

Abstract
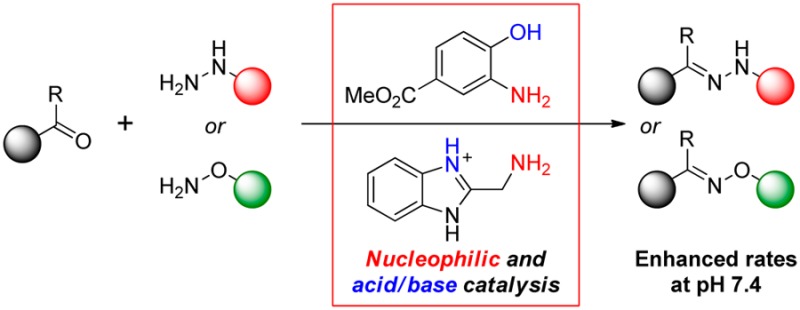
The discovery of two new classes of catalysts for hydrazone and oxime formation in water at neutral pH, namely 2-aminophenols and 2-(aminomethyl)benzimidazoles, is reported. Kinetics studies in aqueous solutions at pH 7.4 revealed rate enhancements up to 7-fold greater than with classic aniline catalysis. 2-(Aminomethyl)benzimidazoles were found to be effective catalysts with otherwise challenging aryl ketone substrates.
Bioconjugation is an essential tool in chemical biology.1 Chemical transformations that are fast, reliable, and biocompatible are key to enable important tasks such as site-selective functionalization of proteins1a and attachment of reporter molecules to biomacromolecules.2
For this reason, numerous bioconjugation techniques have emerged.1b−1d One classical bond-forming reaction is the formation of hydrazone or oxime linkages to aldehyde or ketone moieties. However, the relatively slow rate of these reactions at biological pH has limited the usefulness of this approach.1c,3 In recent years, several groups have used nucleophilic catalysts to facilitate these reactions, including the use of aniline4 and its simple substituted derivatives.5 Nucleophilic catalysis of hydrazone or oxime formation has also found use within other fields of chemistry, such as in dynamic combinatorial chemistry,4a,6 polymer chemistry,7 and the functionalization of carbohydrates via the nonreducing end.5e,5f,8 In recent studies we have focused on elucidating the electronic factors that affect the rate of formation of these linkages and on developing more efficient organocatalysts for these reactions by appending proton donor/acceptor moieties to aniline scaffolds.9 To further improve the utility of hydrazone and oxime conjugation, new catalyst scaffolds would be useful because aniline is cytotoxic and is a relatively inefficient catalyst.10
Detailed studies by Jencks of the mechanism of semicarbazone and oxime formation under aniline catalysis showed that the rate-limiting part of the reaction is condensation of the substrate with the catalyst to form an activated imine, which is subsequently attacked by the α-nucleophile in a rapid transimination reaction.11 Under neutral conditions, Jencks found that the specific rate-determining step in forming the activated imine is dehydration of the intermediate hemiaminal (Figure 1, top).12,13
Figure 1.
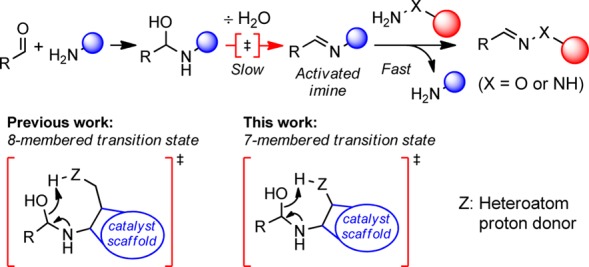
Top: Proposed reaction mechanism for hydrazone and oxime formation under nucleophilic catalysis by an amine at neutral pH.11,12 Bottom: Suggested transition states for the rate-determining dehydration step using catalysts from earlier studies and from this study.
