

Abstract
Growing evidence suggests that histone methyltransferases (HMTs, also known as protein methyltransferases (PMTs)) play an important role in diverse biological processes and human diseases by regulating gene expression and the chromatin state. Therefore, HMTs have been increasingly recognized by the biomedical community as a class of potential therapeutic targets. High quality chemical probes of HMTs, as tools for deciphering their physiological functions and roles in human diseases and testing therapeutic hypotheses, are critical for advancing this promising field. In this review, we focus on the discovery, characterization, and biological applications of chemical probes for HMTs.
Epigenetics refers to mitotically and/or meiotically heritable changes in gene function without changes in DNA sequence.1,2 There has been growing interest in epigenetics due to its fundamental role in diverse biological processes and human diseases.3 Epigenetic regulation of the chromatin state that governs the access to genetic code is mainly controlled by DNA methylation, noncoding RNA, nucleosome remodeling histone variants, as well as post-translational modifications (PTMs) of histones.4 Thus, PTMs of histones play a critical role in chromatin compaction, gene expression, transcriptional regulation, cell differentiation, and other biological processes.3,5−7 The proteins that are directly involved in PTMs of histones can be divided into three categories: the enzymes that create these modifications (the “writers”), the proteins that recognize the modifications (the “readers”), and the enzymes that remove the modifications (the “erasers”). Histone methyltransferases (HMTs), also referred to as protein methyltransferases (PMTs) since they methylate nonhistone proteins as well, are the methyl writers of the histone code.8,9 Histone methylation is one of the most studied post-translational modifications and has been associated with many human diseases.10−12 Therefore, HMTs have been increasingly pursued as potential therapeutic targets over the past decade.
HMTs catalyze the transfer of the methyl group from the cofactor S-5′-adenosyl-L-methionine (SAM) to either lysine or arginine residues of proteins. Therefore, HMTs can be further categorized into: (1) protein lysine methyltransferases (PKMTs) and (2) protein arginine methyltransferases (PRMTs). PKMTs catalyze mono-, di-, and/or trimethylation of lysine residues while PRMTs catalyze mono- and/or asymmetric or symmetric dimethylation of arginine guanidinium groups.13 Even though lysine or arginine methylation does not change the charge state, the increased bulkiness and hydrophobicity of the methylated residues would affect protein–protein interactions and protein recognition, thus gene expression and transcription regulation. To date, more than 50 PKMTs have been identified and characterized.5 All PKMTs except DOT1L (disruptor of telomeric silencing 1-like) contain a conserved, approximately 130 amino acid long SET (Su(var.)3–9 (the suppressor of position-effect variegation 3–9), En(zeste) (an enhancer of the eye color mutant zeste), and Trithorax (the homeotic gene regulator)) domain.14−16 Nine PRMTs have also been identified and characterized.17 All PRMTs do not have a SET domain but contain a conserved core region of about 310 amino acids.18
Potent, selective, and cell-penetrant chemical probes of HMTs would be invaluable tools for elucidating biological functions and disease association of the target HMTs and for assessing the potential of these enzymes as therapeutic targets. For example, well-characterized HMT chemical probes would enable the research community to test biological and disease hypotheses in cell-based and/or animal models with high confidence. Advantages of a small-molecule driven approach include (1) temporal resolution (rapid exposure and elimination of effects), (2) mechanistic flexibility (targeting a specific activity of a protein such as the catalytic activity of an enzyme as opposed to ablating the entire protein with knockout or knockdown), (3) ease of delivery (cell permeable and potentially orally bioavailable), and (4) immediate transition to drug discovery efforts (potentially cutting years off the time between target validation and therapeutic intervention).19 The concept of utilizing chemical probes for therapeutic hypothesis testing and target validation has been successfully applied to multiple drug discovery fields including kinase drug discovery.20 Therefore, it has been anticipated that chemical probes of HMTs will be extremely valuable tools for advancing HMT drug discovery.10,21
The term “chemical probe” has been used quite loosely by the biomedical community, and a lack of consensus on the clear definition of a “chemical probe” exists. In this review, we adopt the chemical probe criteria defined by Frye,22 Workman and Collins,23 and Bunnage et al.24 Therefore, high quality chemical probes are defined here as small-molecule inhibitors that have (1) high in vitro potency with an excellent and well-characterized selectivity profile, (2) a clear mechanism of action (MOA), and (3) robust on-target activities in cells. It is important to note that adequate cell permeability and target engagement in cells should be clearly demonstrated for chemical probes in addition to sufficient in vitro potency and selectivity and clear MOA. These relatively stringent criteria are necessary to associate the observed functional effects in cell-based and/or animal models with the inhibition of the target enzyme(s) by the chemical probes. Selective inhibitors of HMTs, many of which do not meet the chemical probe criteria, have been reviewed previously.25−27 Here, we focus on well-characterized chemical probes of HMTs and describe their discovery, characterization, and biological applications.
Chemical Probes of G9a and GLP
G9a (euchromatic histone-lysine N-methyltransferase 2 (EHMT2) or KMT1C) and GLP (G9a-like protein 1, also known as EHMT1 or KMT1D) are the main lysine methyltransferases that catalyze mono- and dimethylation of H3K9 (histone H3 lysine 9).28,29 They can form a heterodimer and share 80% sequence identity in their respective SET domains.29 Dimethylation of many nonhistone targets30 including the tumor suppressor p53 at lysine 373 has also been shown.31 G9a is overexpressed in a number of cancers,31−35 and knockdown of G9a via RNA interference inhibits prostate cancer,33 leukemia,34 and lung cancer cell growth.35 Loss of G9a results in significant delays in acute myeloid leukemia (AML) progression in mouse models.36 G9a also mediates H3K9me2 patterning during hematopoietic stem and progenitor cells (HSPCs) lineage specification.37 In addition, it has been shown that G9a is required for development of pathogenic T cells and intestinal inflammation.38 Furthermore, G9a has been implicated in maintenance of HIV-1 latency,39 cocaine addiction,40,41 and mental retardation.42 GLP is implicated in a disorder known as Kleefstra syndrome43,44 and is an essential PKMT in the PRDM16 (PR domain containing protein 16) transcriptional complex, which controls brown adipose cell fate and energy homeostasis.45
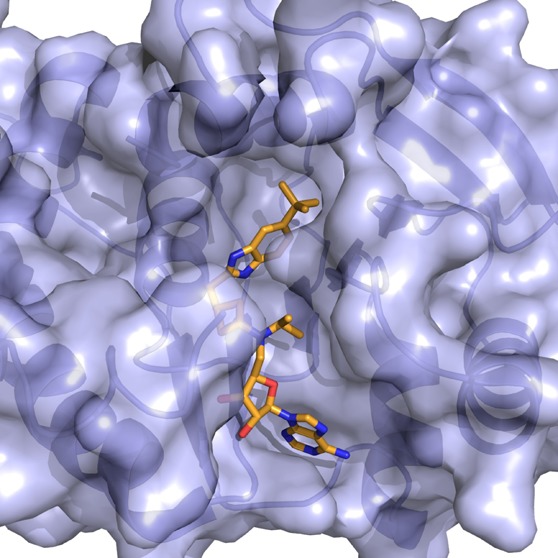
The first selective small-molecule inhibitor of G9a and GLP, BIX01294 (1), was reported by Kubicek et al. in 200746 and was a major advancement in the PKMT inhibitor field (Figure 1). BIX01294 selectively inhibited G9a (IC50 = 1.7 μM) and GLP (IC50 = 38 μM) over several other HMTs tested. It was later determined that BIX01294 displayed similar potency for G9a and GLP, and thus was not selective for G9a over GLP.47 The X-ray crystal structure of GLP in complex with the inhibitor and SAH (S-5′-adenosyl-L-homocysteine) obtained by Chang et al. (PDB code: 3FPD) revealed that BIX01294 bound to the substrate binding groove of GLP,47 consistent with BIX01294 being competitive with the peptide substrate in MOA studies. In addition, BIX01294 was active in cells and can selectively reduce the H3K9me2 (dimethylation of H3K9) mark at global levels and at promoters of G9a target genes.46 However, BIX01294 was toxic at concentrations above 4.1 μM in cellular assays and, consequently, it displayed a poor separation of functional potency and cell toxicity, which limits its use as a chemical probe. Liu et al. explored multiple regions of this quinazoline scaffold and discovered UNC0224 (2) in 2009, which was significantly more potent than BIX01294 in multiple biochemical and biophysical assays (Figure 1).48 UNC0224 possesses a 7-dimethylaminopropoxy chain that interacts with the lysine binding channel,36 which was not exploited by BIX01294. Further structure-based design and synthesis resulted in the discovery of UNC0321 (3), which has a longer chain instead of the three-carbon chain of UNC0224 (Figure 1).49 UNC0321 had a Morrison Ki of 63 pM and was 40- and 250-fold more potent than UNC0224 and BIX01294, respectively. It was more than 1000-fold selective for G9a and GLP over SETD7 (SET domain containing (lysine methyltransferase) 7, also known as KMT7, SET7, SET9, and SET7/9) and SETD8 (SET domain containing (lysine methyltransferase) 8, also known as SET8 or KMT5A) and also selective against a broad range of G-protein coupled receptors (GPCRs), ion channels, and transporters. However, even though UNC0321 was more potent than BIX01294 in biochemical assays, it was less potent in cellular assays most likely due to its relatively high polarity and poor cell membrane permeability.50 By optimizing physicochemical properties while maintaining high in vitro potency of this chemical series, Vedadi et al. discovered the G9a and GLP cellular chemical probe UNC0638 (4) in 2011 (Figure 1), which has a balanced in vitro potency, aqueous solubility, and cell membrane permeability.51
Figure 1.

