

Abstract
JmjC-domain containing histone demethylases (JHDMs) play critical roles in many key cellular processes and have been implicated in multiple disease conditions. Each enzyme within this family is known to have a strict substrate scope, specifically the position of the lysine within the histone and its degree of methylation. While much progress has been made in determining the substrates of each enzyme, new methods with which to systematically profile each histone mark are greatly needed. Novel chemical tools have the potential to fill this role and, furthermore, can be used as probes to answer fundamental questions about these enzymes and serve as potential therapeutic leads. In this work, we first investigated three small-molecule probes differing in the degree of “methylation state” and their differential bindings to JHDM1A (an H3K36me1/2 demethylase) using a fluorescence polarization-based competition assay. We then applied this specificity toward the “methylation state” and combined it with specificity toward lysine position in the design and synthesis of a peptidic probe targeting H3K36me2 JHDMs. The probe is further functionalized with a benzophenone cross-linking moiety and a biotin for affinity purification. Results showed binding of the peptidic probe to JHDM1A and specific enrichment of this protein in the presence of its native histone substrates. Affinity purification pulldown experiments from nuclear lysate coupled with mass spectrometry revealed the capability of the probe to pull out and enrich JHDMs along with other epigenetic proteins and transcriptional regulators.
Epigenetic mechanisms offer a means of secondary control over a cell’s phenotype beyond the basic underlying DNA sequence. One of these mechanisms is the modification of histone protein tails by a variety of covalent attachments. These are known to influence chromatin remodeling and the recruitment or blocking of effector proteins, and in turn influence transcriptional regulation.1,2 One of the most important and well-studied marks is histone methylation, which can occur at different arginine and lysine residues. The regulation of gene transcription by histone methylation has been implicated in many natural biological processes including development and differentiation, DNA replication and repair, normal neuronal functioning, cellular senescence, and the maintenance of proper heterochromatin structure.1−7 Unlike acetylation, which generally represses expression, methylation can be both activating (i.e., histone H3 lysine 4 (H3K4) and H3K36) and repressing (i.e., H3K9 and H3K27), depending on its position.3,4 Once thought irreversible due to similar global turnover rates as histone proteins,8,9 it is now known that two families of enzymes are capable of reversing histone lysine methylations (as of yet, no arginine demethylases have been identified).10 The first family, termed KDM1 (Figure 1A), was characterized in 200411 as flavin adenine dinucleotide (FAD)-dependent monoamine oxidases and contains two proteins that are capable of demethylating mono- and dimethylated H3K4 and H3K9 (H3K4me1/2 and H3K9me1/2).11,12 The second family is divided into seven classes, KDM2–8 (Figure 1A). These were first discovered in 200613 and contain the signature JmjC domain. These JmjC domain-containing histone demethylases (JHDMs) use Fe2+ and α-ketoglutarate (αKG) cofactors, and react with molecular oxygen to produce the highly reactive Fe4+-oxo species, which hydroxylates the C–H bond of the N-methyl group. The resulting hemiaminal deconstructs to formaldehyde and the demethylated product spontaneously under physiological conditions.1 This larger family is more flexible in substrate scope, being able to demethylates lysines in all three possible (mono-, di-, and tri-) methylation states. It is known that these JHDMs often have strict substrate scopes, which include not only the relative position of the lysine substrate but also its methylation state. For example, JHDM1A (a.k.a., KDM2A) demethylates H3K36me1/2 and JMJD2A (a.k.a., KDM4A) can demethylate H3K9me2/3 and H3K36me2/3.3
Figure 1.

Design of class-selective histone demethylase probe for affinity pulldown. (A) Classification of histone lysine demethylases. (B) Structures of bivalent demethylase inhibitor, methylstat acid (1), and its methyl ester, methylstat (2). (C) Structures of methyllysine-αKG mimic conjugates.(D) Structure of a highly functionalized H3K36me2-JHDM-targeting peptidic probe for affinity pulldown.
Aberrant levels of methylation, likely caused by misregulated or impaired demethylases, have been shown to be involved in human diseases14 including X-linked intellectual disability,15,16 and cancer.17−19 These investigations have led to a natural desire for robust and selective chemical tools with which to probe the functions of these enzymes, further elucidate their role in disease states, and eventually serve as leads for therapeutic development. Furthermore, chemical probes could be used to discover the currently unknown demethylases that modify the H3K79me3 and H4K20me3 marks. Histone peptides with specific lysine methylations have been used to discover JHDMs using affinity pulldown. However, this approach suffers from poor affinity of JHDMs with their substrates and undesired binding of methyllysine reader proteins. The substrates of KDMs are usually determined by the ability of a purified protein to demethylate a synthetic histone peptide in biochemical assays.13,20−25 However, this method is sometimes unreliable and can lead to conflicting reports.26,27 Novel methods by which to characterize the substrate scope of individual demethylases are greatly needed.
Inhibitors of JHDMs are often designed to either mimic the structure of the cofactor αKG, or are chelators of cofactor Fe2+.28,29 Although it has long been known that different classes of JHDMs have different substrate scopes and there have been some reports of exploiting the natural selectivity of the histone binding site substrate in order to create more selective inhibitors toward JHDMs,30,31 class-selective chemical probes of JHDMs remain elusive.
Our lab previously developed a novel bivalent JHDM inhibitor, methylstat acid (1, Figure 1B), which combines a lysine-substrate-mimicking motif with a cofactor-mimicking motif that shows in vitro selectivity of JHDMs over other Fe2+ and αKG-dependent hydroxylases. Its methyl ester, methylstat (2, Figure 1B), exhibits selective cellular activity against JHDMs.32 It was noteworthy that methylstat induces cellular hypermethylation of a wide range of histone methylation marks, including H3K79me3 and H4K20me3, suggesting that demethylases modifying these methylation marks also contain JmjC domains. In this work, we first demonstrate that JHDM1A (a.k.a., KDM2A), which is known to preferably modify H3K36me2, selectively binds to a dimethyllysine-αKG mimic conjugate (4, Figure 1C). Next, we synthesized a variant of 4 compatible with solid-phase peptide synthesis (SPPS) and used it to prepare a highly functionalized H3K36me2-JHDM-targeting affinity probe 6 (Figure 1D). This peptidic probe contains a photo-cross-linking moiety (i.e., benzophenone), a biotin for affinity purification, and an H3K36me2 peptide tethered with our αKG mimic. We further demonstrate that this probe binds to JHDM1A and selectively enriches JHDM1A in the presence of its native histone substrates in an affinity purification pulldown experiment utilizing streptavidin-coated magnetic beads. Finally, the native substrate scope of the probe was investigated with pulldown experiments from cell nuclear extract, revealing the specifically pulled-down proteins with mass spectrometry.