Anilines with ortho-proton donors such as carboxylic acid or phosphonic acid groups give considerable rate enhancements over aniline itself in both hydrazone and oxime formation.9c,9d Evidence strongly supports the notion of nucleophilic catalysis combined with general acid/base catalysis, where the best catalysts have a pKa near solution pH.14 We have suggested that intramolecular protonation of the intermediate hemiaminal via an 8-membered ring in the proposed transition state (TS) facilitates the limiting dehydration step (Figure 1, bottom).9c
We hypothesized that catalysts based on a smaller scaffold could potentially lead to increased reaction rates by invoking a more favorable 7-membered intramolecular proton transfer in the TS (Figure 1, bottom).15 To test this, we investigated potential catalysts with three covalent bonds between the nucleophilic amino group and the proton donor and report here the discovery of two new highly active catalyst scaffolds.
To test catalysts, the reaction between p-chlorobenzaldehyde and phenylhydrazine in phosphate-buffered saline at pH 7.4 (PBS) with 10% DMF was followed using UV/vis spectroscopy (Scheme 1). We included the organic cosolvent to ensure solubility of all components, although later tests showed that it was unnecessary, at least in some cases (see below). Nonlinear regression was used to obtain (pseudo)-second-order rate constants (k2) from the data (Tables 1 and 2).
Scheme 1. Model Reaction for Catalyst Screening.
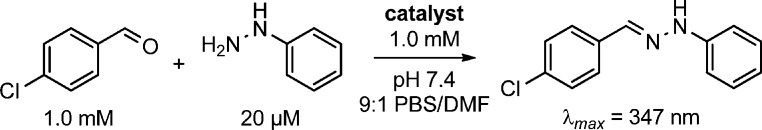
Table 1. Aromatic Amines, Aminophenols, and Aliphatic Amines As Catalysts for Hydrazone Formationa.
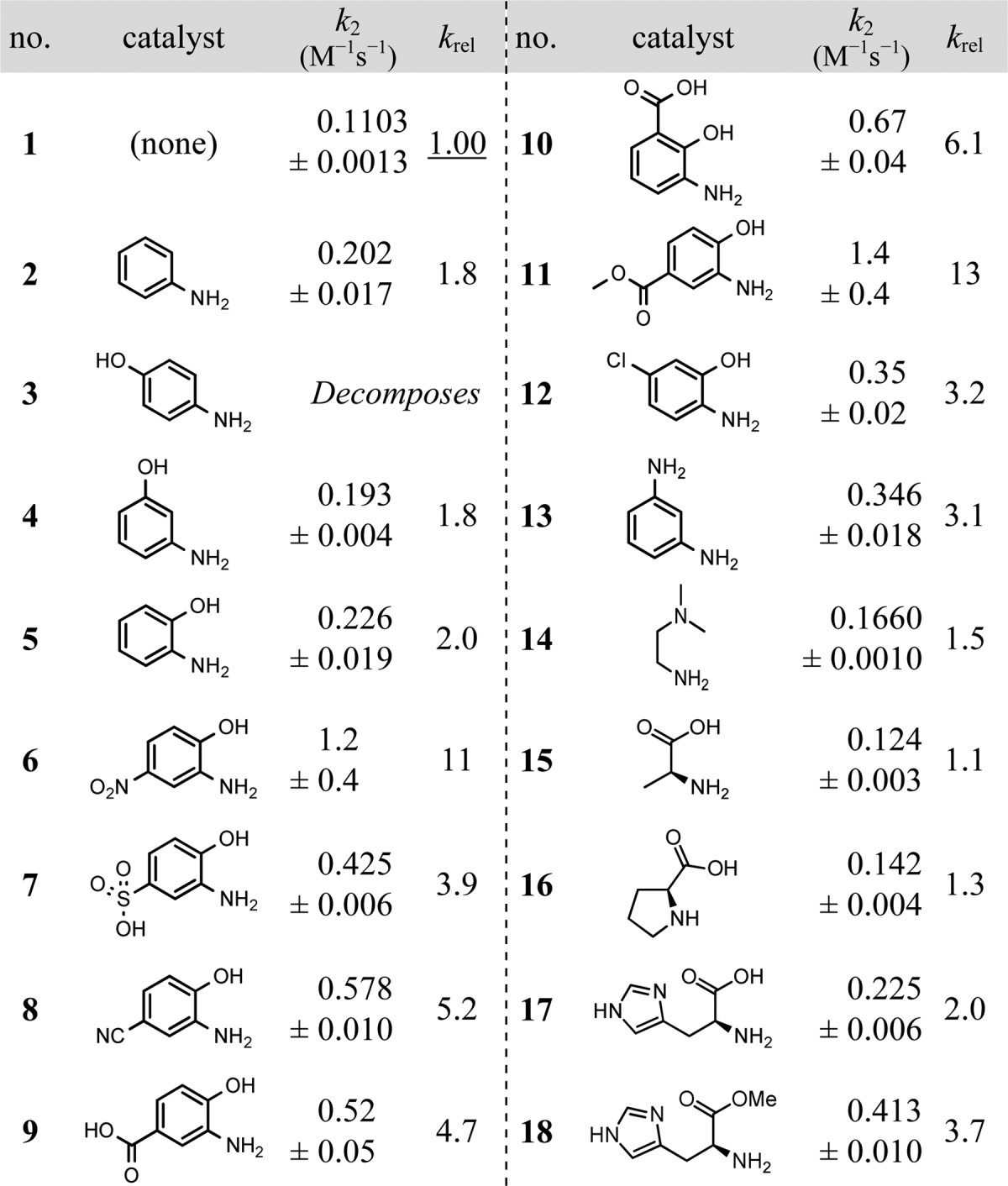