Discovery of chemical probes for G9a and GLP.
UNC0638 was competitive with the peptide substrate and noncompetitive with the cofactor SAM.51 This MOA was confirmed by the X-ray crystal structure of G9a in complex with UNC0638 (PDB code: 3RJW). This inhibitor was more than 200-fold selective for G9a and GLP over 16 other methyltransferases and epigenetic targets. In addition, it was at least 100-fold selective over more than 80 GPCRs, kinases, ion channels, and transporters. UNC0638 (IC50 = 81 ± 9 nM) was more potent and efficacious than BIX01294 (IC50 = 500 ± 43 nM) at reducing global levels of H3K9me2 in MDA-MB-231 cells, a human breast carcinoma cell line, by using an in-cell western assay.51 In addition, it had low cell toxicity, thus an excellent functional potency/toxicity ratio of 138, while BIX01294 had a functional potency/toxicity ratio <6. UNC0638 also displayed a good separation of functional potency and cell toxicity in seven other cell lines. Using quantitative MS-based proteomics, Vedadi et al. confirmed that UNC0638 reduced cellular levels of H3K9me2, but did not change cellular levels of other 20 common histone modifications except H3K14ac, suggesting that cellular actions of UNC0638 are specific and there is a possible cross talk between H3K9me2 and H3K14ac.38 Other on-target activity of UNC0638 in cells include the following: (1) it selectively reduced the H3K9me2 mark at promoters of G9a target genes; (2) it reactivated a silent retroviral vector and G9a target genes in mES cells, and (3) also in mES cells, it reduced the H3K9me2 mark at promoters of G9a target genes and the retroviral long terminal repeat (LTR) region and indirectly induced DNA hypomethylation. UNC0638 as a chemical probe has been used in a number of biological and pharmacological studies. For example, it was recently reported that UNC0638 induced leukemia stem cell differentiation and suppressed the growth of primary human acute myeloid leukemia (AML), suggesting that G9a may be a therapeutic target for blocking the proliferation and self-renewal of AML cells.36 In addition, UNC0638 was reported to induce differentiation of wild-type T cells into regulatory T cells and Th17 cells, suggesting that pharmacological inhibition of G9a could be a strategy for the treatment of colitis.38 Furthermore, it has been shown that adult HSPCs treated with UNC0638 retained stem cell-like phenotypes and function better than the untreated, suggesting a potential application of pharmacological inhibition of G9a in stem cell renewal.37
Although UNC0638 is an excellent chemical probe of G9a and GLP for cell-based studies, it is not suitable for animal studies due to its poor in vivo pharmacokinetic (PK) properties.51 Further optimization of this chemical series resulted in the discovery of UNC0642 (5) as the first in vivo chemical probe of G9a and GLP (Figure 1).52 Similar to UNC0638, UNC0642 had high in vitro potencies for both G9a and GLP (IC50 < 2.5 nM) and was a substrate competitive inhibitor with a Ki of 3.7 ± 1 nM. It was also at least 2000-fold selective for G9a and GLP over 14 other methyltransferases and selective against a broad panel of over 90 kinases, GPCRs, transporters, and ion channels. In a number of tumor and normal cell lines, UNC0642 significantly reduced the H3K9me2 mark and displayed low cell toxicity. Phenotypically, it reduced clonogenicity in PANC-1 cells (a human pancreatic epithelioid carcinoma cell line). Importantly, UNC0642 displayed improved exposure in plasma compared to UNC0638 in mouse PK studies, making it suitable for animal studies as an in vivo chemical probe of G9a and GLP.
BIX01294 was the first chemical probe of G9a and GLP albeit its limited use due to its poor separation of functional potency and cell toxicity. Optimization of this quinazoline scaffold led to the discovery of the cellular chemical probe UNC0638 and in vivo chemical probe UNC0642. These probes were extensively characterized in a battery of biochemical, biophysical, and cellular assays. UNC0638 and UNC0642 not only displayed high in vitro potency and excellent selectivity but also exhibited robust on-target activities in cells. These chemical probes are useful tools for interrogating the role of G9a and GLP in health and disease in cell-based and/or animal studies.
Chemical Probes of EZH2
The recent discovery of several high quality chemical probes for EZH2/EZH1 (enhancer of zeste 2 polycomb repressive complex 2 subunit, also known as KMT6A/enhancer of zeste homologue 1, also known as KMT6B) was another major advancement in the HMT inhibitor field. As we will discuss below, the potential of these methyltransferases as therapeutic targets was brought to the spotlight by very promising results of these chemical probes in cell-based and animal models. The main biological function of polycomb repressive complex 2 (PRC2), a multisubunit protein complex that catalyzes methylation of H3K27 (histone H3 lysine 27), is transcriptional silencing of genes involved in differentiation and development.53−58 The H3K27me3 (trimethylation of H3K27) mark is a very well-known repressive mark.56,59 PRC2 is conserved from Drosophila to humans,53 and EZH2 or EZH1 is the catalytic subunit of the complex. The C-terminal SET domains of EZH2 and EZH1 are essential for the methyltransferase activity and are highly homologous with 96% sequence identity.60 Although EZH2 or EZH1 is the catalytic subunit of PRC2, it does not have enzymatic activity by itself and needs both EED (embryonic ectoderm development) and SUZ12 (suppressor of zeste 12) to gain the methyltransferase activity.61−63 PRC2 complexes containing EZH1 (PRC2–EZH1) or EZH2 (PRC2–EZH2) are involved in the maintenance of H3K27 methylation in cells.56,64,65 Point mutations at tyrosine 641 (Y641F, N, S, H, and C) in the C-terminal SET domain of EZH2 have been identified66 and were observed in 7% of follicular lymphomas and 22% of germinal center B-cell (GCB) and diffuse large B-cell lymphomas (DLBCLs).66−68 While the wild-type EZH2 prefers unmethylated H3K27 as its substrate, the gain-of-function Y641 mutations prefer for H3K27me2 as the substrate.69,70 Thus, the wild-type EZH2 and Y641 mutants work cooperatively, leading to increased levels of H3K27me3 in tumor tissues. Overexpression of EZH2 and/or hypertrimethylation of H3K27 have been associated with a number of human cancers71−73 such as breast,74 prostate,75 lymphoma,76 and leukemia.77
The first selective small-molecule inhibitor of EZH2 was reported by Knutson et al. in 2012.78 High throughput screening (HTS) of a 175 000-member chemical library against the recombinant PRC2 complex led to the identification of the pyridone 6 (IC50 = 620 nM, Ki = 310 nM) as a tractable hit (Figure 2). Optimization of this hit by (1) addition of an amine functionality at the para-position of the phenyl ring, (2) replacing the 5,6-fused pyrrazolapyridine with the indazole, and (3) the addition of a larger lipophilic group at the indazole nitrogen resulted in the first EZH2 chemical probe EPZ005687 (7; Figure 2), which was more potent and soluble than compound 6. EPZ005687 potently inhibited wild-type EZH2 with an IC50 of 54 ± 5 nM and displayed similar potencies for EZH2 Y641 mutants (Y641N, F, S, H, and C). Michaelis Menten kinetic studies indicated that it was competitive with SAM with a Ki of 24 ± 7 nM and noncompetitive with the substrate. EPZ005687 was about 50-fold more selective for EZH2 over EZH1, displayed more than 500-fold selectivity for EZH2 over 14 other methyltransferases, and showed no significant inhibition at 10 μM against most of the 77 GPCRs and ion channels tested.