Results and Discussion
Design and Synthesis of Mono-, Di-, and Trimethyllysine-αKG Mimic Conjugates
We first designed three methyllysine-αKG mimic conjugates differing from each other in the number of methyl groups on the nitrogen of the lysine-mimicking fragment. Each is conjugated, as is methylstat, to a 4-carbon linker and an αKG-mimicking fragment. The synthesis began with a reductive amination of aldehyde 7(33) (Scheme 1) and amine 8(34) followed by acylation of 9 with acid 10 to provide amide 11. The ethyl ester was then removed by basic hydrolysis, and replaced by a tert-butyl ester by reacting with reagent 12.35 Deprotection of the tert-butyldimethylsilyl ether of 13 followed by Swern oxidation of the resulting alcohol 14 yielded aldehyde 15. Reduction amination of 15 with a commercially available carboxybenzyl-protected lysine methyl ester (16) afforded secondary amine 17. This compound was then deprotected using a mixture of trifluoroacetic acid (TFA), triisopropylsilane (TIPS) and water to give 3, the standard condition used for global deprotection in solid-phase peptide synthesis. Alternatively, 17 was modified by the addition of N-methyl groups before deprotection. One additional N-methyl group was installed by reductive amination with formaldehyde to give the tertiary amine 4 after deprotection. The second N-methyl group was installed by reacting the tertiary amine with methyl iodide to give the quaternary ammonium salt 5 after deprotection under similar conditions.
Scheme 1. Synthesis of mono-, di-, and tri-methylated lysine analogs 3–5.

Reagents and conditions: (a) 7 (1.0 equiv), MeOH, 25 °C, 20 min; then NaBH3CN (1.0 equiv/h for 6 h), acetic acid (1 eqiv/h for 6 h), MeOH, 25 °C, 6 h, 51%; (b) 10 (1.5 equiv), oxalyl chloride (2.0 equiv), DMF (0.050 equiv), CH2Cl2, 0–25 °C, 1 h; then 9, Et3N (3.0 equiv), DMAP (0.010 equiv), CH2Cl2, 0 °C, 10 min, 89%; (c) LiOH (1.0 equiv), THF/H2O, 25 °C, 2.5 h, 99%; (d) 12 (3.0 equiv), 1,4-dioxane, 50 °C, 16 h, 68%; (e) TBAF (1.5 equiv), THF, 0–25 °C, 16 h, 96%; (f) oxalyl chloride (2.0 equiv), DMSO (4.0 equiv), CH2Cl2, −78 °C, 30 min.; then 14, CH2Cl2, −78 °C, 15 min.; then Et3N (6.0 equiv), CH2Cl2, −78–25 °C, 15 min, 98%; (g) DIEA (2.0 equiv), 16 (2.0 equiv), MeOH, 25 °C, 2 h; then NaBH3CN (2.0 equiv), acetic acid (1.0 equiv), MeOH, 25 °C, 16 h, 58%; (h) TFA/TIPS/H2O 95:2.5:2.5, 25 °C, 4.5 h, 75%; (i, k) CH2O (2.0 equiv), EtOH, 25 °C, 2 h; then NaBH3CN (2.0 equiv), acetic acid (1.0 equiv), EtOH, 25 °C, 16 h, 38%; (j) TFA/TIPS/H2O 95:2.5:2.5, 25 °C, 4.5 h, 49%; (l) MeI/MeOH 1:1, 25 °C, 2 d, 98%; (m) TFA/TIPS/H2O 95:2.5:2.5, 25 °C, 4.5 h, 99%.
Evaluation of the Binding Affinity of Three Lysine-αKG Mimic Conjugates with JHDM1A
In order to assess the selectivity of the different methylation states and the binding affinity of the three methyllysine αKG mimic conjugates, we utilized a fluorescence polarization (FP)-based JHDM1A competition assay recently developed in our lab.36 JHDM1A is a demethylase known to preferably modify H3K36me2 mark, but not H3K36me3 mark. In addition, it also demethylates H3K36me1 with a slower reaction rate.3 The half-maximum inhibitory concentration (IC50) for each probe was determined by fitting the data using KaleidaGraph (v 4.1.1, Synergy Software) and the dissociation constant (Ki) calculated using an online Ki calculator (Methods).37 As shown in Figure 2, the results showed that the monomethylated compound 3 has a Ki of 0.37 μM, the dimethylated compound 4 has a much lower Ki of 0.12 μM, and the trimethylated compound 5 has the lowest affinity to JHDM1A with a Ki of 3.1 μM. These results clearly mirror the affinities observed in nature by JHDM1A toward native histone methyllysine substrates. While there is a 3-fold decrease in binding affinity between the dimethylated and monomethylated compounds, the difference in binding affinity between the dimethylated and trimethylated compounds is much greater (i.e., 25-fold). These data show for the first time the ability to modulate the binding affinity of small-molecule probes with a JHDM by only modifying their “methylation state”. The ability to differentiate and selectively inhibit specific classes within the JHDM family will benefit from any advantage available to tune inhibitor selectivity. Encouraged by these results, we next decided to design and synthesize a peptidic probe in a “dimethylated” state, in order to achieve the highest affinity toward JHDM1A for selective pulldown experiments.
Figure 2.

Evaluation of the binding affinity of three methyllysine-αKG mimic conjugates with JHDM1A.