Second-order rate constants are given as mean values ± standard deviations (based on triplicate measurements or better). Conditions: 1:9 DMF/water with 123 mM NaCl, 2.4 mM KCl, and 10.7 mM sodium and potassium phosphates (pH 7.4). Concentrations of reactants and catalyst as per Scheme 1.
Table 2. Aminoalkyl-Substituted N-Heterocycles As Catalysts for Hydrazone Formationa.
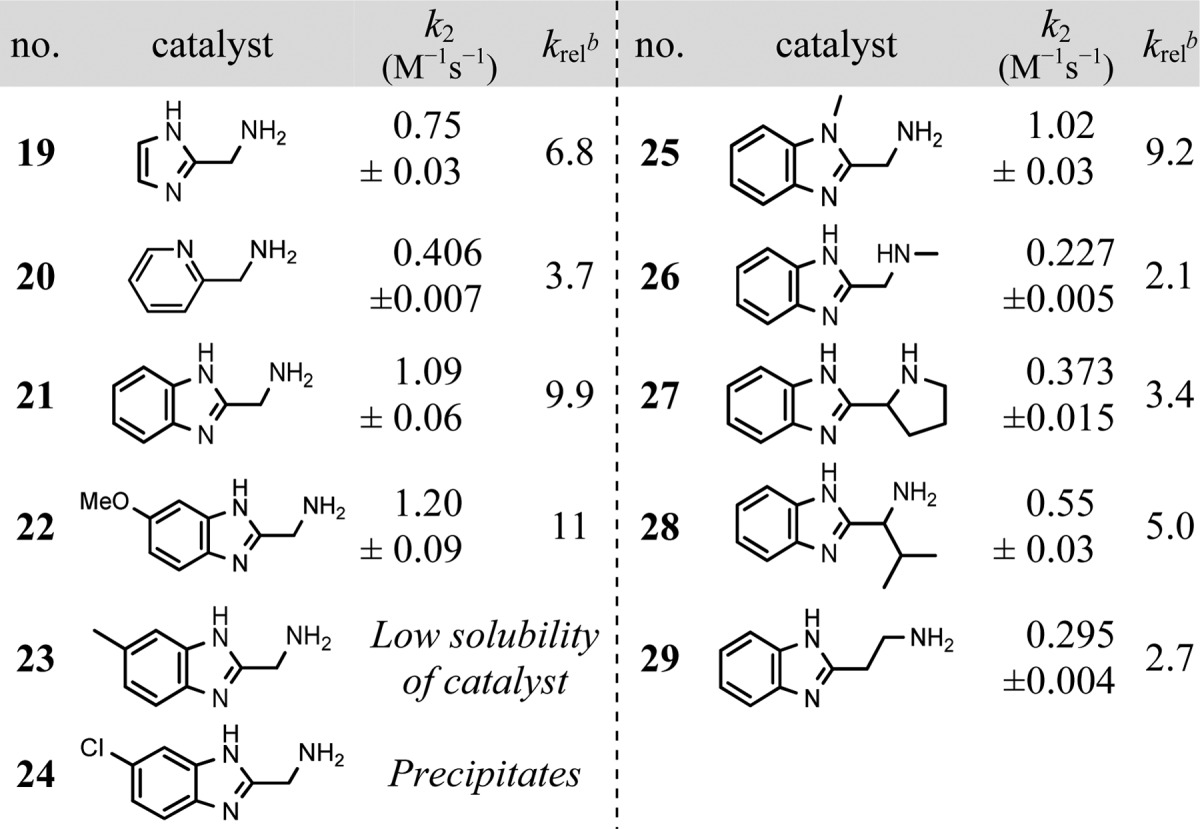
Aniline (2) was screened as a reference (Table 1), and it yielded a 1.8-fold increase in reaction rate at 1.0 mM. Proceeding to new scaffolds, first we studied aminophenols. 4-Aminophenol (3) proved to be unstable, but it was evident that 2-aminophenol (5) is an interesting scaffold. A range of substituted 2-aminophenols were investigated (entries 6–12), and it was found that electron-withdrawing groups para to the hydroxy group (e.g., NO2, CN, COOR) enhanced the catalytic effect. This supports the hypothesis that the system is under general acid/base catalysis, along with the expected nucleophilic catalysis, as adding electron-withdrawing groups to 2-aminophenol (pKa 9.75)16 brings the phenol pKa closer to the pH of the medium. A broader range of other aminophenols were also tested (see Supporting Information (SI)), but p-nitro and p-methoxycarbonyl derivatives (entries 6, 11) yielded the fastest rates. Indeed, the p-methoxycarbonyl derivative (11) yielded rate enhancements 7 times that of aniline and 4-fold better than that of 1,3-diaminobenzene (13), described recently as “highly efficient”.5b
At pH 7.4, simple aliphatic amines, with pKa’s of ca. 9–11, are expected to be poor catalysts in this context due to the non-nucleophilic nature of their ammonium ions. Inspired by the work of Hine on imine formation of protonated diamines,3e,17 2-dimethylaminoethylamine (14) with a pKa value of 7.1318 was investigated, but only a small rate enhancement was observed. All proteinogenic amino acids in principle possess the structural motif sought after in this study, as they all have three covalent bonds between the amine nucleophile and the proton donor (carboxylic acid OH-group). Although α-amino acids are generally zwitterionic at pH 7.4, a small selection was screened. l-Alanine (15) showed practically no catalytic activity (krel = 1.1) while l-proline (16) showed a slightly higher activity (krel = 1.3), possibly due to its higher nucleophilicity.19 However, considerable catalytic activity was found with l-histidine (17) (krel = 2.0) and l-histidine methyl ester (18) (krel = 3.7). Since esterification of the carboxylic acid leads to an increase in reaction rate, we hypothesized that the imidazolium moiety was responsible for the high activity of l-histidine compared to other aliphatic amine compounds. The imidazolium moiety in the methyl ester of histidine has a pKa value of 7.33,20 consistent with the importance of protein histidine units in general acid/base catalysis at physiological pH.21
In light of these results, we examined other aminoalkyl imidazoles (Table 2), with the simplest derivative, 2-(aminomethyl)imidazole (19), giving a 6.8-fold rate enhancement. 2-(Aminomethyl)pyridine (20) with a pKa of 8.622 also showed good catalytic activity for an aliphatic amine, with rate enhancements similar to that of l-histidine methyl ester (entry 18 in Table 1). Significantly, an almost 10-fold increase in reaction rate was seen with 2-(aminomethyl)benzimidazole (21). With a pKa of 7.85,23 this catalyst falls near the ideal range for general acid/base catalysis at pH 7.4.