Figure 2.

Discovery of the EZH2 chemical probes EPZ005687 and EPZ-6438.
Further optimization of EPZ005687 by the same group led to the discovery of EPZ-6438 (8; Figure 2), another EZH2 chemical probe with superior potency and in vivo PK properties.79 This new probe contains the same pyridone core as other EZH2 inhibitors but does not have the indole/indazole motif (Figure 2). EPZ-6438 inhibited wild-type EZH2 with a Ki of 2.5 ± 0.5 nM and known EZH2 mutants with similar potencies. Similar to EPZ005687, EPZ-6438 was competitive with SAM and noncompetitive with the peptide substrate in MOA studies. It was around 35-fold more selective for EZH2 over EZH1 and more than 4500-fold selective for EZH2 over 14 other methyltransferases.
Shortly after EPZ005687 was published, McCabe et al. reported the discovery of another EZH2 chemical probe GSK126 (11; Figure 3).80 Similar to EPZ005687, this discovery began with a tractable hit, GSK-A (9, Kiapp = 700 nM; Figure 3), which was identified by HTS of the corporate compound collection.81 Interestingly, the two screening hits (GSK-A and compound 6) share very high structural similarity (the only difference is one substituent at the 5,6-fused pyrrazolapyridine). Optimization of GSK-A resulted in the discovery of an early lead GSK343 (10; Figure 3)60 and ultimately led to the discovery of GSK126, which is suitable for in vivo studies (see below). GSK126 potently inhibited both wild-type and mutant EZH2 enzymatic activity (Kiapp = 0.5–3 nM).80 It contains the same pyridone functionality as EPZ005687 but has an indole instead of indazole in its core structure (Figure 3). Similar to EPZ005687, GSK126 was competitive with the cofactor SAM and noncompetitive with peptide substrates in MOA studies. It was more than 150-fold more selective for EZH2 over EZH1 and more than 1000-fold more selective for EZH2 over 20 other methyltransferases. In addition, GSK126 did not significantly inhibit a broad panel of kinases, GPCRs, ion channels, and transporters as well as several other chromatin modifying enzymes.
Figure 3.

Discovery of the EZH2 chemical probe GSK126.
In the same year, another EZH2 chemical probe, EI1 (12), was discovered via HTS by Qi et al.82 The same pyridone/indole core was featured in this inhibitor (Figure 4). EI1 was also competitive with the cofactor SAM with a Ki of 13 ± 3 nM and inhibited the enzymatic activity of wild-type EZH2 and Y641F mutant (IC50 =15 ± 2 nM and 13 ± 3 nM, respectively). EI1 was about 90-fold more selective for EZH2 over EZH1 and exceptionally more selective (>10 000 fold) for EZH2 over 10 other methyltransferases.
Figure 4.

Structures of the EZH2/EZH1 chemical probes EI1 and UNC1999 and inactive control UNC2400.
In 2013, Konze et al. discovered an orally bioavailable chemical probe of EZH2 and EZH1, UNC1999 (13; Figure 4).83 This probe was created by combining key structural features of EPZ005687 and GSK126 and extensively exploring the morpholinomethyl group of EPZ005687 to improve physicochemical properties. UNC1999 not only exhibited high in vitro potency for EZH2 (IC50 < 10 nM) but also possesses more desirable lipophilicity (clogP = 3.1), a likely key contributor to the observed oral bioavailability. The MOA studies of UNC1999 indicated that it was a SAM-competitive EZH2 inhibitor with a Ki of 4.6 ± 0.8 nM and was noncompetitive with the peptide substrate. UNC1999 also displayed similar potencies for Y641 mutants. Interestingly, UNC1999 was only about 10- to 15-fold selective for EZH2 over EZH1, in contrast to other EZH2 chemical probes discussed earlier. Therefore, it would be a unique tool in cellular and disease settings where both PRC2–EZH2 and PRC2–EZH1 are significant contributors to H3K27 methylation. UNC1999 was more than 10 000-fold more selective for EZH2 over 15 other methyltransferases and selective over a broad panel of more than 90 kinases, GPCRs, transporters, and ion channels. In addition, Konze et al. generated an inactive control, UNC2400 (14; Figure 4), for cell-based studies.83 UNC2400 is a dimethylated analog of UNC1999 but was more than 1000-fold less potent than UNC1999. The high structural similarity and drastic potency difference between UNC1999 and UNC2400 make them excellent positive and negative control tool compounds.
The EZH2 chemical probes discussed above (EPZ005687, EPZ-6438, GSK126, EI1, and UNC1999) all displayed robust on-target activities in cells. For example, these probes selectively reduced the H3K27me3 mark in a number of EZH2 wild-type and mutant cell lines.78−80,82,83 Phenotypically, EPZ005687, GSK126, EI1, and UNC1999 selectively killed DLBCL cell lines harboring the gain-of-function Y641 mutants. EPZ-6438 displayed antiproliferative effects in SMARCB1-deficient MRT (malignant rhabdoid tumor) cells (G401) but did suppress proliferation of RD cells, which contain wild-type SMARCB1.79 Importantly, GSK126 and EPZ-6438 have been shown to be efficacious in vivo. For example, intraperitoneal (IP) administration of GSK126 led to a drastic reduction in tumor volume and a significant improvement in survival in the more aggressive KARPAS-422 (a DLBCL cell line harboring the EZH2 Y641N mutation) tumor xenograft mouse models.80 After the GSK126 treatment was stopped, complete tumor regression had been observed in high dose treatment groups for several weeks. In addition, oral administration of EPZ-6438 completely eliminated G401 xenografts in severe combined immunodeficiency (SCID) mice.79 The complete tumor eradication was also sustained for multiple weeks after the cessation of the EPZ-6438 treatment. In both in vivo studies, the inhibitors were well tolerated. In 2013, EPZ-6438 became the first EZH2 inhibitor entering human clinical trials.84 GSK126 was another EZH2 chemical probe that was advanced to phase 1 clinical trials for the treatment of GCB–DLBCL.85 The rapid advancement of these chemical probes into clinical evaluation has generated lasting excitement in the biomedical community.
Chemical Probes of DOT1L
DOT1L (disruptor of telomeric silencing 1-like, also known as KMT4) is the only PKMT identified in humans that has a non-SET catalytic domain instead of a SET domain.86−88 DOT1L is responsible for mono-, di-, and trimethylation of H3K79 (histone H3 lysine 79).86,89 Methylation of H3K79 is generally associated with transcriptional activation. It has been linked with DNA repair, embryonic development, cell cycle regulation, hematopoiesis, and cardiac function.90−92 DOT1L interacts with MLL (lysine (K)-specific methyltransferase 2A (KMT2A) or MLL1)) fusion proteins (such as AF4, AF9, AF10, AF6, and ENL) and is recruited to leukemogenic genes such as HOXA9 and MEIS1.90,93−97 DOT1L has been pursued as a potential drug target for the treatment of MLL-rearranged leukemia since the interaction of DOT1L with MLL fusion proteins drives leukemogenesis via abnormal methylation.21,98
The first chemical probe of DOT1L, EPZ004777 (15), was reported by Daigle et al. in 2011 (Figure 5).99 The design of this inhibitor was based on the chemical structures of the cofactor SAM and cofactor product SAH as well as an apo crystal structure of the enzyme active site (PDB code: 1NW3). A series of SAM analogs was synthesized and tested, resulting in the discovery of EPZ004777, which exhibited very high potency (IC50 = 400 ± 100 pM) for DOT1L. Remarkably, EPZ004777 was more than 1000-fold more selective for DOT1L over nine other HMTs despite its structural similarity to SAM. The MOA studies as expected indicated that the inhibitor was SAM competitive.100 EPZ004777 displayed a very long residence time, which was mainly driven by a ligand-induced conformational adaptation of DOT1L.100 In cell-based assays, EPZ004777 reduced global levels of H3K79me2 (dimethylation of H3K79) in a number of leukemia cell lines and did not significantly reduce any other histone marks.99 In addition, EPZ004777 inhibited the expression of HOXA9 and MEIS1 in a concentration-dependent manner. Phenotypically, this inhibitor displayed a drastic antiproliferative effect in MLL-rearranged leukemia cell lines.