Design and Synthesis of SPPS-Compatible Building Blocks
Building blocks 22 and 24 (Scheme 2) were designed to be compatible for SPPS using standard fluorenylmethyloxycarbonyl (Fmoc) solid-phase peptide chemistry. Building block 22 is the biotin affinity tag coupled with a spacer consisting of four monomer units of polyethylene glycol. Its synthesis was accomplished by first activating biotin 18 with N-hydroxysuccinamide 19, facilitated by N,N′-dicyclohexylcarbodiimide (DCC) to give compound 20. Then, this activated ester was joined with commercially available amine 21 and subsequently deprotected using TFA to afford building block 22. The lysine mimic fragment 24 was designed to be in a “dimethylated” state to emulate the selectivity of JHDM1A among compounds 3–5. Its synthesis used the same intermediate 15 in a different reductive amination reaction with commercially available Fmoc-protected lysine 23. Monomethylation was accomplished by another reductive amination with formaldehyde to afford 24.
Scheme 2. Synthesis of Highly Functionalized H3K36me2-JHDM-Targeting Probe 6.

Reagents and conditions: (a) N-hydroxysuccinimide 19 (1.0 equiv), DCC (1.3 equiv), DMF, 70–25 °C, 16 h, 71%; (b) 21 (0.91 equiv), DMF, 25 °C, 16 h, 96%; (c) TFA/DCM 1:1, 0 °C, 30 min, 25 °C 1 h 40 min, 85%. (d) 23 (1.5 equiv), MeOH, 25 °C, 3 h; then NaBH3CN (2.0 equiv), acetic acid (1.0 equiv), 25 °C, 16 h, 59%; (e) CH2O (2.0 equiv), MeOH, 25 °C, 30 min; then NaBH3CN (2.0 equiv), acetic acid (1.0 equiv), MeOH, 25 °C, 30 min; 96%.
Synthesis, Purification, and Characterization of Highly Functionalized H3K36me2-JHDM-Targeting Affinity Probe 6
By utilizing a short histone peptide centered around the H3K36 position, probe 6 was designed to selectively bind to H3K36me2-modifying JHDMs. Photo-cross-linking has been shown to be useful for covalent attachment of a probe to a protein’s surface,38 and its combination with affinity purification has been used to capture histone modifiers.39−42 These advantages allow for irreversible covalent binding and enrichment from complex mixtures. The number of amino acids chosen to neighbor the K36 position is 4 on each side, for a total H3 peptide length of 9 amino acids. This was designed based on the previous report showing that the minimum length of histone peptide required for JMJD2A, another H3K36 demethylase, was 8 amino acids with little variance seen with increasing length.30 The 9 amino acids in the probe would therefore be expected to provide a suitable length to couple the natural selectivity for H3K36-demethylases with the JmjC-targeting dimethyllysine-αKG mimic conjugate fragment in the center.
With building blocks 22 and 24 synthesized, the highly functionalized peptidic probe 6 was constructed on solid phase using standard Fmoc/tBu solid-phase peptide synthesis conditions with commercially available protected amino acids and Wang Resin (w/Nle).43 1,8-Diazabicycloundec-7-ene (DBU) was found to be the optimal base used for the iterative deprotection of Fmoc groups, as the standard base for SPPS, piperidine, underwent an undesired Michael addition with the αKG-mimicking fragment. The peptide was then cleaved from the resin and globally deprotected with TFA/TIPS/H2O (95:2.5:2.5) for 4.5 h. After precipitation in cold diethyl ether, the crude material was further purified by reverse-phase HPLC with a 5–40% acetonitrile gradient over 25 min. The final peptide was confirmed by high-resolution mass spectrometry analysis (Supporting Information Figure 1).
The affinity of the peptidic probe 6 for JHDM1A was determined by the FP competition assay. The IC50 value was determined as 60 μM and the Ki calculated to be 8.6 μM (Figure 3). There is a significant drop of the binding affinity for peptidic probe 6 from that of the compound 4. However, the peptidic probe 6 has almost a 2-fold increased affinity compared to a nonfunctionalized native H3K36me2 histone 20mer peptide, which has a Ki of 15 μM.36 We reasoned that the higher binding affinity of peptidic probe 6, compared with dimethyllysine-αKG mimic conjugate 4, may be due to the unnatural benzophenone-containing moiety and its close position relative to the central methyllysine analog. However, this loss in affinity may be compensated by the irreversible covalent cross-linking of the benzophenone with the targeted JHDM under UV irradiation.
Figure 3.

Evaluation of the binding affinity of peptidic probe 6 with JHDM1A.
Affinity Pulldown of JHDM1A in the Presence of Histone Proteins
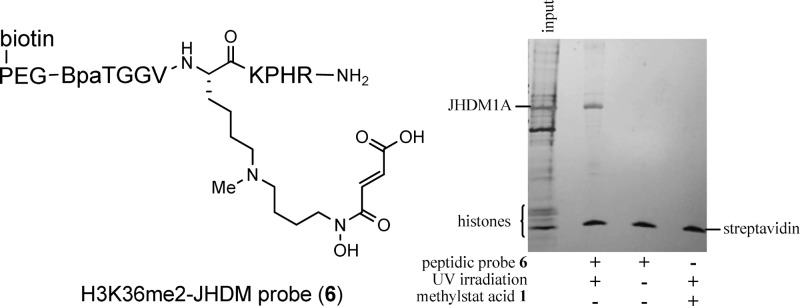
We next investigated whether our peptidic probe could achieve selective affinity pulldown in a more complex system. Our previous biochemical profiling studies of methylstat acid demonstrated that it selectively binds to and inhibits the activity of JHDMs compared with other histone-modifying enzymes, such as the Zn2+-dependent histone lysine deacetylases (HDACs) and the FAD-dependent lysine selective demethylase 1 (LSD1).32 Hence, we believe that the major competitors of our probe 6 with its targeted JHDMs are their native substrates, histone proteins. In order to prove that probe 6 may selectively pulldown H3K36me2-JHDMs, we conducted an affinity pulldown experiment using a mixture of purified JHDM1A and histone proteins extracted from HeLa cells as input. The incubation of the protein mixture and probe 6 was followed by UV irradiation at 350 nm, capture by streptavidin-coated magnetic beads (Invitrogen), and washing to remove nonspecific binding. The results were visualized by electrophoresis followed by silver staining as shown in Figure 4. The first lane shows 20% of the total input proteins. In addition to the JHDM and histones present, there were some nonspecific proteins unavoidably isolated during the histone extraction procedure. After a series of optimization, we found that supplementation of the input with 5.0 μM of NiCl2 significantly improved the pulldown efficiency. Compared with Fe2+, the native cofactor of JHDMs, Ni2+ can stabilize JHDMs without impairing the binding of JHDMs with its ligands.36 We also found that nonspecific binding of the proteins to the beads could be decreased by preincubating the peptidic probe 6 with the magnetic beads followed by addition of the protein mixture. Incubation was performed for 4 h, then UV irradiation at 350 nm for 20 min, both at 4 °C. Comparison of the input and pulldown sample (lanes 1 and 2, Figure 4) shows specific enrichment of JHDM1A over other proteins in the sample with almost no nonspecific capture. In the absence of UV irradiation, or the replacement of the peptidic probe 6 with a JHDM inhibitor, methylstat acid 1, with no photo-cross-linking ability or biotin moiety (lanes 3 and 4, Figure 4), there are complete absence of JHDM1A. It should be noted that the new low molecular weight protein seen in all pulldown lanes is the recombinant streptavidin released from the beads upon denaturation.
