

Abstract
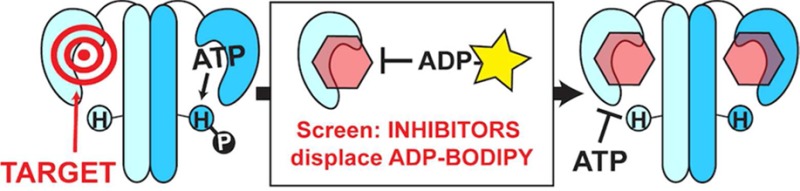
Antibacterial agents that exploit new targets will be required to combat the perpetual rise of bacterial resistance to current antibiotics. We are exploring the inhibition of histidine kinases, constituents of two-component systems. Two-component systems are the primary signaling pathways that bacteria utilize to respond to their environment. They are ubiquitous in bacteria and trigger various pathogenic mechanisms. To attenuate these signaling pathways, we sought to broadly target the histidine kinase family by focusing on their highly conserved ATP-binding domain. Development of a fluorescence polarization displacement assay facilitated high-throughput screening of ∼53 000 diverse small molecules for binding to the ATP-binding pocket. Of these compounds, nine inhibited the catalytic activity of two or more histidine kinases. These scaffolds could provide valuable starting points for the design of broadly effective HK inhibitors, global reduction of bacterial signaling, and ultimately, a class of antibiotics that function by a new mechanism of action.
Alarming levels of antibiotic resistance and the dearth of new drugs to treat bacterial infections1,2 have created an immense public health threat with concerns that a postantibiotic era is imminent. The void in introduction of new antibiotic classes for nearly three decades2,3 has fueled the search for molecules that can impair bacteria by nontraditional mechanisms.3,4 We have focused our efforts on two-component system-mediated cell signaling. The prototypical two-component system (TCS) is composed of a cognate pair of proteins: a membrane-bound histidine kinase (HK) and a response regulator (RR) localized in the cytoplasm. Activated by extracellular signals, the HK autophosphorylates with the γ-phosphate of adenosine triphosphate (ATP) on a conserved histidine residue. The phosphate group is subsequently transferred to the RR, which is typically a transcription factor that alters gene expression (Figure 1a).5,6 In this way, signals that activate the HK are propagated via phosphorylation to elicit intracellular responses.
Figure 1.

Prototypical TCS and its conservation exploited for inhibition. (a) An extracellular signal activates the HK, inducing autophosphorylation and subsequent phosphoryl transfer to the RR to instigate a cellular response. (b) Shading illustrates homology boxes and conservation in the HK ATP-binding domain of HK853 (T. maritima) bound to ADP (PDB 3DGE). N-, G1-, F-, G2-, and G3-boxes are shown in yellow, orange, purple, green, and pink, respectively. An ATP lid connecting the F- and G2-boxes is transparently shaded gray, and examples of residues involved in nucleotide binding are shown in stick form. (c) By targeting a highly conserved domain, our objective is to identify molecules that inhibit multiple HKs in various bacterial organisms by cutting off phosphorylation cascades and reducing signaling.
TCS proteins are attractive drug targets. Absent from human biology,7 TCSs are ubiquitous in the bacterial world, including Gram-positive, Gram-negative,6 and mycobacterial species.8 Significantly, TCSs are widely implicated in survival roles and pathogenic mechanisms, such as nutrient acquisition, sporulation, biofilm formation, and antibiotic resistance.9,10 Early efforts to discover TCS inhibitors in the late 1990s were hindered by the identification of nonspecific molecules.11 Recently, a handful of promising new compounds has emerged.12 Many efforts have targeted variable regions of TCS proteins to exclusively inhibit a single pathway. Instead, we sought to target a domain of high conservation such that one molecule might simultaneously inhibit multiple TCSs per organism and globally reduce TCS-mediated signaling.13 Studies have confirmed the attenuation of infections when single TCSs are mutated.8,14,15 Accordingly, we hypothesize that the inhibition of several would incur greater damage to bacteria, especially as more evidence emerges that some TCSs are intimately coupled.16,17
To identify molecules with broad anti-HK activity, we targeted the ATP-binding domain that is characterized by the Bergerat fold, a sandwich of α helices in one layer and mixed β strands in another, along with a discrete and flexible ATP lid.18−20 Not found in other kinases or the small number of mammalian HKs, the Bergerat fold confers a point of selectively among abundant eukaryotic proteins. Within the ATP-binding domain, homology boxes (G1-, G2-, G3-, F-, and N-boxes) recognize and participate in specific interactions with the nucleotide (Figure 1b). For example, an invariant Asp in the G1-box forms a salt bridge with the N6 exocyclic amino group of ATP; the G1-, F-, and G3-boxes position adenosine; and the N-box contains polar residues that coordinate the phosphate groups and chelate a Mg2+ ion.18,19,21,22 Small molecules exploiting the same conserved residues that bind ATP may enable the targeting of multiple HKs (Figure 1c).13In silico screens targeting the ATP-binding domain from a specific HK have led to the identification of molecules that inhibited these proteins in vitro, possessed antibacterial activity,23,24 and in several cases, they showed promise in animal infection models.25−27 Examples of ATP-competitive compounds that more broadly affect HKs have also been reported. Walkmycin C inhibited three purified HKs and repressed pathogenic phenotypes in Streptococcus mutans,(28) and TEP (3,6-diamino-5-cyano-4-phenylthieno[2,3-b]pyridine-2-carboxylic acid (4-bromo-phenyl)-amide) deactivated four HK proteins with fifty-percent inhibitory concentrations (IC50 values) in the low micromolar range.29 Given the success of these recent studies and the postulation that multitargeted approaches may deter the rapid acquisition of resistance,2,29 we sought to further investigate this strategy.