Figure 5.

Structures of SAM and DOT1L chemical probes.
In 2012, a new chemical probe of DOT1L, SGC0946 (16), with improved potency was reported by Yu et al. (Figure 5).101 The deazaadenosine moiety of EPZ004777 was optimized, resulting in the discovery of SGC0946, which possesses a bromo substitution at the 7 position of the deazaadenosine ring (Figure 5). Although SGC0946 displayed similar potency (IC50 = 0.3 ± 0.1 nM) for DOT1L as EPZ004777 (IC50 = 0.5 ± 0.1 nM) in biochemical assays, it (IC50 = 8.8 ± 1.6 nM) was about 10-fold more potent at reducing H3K79Me2 levels in MCF10A cells than EPZ004777 (IC50 = 84 ± 20 nM) in an in-cell western assay. SGC0946 was highly selective for DOT1L over 13 other methyltransferases. The improved potency of SGC0946 in cellular assays was attributed to a lower off rate together with enhanced cell membrane permeability.
The third chemical probe of DOT1L, EPZ-5676 (17), was reported by Daigle et al. in 2013 (Figure 5).102 This inhibitor was discovered as a result of structure-guided design and optimization studies of the EPZ004777 series.102 EPZ-5676 was highly potent for DOT1L with a Morrison Ki of 0.08 ± 0.03 nM. It was remarkably (more than 37 000-fold) more selective for DOT1L over 15 other methyltransferases. The X-ray crystal structure of the DOT1L–EPZ-5676 complex (PDB code: 4HRA) clearly indicated that (1) EPZ-5676 occupied the cofactor binding pocket and (2) ligand binding induced conformational changes in the protein. EPZ-5676 reduced global levels of H3K79me2 in several cell lines including MV4-11 (a MLL-AF4 expressing acute leukemia cell line) but did not significantly change other histone methylation marks. It also inhibited HOXA9 and MEIS1 mRNA levels in MV4-11 cells. Additionally, EPZ-5676 displayed nanomolar antiproliferative activity against multiple MLL-rearranged cell lines including MV4-11 but weaker potencies against non-MLL-rearranged cell lines.
The DOT1L chemical probes EPZ004777, SGC0946, and EPZ-5676 not only exhibited high in vitro potency, outstanding selectivity, and clear MOA but also displayed robust on-target activities in cells. Significantly, EPZ004777 and EPZ-5676 have demonstrated in vivo efficacy in animal studies. For example, EPZ004777 (by continuous infusion via an implanted osmotic mini-pump) dose-dependently increased survival in NSG (NOD scid gamma) mice injected with MV4-11 cells.99 Similarly, continuous infusion of EPZ-5676 completely eliminated the established MV4-11 tumors in immune-compromised rats.102 The tumor regression was sustained for multiple weeks after the cessation of the treatment, and EPZ-5676 was well tolerated by the test animals. Thus, it was demonstrated for the first time that pharmacological inhibition of DOT1L had antitumor activity in animal models of MLL-rearranged leukemia. On the basis of these promising results, in 2013, EPZ-5676 became the very first HMT inhibitor that entered phase 1 clinical trials for the treatment of AML (acute myeloid leukemia) and ALL (acute lymphoblastic leukemia).103,104 This watershed event highlights the importance of high quality chemical probes.
Chemical Probes of SETD7
SETD7 (SET domain containing (lysine methyltransferase) 7, also known as KMT7, SET7, SET9, and SET7/9) is responsible for monomethylation of H3K4 (histone H3 lysine 4) and many nonhistone proteins (e.g., p53, Rb, Tat, Foxo3, DNMT1).105 It contributes to upregulation of the gene encoding p65 subunit of NF-κB in response to glucose; therefore, SETD8 has been associated with hyperglycemia.106 Despite its association with diverse substrates and molecular pathways, the functional role of this enzyme in disease, and normal biology is largely unknown.
The first chemical probe of SETD7, (R)-PFI-2 (18), was very recently published by Barsyte-Lovejoy et al. (Figure 6).107 HTS of a 150 000-compound collection using a recombinant SETD7 biochemical assay led to the identification of an initial hit (IC50 = 2.1 μM), which was optimized via structure-guided molecular design and synthesis. Further optimization of cell membrane permeability resulted in the discovery of (R)-PFI-2. It was highly potent for SETD7 (IC50 = 2.0 ± 0.2 nM, Kiapp = 0.33 ± 0.04 nM). Its enantiomer, (S)-PFI-2 (19) (Figure 6), was 500-fold less potent and, therefore, was an excellent negative control for chemical biology experiments. (R)-PFI-2 was more than 1000-fold more selective for SETD7 over 19 other methyltransferases and was inactive against 134 additional ion channel, GPCR, and enzyme targets.
Figure 6.

Structures of the SETD7 chemical probe, (R)-PFI-2, and its enantiomer.
The X-ray crystal structure of the SETD7–(R)-PFI-2 complex (PDB code: 4JLG) revealed that the inhibitor occupied the substrate binding groove.107 However, in surface plasmon resonance (SPR) experiments, (R)-PFI-2 bound to SETD7 only in the presence of SAM. These studies suggested that inhibition of SETD7 by (R)-PFI-2 was not purely substrate competitive, and SAM had a significant role in the binding of (R)-PFI-2 to SETD7. Thus, (R)-PFI-2 displayed an unusual cofactor-dependent and substrate-competitive MOA. In a cellular thermal shift assay (CETSA), (R)-PFI-2 increased the stability of SETD7 by 4 °C. The direct binding of (R)-PFI-2 to SETD7 in cells was also demonstrated by the pull-down studies using a biotinylated derivative of (R)-PFI-2. Therefore, (R)-PFI-2 engages its target, SETD7, in cells. Furthermore, this probe concentration-dependently increased nuclear localization of Yes-Associated Protein (YAP), a transcriptional coactivator, and induced expression of YAP target genes in cells. Pharmacological inhibition of SETD7 by (R)-PFI-2 phenocopied genetic deletion of SETD7. In summary, PFI-2 has high in vitro potency and selectivity, clear but unusual MOA, and on-target activities in cells. It will therefore be a very useful tool for investigating the functional role of SETD7 in health and disease.
Conclusion
HMTs, the methyl writers of the histone code, play a critical role in regulating gene expression and the chromatin state and have been implicated in many human diseases including cancer. Over the past decade, there has been rapidly growing interest in pursuing these enzymes as potential therapeutic targets. A systematic coverage of HMTs as a target class with high quality chemical probes would transform this extremely promising field. Well-characterized chemical probes of HMTs would be invaluable tools for investigating biological functions and disease association of the target HMTs and for assessing the potential of these enzymes as drug targets. In this review, we discussed the tremendous progress made in this now very active research area. During the past 3 years, a total of 11 high quality chemical probes (UNC0638 (G9a/GLP), UNC0642 (G9a/GLP), EPZ005687 (EZH2), EPZ-6438 (EZH2), GSK126 (EZH2), EI1 (EZH2), UNC1999 (EZH2/EZH1), EPZ004777 (DOT1L), SGC0946 (DOT1L), EPZ-5676 (DOT1L), and (R)-PFI-2 (SETD7)) has been discovered. These chemical probes have been utilized in a number of cell-based and/or animal-based disease models. In particular, EPZ-5676, EPZ-6438, and GSK126 have demonstrated in vivo efficacy in multiple tumor xenograft models and have been advanced to clinic evaluation, highlighting the importance of chemical probes. Despite the excellent progress that has been made during the relatively short period, we believe there is still much to be accomplished in this exciting field. More than 50 PKMTs and nine PRMTs have been identified. However, high quality chemical probes have been reported for only a small fraction of PKMTs (G9a/GLP, EZH2/EZH1, DOT1L, and SETD7), and no chemical probes have been reported for PRMTs. Creation of first-in-class chemical probes for many PKMTs and PRMTs is urgently needed. It is worth to note that many selective HMT inhibitors reported to date do not meet the chemical probe criteria we outlined in this review. To confidently associate the observed phenotypic effects in cell-based and/or animal models with the inhibition of the target enzyme(s), the inhibitors used in the studies must have high in vitro potency and selectivity, clear MOA, and robust on-target activities in cells. It is therefore a challenge and opportunity for medicinal chemists in the field to optimize the selective HMT inhibitors that do not currently meet the above criteria into high quality chemical probes. It has been an exciting decade for the HMT field. We believe that the momentum of the field is accelerating. It is therefore highly anticipated that many first-in-class chemical probes of HMTs will be discovered in the years to come.
