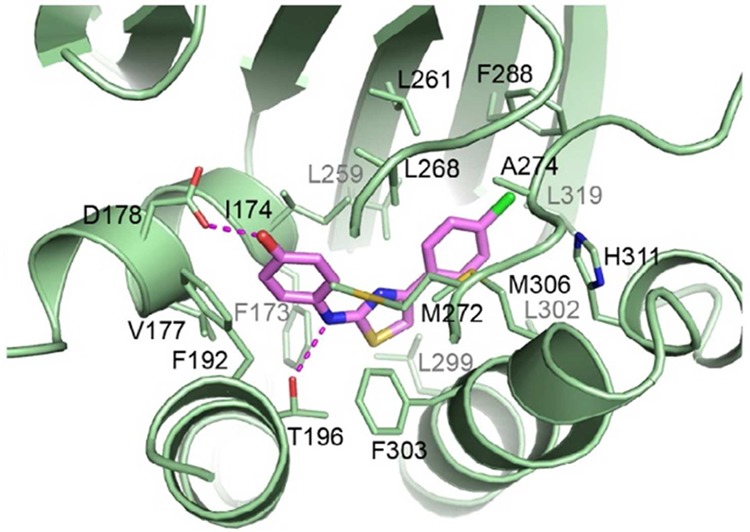
Interactions of SKI-II with SphK1. Reprinted with permission from ref (12). Copyright 2013 Cell Press.
An analogue of SKI-II is ABC294640, which was developed from structure–activity studies. This compound is the most well-studied SphK inhibitor in vivo in that it has been deployed in numerous disease models. ABC294640 suppressed the proliferation of several cancer cell lines in vitro, and several disease models were employed to determine the therapeutic potential of this inhibitor including lupus nephritis,49 osteoarthritis,50 diabetic retinopathy,51 Crohn’s disease,52 rheumatoid arthritis,53 and ulcerative colitis.54ABC294640 has an inhibitory constant of 10 μM and is selective for SphK2 over SphK1 as well as a panel of other kinases.55 It has a half-life of 4.5 h in mice and is orally bioavailable. Biochemical characterization of ABC294640 revealed (i) decreased S1P levels with a corresponding elevation in ceramide levels in vitro, (ii) attenuated expression or activation of STAT3, AKT, ERK,56 and (iii) decreased circulating S1P levels in mice.49 It is not clear whether administration of ABC294640 in mice evokes lower levels of S1P with acute dosing, as the S1P levels were reported after 5 weeks of a 100 mg/kg daily dosing regimen.57 The mechanism whereby ABC294640 promotes cell death has been described to involve apoptotic and autophagy pathways.55,58 However, recent investigation suggests an additional mode of action by inhibiting estrogen receptors in breast cancer cells.59
Although the affinities of the early generation sphingosine kinase inhibitors are 2–3 log orders lower than would be expected of an enzyme inhibitor developed by the pharmaceutical industry, two early generation compounds are in clinical trials for oncology indications. Safingol, in a drug combination, is the subject of two studies that are currently underway or completed (ClinicalTrials.gov identifiers NCT01553071 and NCT00084812).60ABC294640 in being tested in a phase I clinical trial for pancreatic cancer and unspecified solid tumors (NCT01488513) and refractory/relapsed diffuse large B-cell lymphoma (NCT02229981). No results from any of these trials have been reported to date. Although significant patient responses would surely be welcomed by all concerned, the low affinity of safingol and ABC294640 for their putative targets makes assignment of success (or failure) to inhibition of SphKs problematic.