Here, we report HK inhibitors identified with a high-throughput in vitro assay designed to elucidate binding of small molecules to the conserved ATP-binding domain. Hits were assessed for the enzymatic inhibition of three HKs, and select compounds found to inactivate at least two of these proteins were subjected to cell-based assays. The three HKs were chosen to be representative of the 11 subfamilies of HKs. Class 1 comprises the majority of HKs, and members of this group possess all the homology boxes used to describe conservation across the HK superfamily.30,31 As such, we selected two class-1 HKs and an extensively studied member of class 9. Assay development and screening employed HK853 (Thermotoga maritima), a class-1 HK that can readily be produced in sufficient quantities for a large screen and is stable and active over extended periods.32−35 Subsequent secondary screening included VicK and CheA.36a,37a VicK (Streptococcus pneumoniae; homologous to WalK or YycG) is a class-1 HK and comprises an essential TCS in low-GC Gram-positive bacteria. For its TCS essentiality and implications in peptidoglycan synthesis, VicK alone has been considered an antibacterial target.30,36,37 The chemotactic HK, CheA (Escherichia coli; class 9), was included due to the distinct organization of its domains and the prevalence of this protein in TCS literature.30 While many of CheA’s ATP-binding residues are conserved in regards to the HK superfamily, enough variation exists to make CheA unique (e.g., N- and F-boxes; Supporting Information Figure 1).30,38 Thus, molecules capable of inhibiting two or three of these proteins, nine of which were identified in this work, are likely to be general scaffolds useful for the development of wide-scale HKs inhibitors in numerous bacterial species.
Results and Discussion
Development of Fluorescence Polarization Assay
Although previous in silico screens targeted the conserved ATP-binding pocket, follow-up experiments only characterized inhibition of one HK.23,24,27 Conversely, studies that identified molecules with inhibitory activity against multiple HKs used methods (i.e., differential growth assays, phosphorylation) that did not explicitly elucidate that nucleotide binding to the ATP-binding domain was disrupted.28,29 Therefore, we pursued the development of a straightforward assay that would indicate binding of small molecules in the active site of HK853, with the ultimate goal of validating the identified leads with multiple HKs. We sought to generate a fluorescence polarization (FP) assay as these are readily translated into high-throughput format (Figure 2a). Thus, we synthesized a fluorescent, nonhydrolyzable adenosine diphosphate (ADP) probe, ADP-BODIPY (1) (Figure 2b), which was competitive with the activity-based probe BODIPY-FL-ATPγS (B-ATPγS; Supporting Information Figure 2).34 The Kd 6.79 ± 0.11 μM for the HK–probe complex is in range with reported HK–ATP affinity values of low to midmicromolar (Figure 2c).34,39,40 Optimized concentrations of individual components were determined to be 10 nM ADP-BODIPY (1) for minimal FP variation (Supporting Informaiton Figure 3) and 25 μM HK853 to achieve 80% probe binding, providing an ample window of signal to monitor probe displacement (Supporting Information Figure 4). Using these conditions, the displacement of ADP-BODIPY (1) was visualized as a dose-dependent decrease in FP, as illustrated in competition assays with nucleotides ATP (2), ADP (3), and nonhydrolyzable analogue AMP-PNP (4) (Figure 2d). The IC50 values were determined to be 23.0 ± 1.1 μM, 27.7 ± 0.9 μM, and 38.1 ± 1.4 μM, respectively. In addition, the assay was tolerant to both Triton X-100 (Supporting Information Figure 5) and dimethyl sulfoxide (DMSO; Supporting Information Figure 6), both of which were important for compound solubility in screening.
Figure 2.

FP assay. (a) Displacement assay designed to identify compounds that specifically affect binding of native substrate to the conserved ATP-binding domain. (b) FP probe, ADP-BODIPY (1), used in screening. (c) Binding curve for 0–200 μM HK853 with 10 nM (1) (Kd = 6.79 ± 0.11 μM). (d) Competition of 25 μM HK853 and 10 nM (1) with 0–1000 μM nucleotides.
High-Throughput Primary Screening of 53 311 Compounds
A pilot screen containing 3391 known bioactive molecules was performed in 384-well plate format. Hit rates were 4–11% for individual collections in the pilot screen, and the assay’s excellent performance in 384-well plates (Z′ = 0.85) prompted further miniaturization to 1536-well plates (Z′ = 0.73). This format was used to screen a total of 49 920 diverse and target-based small molecules, resulting in 206 hit compounds (0.41% hit rate) that were confirmed by examining autofluorescence, ADP-BODIPY (1) quenching, reproducibility, and dose–response activity. Ultimately, 115 compounds were purchased for secondary screening.
Aggregation Analysis of 115 Compounds
One of the largest setbacks in previous HK inhibitor initiatives was the discovery of molecules that inhibited by aggregation.11 Due to the hydrophobic nature of many of the molecules, all 115 compounds were screened for their propensity to aggregate HK853 under nondenaturing conditions by native polyacrylamide gel electrophoresis (native-PAGE) and silver staining. Disappearance of the dimeric HK853 gel band was indicative of higher-ordered oligomers, establishing a “cut-off” concentration for each compound (Supporting Information Figures 7 and 8).
Screening of Three Proteins for Inhibition of Autophosphorylation
All 115 compounds were tested at subaggregatory concentrations for the dose-dependent inhibition of three proteins—HK853, Vick, and CheA—to identify small molecules that target multiple HKs. Since the HTS measured binding, demonstrating the inhibition of autophosphorylation was essential. To attain appropriate signal windows for quantitating dose-dependent inhibition, HK853 activity was measured using B-ATPγS34 and VicK and CheA with ATP [γ-33P]. Based on HK853 % inhibition (half-maximal and total), 69 compounds were further evaluated for VicK inhibition, of which 28 were finally tested with CheA. Nine of the original 115 follow-up compounds were chosen as leads, six that inhibited all three HKs and three that inhibited both HK853 and VicK, all representing promising scaffolds for general HK inhibitors.
Lead Compounds Containing Adenine Scaffold
Four of the identified lead compounds (5, 6, 7, 8; Supporting Information Figures 9–12) active against all three HKs share an adenine core (Figure 3a). It is not surprising that molecules mirroring the native substrate would inhibit an ATP-binding enzyme. However, their activity is curious when compared to the nucleotides ADP (3) and adenosine monophosphate (AMP) (9) (Figure 3b; Supporting Information Figures 13–14). The presence of the β-phosphate on ADP (3) results in more than 300-fold increased potency over AMP (9) (Table 1). However, the lead compounds—void of phosphates and the ribose ring altogether—are more potent than AMP. An overlay of the HK853 dose–response curves (DRCs) illustrates this well, where the DRCs for lead compounds 5–8 fall between those of ADP (3) and AMP (9) (Figure 3c). Additionally, adenine’s exocyclic primary amine is a key interaction for nucleotide binding in the HK active site through formation of a salt bridge with the invariant Asp in the G1-box (Figure 1b, orange).35 Not only do the lead compounds lack the phosphates, but they are also modified at the amino position, which could affect Asp binding and increase steric hindrance. When we compared the inhibitory activity of the leads to adenine (10) (Supporting Information Figure 15), which contains the native N6 primary amine but similarly lacks the ribose ring and phosphates, we found that the lead compounds with larger N6 modifications were unexpectedly more effective at inhibiting HK853. Together, the comparison of DRCs for purine-like leads suggests that 5–8 may bind the ATP-binding pocket in a different orientation than adenine and that the enzymes are highly discerning about what moieties can be added at this position as a large number of related molecules did not inhibit multiple HKs (Supporting Information Table 1, compounds S4, S27–38).