Acknowledgments
This work was supported by grant R01GM103893 (to J.J.) from the U.S. National Institutes of Health. H.Ü.K. was supported by a postdoctoral fellowship from the Structural Genomics Consortium. We also thank Dr. Nicolas Babault for generating the TOC Graphic.
Glossary
Keywords
- Epigenetics
Epigenetics is referred to heritable changes in gene expression or phenotype without changes in DNA sequence
- HMTs
Histone methyltransferases are the enzymes that responsible for methylation of histone and nonhistone proteins
- PKMTs
Protein lysine methyltransfearases catalyze mono, di, and/or trimethylation of lysine residues
- PRMTs
Protein aginine methyltransfearases catalyze mono and/or asymmetric or symmetric dimethylation of arginine residues
- Chemical probes
Potent, selective, and cell-penetrant small-molecule inhibitors that used as tools for elucidating biological functions and disease association of the target proteins and for testing therapeutic hypotheses
- G9a/GLP
Euchromatic histone-lysine N-methyltransferase 2 (G9a) and G9a–like protein 1 (GLP) are the main lysine methyltransferases that catalyze mono- and dimethylation of histone H3 lysine 9
- EZH2/EZH1
Enhancer of zeste 2 or 1 is the catalytic subunit of polycomb repressive complex 2 (PRC2) that catalyzes methylation of histone H3 lysine 27
- DOT1L
Disruptor of telomeric silencing 1-like catalyzes mono, di, and trimethylation of histone H3 lysine 79 and is the only PKMT identified in humans with a non-SET catalytic domain
- SETD7
SET domain containing (lysine methyltransferase) 7 is responsible for monomethylation of H3 lysine 4 as well as many nonhistone proteins
The authors declare no competing financial interest
Figure 4 has been updated. The revised version was re-posted on November 26, 2014.
Funding Statement
National Institutes of Health, United States
References
- Berger S. L.; Kouzarides T.; Shiekhattar R.; Shilatifard A. (2009) An operational definition of epigenetics. Genes Dev. 23, 781–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. A.; Baylin S. B. (2007) The epigenomics of cancer. Cell 128, 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein B. E.; Meissner A.; Lander E. S. (2007) The mammalian epigenome. Cell 128, 669–681. [DOI] [PubMed] [Google Scholar]
- Allis C. D., Jenuwein T., and Reinberg D. (2007) Epigenetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Kouzarides T. (2007) Chromatin modifications and their function. Cell 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Gelato K. A.; Fischle W. (2008) Role of histone modifications in defining chromatin structure and function. Biol. Chem. 389, 353–363. [DOI] [PubMed] [Google Scholar]
- Grewal S. I.; Moazed D. (2003) Heterochromatin and epigenetic control of gene expression. Science 301, 798–802. [DOI] [PubMed] [Google Scholar]
- Jenuwein T.; Allis C. D. (2001) Translating the histone code. Science 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- Strahl B. D.; Allis C. D. (2000) The language of covalent histone modifications. Nature 403, 41–45. [DOI] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Bountra C.; Fish P. V.; Lee K.; Schapira M. (2012) Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discovery 11, 384–400. [DOI] [PubMed] [Google Scholar]
- Helin K.; Dhanak D. (2013) Chromatin proteins and modifications as drug targets. Nature 502, 480–488. [DOI] [PubMed] [Google Scholar]
- Zagni C.; Chiacchio U.; Rescifina A. (2013) Histone methyltransferase inhibitors: novel epigenetic agents for cancer treatment. Curr. Med. Chem. 20, 167–185. [DOI] [PubMed] [Google Scholar]
- Martin C.; Zhang Y. (2005) The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 6, 838–849. [DOI] [PubMed] [Google Scholar]
- Jenuwein T.; Laible G.; Dorn R.; Reuter G. (1998) SET domain proteins modulate chromatin domains in eu- and heterochromatin. Cell. Mol. Life Sci. 54, 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumbusch L. O.; Thorstensen T.; Krauss V.; Fischer A.; Naumann K.; Assalkhou R.; Schulz I.; Reuter G.; Aalen R. B. (2001) The Arabidopsis thaliana genome contains at least 29 active genes encoding SET domain proteins that can be assigned to four evolutionarily conserved classes. Nucleic Acids Res. 29, 4319–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X.; Collins R. E.; Zhang X. (2005) Structural and sequence motifs of protein (histone) methylation enzymes. Annu. Rev. Biophys. Biomol. Struct. 34, 267–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lorenzo A.; Bedford M. T. (2011) Histone arginine methylation. FEBS Lett. 585, 2024–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Zhou L.; Cheng X. (2000) Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. EMBO J. 19, 3509–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss W. A.; Taylor S. S.; Shokat K. M. (2007) Recognizing and exploiting differences between RNAi and small-molecule inhibitors. Nat. Chem. Biol. 3, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Yang P. L.; Gray N. S. (2009) Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A.; Solomon M. E.; Richon V. M. (2009) Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discovery 8, 724–732. [DOI] [PubMed] [Google Scholar]
- Frye S. V. (2010) The art of the chemical probe. Nat. Chem. Biol. 6, 159–161. [DOI] [PubMed] [Google Scholar]
- Workman P.; Collins I. (2010) Probing the probes: fitness factors for small molecule tools. Chem. Biol. 17, 561–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnage M. E.; Chekler E. L.; Jones L. H. (2013) Target validation using chemical probes. Nat. Chem. Biol. 9, 195–199. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Patel D. J. (2013) Small molecule epigenetic inhibitors targeted to histone lysine methyltransferases and demethylases. Q. Rev. Biophys. 46, 349–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweis R. F.; Michaelides M. R. (2013) Recent Advances in Small-Molecule Modulation of Epigenetic Targets: Discovery and Development of Histone Methyltransferase and Bromodomain Inhibitors. Annu. Rep. Med. Chem. 48, 185–203. [Google Scholar]
- Finley A.; Copeland R. A. (2014) Small Molecule Control of Chromatin Remodeling. Chem. Biol. 21, 1196–1210. [DOI] [PubMed] [Google Scholar]
- Tachibana M.; Sugimoto K.; Nozaki M.; Ueda J.; Ohta T.; Ohki M.; Fukuda M.; Takeda N.; Niida H.; Kato H.; Shinkai Y. (2002) G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 16, 1779–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M.; Ueda J.; Fukuda M.; Takeda N.; Ohta T.; Iwanari H.; Sakihama T.; Kodama T.; Hamakubo T.; Shinkai Y. (2005) Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 19, 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P.; Dhayalan A.; Murakami M.; Zhang X.; Tamas R.; Jurkowska R.; Komatsu Y.; Shinkai Y.; Cheng X.; Jeltsch A. (2008) Protein lysine methyltransferase G9a acts on non-histone targets. Nat. Chem. Biol. 4, 344–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Dorsey J.; Chuikov S.; Zhang X.; Jenuwein T.; Reinberg D.; Berger S. L. (2010) G9A and GLP methylate lysine 373 in the tumor suppressor p53. J. Biol. Chem. 285, 9636–9641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y.; Shen L.; Suzuki S.; Kurokawa T.; Masuko K.; Tanaka Y.; Kato H.; Mizuno Y.; Yokoe M.; Sugauchi F.; Hirashima N.; Orito E.; Osada H.; Ueda R.; Guo Y.; Chen X.; Issa J. P.; Sekido Y. (2007) Alterations of DNA methylation and histone modifications contribute to gene silencing in hepatocellular carcinomas. Hepatol. Res. 37, 974–983. [DOI] [PubMed] [Google Scholar]
- Kondo Y.; Shen L.; Ahmed S.; Boumber Y.; Sekido Y.; Haddad B. R.; Issa J. P. (2008) Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS One 3, e2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyama S.; Nitta E.; Yoshino T.; Kako S.; Watanabe-Okochi N.; Shimabe M.; Imai Y.; Takahashi K.; Kurokawa M. (2010) EVI-1 interacts with histone methyltransferases SUV39H1 and G9a for transcriptional repression and bone marrow immortalization. Leukemia 24, 81–88. [DOI] [PubMed] [Google Scholar]