Figure 4.

Selective affinity pulldown of JHDM1A in the presence of histone proteins.
Enrichment Profiling of Nuclear Proteins Extracted from Mammalian Cells
Finally, we investigated whether peptidic probe 6 had the ability to pulldown native histone demethylases, in particular the H3K36me2 JHDMs (certain members of subfamilies KDM2, KDM4, and KDM8, as well as PHF8), from a complex nuclear lysate extracted from HeLa cells. To differentiate specific from nonspecifically captured proteins, a series of three pulldowns were performed and profiled by high resolution Orbitrap mass spectrometry (Methods). Peptidic probe 6 (20 μM) was added to nuclear extract to pull down endogenous demethylases and associated complexes (“endogenous”, Table 1). To control for proteins nonspecifically associating with the beads, the pull down was performed with nuclear extract without addition of probe 6 (“negative”), while the final experiment contained 20 μM probe 6, with recombinant human JMJD2A (KDM4A) spiked into the lysate at a concentration of 25 nM, >10 times the estimated native concentration by Western blot (“spiked-in”). JMJD2A was chosen instead of JHDM1A because the former is known to be highly expressed in HeLa cells.44 This third pulldown was used as a positive control and to determine whether probe 6 was capable of pulling down one of our targeted JHDMs in the presence of a complex environment regardless of native abundance. Proteins were considered specifically enriched if there was a spectral count of one or more assigned in both the positive and spiked-in pulldowns, while absent (or significantly less) in the negative pulldown.
Table 1. Epigenetic Proteins Specifically Pulled down by Peptidic Probe 6a.
| Uniprot accession number | gene name | epigenetic protein family | endogenousb | negativec | spiked-ind |
|---|---|---|---|---|---|
| Q15652 | JMJD1C/KDM3C | histone lysine demethylase | 6 | 0 | 3 |
| O75164 | JMJD2A/KDM4A | histone lysine demethylase | 0 | 0 | 5 |
| Q9UPP1 | PHF8/KDM7A | histone lysine demethylase | 1 | 0 | 1 |
| Q9HCK8 | CHD8 | chromodomain-containing protein | 2 | 0 | 3 |
| Q9P2D1 | CHD7 | chromodomain-containing protein | 5 | 0 | 1 |
| Q13547/Q92769 | HDAC1/2 | histone lysine deacetylase | 8 | 0 | 3 |
The spectral count is listed for three pulldown experiments.
The endogenous experiment corresponded to the addition of the peptidic probe 6.
The negative control corresponded to the absence of peptidic probe 6.
The spiked-in experiment corresponded to endogenous conditions with the addition of recombinant KDM4A to the nuclear extract.
It is known that several HDMs perform their functions as part of multiprotein complexes. For example, LSD1 of the KDM1 subfamily is found in complexes including HDAC/CoREST, HMG20B, BHC80, noncoding scaffold RNAs, and the NuRD complex. Other HDMs including those of the KDM5 subfamily have been shown to be members of chromatin complexes as well, such as JARID1A with the PRC2 Polycomb repressive complex and JARID1C with others including components such as HDAC1/2/REST or with methyltransferase G9a.14 It is very likely that other HDMs are integral members of multiprotein complexes responsible for gene expression yet to be elucidated. Because of both the relative size of probe 6 and the nondiscriminating nature of the adjacent benzophenone cross-linker, we anticipated the possibility of pulling down not only targeted JHDMs but also close proximity binders and interacting members of complexes. Table 1 summarizes only those proteins specifically pulled down that are epigenetic modifiers/readers.
The most enriched was the demethylase JMJD1C of the KDM3 subfamily. This protein, while containing a JmjC domain, currently has no known histone substrate. While there was success in pulling down JMJD2A in the positive control spiked lysate sample, we did not detect it in unenriched lysates, perhaps due to sensitivity limitations of the mass spectrometry experiment. We conclude that, while the probe is successful at targeting this protein in a complex environment, the native abundance was unfortunately below the limit of detection. The final demethylase pulled down by probe 6 was the KDM7 subfamily member PHF8, a known demethylase for the H3K36me2 mark. Its catalytic domain is highly structural similar to JHDM1A/KDM2A, which could explain activity toward the H3K36 position.29
The histone deacetylases HDAC1/2 were also enriched compared to the negative control. These proteins are known to participate in large protein complexes involved in transcriptional repression. Probe 6 mimics the H3K36 dimethyl mark (an activating mark itself); thus, we anticipate that complexes associated with demethylases targeting this mark would likely represent repressive epigenetic modifier complexes. Class I deacetylases HDAC1 and 2 are known to interact with Nuclear Receptor Corepressor 2 (NCOR2) via the Sin3A-HDAC complex.45 Interestingly, while NCOR2 was detected in all samples, the spectral count was increased in comparison to negative conditions. This protein represses gene transcription by promoting chromatin condensation via deacetylase activity. JMJD2A has also been shown to interact with HDACs and the Nuclear receptor corepressor to inhibit gene expression.46−48 Chromodomain-helicase-DNA binding proteins 7 and 8 (CHD7 and CHD8) were also specifically pulled down. These are transcriptional regulators and possible readers of methylated histone lysines. Other specifically pulled-down proteins involved in transcriptional regulation included Polycomb group RING protein 5 (a component of the Polycomb group PcG multiprotein repressive complex), Kruppel-like factor 4 (a transcription factor shown to interact with HDAC2),49 and cyclin-dependent kinase 9 (a component of the P-TEFb factor which activates RNA Pol II for transcription). For a complete list of all proteins specifically pulled down or enriched, see the Supporting Information Table.