Second-Generation SphK1 Inhibitors
A second wave of SphK inhibitors included a set of compounds discovered in Pharma industry discovery programs. As is expected, these molecules are more drug-like than earlier inhibitors. These compounds, which were published in academic journals and/or the patent literature, are focused on the SphK1 isoform. The compounds discovered at Genzyme are featured in two reports by Xiang et al.61,62 as well as a patent application.71Genzyme 51 (Figure 5) emerged as one of their best inhibitors of SphK1, with a measured IC50 value of 58 nM, and no inhibition was observed with SphK2 at a concentration of 10 μM. Pharmacokinetic profiling of this compound revealed 18% oral bioavailability and a 7.6 h half-life in rat plasma. However, whether any of these compounds affect Sph and S1P levels in blood was not reported. A more recent addition to the catalog of SphK1 inhibitors was disclosed by a group from Amgen.63 Hampered by the lack of hits from an in-house high-throughput screening campaign, this group improved dual SphK inhibitors using structure-based design. Using the crystal structure of human SphK1 with bound SKI-II, several derivatives such as Amgen 82 emerged as dual inhibitors (Figure 5). This compound inhibits human SphK1 and SphK2 with IC50 values of 20 and 114 nM, respectively. X-ray crystal structure analysis of Amgen 82 bound to SphK1 shows binding in the hydrophobic site of Sph similar to that for SKI-II (Figure 6).63 In addition, the polar headgroup (hydroxylated piperidine) mimics the interactions observed when Sph is bound to SphK1: Asp178 forms a bipartite hydrogen bond to the ring nitrogen of piperidine and exocyclic hydroxyl group. This hydroxyl group then forms a network of hydrogen bonds to neighboring amino acids (Ser168, G342, A339) mediated by a water molecule. The second hydroxyl group on the 4 position of piperidine contacts Asp81, the key active site residue that acts as a base to activate the 1-OH of sphingosine, via a hydrogen bond. To date, Amgen 82 is the most potent, dual SphK inhibitor with an excellent pharmacokinetic profile and has been used as a chemical biology tool to investigate the consequence of inhibiting both enzymes in vivo. In a panel of cancer cell lines, Amgen 82 was shown to attenuate levels of S1P, as expected. Unlike results with earlier inhibitors, no appreciable effect on cell viability was observed when administered at therapeutic concentrations; only at significantly higher concentration is cell death seen, but this has been attributed to detergent-like physicochemical properties of this and related compounds. In athymic nude mice, daily dosing over 7 days at 100 mg/kg by oral gavage resulted in approximately 70% lower plasma S1P compared to vehicle.64 The decreased S1P level observed during the duration of the study (days 1 to 7) served as a pharmacodynamic marker of SphK inhibition. To further corroborate SphK inhibition, a decrease in VEGF-mediated vascular permeability in mice by following Evans blue dye extravasation was observed. With in vivo evidence supporting SphK inhibition in hand, investigations that relate lower circulating S1P levels to disease states (for example, cancer) can be tested. One long-standing hypothesis in the field suggests the idea of a sphingolipid rheostat, wherein the balance of sphingolipid levels within a cell determines its fate: whether to survive or undergo apoptosis. Hence, tipping the balance toward Sph/ceramide, via inhibition of SphK, would be a useful therapy for hyperproliferative diseases. In a tumor xenograph model in mice, treatment with Amgen 82 at 300 mg/kg once a day dose did not have a statistically significant effect on tumor volume compared to control. Thus, on the basis of both in vitro and in vivo work, these studies suggest that simply decreasing S1P levels in circulation plays a minor role, if any, toward apoptosis and cancer cell proliferation in this model. However, there are some limitations to using Amgen 82 in mouse models. For example, Amgen 82 has no inhibitory activity against mSphK2 (i.e., >10 μM);64 thus, in the study described above, the blockade of S1P generation may be due only to SphK1 inhibition. Whether additional SphK2 inhibition is necessary for beneficial effects remain to be seen. Furthermore, it is unclear if Amgen 82 has an effect on the level of sphingosine or ceramide because this data is not reported; hence, relating S1P and sphingosine/ceramide in the rheostat theory is inconclusive.
Figure 5.

Second-generation sphingosine kinase inhibitors.
Figure 6.
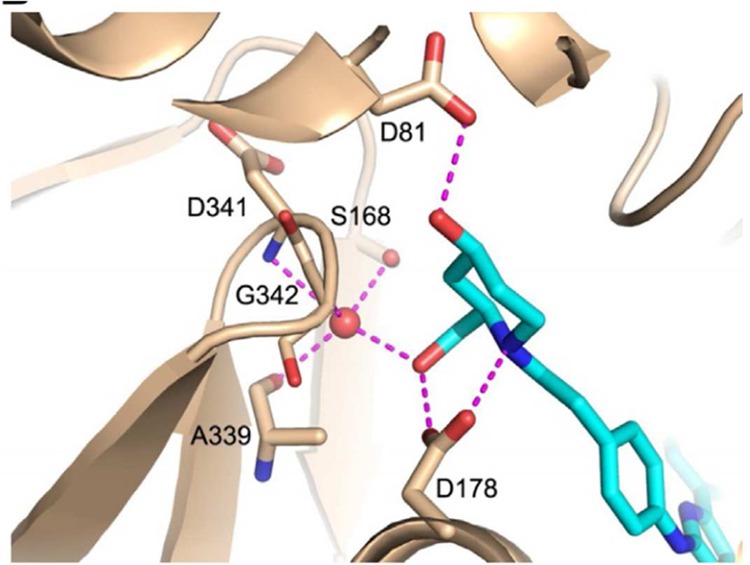
Hydrogen-bond interaction of Amgen 82 with amino acid residues of SphK1. Reprinted with permission from ref (63). Copyright 2013 Elsevier.