Figure 3.

Lead compounds with adenine core inhibit HK853, VicK, and CheA. (a) Molecules 5, 6, 7, 8 vary by modifications to the exocyclic, N6-amine of adenine. (b) Inhibition of HK853, VicK, and CheA with phosphate-containing ADP (3) and AMP (9). (c) Comparison of purine-containing compounds for HK853 inhibition.
Table 1. In Vitro Inhibition of Three HKs by Leads and Related Compounds.
| IC50 values (μM)a (95% confidence interval), n = 2 | |||
|---|---|---|---|
| compd. | HK853 | VicK | CheA |
| ADP (3) | 2.57 (2.30–2.87) | 4.81 (3.96–5.85) | 21.5 (17.7–26.1) |
| 5 | 49.6 (37.5–65.6) | 372 (306–453)b | 268 (180–399)b |
| 6 | 95.3 (69.0–131.5) | 1190 (1050–1350)b | 709 (655–768)b |
| 7 | 145 (82–254) | 1310 (875–1960)b | 478 (399–572)b |
| 8 | 131 (111–154)b | 367 (328–410)b | 413 (353–483)b |
| AMP (9)c | 1030 (830–1270) | 7150 (4820–10600) | 7640 (6230–9360) |
| adenine (10) | 463 (279–767)b | d | d |
| 11 | 1.21 (1.03–1.42) | 75.0 (69.9–80.5)b | e |
| 12 | 7.15 (6.73–7.59) | 618 (517–740)b | 1340 (1120–1600)b |
| 13 | 28.4 (24.4–33.2)b | 171 (127–230)b | e |
| 14 | 3.25 (2.59–4.06) | 26.4 (21.1–33.0) | e |
| 15 | 15.1 (10.5–21.8) | 216 (133–353) | 111 (84–146) |
Concentration at which phosphorylation decreased by 50% in competition assays according to eq 1.
Dose–response curve did not plateau at complete inhibition; IC50 was estimated by constraining bottom of curve to 0% activity.
AMP was tested at higher concentrations to reach complete inhibition as it was not restricted by % DMSO.
Not tested.
No inhibition observed.
Lead Compounds with Diverse Structures
The remaining leads possess diverse structural features. Compounds 11, 13, and 14 were active against HK853 and VicK, and 12 and 15 were also active against CheA (Figure 4a; Supporting Information Figures 16–20). Moreover, potency of 11–15 is greater than the adenine-like leads, with HK853 inhibition more closely reflecting that of ADP (Figure 4b, Table 1). To confirm 11–15 were not inhibiting nonspecifically by forming colloidal aggregates,41 dose-inhibition assays were repeated with HK853 in the presence of a high concentration of bovine serum albumin (BSA) (Supporting Information Figure 21).
Figure 4.
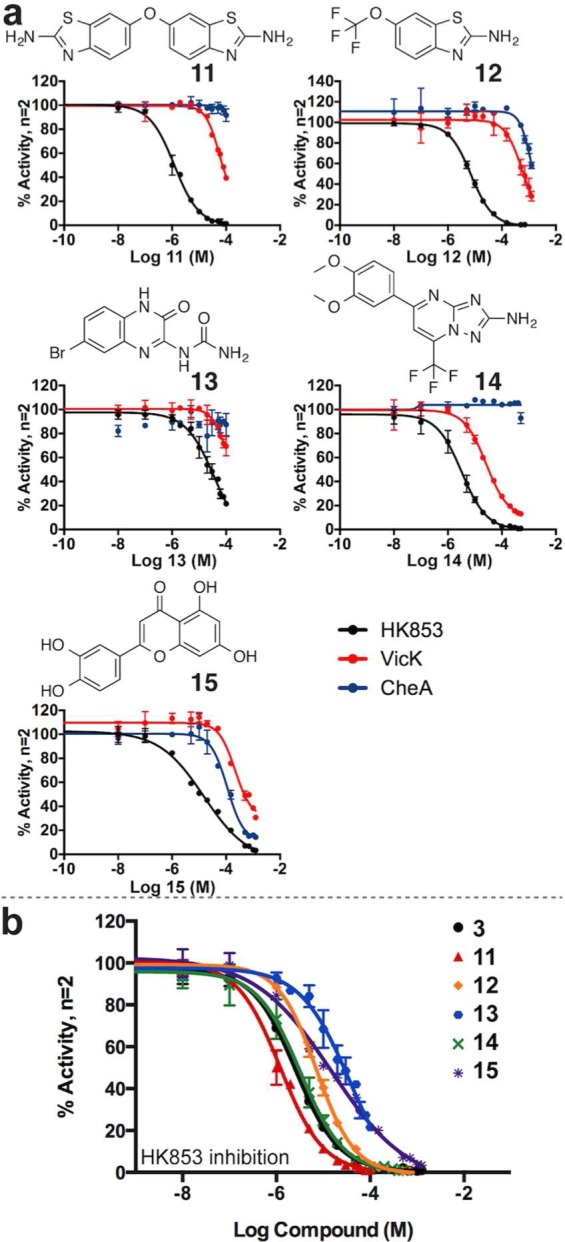
Lead compounds with diverse structures inhibit HK proteins. (a) DRCs for leads 11, 12, 13, 14, and 15 with HK853, VicK, and CheA. (b) Comparison of leads with ADP (3) for HK853 inhibition.