Overall, peptidic probe 6 has shown capability of pulling out JmjC-domain containing proteins, one of which has specific activity toward the H3K36me2 mark. With increased concentration, the spiked-in targeted JMJD2A (KDM4A) was successfully pulled out and identified as well. The other proteins specifically isolated contain known demethylase interactors, large numbers of transcriptional regulators, and other proteins that could serve as candidates of protein-demethylase interactors in future study.
Conclusion
This work first describes the design and synthesis of three methyllysine-αKG conjugates differing in the degree of “methylation state” and evaluation of affinity to JHDM1A by fluorescence polarization. Because of the strict substrate scope of JHDMs observed in nature (both of lysine position and degree of methylation), we investigated whether affinities of small-molecule probes could be tuned by modifying the “methylation state” of these probes. It is known that JHDM1A selectively demethylates the H3K36me1/2 marks but not the H3K36me3 mark.3 Our FP assay results clearly mirror this pattern with the dimethyllysine-αKG mimic conjugate having almost 25-fold greater affinity than the trimethyl version. We next applied this knowledge in the design of a peptidic probe, combining the specificity of lysine position (i.e., with an H3 peptide adjacent to the K36 position) with the specificity of the methylation state (i.e., the K36 replacement lysine-αKG mimic conjugate in a “dimethylated” state). This probe was further functionalized with a photo-cross-linking moiety (i.e., benzophenone), and a biotin for affinity purification. Evaluation by FP showed binding of this probe to JHDM1A. Next, we employed this probe in an affinity pulldown experiment of JHDM1A in the presence of its native histone substrates. The results show the ability of the probe to selectively enrich JHDM1A on streptavidin-coated magnetic beads, while negative controls confirm the biotin and UV-cross-linking motifs are essential. Finally, the probe was employed in enrichment pulldowns from HeLa nuclear lysate in order to define its true substrate scope. The specific proteins identified by mass spectrometry include two JmjC-domain containing demethylases, one of which (PHF8) has been proven to demethylate H3K36me2. With increased concentration, JMJD2A was also identified. Other specifically pulled down/enriched proteins include other epigenetic modifiers, regulators of gene expression, as well as known and prospective demethylase interactor proteins.
This preliminary study was an exploration into the essential problem of identifying inhibitors that can specifically differentiate between members of the JHDM family. This approach has the potential to be a systematic method by which to target and identify individual JHDMs in complex environments and to identify and confirm their substrate scopes. In addition, simply changing the peptide sequence can then target different members of the JHDM family, and be used to discover novel JHDMs. Mass-spectrometry based identification could allow a methodical profiling of tissue-specific, cell type-specific and even disease state-specific extant JHDMs and their associated interactors, which would be extremely useful in determining the roles of these proteins in cells, their presence in complexes, their involvement in histone crosstalk, and the deleterious effects of their misregulation. Future work in our lab aims to continually improve the specificity and affinity of the probes. Application of this approach to identify JHDMs that modify H4K20me3 and H3K79me3 are also ongoing and will be reported in due course.
Methods
Chemical Synthesis
The detailed synthetic procedures and characterization of unknown compounds are presented in the Supporting Information along with high-resolution mass spectrometry data of the purified peptidic probe.
Protein Purification
Recombinant JHDM1A and JMJD2A were expressed and purified as previously described.32,36
Fluorescence Polarization Assays for JHDM1A
The fluorescence polarization (FP)-based assays were performed as previously described.36 The binding curves were generated using KaleidaGraph (v 4.1.1, Synergy Software) and the IC50 values derived were used to calculate the dissociation constants (Ki) using the online Ki calculator37 with the following equation:
| 1 |
where [I]50 and [L]50 are free inhibitor concentration and free ligand concentration at 50% inhibition, [P]0 is free protein concentration at 0% inhibition, and the Kd is the dissociation constant between the protein and fluorophore. We assume I inhibits binding of the fluorophore to protein competitively with a 1:1 binding stoichiometry.
Cell Culture
HeLa cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS), 1% penicillin/streptomycin and were grown in 10 cm treated dishes (Corning) at 37 °C in 5%/95% CO2/air.
Histone Extraction
Cells were harvested and then lysed at a density of 107 cells/mL in triton extraction buffer (TEB: PBS containing 0.5% triton X (v/v), 0.02% NaN3 (w/v), 1 protease inhibitor tablet (Roche)/10 mL buffer). After centrifugation and washing with half volume of TEB, the cells were resuspended in 0.2 N HCl at a density of 4 × 104 cells/mL and histones were extracted overnight at 4 °C. Before use, the water was removed by lyophilization and the proteins were reconstituted in affinity purification pulldown buffer (25 mM TRIS, 150 mM NaCl, 5 μM NiCl2, pH 7.5).
Affinity Pulldown of JHDM1A with Magnetic Beads
The pulldown experiments were performed in the presence of 5 μM JHDM1A protein, equal amounts of histone proteins quantified with ImageJ, and 20 μM probe 6 in a total volume of 200 μL buffer (25 mM TRIS, 100 mM NaCl, 5 μM NiCl2, pH 7.5). First, 25 μL of Dynabeads MyOne Streptavidin C1 (Invitrogen) was prepared and washed according to the manufacturer’s instructions. Then, 200 μL buffer was added to the beads, followed by the peptidic probe 6. After a 30 min incubation at 4 °C, the protein mixture of JHDM1A and histones were added and incubated for 4 h at 4 °C. Next, UV-irradiation was performed at 350 nm for 20 min with three external low-pressure mercury lamps (Southern New England Ultraviolet Co.).50 The beads were then collected with a magnetic stand and the supernatant was removed. The beads were washed 7 times with wash buffer (10 mM HEPES, 1 M NaCl, 1% triton X (v/v), pH 7.5) and then 3 times with water. Negative control experiments were carried out in the absence of UV irradiation or the replacement of peptidic probe 6, with methylstat acid 1.