A related, single compound, PF543, bearing a hydroxymethylpyrrolidine headgroup (Figure 5), was recently reported by a group from Pfizer.65PF543 is the most potent (Ki = 4 nM) and selective SphK1 inhibitor described to date. PF543 is competitive with Sph and is greater than 100-fold selective over SphK2. Similar to the aforementioned Amgen compound, inhibition of SphK1 with PF543 in cultured cancer cells did not affect cell viability. Interestingly, a recent study reported that blood S1P level is increased (correlates with elevated SphK1) in mice and humans with sickle cell disease. When PF543 was deployed both in in vitro and in vivo studies to inhibit SphK1, reduced sickling of red blood cells was observed, suggesting a possible therapeutic indication for sickle cell disease.36 The use of an osmotic pump to deliver this compound in mice indicates a poor pharmacokinetic profile and highlights the need for in vivo stable SphK1 selective inhibitors. Nevertheless, PF-543 provides an outstanding tool (it is available commercially) for interrogating cultured cells.66
Another sphingosine analogue, K145, which features a thiazolidine-2,4-dione, has been recently reported as a selective SphK2 inhibitor.67 Biochemical characterization of K145 reveals a Ki of 6.4 μM for SphK2 without any inhibition of SphK1 and ceramide kinase (up to 10 μM). To further confirm SphK2 inhibition, compound treatment of U937 cells caused a slight decrease in S1P levels as well as FTY720-phosphate, a SphK2 selective substrate. In contrast with Amgen 82 and PF543, in vitro studies with U937 cells revealed an antiproliferative effect in a dose-dependent manner, possibly through inhibition of phosphorylation of downstream ERK and Akt signaling pathways. In vivo studies with K145 suggest oral bioavailability, as monitored by decrease in tumor volume, which also correlates with a decrease in tumor S1P level. While it is unknown whether sphingosine levels were affected by inhibitor treatment, K145 appears to be as a good starting point for further improvement.
A unique scaffold of SphK inhibitors contains a carboximidamide group. These compounds arose from a campaign to develop sphingosine kinase substrates, i.e., analogues of FTY720. Compound 1a (Figure 5) is an amidine-containing SphK1 inhibitor that, although relatively short-lived in vivo, was sufficiently persistent to document that acute SphK1 inhibition results in a rapid decrease in circulating S1P.27 Although 1a is potent as a SphK1 inhibitor (Ki = 100 nM), it is rapidly cleared in vivo when administered i.v. (half-life < 10 min), which, in turn, results in rapid restoration of basal S1P levels. Like PF-543, compound 1a is competitive with Sph and does not affect cell viability at concentrations that achieve an effective SphK1 blockade. Docking of compound 1a and analogues into a model of SphK1 suggests that the carboximidamide functionality chelates the gamma phosphate of ATP.68
Building from compound 1a, the amidine group was replaced with a guanidine warhead in SLR080811.(69) This chemical manipulation resulted in an improved compound with a much more favorable half-life (∼5 h) in mice. Surprisingly, SLR080811 had a reversed selectivity with SphK2 compared to that of 1a, with a Ki of 1 μM toward SphK2 (10-fold selective). In leukemia cells, SLR080811 lowered S1P levels, as expected of SphK inhibitors. As both SphK1 and 2 are present in this cell line, inhibition of SphK2 was confirmed by monitoring the decrease in phosphorylation level of exogenously added FTY720 (a SphK2 specific substrate). Surprisingly, when SLR080811 was administered to mice, the blockade of SphK2 triggered a rapid increase in blood S1P level. This result is in sharp contrast with in vivo studies using SphK1 selective or SphK1/2 dual inhibitors or ABC294640 (SphK2 selective inhibitor) where the concentration of circulating S1P decreased.27,49,63 While the mechanism by which SphK2 inhibition raises S1P level in vivo is currently unknown and is being investigated, the small molecule inhibition of SphK2 recapitulates the increased S1P levels observed in SphK2 knockout mice.
Conclusions and Future Directions
The physiological role of S1P is complex, and SphKs play a pivotal role in the S1P signaling axis. Numerous in vitro and in vivo studies indicate targeting SphKs for the potential treatment of many other diseases. However, most of these data point toward SphK1, which has been the more commonly studied isoform. To validate SphK1 for its therapeutic potential, small molecule inhibitors are necessary. The first-generation inhibitors are moderately potent and nonselective and therefore the results obtained from their application may not be a direct result of modulating SphK1 activity but rather a consequence of off-target effects. Thus, one of the main challenges for researchers in the field is the development of potent and selective inhibitors that can mount a sustained blockade of SphK1 activity in vivo. It has been over a decade since the first inhibitor was reported; only recently have potent and selective SphK1 inhibitors that hold promise appeared. These include (1) Genzyme 51, which has good pharmacokinetic properties, but measurement of biomarkers to confidently confirm target engagement in vivo have not yet been reported, and (2) PF543, which is the most potent and selective SphK1 inhibitor published to date but apparently is not ideal for in vivo studies. The recently disclosed SphK dual inhibitor, Amgen 82, will be one of many valuable tools to elucidate the physiological function of these enzymes. Thus far, this compound is the best biochemically characterized inhibitor in vitro and in vivo and has already demonstrated that inhibiting SphKs to consequently decrease S1P concentration has no effect on tumor cell viability. The publication of X-ray crystal structures of SphK1 is an important landmark in the study of the enzyme, and its availability should accelerate the development of additional inhibitors.