As HK853 and VicK are from the largest class of HKs, compounds inhibiting two of our test HKs hold promise for inhibiting a large subset of bacterial HKs. An aminobenzothiazole scaffold is present in both leads 11 and 12 with the former appearing “dual-headed,” containing two of these components. Interestingly, 11 exhibited increased potency in comparison to 12 in the HK inhibition assays, perhaps suggesting that the aminobenzothiazole scaffold could be important in the development of HK inhibitors. Thirteen compounds containing an aminobenzothiazole were found to only inhibit one of the three targeted proteins and will provide important information for future design of global HK inhibitors (Supporting Information Table 1, compounds S1–3, S5–8, S12–14, S44–46). Compounds 11–14 all possess a nitrogen-containing functionality with the potential to interact with polar residues in the active site. We previously reported that molecules containing a guanidine moiety bind to this site, participating in critical interactions with the invariant Asp deep in the pocket, a group that could potentially also be accessed by 11–14.35 Related compounds that were less active against VicK were devoid of such a group (Supporting Information Table 1, compounds S16, S44–47, S54–64).
Compounds 11, 13, and 14 (exact structures) have no previously reported bioactivity. However, 12 and 15 are the known bioactive compounds riluzole and luteolin, respectively. Riluzole (12) is used in the treatment of amyotrophic lateral sclerosis (ALS),42 and luteolin (15) is a flavonoid with a range of proposed mechanisms, including antioxidant and anti-inflammatory activity.43 Using standard broth microdilution assays, 11 and 12 inhibited the growth of Bacillus subtilis 3610, a Gram-positive wild-type strain, and E. coli DC2, a hypersensitive Gram-negative strain (Table 2; Supporting Information Figures 22–24).44 Unfortunately, when the compounds were subsequently assessed for toxicity against Vero 76 cells (African green monkey epithelial cells), these compounds were cytotoxic at the concentrations they exhibited antibacterial effects (Supporting Information Figure 25). Luteolin, 15, was one of the two compounds that did not contain an adenine core that inhibited all three HKs in our follow-up assays. This molecule inhibited the growth of E. coli DC2 without cytotoxicity to Vero 76 cells (Table 2; Supporting Information Figure 25). Lead 15 is structurally similar to the flavonoid genistein that was previously reported to inhibit a yeast HK.45 Genistein, too, was a hit in our HTS but only showed slight inhibition of HK853 activity in follow-up assays (Supporting Information Table 1, S20 along with related analogs S21 and S26). Although inhibition of several other targets by 15 (PubChem database) suggests that it may not provide selective inhibition of HKs, it would still be valuable to understand its mechanism of binding in the ATP-binding pocket of HKs since it inhibited all three of our tested proteins.
Table 2. Whole-Cell Biological Assessment of Lead Compoundsa.
| MIC valuesb (μg mL–1), n = 2 |
CC50c (μg mL–1), n = 2 | ||
|---|---|---|---|
| compd. | E. coli DC2 | B. subtilis 3610 | Vero 76 Cells |
| DMSO | 10–20% (v/v) | 20% (v/v) | >0.5% (v/v) |
| ampicillin | 0.25 | d | d |
| chloramphenicol | 0.5–1 | 2–4 | d |
| penicillin | d | ≤0.06–0.5 | d |
| vancomycin | d | 0.5 | d |
| 11 | 32–64 | 49–64 | 42 |
| 12 | 128 | 293e | 58 |
| 13 | >128 | >128 | >128 |
| 14 | >128 | >128 | >128 (16)f |
| 15 | 8 | >128 | >128 |
See Supporting Information for plate pictures and additional controls.
Lowest concentration that inhibited visible growth as observed with the unaided eye.
Concentration at which cell viability was decreased by 50%. Final concentration of DMSO ≤ 0.5% (v/v).
Not tested.
12 was tested at higher concentrations because of its structural similarity to 11, which inhibited growth.
An insufficient quantity of molecule 14 was available for these experiments. A similar compound (16; missing a methoxy group) was examined to provide preliminary information about the potential toxicity of this scaffold (Supporting Information Figure 25).
The Bergerat fold in the ATP-binding domain of HKs is not present in the abundant Ser/Thr and Tyr kinases, providing a specific kinase target within a human host. However, the GHKL protein class (DNA gyrase, heat shock protein 90 (Hsp90), HKs, and MutL), in conjunction with mitochondrial proteins pyruvate dehydrogenase kinase and branched-chain α-ketoacid dehydrogenase kinase, all share the Bergerat fold.13,18,19 Indeed, a survey of the literature reveals similarities between our lead compounds and those being used to inhibit GHL proteins. For example, the purine-like nature of lead 11 mirrors that of Hsp90 inhibitors Debio 0932 and PU-H71 (Supporting Information Figure 26a).46 Co-crystal structures of Hsp90 with PU-H7147 and a triazoloquinazoline48 illustrated that similar exocyclic amino groups bind to the conserved Asp, again indicating the importance of this residue. Furthermore, the aminothiazole of 11 and 12, the urea of 13, and the aminotriazole of 14 are consistent components of GHL inhibitory compounds, and several also appear in other HK studies (Supporting Information Figure 26b).23,27,46,48−51 Evidence of these successes in proteins with similar ATP-binding domains validate the scaffolds identified in our studies and will provide guidance in future inhibitor development.
Eventually, the challenge of achieving selectivity for HKs among the GHKL proteins will need to be addressed. However, precedence for this has already been reported. Tso et al. analyzed the structure–activity relationship (SAR) of a known Hsp90 inhibitor and effectively shifted its specificity from Hsp90 to pyruvate dehydrogenase kinase.52 Additionally, study of four Hsp90 inhibitors for binding to the PhoQ (an HK) ATP-binding domain resulted in only one that bound, radicicol, but with >30-fold difference in selectivity between Hsp90 and PhoQ.19 A perusal of compounds included in our HTS revealed that the Hsp90 inhibitors geldanamyin, 17-AAG, and radicicol were included in the primary screening but failed to displace ADP-BODIPY (1). These examples and knowledge of residue-specific variations between the GHKL proteins will facilitate the tuning of HK specificity in future inhibitor development.13 Our preliminary cytotoxicity screen alerts us to the need for the careful monitoring of toxic effects. Optimistically, the use of SAR to reduce toxicity has been accomplished through optimization of thiazolidione molecules targeting YycG.27 In addition, compounds with similar structures to our leads provide initial SAR for future lead optimization (Supporting Information Table 1).