- Watanabe H.; Soejima K.; Yasuda H.; Kawada I.; Nakachi I.; Yoda S.; Naoki K.; Ishizaka A. (2008) Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell Int. 8, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnertz B.; Pabst C.; Su L.; Miller M.; Liu F.; Yi L.; Zhang R.; Krosl J.; Yung E.; Kirschner J.; Rosten P.; Underhill T. M.; Jin J.; Hebert J.; Sauvageau G.; Humphries R. K.; Rossi F. M. (2014) The methyltransferase G9a regulates HoxA9-dependent transcription in AML. Genes Dev. 28, 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Skutt-Kakaria K.; Davison J.; Ou Y. L.; Choi E.; Malik P.; Loeb K.; Wood B.; Georges G.; Torok-Storb B.; Paddison P. J. (2012) G9a/GLP-dependent histone H3K9me2 patterning during human hematopoietic stem cell lineage commitment. Genes Dev. 26, 2499–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antignano F.; Burrows K.; Hughes M. R.; Han J. M.; Kron K. J.; Penrod N. M.; Oudhoff M. J.; Wang S. K.; Min P. H.; Gold M. J.; Chenery A. L.; Braam M. J.; Fung T. C.; Rossi F. M.; McNagny K. M.; Arrowsmith C. H.; Lupien M.; Levings M. K.; Zaph C. (2014) Methyltransferase G9A regulates T cell differentiation during murine intestinal inflammation. J. Clin. Invest. 124, 1945–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K.; Togami H.; Okamoto T. (2010) Involvement of histone H3 Lysine 9 (H3K9) methyl transferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 285, 16538–16545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I.; Covington H. E. III; Dietz D. M.; LaPlant Q.; Renthal W.; Russo S. J.; Mechanic M.; Mouzon E.; Neve R. L.; Haggarty S. J.; Ren Y.; Sampath S. C.; Hurd Y. L.; Greengard P.; Tarakhovsky A.; Schaefer A.; Nestler E. J. (2010) Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science 327, 213–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington H. E. III; Maze I.; Sun H.; Bomze H. M.; DeMaio K. D.; Wu E. Y.; Dietz D. M.; Lobo M. K.; Ghose S.; Mouzon E.; Neve R. L.; Tamminga C. A.; Nestler E. J. (2011) A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron 71, 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer A.; Sampath S. C.; Intrator A.; Min A.; Gertler T. S.; Surmeier D. J.; Tarakhovsky A.; Greengard P. (2009) Control of Cognition and Adaptive Behavior by the GLP/G9a Epigenetic Suppressor Complex. Neuron 64, 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleefstra T.; Brunner H. G.; Amiel J.; Oudakker A. R.; Nillesen W. M.; Magee A.; Genevieve D.; Cormier-Daire V.; van Esch H.; Fryns J. P.; Hamel B. C.; Sistermans E. A.; de Vries B. B.; van Bokhoven H. (2006) Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am. J. Hum. Genet. 79, 370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleefstra T.; van Zelst-Stams W. A.; Nillesen W. M.; Cormier-Daire V.; Houge G.; Foulds N.; van Dooren M.; Willemsen M. H.; Pfundt R.; Turner A.; Wilson M.; McGaughran J.; Rauch A.; Zenker M.; Adam M. P.; Innes M.; Davies C.; Lopez A. G.; Casalone R.; Weber A.; Brueton L. A.; Navarro A. D.; Bralo M. P.; Venselaar H.; Stegmann S. P.; Yntema H. G.; van Bokhoven H.; Brunner H. G. (2009) Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of EHMT1 haploinsufficiency to the core phenotype. J. Med. Genet. 46, 598–606. [DOI] [PubMed] [Google Scholar]
- Ohno H.; Shinoda K.; Ohyama K.; Sharp L. Z.; Kajimura S. (2013) EHMT1 controls brown adipose cell fate and thermogenesis through the PRDM16 complex. Nature 504, 163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicek S.; O’Sullivan R. J.; August E. M.; Hickey E. R.; Zhang Q.; Teodoro M. L.; Rea S.; Mechtler K.; Kowalski J. A.; Homon C. A.; Kelly T. A.; Jenuwein T. (2007) Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 25, 473–481. [DOI] [PubMed] [Google Scholar]
- Chang Y.; Zhang X.; Horton J. R.; Upadhyay A. K.; Spannhoff A.; Liu J.; Snyder J. P.; Bedford M. T.; Cheng X. (2009) Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat. Struct. Mol. Biol. 16, 312–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Chen X.; Allali-Hassani A.; Quinn A. M.; Wasney G. A.; Dong A.; Barsyte D.; Kozieradzki I.; Senisterra G.; Chau I.; Siarheyeva A.; Kireev D. B.; Jadhav A.; Herold J. M.; Frye S. V.; Arrowsmith C. H.; Brown P. J.; Simeonov A.; Vedadi M.; Jin J. (2009) Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a. J. Med. Chem. 52, 7950–7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Chen X.; Allali-Hassani A.; Quinn A. M.; Wigle T. J.; Wasney G. A.; Dong A.; Senisterra G.; Chau I.; Siarheyeva A.; Norris J. L.; Kireev D. B.; Jadhav A.; Herold J. M.; Janzen W. P.; Arrowsmith C. H.; Frye S. V.; Brown P. J.; Simeonov A.; Vedadi M.; Jin J. (2010) Protein lysine methyltransferase G9a inhibitors: design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines. J. Med. Chem. 53, 5844–5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Barsyte-Lovejoy D.; Allali-Hassani A.; He Y.; Herold J. M.; Chen X.; Yates C. M.; Frye S. V.; Brown P. J.; Huang J.; Vedadi M.; Arrowsmith C. H.; Jin J. (2011) Optimization of Cellular Activity of G9a Inhibitors 7-Aminoalkoxy-quinazolines. J. Med. Chem. 54, 6139–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedadi M.; Barsyte-Lovejoy D.; Liu F.; Rival-Gervier S.; Allali-Hassani A.; Labrie V.; Wigle T. J.; DiMaggio P. A.; Wasney G. A.; Siarheyeva A.; Dong A.; Tempel W.; Wang S.-C.; Chen X.; Chau I.; Mangano T.; Huang X.-P.; Simpson C. D.; Pattenden S. G.; Norris J. L.; Kireev D. B.; Tripathy A.; Edwards A.; Roth B. L.; Janzen W. P.; Garcia B. A.; Petronis A.; Ellis J.; Brown P. J.; Frye S. V.; Arrowsmith C. H.; Jin J. (2011) A Chemical Probe Selectively Inhibits G9a and GLP Methyltransferase Activity in Cells. Nat. Chem. Biol. 7, 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Barsyte-Lovejoy D.; Li F.; Xiong Y.; Korboukh V.; Huang X. P.; Allali-Hassani A.; Janzen W. P.; Roth B. L.; Frye S. V.; Arrowsmith C. H.; Brown P. J.; Vedadi M.; Jin J. (2013) Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J. Med. Chem. 56, 8931–8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Meara M. M.; Simon J. A. (2012) Inner workings and regulatory inputs that control Polycomb repressive complex 2. Chromosoma 121, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R.; Wang L.; Wang H.; Xia L.; Erdjument-Bromage H.; Tempst P.; Jones R. S.; Zhang Y. (2002) Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Kuzmichev A.; Nishioka K.; Erdjument-Bromage H.; Tempst P.; Reinberg D. (2002) Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 16, 2893–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R.; Reinberg D. (2011) The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czermin B.; Melfi R.; McCabe D.; Seitz V.; Imhof A.; Pirrotta V. (2002) Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111, 185–196. [DOI] [PubMed] [Google Scholar]
- Muller J.; Hart C. M.; Francis N. J.; Vargas M. L.; Sengupta A.; Wild B.; Miller E. L.; O’Connor M. B.; Kingston R. E.; Simon J. A. (2002) Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111, 197–208. [DOI] [PubMed] [Google Scholar]
- Zee B. M.; Levin R. S.; Xu B.; LeRoy G.; Wingreen N. S.; Garcia B. A. (2010) In vivo residue-specific histone methylation dynamics. J. Biol. Chem. 285, 3341–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S. K.; Tian X.; LaFrance L. V.; Duquenne C.; Suarez D. P.; Newlander K. A.; Romeril S. P.; Burgess J. L.; Grant S. W.; Brackley J. A.; Graves A. P.; Scherzer D. A.; Shu A.; Thompson C.; Ott H. M.; Aller G. S. V.; Machutta C. A.; Diaz E.; Jiang Y.; Johnson N. W.; Knight S. D.; Kruger R. G.; McCabe M. T.; Dhanak D.; Tummino P. J.; Creasy C. L.; Miller W. H. (2012) Identification of Potent, Selective, Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS Med. Chem. Lett. 3, 1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R.; Zhang Y. (2004) SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 15, 57–67. [DOI] [PubMed] [Google Scholar]