Based on the latter finding, some derivatives of 2-(aminomethyl)benzimidazole were investigated to elucidate the electronic and structural requirements for this new catalyst scaffold (entries 22–29). While solubility issues prevented the proper measurement of derivatives bearing methyl or chloro substituents in the 5-position (entries 23, 24), the 5-methoxy derivative (22) gave a slightly higher rate constant than the parent compound (21). Studies of the two N-methyl derivatives (25 and 26) suggest that the mechanism of action for 2-(aminomethyl)benzimidazole catalysts involves imine formation on the amino group, rather than iminium formation on the imidazole moiety, since methylation of the imidazole-N has only a small effect on the rate. Employing a more nucleophilic secondary amine, as in the 2-pyrrolidinyl derivative (27), did improve the catalyst slightly compared to the N-methyl derivative (26), but it did not fully restore the reactivity seen in the parent compound (21). Adding an isopropyl group to the bridging methylene lowers the activity only slightly (entry 28). Using an ethylamino group instead of the methylamino group confers more flexibility to the catalyst, but this also lowers the activity (entry 29), consistent with the notion that smaller ring sizes are favorable for intramolecular catalysis.
The finding of these new, active catalyst scaffolds prompted us to investigate their reaction scope with a range of aldehydes and ketones. We tested the best catalysts identified here with 10 different electrophiles, employing phenylhydrazine as the standard nucleophile (Scheme 2). Rate constants for all substrates were measured without a catalyst and under aniline catalysis for comparison (Table 3). We also measured rate constants with 2-amino-5-methylphenylphosphonic acid (denoted PA in Table 3), a catalyst previously reported by our group.9d
Scheme 2. General Reaction for Scope Investigation.
Table 3. Scope of Catalysis of Hydrazone Formation between Phenylhydrazine and a Range of Aldehydes and Ketonesa.
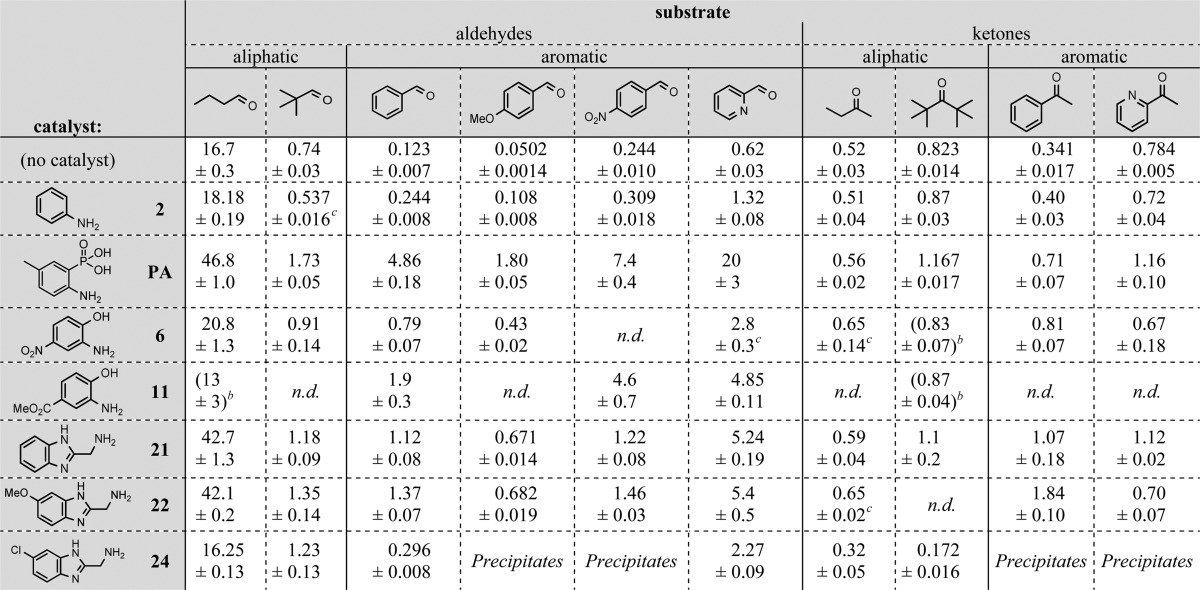
Second-order rate constant (M–1 s–1) given as mean value ± standard deviation (based on triplicate measurements). Conditions as per Table 1 and Scheme 2.