Conclusions
Our chief objective was to identify scaffolds and compounds that generally target the conserved ATP-binding domain of HKs. In this paper, we have described the development and execution of a HTS using an FP displacement assay to elucidate binding to the active site. Follow-up studies guided us to nine compounds that inhibit multiple HKs. Four leads containing an adenine scaffold may have implications for targetable inhibitor space in the ATP-binding pocket as they bind with similar or better potency than nucleotides featuring the native phosphate and exocyclic amine. The other five molecules possess distinct structures with greater potency. While the most potent compounds also demonstrated cytotoxicity, they expand our knowledge of small-molecule inhibition of HKs. Excitingly, leads were effective in multiple proteins, two of which comprise a large division of the HK superfamily and are a significant advancement toward general HK inhibition. The proposal for a multitargeted, TCS-mediated antibiotic may find inspiration from these molecules to facilitate progress in the development of antibacterials with a novel mechanism of action.
Methods
General methods, protein overexpression and purification, ADP-BODIPY synthesis, FP assay optimization, high-throughput screening details, cytotoxicity screening, and further experimental details are described in the Supporting Information.
Reaction Buffer
Used in all assays, the reaction buffer was composed of 50 mM Tris-HCl, pH 7.8, 200 mM KCl, 5 mM MgCl2.
Data Analysis
Integrated density measurements of in-gel fluorescence and phosphorescence were performed in ImageJ.53 Data were prepared and analyzed in GraphPad Prism (version 6.0 for Mac, GraphPad Software, San Diego, California, U.S.A., www.graphpad.com). For all DRCs (control FP competition and activity assays), data were fit to a four-parameter logistic equation,
| 1 |
where y is the response, Bottom and Top are plateaus in the units of the y-axis, x is the log of the molar concentration of inhibitor, HillSlope is the slope of the curve, and IC50 is the concentration of compound required for 50% inhibition (a response half way between Bottom and Top).
Some compounds exhibited incomplete DRCs because going to higher concentrations would increase DMSO or aggregation. Visually, this meant there was no curve plateau for the “Bottom” value. However, IC50 values were desirable for purposes of comparison to other compounds. As a result, IC50 values were estimated by constraining the bottom of the curve to “0.”
For all plotted data (i.e., FP graphs, DRCs, and bar graphs), including that in the Supporting Information, error bars represent standard deviations from the number of trials indicated in the figures.
ADP-BODIPY-HK853 Binding
HK853 was concentrated, exchanged into the reaction buffer, and filtered (0.22 μM). The concentration was determined using a Nanodrop Spectrophotometer. In 96-well, polystyrene, flat-bottom, nonbinding, black plates (Greiner Bio-One), 0–200 μM HK853 was mixed with 10 nM ADP-BODIPY (1) to a final volume of 50 μL in reaction buffer. Analogous assays were set up with only HK853 to assess background and others with a saturating concentration of 5 mM ADP for the displacement of all ADP-BODIPY (1) from the active site to determine nonspecific FP.54 Plates were shaken for 5 min and incubated at room temperature (RT) for 25 min. Parallel and perpendicular fluorescence intensities (FIs) were read at RT on a BioTek Synergy H1Microplate Reader. An excitation filter of 485/20 nm and an emission filter of 528/20 nm were used for the detection of BODIPY fluorescence. Intensities from protein alone (no ADP-BODIPY (1)) were subtracted out as background. FP values were calculated using
| 2 |
where I∥ and I⊥ are parallel and perpendicular FIs from the microplate reader, respectively, and G is the G-factor,55 which was 0.87. Wells containing saturating ADP concentrations were used to determine FP due to nonspecific binding of ADP-BODIPY (1) to HK853, and thus specific binding by subtracting these values from the total measured FP.54 All assays were performed in triplicate, and average FP values were plotted in GraphPad Prism. The concentration of HK853 was high enough to ensure minimal depletion after ADP-BODIPY (1) binding, and data were fit to a single-site binding model for determination of the equilibrium dissociation constant, or Kd value.54 The fraction of ADP-BODIPY (1) bound as HK853 increased was calculated to determine the HK853 concentration at which >80% of the probe was bound.54,55
Control FP Competition Assays
Control DRCs were performed with the nucleotides ATP, ADP, and AMP-PNP. In 96-well plates, 25 μM HK853 was mixed with 10 nM ADP-BODIPY (1) and 0–1000 μM competing nucleotide, >100-fold higher than the Kd.54 To mimic potential HTS conditions, 5% (v/v) DMSO was also added to wells. Assays and FP readings were performed in triplicate as above, and average FP values were plotted in GraphPad Prism with respect to the log of the molar concentration of competitor. IC50 values were determined using eq 1.
HK853 Aggregation Analysis
To analyze the propensity for test compounds to cause aggregation, each was mixed at eight concentrations (0–1250 μM) with purified 0.44 μM HK853 in 25 μL of 20 mM HEPES buffer (5% (v/v) DMSO final). After incubating at RT for 30 min, 8.6 μL native-PAGE sample loading buffer was added, and 15 μL was loaded onto a 7.5% polyacrylamide gel (160 ng HK853 per lane). Proteins were resolved by native-PAGE and silver staining. Compound-induced aggregation was detected by the disappearance of the dimeric HK853 band. NH125 was used as a positive aggregation control.35
Inhibition of HK853 Activity
B-ATPγS competition screening with all 115 compounds was performed at concentrations that did not cause aggregation.34 Triton X-100 was premixed with reaction buffer to yield 0.1% (v/v) in final 25-μL reactions. In reaction buffer, 0.46 μM HK853 was preincubated with test compounds in 24 μL for 30 min. The addition of 1 μL B-ATPγS brought the final 25-μL reactions to 0.44 μM HK853 and 2 μM B-ATPγS in the presence of competitors and 5% DMSO. Samples were mixed and incubated in the dark at RT for 1 h before quenching with 8.6 μL 2× SDS-PAGE sample loading buffer and loading 10 μL on a 10% stacking gel. After SDS-PAGE, in-gel fluorescence detection elucidated HK853 activity, and silver staining of the gels ensured even protein loading. Integrated density values of the fluorescent gel bands were normalized as “% Activity” with respect to a control that contained no inhibitor. Data were plotted in GraphPad Prism with relation to the log of molar inhibitor to determine IC50 values (eq 1).
Inhibition of VicK Activity
Sixty-nine compounds were tested for inhibition of VicK in competitive ATP [γ-33P] assays. VicK was preincubated with test compounds in 8 μL for 30 min at RT. Then, 2 μL of a radioactive ATP mix was added. The final 10-μL reactions contained 2 μM VicK, 3 μM ATP (20 Ci mmol–1), test compounds (5% DMSO), and 0.1% Triton X-100 (premixed in reaction buffer). Reactions were incubated for 30 min at RT and quenched with 10 μL 2× SDS-PAGE sample loading buffer prior to loading onto 10% stacking gels. Integrated density values of the radioactive gel bands were normalized as “% Activity” with respect to a control that contained no inhibitor. Data were plotted in GraphPad Prism with relation to the log of molar inhibitor to determine IC50 values (eq 1).