Nuclear Protein Extraction from HeLa Cells
The cells were collected by centrifugation and washed with cold PBS. The pellet was then resuspended in 5 times the packed cell volume of lysis buffer (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, pH 7.9) supplemented with a protease inhibitor tablet (Roche). The cells were allowed to swell on ice for 20 min, then centrifuged, and the supernatant was removed. The pellet was resuspended in 2 times the packed cell volume and then dounced 10 times with a glass dounce homogenizer. The homogenate was centrifuged and the supernatant was removed, repeating several times with fresh lysis buffer. The nuclear pellet was frozen and stored at −80 °C until use. After thawing, the pellet was resuspended in 0.9 times the packed nuclear volume in the second lysis buffer (20 mM HEPES, 0.4 M NaCl, 1.5 mM MgCl2, 20% glycerol (v/v), pH 7.9) supplemented with a protease inhibitor tablet (Roche). The suspension was dounced 20 times with the glass dounce homogenizer. After incubation at 4 °C for 30 min on a rotator, the sample was centrifuged at high speed (12 000 rpm) for 30 min. The supernatant was removed and then dialyzed into a glycerol free, lower salt buffer (20 mM HEPES, 100 mM KCl, 2 mM MgCl2, pH 7.9) supplemented with a protease inhibitor tablet (Roche). The lysate was aliquoted, flash frozen in liquid nitrogen, and stored at −80 °C until needed.
Affinity Pulldown from Nuclear Lysates
Three pulldown experiments were performed as previously described in the presence of 2 mL of nuclear lysate with 20 μM added peptidic probe 6 for the positive sample, without 6 for the negative control, or with 20 μM 6 in the presence of spiked-in JMJD2A (KDM4A) at a concentration of 25 nM (>10 times the estimated native concentration by Western blot). After washing the beads as before, the samples were subjected to an on-beads tryptic digest and processed for LC/MS/MS analysis. Briefly, the cysteine disulfides were reduced using 4 mM dithiothreitol and alkylated using 14 mM iodoacetamide. An ammonium bicarbonate solution was added to the samples to a final concentration of 100 mM supplemented with 1 mM calcium chloride. Sequencing grade modified trypsin was added according to the manufacturer’s instructions (Promega), and the samples were digested overnight at 37 °C. The samples were acidified with trifluoroacetic acid and then the peptides were desalted using Pepclean C18 spin columns (Pierce) according to the manufacturer’s instructions. After elution, the peptides were concentrated by speedvac, then reconstituted in 8 μL of 0.1% formic acid and sonicated for 5 min prior to analysis.
Mass Spectrometry Analysis
The resulting peptide mixtures were analyzed by reversed-phase chromatography using a Waters nanoAcquity UPLC pump coupled online to an LTQ/Orbitrap mass spectrometer. Peptides were loaded onto a Symmetry 180 μm × 2 cm C18 trap column, which was washed and placed in-line with a Waters BEH130 C18 analytical column (75 μm × 25 cm). As the peptides elute off the column they are ionized into the gas phase of the mass spectrometer. The peptide ion masses are measured with resolution of 60 000 at m/z 400 by the Orbitrap, followed by MS/MS fragmentation of the six most intense precursors with charge state ≥2 above an intensity threshold of 10 000 in the LTQ ion trap. Dynamic exclusion was used to avoid repeat sequencing, with a repeat count of one and exclusion duration of 30 s for precursors within 10 ppm. The MS/MS spectra were extracted and searched against the human Uniprot (release 2013_01) using the MASCOT search engine, version 2.2 (Matrix Science) with a mass tolerance on precursor ions of 25 ppm and 0.5 Da for fragment ions. The following variable modifications were considered: carbamidomethyl-Cys, methionine oxidation, and pyro-glutamic acid for N-terminal Gln. Peptide identifications were accepted at a 1% FDR threshold based on a reversed protein database search. Protein assembly and label-free quantification by spectral counting was performed using Isoform Resolver.51
Acknowledgments
The authors thank Prof. T. Koch (Department of Chemistry and Biochemistry, University of Colorado Boulder) for useful discussions and use of the UV apparatus. The authors also thank the National Institutes of Health for providing funding under grant no. R01-GM098390 and the Signaling and Cellular Regulation Training Grant (T32 GM08759) for supporting L.J.M.