In contrast with SphK1, studies on SphK2 inhibitors are scarce. However, in the past few years, momentum is building for such studies, and roles for this enzyme are starting to be unraveled.70 Much of what is known about this enzyme is that it is responsible for intracellular S1P generation so as to act an epigenetic factor to control transcription or promote apoptosis depending on its localization within the cell. Akin to SphK1, in vivo active small molecule SphK2 selective inhibitors are necessary to decipher its role in physiology and disease states where S1P signaling might be awry.
Unfortunately, current SphK2 inhibitors are moderately potent and only about 10-fold selective. The first SphK2 selective inhibitor described, ABC294640, has been tested in a variety of animal models of disease with some success, but some of the effects observed in in vivo studies may be attributed to off-target effects of this low-potency compound. SLR080811 is a second-generation SphK2 inhibitor that is about 10-fold more potent than ABC294640 but no more selective. A curious aspect of SphK2 is that its absence in mice results in circulating S1P levels that are three times normal (SphK1 ablation results in one-half normal levels). SLR080811 treatment recapitulates this phenomenon. If the propensity to raise S1P levels is a general property of SphK2 inhibitors and if this effect extends beyond rodent species, then application of selective SphK inhibitors could be used to raise or lower circulating S1P levels. Proof of the therapeutic benefit, if any, of either maneuver awaits further improvement of SphK inhibitors.
Acknowledgments
We acknowledge financial support by the National Institutes of Health (grant nos. R01 GM104366 and R01 GM067958).
The authors declare the following competing financial interest(s): The authors are among the co-founders of SphynKx Therapeutics LLC, which was created to commercialize S1P-related discoveries, including SphK inhibitors, discovered and characterized in their laboratories. Compounds 1a and SLR080811 are included in patent applications that are licensed to SphynKx.
Funding Statement
National Institutes of Health, United States
References
- Kunkel G. T.; Maceyka M.; Milstien S.; Spiegel S. (2013) Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug Discovery 12, 688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigaud M.; Guerini D.; Billich A.; Bassilana F.; Brinkmann V. (2014) Second generation S1P pathway modulators: research strategies and clinical developments. Biochim. Biophys. Acta 1841, 745–758. [DOI] [PubMed] [Google Scholar]
- Brinkmann V. (2007) Sphingosine 1-phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol. Ther. 115, 84–105. [DOI] [PubMed] [Google Scholar]
- Brinkmann V.; Davis M. D.; Heise C. E.; Albert R.; Cottens S.; Hof R.; Bruns C.; Prieschl E.; Baumruker T.; Hiestand P.; Foster C. A.; Zollinger M.; Lynch K. R. (2002) The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J. Biol. Chem. 277, 21453–21457. [DOI] [PubMed] [Google Scholar]
- Mandala S.; Hajdu R.; Bergstrom J.; Quackenbush E.; Xie J.; Milligan J.; Thornton R.; Shei G. J.; Card D.; Keohane C.; Rosenbach M.; Hale J.; Lynch C. L.; Rupprecht K.; Parsons W.; Rosen H. (2002) Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 296, 346–349. [DOI] [PubMed] [Google Scholar]
- Brinkmann V.; Billich A.; Baumruker T.; Heining P.; Schmouder R.; Francis G.; Aradhye S.; Burtin P. (2010) Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discovery 9, 883–897. [DOI] [PubMed] [Google Scholar]
- Liu H.; Sugiura M.; Nava V. E.; Edsall L. C.; Kono K.; Poulton S.; Milstien S.; Kohama T.; Spiegel S. (2000) Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J. Biol. Chem. 275, 19513–19520. [DOI] [PubMed] [Google Scholar]
- Zemann B.; Urtz N.; Reuschel R.; Mechtcheriakova D.; Bornancin F.; Badegruber R.; Baumruker T.; Billich A. (2007) Normal neutrophil functions in sphingosine kinase type 1 and 2 knockout mice. Immunol. Lett. 109, 56–63. [DOI] [PubMed] [Google Scholar]
- Allende M. L.; Sasaki T.; Kawai H.; Olivera A.; Mi Y. D.; van Echten-Deckert G.; Hajdu R.; Rosenbach M.; Keohane C. A.; Mandala S.; Spiegel S.; Proia R. L. (2004) Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J. Biol. Chem. 279, 52487–52492. [DOI] [PubMed] [Google Scholar]