Inhibition of CheA Activity
Twenty-eight compounds were pursued for inhibition of CheA by competitive ATP [γ-33P] assays, which were the same as for VicK except a mixture of 2 μM CheA and 6 μM CheW was used.
Antimicrobial Testing
Using standard broth microdilution assays,56,57 leads 11–15 were assessed for antimicrobial activity. In 96-well, round-bottom microtiter plates, serial dilutions of leads in sterile Mueller-Hinton Broth (MHB) 2 were inoculated with 5 × 105 cfu mL–1E. coli DC2 or B. subtilis 3610. Wells contained ≤5% DMSO. Each plate included growth controls, sterility controls, DMSO controls, and controls with known antibiotics. At the time of the assay, dilutions of the growth controls were plated on MHB 2 agar to confirm that the correct number of colony-forming units were used. Agar and microtiter plates were incubated for 20 h at 37 °C. The minimal inhibitory concentration (MIC) was concluded as the lowest concentration of compound that inhibited growth as observed with the unaided eye.
Acknowledgments
We are especially grateful to M. Larsen and S. Vander Roest for screening guidance, implementation of the HTS at the University of Michigan’s Center for Chemical Genomics, and helpful comments on the manuscript. We thank A. Zlotnick and L. Li for use and help with a microplate reader for assay development. We are thankful to Y. Yu and Y. Gao for advice and facilitating the cytotoxicity assays and R. Godsey for assistance in compound characterization. Indiana University’s Physical Biochemistry Instrumentation Facility and the Indiana Molecular Biology Institute provided essential instrumentation. We thank L. Kiessling and E. Underbakke for strains overexpressing CheA and CheW and M. Winkler for an overexpression strain for the VicK construct. B. subtilis 3610 was a gift from D. Kearns. This work was supported by the National Institutes of Health (NIH) DP2OD008592, a Pew Biomedical Scholar Award (E.E.C.), an Indiana University Dean’s Fellowship (E.E.C.), and an Indiana University Quantitative and Chemical Biology training fellowship (K.E.W.).
Supporting Information Available
Methods and results for the following: ADP-BODIPY synthesis, protein production, fluorescence polarization assay, high-throughout primary screen, small-molecule screening libraries, secondary screens, screen statistics, antimicrobial testing, cytotoxicity screening, lead compound information. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ Department of Chemistry, University of Minnesota, 207 Pleasant St. SE, Minneapolis, MN 55455, United States.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Centers for Disease Control and Prevention (2013) Antibiotic Resistance Threats in the United States, CDC, Atlanta, GA. Available Online: http://www.cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf.
- Silver L. L. (2011) Challenges of antibacterial discovery. Clin. Microbiol. Rev. 24, 71–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clatworthy A. E.; Pierson E.; Hung D. T. (2007) Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 3, 541–548. [DOI] [PubMed] [Google Scholar]
- Nathan C. (2012) Fresh approaches to anti-infective therapies. Sci. Transl. Med. 4, 140sr142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock A. M.; Robinson V. L.; Goudreau P. N. (2000) Two-component signal transduction. Annu. Rev. Biochem. 69, 183–215. [DOI] [PubMed] [Google Scholar]
- Stock J. B.; Stock A. M.; Mottonen J. M. (1990) Signal transduction in bacteria. Nature 344, 395–400. [DOI] [PubMed] [Google Scholar]
- Thomason P.; Kay R. (2000) Eukaryotic signal transduction via histidine-aspartate phosphorelay. J. Cell Sci. 113, 3141–3150. [DOI] [PubMed] [Google Scholar]
- Bretl D. J.; Demetriadou C.; Zahrt T. C. (2011) Adaptation to environmental stimuli within the host: Two-component signal transduction systems of Mycobacterium tuberculosis. Microbiol. Mol. Biol. R. 75, 566–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh Y.; Eguchi Y.; Watanabe T.; Okamoto S.; Doi A.; Utsumi R. (2010) Two-component signal transduction as potential drug targets in pathogenic bacteria. Curr. Opin. Microbiol. 13, 232–239. [DOI] [PubMed] [Google Scholar]
- Stephenson K.; Hoch J. A. (2002) Virulence- and antibiotic resistance-associated two-component signal transduction systems of Gram-positive pathogenic bacteria as targets for antimicrobial therapy. Pharmacol. Therapeut. 93, 293–305. [DOI] [PubMed] [Google Scholar]
- Stephenson K.; Yamaguchi Y.; Hoch J. A. (2000) The mechanism of action of inhibitors of bacterial two-component signal transduction systems. J. Biol. Chem. 275, 38900–38904. [DOI] [PubMed] [Google Scholar]
- Worthington R. J.; Blackledge M. S.; Melander C. (2013) Small-molecule inhibition of bacterial two-component systems to combat antibiotic resistance and virulence. Future Med. Chem. 5, 1265–1284. [DOI] [PubMed] [Google Scholar]
- Wilke K. E.; Carlson E. E. (2013) All signals lost. Sci. Transl. Med. 5, 203ps212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman A. L.; Kulasekara B.; Rietsch A.; Boyd D.; Smith R. S.; Lory S. (2004) A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev. Cell 7, 745–754. [DOI] [PubMed] [Google Scholar]