Glossary
Abbreviations
- FP
fluorescence polarization
- HPLC
high performance liquid chromatography
- IC50
half-maximum inhibitory concentration
- Ki
inhibitor dissociation constant
- SPPS
solid phase peptide synthesis
Supporting Information Available
Supplemental detailed synthetic methods, compound characterization, and high resolution mass spectrometry data. A list of enriched proteins identified by mass spectrometry and associated data in the supplemental table. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cloos P. A. C.; Christensen J.; Agger K.; Helin K. (2008) Erasing the methyl mark: Histone demethylases at the center of cellular differentiation and disease. Genes Dev. 22, 1115–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. (2007) Chromatin modifications and their function. Cell 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Klose R. J.; Zhang Y. (2007) Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 8, 307–318. [DOI] [PubMed] [Google Scholar]
- Shi Y. (2007) Histone lysine demethylases: Emerging roles in development, physiology, and disease. Nat. Rev. Genet. 8, 829–833. [DOI] [PubMed] [Google Scholar]
- Ciccone D. N.; Su H.; Hevi S.; Gay F.; Lei H.; Bajko J.; Xu G.; Li E.; Chen T. (2009) KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 461, 415–418. [DOI] [PubMed] [Google Scholar]
- Agger K.; Christensen J.; Cloos P. A. C.; Helin K. (2008) The emerging functions of histone demethylases. Curr. Opin. Genet. Dev. 18, 159–168. [DOI] [PubMed] [Google Scholar]
- Nottke A.; Colaiácovo M. P.; Shi Y. (2009) Developmental roles of the histone lysine demethylases. Development 136, 879–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byvoet P.; Shepherd G. R.; Hardin J. M.; Noland B. J. (1972) The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Arch. Biochem. Biophys. 148, 558–567. [DOI] [PubMed] [Google Scholar]
- Duerre J. A.; Lee C. T. (1974) In vivo methylation and turnover of rat brain histones. J. Neurochem. 23, 541–547. [DOI] [PubMed] [Google Scholar]
- Thompson P. R.; Fast W. (2006) Histone citrullination by protein arginine deiminase: Is arginine methylation a green light or a roadblock?. ACS Chem. Biol. 1(7), 433–441. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Lan F.; Matson C.; Mulligan P.; Whetstine J. R.; Cole P. A.; Casero R. A.; Shi Y. (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953. [DOI] [PubMed] [Google Scholar]
- Wissmann M.; Yin N.; Mueller J. M.; Greschik H.; Fodor B. D.; Jenuwein T.; Vogler C.; Schneider R.; Guenther T.; Buettner R.; Metzger E.; Scheule R. (2007) Co-operative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 9, 347–353. [DOI] [PubMed] [Google Scholar]
- Tsukada Y.; Fang J.; Erdjument-Bromage H.; Warren M. E.; Borchers C. H.; Tempst P.; Zhang Y. (2006) Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816. [DOI] [PubMed] [Google Scholar]
- Johansson C.; Tumber A.; Che K.; Cain P.; Nowak R.; Gileadi C.; Oppermann U. (2014) The roles of Jumonji-type oxygenases in human disease. Epigenomics 6, 89–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen L. R.; Amende M.; Gurok U.; Moser B.; Gimmel V.; Tzschach A.; Janecke A. R.; Tariverdian G.; Chelly J.; Fryns J. P.; Van Esch H.; Kleefstra T.; Hamel B.; Moraine C.; Gecz J.; Turner G.; Reinhardt R.; Kalscheuer V. M.; Ropers H. H.; Lenzner S. (2005) Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am. J. Hum. Genet. 76, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnier F.; Holbert S.; Ronce N.; Faravelli F.; Lenzner S.; Schwartz C. E.; Lespinasse J.; Esch H. V.; Lacombe D.; Goizet C.; Tuy F. P. D.; Bokhoven H. V.; Fryns J. P.; Chelly H. H.; Ropers C.; Moraine C.; Hamel B. C. J.; Briault S. (2005) Mutations in PHF8 are associated with X-linked mental retardation and cleft lip/cleft palate. J. Med. Genet. 42, 780–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotili D.; Mai A. (2011) Targeting histone demethylases: A new avenue for the fight against cancer. Genes Cancer 2, 663–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varier R. A.; Timmers H. T. M. (2011) Histone lysine methylation and demethylation pathways in cancer. Biochem. Biophys. Acta 1815, 75–89. [DOI] [PubMed] [Google Scholar]
- Grant S. (2009) Targeting histone demethylases in cancer therapy. Clin. Cancer Res. 15, 7111–7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J.; Agger K.; Cloos P. A.; Pasini D.; Rose S.; Sennels L.; Rappsilber J.; Hansen K. H.; Salcini A. E.; Helin K. (2007) RBP2 belongs to a family of demethylases, specific for tri- and dimethylated lysine 4 on histone 3. Cell 128, 1063–1076. [DOI] [PubMed] [Google Scholar]
- Iwase S.; Lan F.; Bayliss P.; de la Torre-Ubieta L.; Huarte M.; Qi H. H.; Whetstine J. R.; Bonni A.; Roberts T. M.; Shi Y. (2007) The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell 128, 1077–1088. [DOI] [PubMed] [Google Scholar]
- Yamane K.; Toumazou C.; Tsukada Y.; Erdjument-Bromage H.; Tempst P.; Wong J.; Zhang Y. (2006) JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 125, 483–495. [DOI] [PubMed] [Google Scholar]
- De Santa F.; Totaro M. G.; Prosperini E.; Notarbartolo S.; Testa G.; Natoli G. (2007) The histone H3 lysine 27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 130, 1083–1094. [DOI] [PubMed] [Google Scholar]
- Hong S.; Cho Y. W.; Yu L. R.; Yu H. (2007) Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Prot. Natl. Acad. Sci. U.S.A. 104, 18439–18444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose R. J.; Yan Q.; Tothova Z.; Yamane K.; Erdjument-Bromage H.; Tempst P.; Gilland D. G.; Zhang Y.; Kaelin W. G. Jr. (2007) The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell 128, 889–900. [DOI] [PubMed] [Google Scholar]
- Qi H. H.; Sarkissian M.; Hu G. Q.; Wang Z.; Bhattacharjee A.; Gordon D. B.; Gonzales M.; Lan F.; Ongusaha P. P.; Huarte M.; Yaghi N. K.; Lim H.; Garcia B. A.; Brizuela L.; Zhao K.; Roberts T. M.; Shi Y. (2010) Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature 466, 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Tanasa B.; Tyurina O. V.; Zhou T. Y.; Gassmann R.; Liu W. T.; Ohgi K. A.; Benner C.; Garcia-Bassets I.; Aggarwal A. K.; Desai A.; Dorrestein P. C.; Glass C. K.; Rosenfield M. G. (2010) PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature 466, 508–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T.; Miyata N. (2011) Lysine demethylase inhibitors. J. Med. Chem. 54, 8236–8250. [DOI] [PubMed] [Google Scholar]