- Ketel C. S.; Andersen E. F.; Vargas M. L.; Suh J.; Strome S.; Simon J. A. (2005) Subunit contributions to histone methyltransferase activities of fly and worm polycomb group complexes. Mol. Cell. Biol. 25, 6857–6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini D.; Bracken A. P.; Jensen M. R.; Lazzerini Denchi E.; Helin K. (2004) Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 23, 4061–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X.; Liu Y.; Hsu Y.-J.; Fujiwara Y.; Kim J.; Mao X.; Yuan G.-C.; Orkin S. H. (2008) EZH1Mediates Methylation on Histone H3 Lysine 27 and Complements EZH2 in Maintaining Stem Cell Identity and Executing Pluripotency. Mol. Cell 32, 491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R.; Li G.; Sarma K.; Blais A.; Zavadil J.; Woodcock C. L.; Dynlacht B. D.; Reinberg D. (2008) Ezh1 and Ezh2Maintain Repressive Chromatin through Different Mechanisms. Mol. Cell 32, 503–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin R. D.; Johnson N. A.; Severson T. M.; Mungall A. J.; An J.; Goya R.; Paul J. E.; Boyle M.; Woolcock B. W.; Kuchenbauer F.; Yap D.; Humphries R. K.; Griffith O. L.; Shah S.; Zhu H.; Kimbara M.; Shashkin P.; Charlot J. F.; Tcherpakov M.; Corbett R.; Tam A.; Varhol R.; Smailus D.; Moksa M.; Zhao Y.; Delaney A.; Qian H.; Birol I.; Schein J.; Moore R.; Holt R.; Horsman D. E.; Connors J. M.; Jones S.; Aparicio S.; Hirst M.; Gascoyne R. D.; Marra M. A. (2010) Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 42, 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin R. D.; Mendez-Lago M.; Mungall A. J.; Goya R.; Mungall K. L.; Corbett R. D.; Johnson N. A.; Severson T. M.; Chiu R.; Field M.; Jackman S.; Krzywinski M.; Scott D. W.; Trinh D. L.; Tamura-Wells J.; Li S.; Firme M. R.; Rogic S.; Griffith M.; Chan S.; Yakovenko O.; Meyer I. M.; Zhao E. Y.; Smailus D.; Moksa M.; Chittaranjan S.; Rimsza L.; Brooks-Wilson A.; Spinelli J. J.; Ben-Neriah S.; Meissner B.; Woolcock B.; Boyle M.; McDonald H.; Tam A.; Zhao Y. J.; Delaney A.; Zeng T.; Tse K.; Butterfield Y.; Birol I.; Holt R.; Schein J.; Horsman D. E.; Moore R.; Jones S. J. M.; Connors J. M.; Hirst M.; Gascoyne R. D.; Marra M. A. (2011) Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 476, 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr J. G.; Stojanov P.; Lawrence M. S.; Auclair D.; Chapuy B.; Sougnez C.; Cruz-Gordillo P.; Knoechel B.; Asmann Y. W.; Slager S. L.; Novak A. J.; Dogan A.; Ansell S. M.; Link B. K.; Zou L. H.; Gould J.; Saksena G.; Stransky N.; Rangel-Escareno C.; Fernandez-Lopez J. C.; Hidalgo-Miranda A.; Melendez-Zajgla J.; Hernandez-Lemus E.; Celis A. S. C. Y.; Imaz-Rosshandler I.; Ojesina A. I.; Jung J.; Pedamallu C. S.; Lander E. S.; Habermann T. M.; Cerhan J. R.; Shipp M. A.; Getz G.; Golub T. R. (2012) Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. U. S. A. 109, 3879–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap D. B.; Chu J.; Berg T.; Schapira M.; Cheng S. W.; Moradian A.; Morin R. D.; Mungall A. J.; Meissner B.; Boyle M.; Marquez V. E.; Marra M. A.; Gascoyne R. D.; Humphries R. K.; Arrowsmith C. H.; Morin G. B.; Aparicio S. A. (2011) Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 117, 2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneeringer C. J.; Scott M. P.; Kuntz K. W.; Knutson S. K.; Pollock R. M.; Richon V. M.; Copeland R. A. (2010) Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. U. S. A. 107, 20980–20985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken A. P.; Pasini D.; Capra M.; Prosperini E.; Colli E.; Helin K. (2003) EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 22, 5323–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon J. A.; Lange C. A. (2008) Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 647, 21–29. [DOI] [PubMed] [Google Scholar]
- Chi P.; Allis C. D.; Wang G. G. (2010) Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 10, 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleer C. G.; Cao Q.; Varambally S.; Shen R.; Ota I.; Tomlins S. A.; Ghosh D.; Sewalt R. G.; Otte A. P.; Hayes D. F.; Sabel M. S.; Livant D.; Weiss S. J.; Rubin M. A.; Chinnaiyan A. M. (2003) EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 100, 11606–11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varambally S.; Dhanasekaran S. M.; Zhou M.; Barrette T. R.; Kumar-Sinha C.; Sanda M. G.; Ghosh D.; Pienta K. J.; Sewalt R. G.; Otte A. P.; Rubin M. A.; Chinnaiyan A. M. (2002) The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419, 624–629. [DOI] [PubMed] [Google Scholar]
- Velichutina I.; Shaknovich R.; Geng H.; Johnson N. A.; Gascoyne R. D.; Melnick A. M.; Elemento O. (2010) EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood 116, 5247–5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih A. H.; Abdel-Wahab O.; Patel J. P.; Levine R. L. (2012) The role of mutations in epigenetic regulators in myeloid malignancies. Nat. Rev. Cancer 12, 599–612. [DOI] [PubMed] [Google Scholar]
- Knutson S. K.; Wigle T. J.; Warholic N. M.; Sneeringer C. J.; Allain C. J.; Klaus C. R.; Sacks J. D.; Raimondi A.; Majer C. R.; Song J.; Scott M. P.; Jin L.; Smith J. J.; Olhava E. J.; Chesworth R.; Moyer M. P.; Richon V. M.; Copeland R. A.; Keilhack H.; Pollock R. M.; Kuntz K. W. (2012) A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 8, 890–896. [DOI] [PubMed] [Google Scholar]
- Knutson S. K.; Warholic N. M.; Wigle T. J.; Klaus C. R.; Allain C. J.; Raimondi A.; Porter Scott M.; Chesworth R.; Moyer M. P.; Copeland R. A.; Richon V. M.; Pollock R. M.; Kuntz K. W.; Keilhack H. (2013) Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. U. S. A. 110, 7922–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe M. T.; Ott H. M.; Ganji G.; Korenchuk S.; Thompson C.; Van Aller G. S.; Liu Y.; Graves A. P.; Iii A. D.; Diaz E.; Lafrance L. V.; Mellinger M.; Duquenne C.; Tian X.; Kruger R. G.; McHugh C. F.; Brandt M.; Miller W. H.; Dhanak D.; Verma S. K.; Tummino P. J.; Creasy C. L. (2012) EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112. [DOI] [PubMed] [Google Scholar]
- Diaz E.; Machutta C. A.; Chen S.; Jiang Y.; Nixon C.; Hofmann G.; Key D.; Sweitzer S.; Patel M.; Wu Z.; Creasy C. L.; Kruger R. G.; LaFrance L.; Verma S.; Pappalardi M. B.; Le B.; Van Aller G. S.; McCabe M. T.; Tummino P. J.; Pope A. J.; Thrall S. H.; Schwartz B.; Brandt M. (2012) Development and Validation of Reagents and Assays for EZH2 Peptide and Nucleosome High-Throughput Screens. J. Biomol. Screening 17, 1279–1292. [DOI] [PubMed] [Google Scholar]
- Qi W.; Chan H.; Teng L.; Li L.; Chuai S.; Zhang R.; Zeng J.; Li M.; Fan H.; Lin Y.; Gu J.; Ardayfio O.; Zhang J.-H.; Yan X.; Fang J.; Mi Y.; Zhang M.; Zhou T.; Feng G.; Chen Z.; Li G.; Yang T.; Zhao K.; Liu X.; Yu Z.; Lu C. X.; Atadja P.; Li E. (2012) Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl. Acad. Sci. U. S. A. 109, 21360–21365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konze K. D.; Ma A.; Li F.; Barsyte-Lovejoy D.; Parton T.; MacNevin C. J.; Liu F.; Gao C.; Huang X. P.; Kuznetsova E.; Rougie M.; Jiang A.; Pattenden S. G.; Norris J. L.; James L. I.; Roth B. L.; Brown P. J.; Frye S. V.; Arrowsmith C. H.; Hahn K. M.; Wang G. G.; Vedadi M.; Jin J. (2013) An Orally Bioavailable Chemical Probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem. Biol. 8, 1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ClinicalTrials.gov Identifier: NCT01897571. Study of E7438 (EZH2 Histone Methyl Transferase [HMT] Inhibitor) as a Single Agent in Subjects With Advanced Solid Tumors or With B Cell Lymphomas.