- Kraus D.; Herbert S.; Kristian S. A.; Khosravi A.; Nizet V.; Gotz F.; Peschel A. (2008) The GraRS regulatory system controls Staphylococcus aureus susceptibility to antimicrobial host defenses. BMC Microbiol. 8, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova O. E.; Sauer K. (2009) A novel signaling network essential for regulating Pseudomonas aeruginosa biofilm development. PLoS Pathog. 5, e1000668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi Y.; Okada T.; Minagawa S.; Oshima T.; Mori H.; Yamamoto K.; Ishihama A.; Utsumi R. (2004) Signal transduction cascade between EvgA/EvgS and PhoP/PhoQ two-component systems of Escherichia coli. J. Bacteriol. 186, 3006–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R.; Inouye M. (2000) GHKL, an emergent ATPase/kinase superfamily. Trends Biochem. Sci. 25, 24–28. [DOI] [PubMed] [Google Scholar]
- Guarnieri M. T.; Zhang L. D.; Shen J. P.; Zhao R. (2008) The Hsp90 inhibitor radicicol interacts with the ATP-binding pocket of bacterial sensor kinase PhoQ. J. Mol. Biol. 379, 82–93. [DOI] [PubMed] [Google Scholar]
- Tanaka T.; Saha S. K.; Tomomori C.; Ishima R.; Liu D.; Tong K. I.; Park H.; Dutta R.; Qin L.; Swindells M. B.; Yamazaki T.; Ono A. M.; Kainosho M.; Inouye M.; Ikura M. (1998) NMR structure of the histidine kinase domain of the E. coli osmosensor EnvZ. Nature 396, 88–92. [DOI] [PubMed] [Google Scholar]
- Marina A.; Mott C.; Auyzenberg A.; Hendrickson W. A.; Waldburger C. D. (2001) Structural and mutational analysis of the PhoQ histidine kinase catalytic domain. Insight into the reaction mechanism. J. Biol. Chem. 276, 41182–41190. [DOI] [PubMed] [Google Scholar]
- Xie W.; Dickson C.; Kwiatkowski W.; Choe S. (2010) Structure of the cytoplasmic segment of histidine kinase receptor QseC, a key player in bacterial virulence. Protein Peptide Lett. 17, 1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R. Z.; Zheng L. K.; Liu H. Y.; Pan B.; Hu J.; Zhu T.; Wang W.; Jiang D. B.; Wu Y.; Wu Y. C.; Han S. Q.; Qu D. (2012) Thiazolidione derivatives targeting the histidine kinase YycG are effective against both planktonic and biofilm-associated Staphylococcus epidermidis. Acta Pharmacol. Sin. 33, 418–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z.; Zhang J.; Xu B.; Chen L.; Wu Y.; Yang X.; Shen X.; Molin S.; Danchin A.; Jiang H.; Qu D. (2006) Structure-based discovery of inhibitors of the YycG histidine kinase: New chemical leads to combat Staphylococcus epidermidis infections. BMC Microbiol. 6, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N.; Wang F.; Niu S.; Cao J.; Wu K.; Li Y.; Yin N.; Zhang X.; Zhu W.; Yin Y. (2009) Discovery of novel inhibitors of Streptococcus pneumoniae based on the virtual screening with the homology-modeled structure of histidine kinase (VicK). BMC Microbiol. 9, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X.; Zhang J.; Chen M.; Wu Y.; Wang X.; Chen J.; Zhang J.; Shen X.; Qu D.; Jiang H. (2011) The effect of the potential PhoQ histidine kinase inhibitors on Shigella flexneri virulence. PLoS One 6, e23100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Zhao D.; Chang J.; Yan L.; Zhao F.; Wu Y.; Xu T.; Gong T.; Chen L.; He N.; Wu Y.; Han S.; Qu D. (2014) Efficacy of novel antibacterial compounds targeting histidine kinase YycG protein. Appl. Microbiol. Biot. 98, 6003–6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi Y.; Kubo N.; Matsunaga H.; Igarashi M.; Utsumi R. (2011) Development of an antivirulence drug against Streptococcus mutants: Repression of biofilm formation, acid tolerance, and competence by a histidine kinase inhibitor, Walkmycin C. Antimicrob. Agents Ch. 55, 1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour R.; Foster J. E.; Sheng Q.; McClain J. R.; Riley A.; Sun P. M.; Ng W. L.; Yan D.; Nicas T. I.; Henry K.; Winkler M. E. (2005) New class of competitive inhibitor of bacterial histidine kinases. J. Bacteriol. 187, 8196–8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grebe T. W.; Stock J. B. (1999) The histidine protein kinase superfamily. Adv. Microb. Physiol. 41, 139–227. [DOI] [PubMed] [Google Scholar]
- Wolanin P. M.; Thomason P. A.; Stock J. B. (2002) Histidine protein kinases: Key signal transducers outside the animal kingdom. Genome Biol. 3, 3013.1–3013.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casino P.; Rubio V.; Marina A. (2009) Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell 139, 325–336. [DOI] [PubMed] [Google Scholar]
- Marina A.; Waldburger C. D.; Hendrickson W. A. (2005) Structure of the entire cytoplasmic portion of a sensor histidine-kinase protein. EMBO J. 24, 4247–4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke K. E.; Francis S.; Carlson E. E. (2012) Activity-based probe for histidine kinase signaling. J. Am. Chem. Soc. 134, 9150–9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis S.; Wilke K. E.; Brown D. E.; Carlson E. E. (2013) Mechanistic insight into inhibition of two-component system signaling. Med. Chem. Commun. 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutu A. D.; Wayne K. J.; Sham L. T.; Winkler M. E. (2010) Kinetic characterization of the WalRKSpn (VicRK) two-component system of Streptococcus pneumoniae: Dependence of WalKSpn (VicK) phosphatase activity on its PAS domain. J. Bacteriol. 192, 2346–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underbakke E. S.; Zhu Y.; Kiessling L. L. (2011) Protein footprinting in a complex milieu: Identifying the interaction surfaces of the chemotaxis adaptor protein CheW. J. Mol. Biol. 409, 483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrac S., and Msadek T. (2008) Tearing down the wall: Peptidoglycan metabolism and the WalK/WalR (YycG/YycF) essential two-component system, in Bacterial Signal Transduction: Networks and Drug Targets (Utsumi R., Ed.), pp 214–228, Springer, New York. [Google Scholar]