- Mizugishi K.; Yamashita T.; Olivera A.; Miller G. F.; Spiegel S.; Proia R. L. (2005) Essential role for sphingosine kinases in neural and vascular development. Mol. Cell. Biol. 25, 11113–11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plano D.; Amin S.; Sharma A. K. (2014) Importance of sphingosine kinase (SphK) as a target in developing cancer therapeutics and recent developments in the synthesis of novel SphK inhibitors. J. Med. Chem. 57, 5509–5524. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Min X.; Xiao S. H.; Johnstone S.; Romanow W.; Meininger D.; Xu H.; Liu J.; Dai J.; An S.; Thibault S.; Walker N. (2013) Molecular basis of sphingosine kinase 1 substrate recognition and catalysis. Structure 21, 798–809. [DOI] [PubMed] [Google Scholar]
- Nagahashi M.; Kim E. Y.; Yamada A.; Ramachandran S.; Allegood J. C.; Hait N. C.; Maceyka M.; Milstien S.; Takabe K.; Spiegel S. (2013) Spns2, a transporter of phosphorylated sphingoid bases, regulates their blood and lymph levels, and the lymphatic network. FASEB J. 27, 1001–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviad E. L.; Albee L.; Pankova-Kholmyansky I.; Epstein S.; Park H.; Merrill A. H. Jr.; Futerman A. H. (2008) Characterization of ceramide synthase 2: tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J. Biol. Chem. 283, 5677–5684. [DOI] [PubMed] [Google Scholar]
- Hait N. C.; Allegood J.; Maceyka M.; Strub G. M.; Harikumar K. B.; Singh S. K.; Luo C.; Marmorstein R.; Kordula T.; Milstien S.; Spiegel S. (2009) Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325, 1254–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez S. E.; Harikumar K. B.; Hait N. C.; Allegood J.; Strub G. M.; Kim E. Y.; Maceyka M.; Jiang H.; Luo C.; Kordula T.; Milstien S.; Spiegel S. (2010) Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465, 1084–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strub G. M.; Paillard M.; Liang J.; Gomez L.; Allegood J. C.; Hait N. C.; Maceyka M.; Price M. M.; Chen Q.; Simpson D. C.; Kordula T.; Milstien S.; Lesnefsky E. J.; Spiegel S. (2011) Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 25, 600–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk J. E.; McStay G. P.; Bharti A.; Kuwana T.; Clarke C. J.; Siskind L. J.; Obeid L. M.; Green D. R. (2012) Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148, 988–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K.; Malchinkhuu E.; Horiuchi Y.; Mogi C.; Tomura H.; Tosaka M.; Yoshimoto Y.; Kuwabara A.; Okajima F. (2007) Critical role of ABCA1 transporter in sphingosine 1-phosphate release from astrocytes. J. Neurochem. 103, 2610–2619. [DOI] [PubMed] [Google Scholar]
- Takabe K.; Kim R. H.; Allegood J. C.; Mitra P.; Ramachandran S.; Nagahashi M.; Harikumar K. B.; Hait N. C.; Milstien S.; Spiegel S. (2010) Estradiol induces export of sphingosine 1-phosphate from breast cancer cells via ABCC1 and ABCG2. J. Biol. Chem. 285, 10477–10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza A.; Breart B.; Ramos-Perez W. D.; Pitt L. A.; Gobert M.; Sunkara M.; Lafaille J. J.; Morris A. J.; Schwab S. R. (2012) The transporter Spns2 is required for secretion of lymph but not plasma sphingosine-1-phosphate. Cell Rep. 2, 1104–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatomi Y.; Igarashi Y.; Yang L.; Hisano N.; Qi R.; Asazuma N.; Satoh K.; Ozaki Y.; Kume S. (1997) Sphingosine 1-phosphate, a bioactive sphingolipid abundantly stored in platelets, is a normal constituent of human plasma and serum. J. Biochem. 121, 969–973. [DOI] [PubMed] [Google Scholar]
- Sensken S. C.; Bode C.; Nagarajan M.; Peest U.; Pabst O.; Graler M. H. (2010) Redistribution of sphingosine 1-phosphate by sphingosine kinase 2 contributes to lymphopenia. J. Immunol. 184, 4133–4142. [DOI] [PubMed] [Google Scholar]
- Venkataraman K.; Lee Y.-M.; Michaud J.; Thangada S.; Ai Y.; Bonkovsky H. L.; Parikh N. S.; Habrukowich C.; Hla T. (2008) Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ. Res. 102, 669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peest U.; Sensken S. C.; Andreani P.; Hanel P.; Van Veldhoven P. P.; Graler M. H. (2008) S1P-lyase independent clearance of extracellular sphingosine 1-phosphate after dephosphorylation and cellular uptake. J. Cell. Biochem. 104, 756–772. [DOI] [PubMed] [Google Scholar]