- ClinicalTrials.gov Identifier: NCT02082977. A Study to Investigate the Safety, Pharmacokinetics, Pharmacodynamics and Clinical Activity of GSK2816126 in Subjects With Relapsed/Refractory Diffuse Large B Cell and Transformed Follicular Lymphoma.
- Feng Q.; Wang H.; Ng H. H.; Erdjument-Bromage H.; Tempst P.; Struhl K.; Zhang Y. (2002) Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol. 12, 1052–1058. [DOI] [PubMed] [Google Scholar]
- Min J.; Feng Q.; Li Z.; Zhang Y.; Xu R. M. (2003) Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell 112, 711–723. [DOI] [PubMed] [Google Scholar]
- Schubert H. L.; Blumenthal R. M.; Cheng X. D. (2003) Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci. 28, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederiks F.; Tzouros M.; Oudgenoeg G.; van Welsem T.; Fornerod M.; Krijgsveld J.; van Leeuwen F. (2008) Nonprocessive methylation by Dot1 leads to functional redundancy of histone H3K79 methylation states. Nat. Struct. Mol. Biol. 15, 550–557. [DOI] [PubMed] [Google Scholar]
- Nguyen A. T.; Zhang Y. (2011) The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 25, 1345–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A. T.; He J.; Taranova O.; Zhang Y. (2011) Essential role of DOT1L in maintaining normal adult hematopoiesis. Cell Res. 21, 1370–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A. T.; Xiao B.; Neppl R. L.; Kallin E. M.; Li J. A.; Chen T. P.; Wang D. Z.; Xiao X. A.; Zhang Y. (2011) DOT1L regulates dystrophin expression and is critical for cardiac function. Genes Dev. 25, 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A. T.; Taranova O.; He J.; Zhang Y. (2011) DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood 117, 6912–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitoun E.; Oliver P. L.; Davies K. E. (2007) The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum. Mol. Genet. 16, 92–106. [DOI] [PubMed] [Google Scholar]
- Mueller D.; Bach C.; Zeisig D.; Garcia-Cuellar M. P.; Monroe S.; Sreekumar A.; Zhou R.; Nesvizhskii A.; Chinnaiyan A.; Hess J. L.; Slany R. K. (2007) A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 110, 4445–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A.; Lin M.; Naresh A.; Kitabayashi I.; Cleary M. L. (2010) A Higher-Order Complex Containing AF4 and ENL Family Proteins with P-TEFb Facilitates Oncogenic and Physiologic MLL-Dependent Transcription. Cancer Cell 17, 198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas D.; Milne T. A.; Basrur V.; Kim J.; Elenitoba-Johnson K. S. J.; Allis C. D.; Roeder R. G. (2011) Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes. Proc. Natl. Acad. Sci. U. S. A. 108, 15751–15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglin J. L.; Song Y. (2013) A medicinal chemistry perspective for targeting histone H3 lysine-79 methyltransferase DOT1L. J. Med. Chem. 56, 8972–8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle S. R.; Olhava E. J.; Therkelsen C. A.; Majer C. R.; Sneeringer C. J.; Song J.; Johnston L. D.; Scott M. P.; Smith J. J.; Xiao Y.; Jin L.; Kuntz K. W.; Chesworth R.; Moyer M. P.; Bernt K. M.; Tseng J. C.; Kung A. L.; Armstrong S. A.; Copeland R. A.; Richon V. M.; Pollock R. M. (2011) Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavapathruni A.; Jin L.; Daigle S. R.; Majer C. R.; Therkelsen C. A.; Wigle T. J.; Kuntz K. W.; Chesworth R.; Pollock R. M.; Scott M. P.; Moyer M. P.; Richon V. M.; Copeland R. A.; Olhava E. J. (2012) Conformational adaptation drives potent, selective and durable inhibition of the human protein methyltransferase DOT1L. Chem. Biol. Drug Des. 80, 971–980. [DOI] [PubMed] [Google Scholar]
- Yu W.; Chory E. J.; Wernimont A. K.; Tempel W.; Scopton A.; Federation A.; Marineau J. J.; Qi J.; Barsyte-Lovejoy D.; Yi J.; Marcellus R.; Iacob R. E.; Engen J. R.; Griffin C.; Aman A.; Wienholds E.; Li F.; Pineda J.; Estiu G.; Shatseva T.; Hajian T.; Al-awar R.; Dick J. E.; Vedadi M.; Brown P. J.; Arrowsmith C. H.; Bradner J. E.; Schapira M. (2012) Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 3, 1288. [DOI] [PubMed] [Google Scholar]
- Daigle S. R.; Olhava E. J.; Therkelsen C. A.; Basavapathruni A.; Jin L.; Boriack-Sjodin P. A.; Allain C. J.; Klaus C. R.; Raimondi A.; Scott M. P.; Waters N. J.; Chesworth R.; Moyer M. P.; Copeland R. A.; Richon V. M.; Pollock R. M. (2013) Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 122, 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ClinicalTrials.gov Identifier: NCT02141828. Dose Escalation Study of EPZ-5676 in Pediatric Patients With Leukemias Bearing a Rearrangement of the MLL Gene.
- ClinicalTrials.gov Identifier: NCT01684150. A First-in-Human Phase 1 and Expanded Cohort Study of EPZ-5676 in Advanced Hematologic Malignancies, Including Acute Leukemia With Rearrangement of the MLL Gene.
- Pradhan S.; Chin H. G.; Esteve P. O.; Jacobsen S. E. (2009) SET7/9 mediated methylation of non-histone proteins in mammalian cells. Epigenetics 4, 383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasacchio D.; Okabe J.; Tikellis C.; Balcerczyk A.; George P.; Baker E. K.; Calkin A. C.; Brownlee M.; Cooper M. E.; El-Osta A. (2009) Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 58, 1229–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsyte-Lovejoy D.; Li F.; Oudhoff M. J.; Tatlock J. H.; Dong A.; Zeng H.; Wu H.; Freeman S. A.; Schapira M.; Senisterra G. A.; Kuznetsova E.; Marcellus R.; Allali-Hassani A.; Kennedy S.; Lambert J. P.; Couzens A. L.; Aman A.; Gingras A. C.; Al-Awar R.; Fish P. V.; Gerstenberger B. S.; Roberts L.; Benn C. L.; Grimley R. L.; Braam M. J.; Rossi F. M.; Sudol M.; Brown P. J.; Bunnage M. E.; Owen D. R.; Zaph C.; Vedadi M.; Arrowsmith C. H. (2014) (R)-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proc. Natl. Acad. Sci. U. S. A. 111, 12853–12858. [DOI] [PMC free article] [PubMed] [Google Scholar]