- Winkler M. E.; Hoch J. A. (2008) Essentiality, bypass, and targeting of the YycFG (VicRK) two-component regulatory system in Gram-positive bacteria. J. Bacteriol. 190, 2645–2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilwes A. M.; Quezada C. M.; Croal L. R.; Crane B. R.; Simon M. I. (2001) Nucleotide binding by the histidine kinase CheA. Nat. Struct. Biol. 8, 353–360. [DOI] [PubMed] [Google Scholar]
- Stewart R. C.; VanBruggen R.; Ellefson D. D.; Wolfe A. J. (1998) TNP-ATP and TNP-ADP as probes of the nucleotide binding site of CheA, the histidine protein kinase in the chemotaxis signal transduction pathway of Escherichia coli. Biochemistry 37, 12269–12279. [DOI] [PubMed] [Google Scholar]
- Casino P.; Miguel-Romero L.; Marina A. (2014) Visualizing autophosphorylation in histidine kinases. Nature Commun. 5, 3258. 10.1038/ncomms4258. [DOI] [PubMed] [Google Scholar]
- McGovern S. L.; Caselli E.; Grigorieff N.; Shoichet B. K. (2002) A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 45, 1712–1722. [DOI] [PubMed] [Google Scholar]
- Bensimon G.; Lacomblez L.; Meininger V.; Bouche P.; Delwaide C.; Couratier P.; Blin O.; Viader F.; Peyrostpaul H.; David J.; Maloteaux J. M.; Hugon J.; Laterre E. C.; Rascol A.; Clanet M.; Vallat J. M.; Dumas A.; Serratrice G.; Lechevallier B.; Peuch A. J.; Nguyen T.; Shu C.; Bastien P.; Papillon C.; Durrleman S.; Louvel E.; Guillet P.; Ledoux L.; Orvoenfrija E.; Dib M. (1994) A controlled trial of riluzole in amyotrophic-lateral-sclerosis. N. Engl. J. Med. 330, 585–591. [DOI] [PubMed] [Google Scholar]
- López-Lázaro M. (2009) Distribution and biological activities of the flavonoid luteolin. Mini-Rev. Med. Chem. 9, 31–59. [DOI] [PubMed] [Google Scholar]
- Clark D. (1984) Novel antibiotic hypersensitive mutants of Escherichia coli. Genetic mapping and chemical characterization. FEMS Microbiol. Lett. 21, 189–195. [Google Scholar]
- Huang J.; Nasr M.; Kim Y.; Matthews H. R. (1992) Genistein inhibits protein histidine kinase. J. Biol. Chem. 267, 15511–15515. [PubMed] [Google Scholar]
- Sidera K.; Patsavoudi E. (2014) Hsp90 inhibitors: Current development and potential in cancer therapy. Recent Pat. Anti-Cancer Drug Discovery 9, 1–20. [PubMed] [Google Scholar]
- Immormino R. M.; Kang Y.; Choisis G.; Gewirth D. T. (2006) Structural and quantum chemical studies of 8-aryl-sulfanyl adenine class Hsp90 inhibitors. J. Med. Chem. 49, 4953–4960. [DOI] [PubMed] [Google Scholar]
- Casale E.; Amboldi N.; Brasca M. G.; Caronni D.; Colombo N.; Dalvit C.; Felder E. R.; Fogliatto G.; Galvani A.; Isacchi A.; Polucci P.; Riceputi L.; Sola F.; Visco C.; Zuccotto F.; Casuscelli F. (2014) Fragment-based hit discovery and structure-based optimization of aminotriazoloquinazolines as novel Hsp90 inhibitors. Bioorgan. Med. Chem. 22, 4135–4150. [DOI] [PubMed] [Google Scholar]
- Kale R. R.; Kale M. G.; Waterson D.; Raichurkar A.; Hameed S. P.; Manjunatha M. R.; Kishore Reddy B. K.; Malolanarasimhan K.; Shinde V.; Koushik K.; Jena L. K.; Menasinakai S.; Humnabadkar V.; Madhavapeddi P.; Basavarajappa H.; Sharma S.; Nandishaiah R.; Mahesh Kumar K. N.; Ganguly S.; Ahuja V.; Gaonkar S.; Naveen Kumar C. N.; Ogg D.; Boriack-Sjodin P. A.; Sambandamurthy V. K.; de Sousa S. M.; Ghorpade S. R. (2014) Thiazolopyridone ureas as DNA gyrase B inhibitors: Optimization of antitubercular activity and efficacy. Bioorg. Med. Chem. Lett. 24, 870–879. [DOI] [PubMed] [Google Scholar]
- Basarab G. S.; Manchester J. I.; Bist S.; Boriack-Sjodin P. A.; Dangel B.; Illingworth R.; Sherer B. A.; Sriram S.; Uria-Nickelsen M.; Eakin A. E. (2013) Fragment-to-hit-to-lead discovery of a novel pyridylurea scaffold of ATP competitive dual targeting type II topoisomerase inhibiting antibacterial agents. J. Med. Chem. 56, 8712–8735. [DOI] [PubMed] [Google Scholar]
- Charifson P. S.; Grillot A.-L.; Grossman T. H.; Parsons J. D.; Badia M.; Bellon S.; Deininger D. D.; Drumm J. E.; Gross C. H.; LeTiran A.; Liao Y.; Mani N.; Nicolau D. P.; Perola E.; Ronkin S.; Shannon D.; Swenson L. L.; Tang Q.; Tessier P. R.; Tian S.-K.; Trudeau M.; Wang T.; Wei Y.; Zhang H.; Stamos D. (2008) Novel dual-targeting benzimidazole urea inhibitors of DNA gyrase and topoisomerase IV possessing potent antibacterial activity: Intelligent design and evolution through the judicious use of structure-guided design and stucture–activity relationships. J. Med. Chem. 51, 5243–5263. [DOI] [PubMed] [Google Scholar]
- Tso S. C.; Qi X.; Gui W. J.; Wu C. Y.; Chuang J. L.; Wernstedt-Asterholm I.; Morlock L. K.; Owens K. R.; Scherer P. E.; Williams N. S.; Tambar U. K.; Wynn R. M.; Chuang D. T. (2014) Structure-guided development of specific pyruvate dehydrogenase kinase inhibitors targeting the ATP-binding pocket. J. Biol. Chem. 289, 4432–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband W. S. (1997–2014) ImageJ; U.S. National Institutes of Health, Bethesda, MD. [Google Scholar]
- Rossi A. M.; Taylor C. W. (2011) Analysis of protein–ligand interactions by fluorescence polarization. Nat. Protoc. 6, 365–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld D. S., Farmen M. W., Kahl S. D., Kriauciunas A., McKnight K. L., Montrose C., and Weidner J. R. (2012) Receptor binding assays for HTS and drug discovery, in Assay Guidance Manual (Sittampalam S., Ed.), Eli Lilly & Company, Indianapolis, IN. [Google Scholar]
- Andrews J. M. (2001) Determination of minimum inhibitory concentrations. J. Antimicrob. Chemoth. 48(Suppl. SI), 5–16. [DOI] [PubMed] [Google Scholar]
- Wiegand I.; Hilpert K.; Hancock R. E. (2008) Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3, 163–175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.