- Lohse B.; Kristensen J. L.; Kristensen L. H.; Agger K.; Helin K.; Gajhede M.; Clausen R. P. (2011) Inhibitors of histone demethylases. Bioorg. Med. Chem. 19, 3625–3636. [DOI] [PubMed] [Google Scholar]
- Lohse B.; Nielsen A. L.; Kristensen J. B. L.; Helgstrand C.; Cloos P. A. C.; Olsen L.; Gajhede M.; Clausen R. P.; Kristensen J. L. (2011) Targeting histone lysine demethylases by truncating the histone 3 tail to obtain selective substrate-based inhibitors. Angew. Chem., Int. Ed. 50, 9100–9103. [DOI] [PubMed] [Google Scholar]
- Woon E. C. Y.; Tumber A.; Kawamura A.; Hillringhaus L.; Ge W.; Rose N. R.; Ma J. H. Y.; Chan M. C.; Walport L. J.; Che K. H.; Ng S. S.; Marsden B. D.; Oppermann U.; McDonough M. A.; Schofield C. J. (2012) Linking of 2-oxoglutarate and substrate binding sites enables potent and highly selective inhibition of JmjC histone demethylases. Angew. Chem., Int. Ed. 51, 1631–1634. [DOI] [PubMed] [Google Scholar]
- Luo X.; Liu Y.; Kubicek S.; Myllyharju J.; Tumber A.; Ng S.; Che K. H.; Podoll J.; Heightman T. D.; Oppermann U.; Schreiber S. L.; Wang X. (2011) A selective inhibitor and probe of the cellular functions of jumonji C domain-containing histone demethylases. J. Am. Chem. Soc. 133, 9451–9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Zou G.; Zhao G. (2013) Asymmetric intramolecular oxa-michael reactions to tetrahydrofurans/2H-pyrans catalyzed by primary-secondary diamines. ACS Catal. 3, 1356–1359. [Google Scholar]
- Ishikawa T.; Iizawa Y.; Okonogi K.; Miyake A. (2000) Studies on anti-MRSA parenteral cephalosporins. I. Synthesis and antibacterial activity of 7 β-[2-(5-amino-1,2,4-thiadiazol-3-yl)-2(Z)-hydroxyiminoacetam-ido]-3-(substituted imidazo[1,2-b]-pyridazinium-1-yl)methyl-3-cephem-4-carboxylates and related compounds. J. Antibiot. 53, 1053–1070. [DOI] [PubMed] [Google Scholar]
- Cambeiro X. C.; Martín-Rapún R.; Miranda P. O.; Sayalero S.; Alza E.; Llanes P.; Pericàs M. A. (2011) Continuous-flow enantioselective α-aminoxylation of aldehydes catalyzed by a polystyrene-immobilized hydroxyproline. Beilstein J. Org. Chem. 7, 1486–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W.; Podoll J. D.; Dong X.; Tumber A.; Oppermann U.; Wang X. (2013) Quantitative analysis of histone demethylase probes using fluorescence polarization. J. Med. Chem. 56, 5198–5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolovska-Coleska Z.; Wang R. X.; Fang X. L.; Pan H. G.; Tomita Y.; Li P.; Roller P. P.; Krajewski K.; Saito N. G.; Stuckey J. A.; Wang S. (2004) Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal. Biochem. 332, 261–273. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy M.; Dugan A.; Nwokoye A.; Fung Y. H.; Lancia J. K.; Majmudar C. Y.; Mapp A. K. (2011) Caught in the act: Covalent cross-linking captures activator–coactivator interactions in vivo. ACS Chem. Biol. 6, 1321–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotili D.; Altun M.; Kawamura A.; Wolf A.; Fischer R.; Leung I. K. H.; Mackeen M. M.; Tian Y.; Ratcliffe P. J.; Mai A.; Kessler B. M.; Schofield C. J. (2011) A photoreactive small-molecule probe for 2-oxoglutarate oxygenases. Chem. Biol. 18, 642–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack J. L.; Causey C. P.; Luo Y.; Thompson P. R. (2011) Development and use of clickable activity based protein profiling agents for protein arginine deiminase 4. ACS Chem. Biol. 6, 466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salisbury C. M.; Cravatt B. F. (2007) Activity-based probes for proteomic profiling of histone deacetylase complexes. Proc. Natl. Acad. Sci. U.S.A. 104(4), 1171–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salisbury C. M.; Cravatt B. F. (2008) Optimization of activity-based probes for proteomic profiling of histone deacetylase complexes. J. Am. Chem. Soc. 130, 2184–2194. [DOI] [PubMed] [Google Scholar]
- Coin I.; Beyermann M.; Bienert M. (2007) Solid-phase peptide synthesis: From standard procedures to the synthesis of difficult sequences. Nat. Protoc. 2, 3247–3256. [DOI] [PubMed] [Google Scholar]
- Kawazu M.; Saso K.; Tong K. I.; McQuire T.; Goto K.; Son D.; Wakeham A.; Miyagishi M.; Mak T. W.; Okada H. (2011) Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS One 6(3), e17830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill C.; Qutob M. S.; Yee S. P.; Torchia J. (2000) A novel nuclear receptor corepressor complex, N-CoR, contains components of the mammalian SWI/SNF complex and the corepressor KAP-1. J. Biol. Chem. 275(51), 40463–70. [DOI] [PubMed] [Google Scholar]
- Gray S. G.; Iglesias A. H.; Lizcano F.; Villanueva R.; Camelo S.; Jingu H.; The B. T.; Koibuchi N.; Chin W. W.; Kokkotou E.; Dangond F. (2005) Functional characterization of JMJD2A, a histone deacetylase- and retinoblastoma-binding protein. J. Biol. Chem. 280, 28507–28518. [DOI] [PubMed] [Google Scholar]
- Yoon H. G.; Chan D. W.; Huang Z. Q.; Li J.; Fondell J. D.; Qin J.; Wong J. (2003) Purification and functional characterization of the human N-CoR complex: the roles of HDAC3, TBL1 and TBLR1. EMBO J. 22, 1336–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.; Yoon H. G.; Wong J. (2005) JMJD2A is a novel N-CoR-interacting protein and is involved in repression of the human transcription factor achaete scute-like homologue 2 (ASCL2/Hash2). Mol. Cell. Biol. 25, 6404–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F.; Han M.; Zheng B.; Wang C.; Zhang R.; Zhang X. H.; Wen J. K. (2009) All-trans retinoic acid increases KLF4 acetylation by inducing HDAC2 phosphorylation and its dissociation from KLF4 in vascular smooth muscle cells. Biochm. Biophys. Res. Commun. 387(1), 13–8. [DOI] [PubMed] [Google Scholar]
- Koch T. H.; Brown D. A. (1971) Photochemical valence isomerization of a conjugated imino ether. J. Org. Chem. 36, 1934–1937. [Google Scholar]
- Meyer-Arendt K.; Old W. M.; Houel S.; Renganathan K.; Eichelberger B.; Resing K. A.; Ahn N. G. (2011) Isoform resolver: A peptide-centric algorithm for protein inference. J. Proteome Res. 10(7), 3060–3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.