- Salous A. K.; Panchatcharam M.; Sunkara M.; Mueller P.; Dong A.; Wang Y.; Graf G. A.; Smyth S. S.; Morris A. J. (2013) Mechanism of rapid elimination of lysophosphatidic acid and related lipids from the circulation of mice. J. Lipid Res. 54, 2775–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharel Y.; Mathews T. P.; Gellett A. M.; Tomsig J. L.; Kennedy P. C.; Moyer M. L.; Macdonald T. L.; Lynch K. R. (2011) Sphingosine kinase type 1 inhibition reveals rapid turnover of circulating sphingosine 1-phosphate. Biochem. J. 440, 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemann B.; Kinzel B.; Muller M.; Reuschel R.; Mechtcheriakova D.; Urtz N.; Bornancin F.; Baumruker T.; Billich A. (2006) Sphingosine kinase type 2 is essential for lymphopenia induced by the immunomodulatory drug FTY720. Blood 107, 1454–1458. [DOI] [PubMed] [Google Scholar]
- Olivera A.; Mizugishi K.; Tikhonova A.; Ciaccia L.; Odom S.; Proia R. L.; Rivera J. (2007) The sphingosine kinase-sphingosine-1-phosphate axis is a determinant of mast cell function and anaphylaxis. Immunity 26, 287–297. [DOI] [PubMed] [Google Scholar]
- Marsolais D.; Rosen H. (2009) Chemical modulators of sphingosine-1-phosphate receptors as barrier-oriented therapeutic molecules. Nat. Rev. Drug Discovery 8, 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Dudek S. M. (2009) Regulation of vascular permeability by sphingosine 1-phosphate. Microvasc. Res. 77, 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckhaberle E.; Rody A.; Engels K.; Gaetje R.; von Minckwitz G.; Schiffmann S.; Grosch S.; Geisslinger G.; Holtrich U.; Karn T.; Kaufmann M. (2008) Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res. Treat. 112, 41–52. [DOI] [PubMed] [Google Scholar]
- Kawamori T.; Kaneshiro T.; Okumura M.; Maalouf S.; Uflacker A.; Bielawski J.; Hannun Y. A.; Obeid L. M. (2009) Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J. 23, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne S.; Edwards J.; Ohotski J.; Pyne N. J. (2012) Sphingosine 1-phosphate receptors and sphingosine kinase 1: novel biomarkers for clinical prognosis in breast, prostate, and hematological cancers. Front. Oncol. 2, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Guan H. Y.; Gong L. Y.; Song L. B.; Zhang N.; Wu J.; Yuan J.; Zheng Y. J.; Huang Z. S.; Li M. (2008) Clinical significance of sphingosine kinase-1 expression in human astrocytomas progression and overall patient survival. Clin. Cancer Res. 14, 6996–7003. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Berka V.; Song A.; Sun K.; Wang W.; Zhang W.; Ning C.; Li C.; Zhang Q.; Bogdanov M.; Alexander D. C.; Milburn M. V.; Ahmed M. H.; Lin H.; Idowu M.; Zhang J.; Kato G. J.; Abdulmalik O. Y.; Zhang W.; Dowhan W.; Kellems R. E.; Zhang P.; Jin J.; Safo M.; Tsai A. L.; Juneja H. S.; Xia Y. (2014) Elevated sphingosine-1-phosphate promotes sickling and sickle cell disease progression. J. Clin. Invest. 124, 2750–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buehrer B. M.; Bell R. M. (1992) Inhibition of sphingosine kinase in vitro and in platelets. Implications for signal transduction pathways. J. Biol. Chem. 267, 3154–3159. [PubMed] [Google Scholar]
- Coward J.; Ambrosini G.; Musi E.; Truman J. P.; Haimovitz-Friedman A.; Allegood J. C.; Wang E.; Merrill A. H. Jr.; Schwartz G. K. (2009) Safingol (l-threo-sphinganine) induces autophagy in solid tumor cells through inhibition of PKC and the PI3-kinase pathway. Autophagy 5, 184–193. [DOI] [PubMed] [Google Scholar]
- Melendez A. J.; Carlos-Dias E.; Gosink M.; Allen J. M.; Takacs L. (2000) Human sphingosine kinase: molecular cloning, functional characterization and tissue distribution. Gene 251, 19–26. [DOI] [PubMed] [Google Scholar]
- Paugh S. W.; Paugh B. S.; Rahmani M.; Kapitonov D.; Almenara J. A.; Kordula T.; Milstien S.; Adams J. K.; Zipkin R. E.; Grant S.; Spiegel S. (2008) A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood 112, 1382–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov D.; Allegood J. C.; Mitchell C.; Hait N. C.; Almenara J. A.; Adams J. K.; Zipkin R. E.; Dent P.; Kordula T.; Milstien S.; Spiegel S. (2009) Targeting sphingosine kinase 1 inhibits Akt signaling, induces apoptosis, and suppresses growth of human glioblastoma cells and xenografts. Cancer Res. 69, 6915–6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French K. J.; Schrecengost R. S.; Lee B. D.; Zhuang Y.; Smith S. N.; Eberly J. L.; Yun J. K.; Smith C. D. (2003) Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 63, 5962–5969. [PubMed] [Google Scholar]
- French K. J.; Upson J. J.; Keller S. N.; Zhuang Y.; Yun J. K.; Smith C. D. (2006) Antitumor activity of sphingosine kinase inhibitors. J. Pharmacol. Exp. Ther. 318, 596–603. [DOI] [PubMed] [Google Scholar]
- Lim K. G.; Tonelli F.; Li Z.; Lu X.; Bittman R.; Pyne S.; Pyne N. J. (2011) FTY720 analogues as sphingosine kinase 1 inhibitors: enzyme inhibition kinetics, allosterism, proteasomal degradation, and actin rearrangement in MCF-7 breast cancer cells. J. Biol. Chem. 286, 18633–18640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maines L. W.; Fitzpatrick L. R.; French K. J.; Zhuang Y.; Xia Z. P.; Keller S. N.; Upson J. J.; Smith C. D. (2008) Suppression of ulcerative colitis in mice by orally available inhibitors of sphingosine kinase. Dig. Dis. Sci. 53, 997–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loveridge C.; Tonelli F.; Leclercq T.; Lim K. G.; Long J. S.; Berdyshev E.; Tate R. J.; Natarajan V.; Pitson S. M.; Pyne N. J.; Pyne S. (2010) The sphingosine kinase 1 inhibitor 2-(p-hydroxyanilino)-4-(p-chlorophenyl)thiazole induces proteasomal degradation of sphingosine kinase 1 in mammalian cells. J. Biol. Chem. 285, 38841–38852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren S.; Xin C.; Pfeilschifter J.; Huwiler A. (2010) A novel mode of action of the putative sphingosine kinase inhibitor 2-(p-hydroxyanilino)-4-(p-chlorophenyl) thiazole (SKI II): induction of lysosomal sphingosine kinase 1 degradation. Cell. Physiol. Biochem. 26, 97–104. [DOI] [PubMed] [Google Scholar]
- Cingolani F.; Casasampere M.; Sanllehi P.; Casas J.; Bujons J.; Fabrias G. (2014) Inhibition of dihydroceramide desaturase activity by the sphingosine kinase inhibitor SKI II. J. Lipid Res. 55, 1711–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider A. J.; Ruiz P.; Obeid L. M.; Oates J. C. (2013) Inhibition of sphingosine kinase-2 in a murine model of lupus nephritis. PLoS One 8, e53521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick L. R.; Green C.; Maines L. W.; Smith C. D. (2011) Experimental osteoarthritis in rats is attenuated by ABC294640, a selective inhibitor of sphingosine kinase-2. Pharmacology 87, 135–143. [DOI] [PubMed] [Google Scholar]
- Maines L. W.; French K. J.; Wolpert E. B.; Antonetti D. A.; Smith C. D. (2006) Pharmacologic manipulation of sphingosine kinase in retinal endothelial cells: implications for angiogenic ocular diseases. Invest. Ophthalmol. Visual Sci. 47, 5022–5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maines L. W.; Fitzpatrick L. R.; Green C. L.; Zhuang Y.; Smith C. D. (2010) Efficacy of a novel sphingosine kinase inhibitor in experimental Crohn’s disease. Inflammopharmacology 18, 73–85. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick L. R.; Green C.; Frauenhoffer E. E.; French K. J.; Zhuang Y.; Maines L. W.; Upson J. J.; Paul E.; Donahue H.; Mosher T. J.; Smith C. D. (2011) Attenuation of arthritis in rodents by a novel orally-available inhibitor of sphingosine kinase. Inflammopharmacology 19, 75–87. [DOI] [PubMed] [Google Scholar]
- Maines L. W.; Fitzpatrick L. R.; French K. J.; Zhuang Y.; Xia Z.; Keller S. N.; Upson J. J.; Smith C. D. (2008) Suppression of ulcerative colitis in mice by orally available inhibitors of sphingosine kinase. Dig. Dis. Sci. 53, 997–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French K. J.; Zhuang Y.; Maines L. W.; Gao P.; Wang W.; Beljanski V.; Upson J. J.; Green C. L.; Keller S. N.; Smith C. D. (2010) Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J. Pharmacol. Exp. Ther. 333, 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P.; Peterson Y. K.; Smith R. A.; Smith C. D. (2012) Characterization of isoenzyme-selective inhibitors of human sphingosine kinases. PLoS One 7, e44543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beljanski V.; Lewis C. S.; Smith C. D. (2011) Antitumor activity of sphingosine kinase 2 inhibitor ABC294640 and sorafenib in hepatocellular carcinoma xenografts. Cancer Biol. Ther. 11, 524–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beljanski V.; Knaak C.; Smith C. D. (2010) A novel sphingosine kinase inhibitor induces autophagy in tumor cells. J. Pharmacol. Exp. Ther. 333, 454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoon J. W.; White M. D.; Meacham W. D.; Slaughter E. M.; Muir S. E.; Elliott S.; Rhodes L. V.; Ashe H. B.; Wiese T. E.; Smith C. D.; Burow M. E.; Beckman B. S. (2010) Antiestrogenic effects of the novel sphingosine kinase-2 inhibitor ABC294640. Endocrinology 151, 5124–5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson M. A.; Carvajal R. D.; Merrill A. H. Jr.; Gonen M.; Cane L. M.; Schwartz G. K. (2011) A phase I clinical trial of safingol in combination with cisplatin in advanced solid tumors. Clin. Cancer Res. 17, 2484–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y. B.; Hirth B.; Kane J. L.; Liao J. K.; Noson K. D.; Yee C.; Asmussen G.; Fitzgerald M.; Klaus C.; Booker M. (2010) Discovery of novel sphingosine kinase-1 inhibitors. Part 2. Bioorg. Med. Chem. Lett. 20, 4550–4554. [DOI] [PubMed] [Google Scholar]
- Xiang Y.; Asmussen G.; Booker M.; Hirth B.; Kane J. L. Jr.; Liao J.; Noson K. D.; Yee C. (2009) Discovery of novel sphingosine kinase 1 inhibitors. Bioorg. Med. Chem. Lett. 19, 6119–6121. [DOI] [PubMed] [Google Scholar]
- Xiang Y., Hirth B., Kane J. L., Liao J., Noson K., and Yee C. (2010) Patent WO/2010/033701.
- Gustin D. J.; Li Y.; Brown M. L.; Min X.; Schmitt M. J.; Wanska M.; Wang X.; Connors R.; Johnstone S.; Cardozo M.; Cheng A. C.; Jeffries S.; Franks B.; Li S.; Shen S.; Wong M.; Wesche H.; Xu G.; Carlson T. J.; Plant M.; Morgenstern K.; Rex K.; Schmitt J.; Coxon A.; Walker N.; Kayser F.; Wang Z. (2013) Structure guided design of a series of sphingosine kinase (SphK) inhibitors. Bioorg. Med. Chem. Lett. 23, 4608–4616. [DOI] [PubMed] [Google Scholar]
- Rex K.; Jeffries S.; Brown M. L.; Carlson T.; Coxon A.; Fajardo F.; Frank B.; Gustin D.; Kamb A.; Kassner P. D.; Li S.; Li Y.; Morgenstern K.; Plant M.; Quon K.; Ruefli-Brasse A.; Schmidt J.; Swearingen E.; Walker N.; Wang Z.; Watson J. E.; Wickramasinghe D.; Wong M.; Xu G.; Wesche H. (2013) Sphingosine kinase activity is not required for tumor cell viability. PLoS One 8, e68328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnute M. E.; McReynolds M. D.; Kasten T.; Yates M.; Jerome G.; Rains J. W.; Hall T.; Chrencik J.; Kraus M.; Cronin C. N.; Saabye M.; Highkin M. K.; Broadus R.; Ogawa S.; Cukyne K.; Zawadzke L. E.; Peterkin V.; Iyanar K.; Scholten J. A.; Wendling J.; Fujiwara H.; Nemirovskiy O.; Wittwer A. J.; Nagiec M. M. (2012) Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem. J. 444, 79–88. [DOI] [PubMed] [Google Scholar]
- Lynch K. R. (2012) Building a better sphingosine kinase-1 inhibitor. Biochem. J. 444, e1–2. [DOI] [PubMed] [Google Scholar]
- Liu K.; Guo T. L.; Hait N. C.; Allegood J.; Parikh H. I.; Xu W.; Kellogg G. E.; Grant S.; Spiegel S.; Zhang S. (2013) Biological characterization of 3-(2-amino-ethyl)-5-[3-(4-butoxyl-phenyl)-propylidene]-thiazolidine-2,4-dione (K145) as a selective sphingosine kinase-2 inhibitor and anticancer agent. PLoS One 8, e56471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy A. J.; Mathews T. P.; Kharel Y.; Field S. D.; Moyer M. L.; East J. E.; Houck J. D.; Lynch K. R.; Macdonald T. L. (2011) Development of amidine-based sphingosine kinase 1 nanomolar inhibitors and reduction of sphingosine 1-phosphate in human leukemia cells. J. Med. Chem. 54, 3524–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharel Y.; Raje M.; Gao M.; Gellett A. M.; Tomsig J. L.; Lynch K. R.; Santos W. L. (2012) Sphingosine kinase type 2 inhibition elevates circulating sphingosine 1-phosphate. Biochem. J. 447, 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer H. A.; Pitson S. M. (2013) Roles, regulation and inhibitors of sphingosine kinase 2. FEBS J. 280, 5317–5336. [DOI] [PubMed] [Google Scholar]