Values in parentheses are normalized to 1.0 mM catalyst concentration by extrapolation from lower catalyst concentrations to avoid detector saturation caused by spectral overlap (see SI).
From duplicate measurements. n.d., no data.
Comparing reaction rates of aliphatic vs aromatic aldehydes, we confirmed previous findings that aliphatics react much more rapidly.9a,9b Even bulky aliphatic aldehydes react much faster than aromatic aldehydes, and rate enhancements when adding catalysts are relatively low (up to 2.8-fold), suggesting that the catalysts have less impact when background hydrazone formation is fast. Significantly higher rate enhancements are seen for the aromatic aldehydes (up to 19-fold for new catalysts). The best new catalysts are roughly on par with the previously reported phosphonic acid catalyst (PA) when using aliphatic aldehyde substrates, but for aromatic aldehydes the phosphonic acid derivative is generally better (1.6–3.7-fold faster than the best new catalyst).
In contrast to aldehydes, ketones react at similar rates whether they are aromatic or aliphatic, and efficient catalysts for these substrates have previously failed to emerge, either from studies of the classic aniline catalyst or more recent investigations.9c,9d,24 We reasoned that employing primary aliphatic amine nucleophiles such as 2-(aminomethyl)benzimidazoles might solve this problem by presenting less steric bulk. In fact, 2-(aminomethyl)-5-methoxybenzimidazole (22) gives a 5.4-fold increase of the reaction rate of phenylhydrazine with acetophenone at 1.0 mM catalyst concentration, a hitherto unseen rate enhancement for the catalysis of hydrazone formation on ketones under physiological conditions.24 With ketone substrates, the 2-(aminomethyl)benzimidazoles 21 and 22 are better than or equivalent to the previously reported phosphonic acid catalyst (PA). This is significant because aryl ketones are common reactants in bioconjugations.1b−1d,2a,25
In order to test the applicability of these catalysts in purely aqueous conditions without cosolvent, the reaction between benzaldehyde and phenylhydrazine in phosphate-buffered saline at pH 7.4 was performed under identical conditions to Scheme 1 without addition of DMF. Rate enhancements for the three employed catalysts, aniline (2) (2.2-fold), 2-amino-4-nitrophenol (6) (4.8-fold), and 2-(aminomethyl)benzimidazole (21) (9.0-fold), were very similar to those reported in Tables 1 and 2 (see SI for details). This establishes that the cosolvent DMF does not dramatically influence the catalytic effect and is not needed for the solubility of these catalysts. This suggests their further use in biological applications.
Oxime formation is considerably slower than hydrazine formation,3a,4a,9b but oximes are more resistant to hydrolysis than hydrazones.3d Accordingly, there is a demand for catalysts of this related reaction. Therefore, we investigated our most efficient new scaffold as a catalyst for the oxime formation between 2-(dimethylamino)ethoxyamine and 4-nitrobenzaldehyde. We found that 2-(aminomethyl)benzimidazole (21) was effective in catalyzing this reaction, giving a 5.2-fold rate enhancement. This was over twice the rate afforded by aniline (see SI for details).
In summary, we have expanded the collection of catalysts for hydrazone and oxime formation by introducing two new subclasses of catalyst scaffolds, namely the 2-aminophenols and the 2-(aminomethyl)benzimidazoles. Both new scaffolds are broadly active at neutral pH, and the latter class of catalysts was shown to have high activity with aromatic aldehyde and ketone substrates. With second-order rate constants in the 0.1–50 M–1 s–1 range using a relatively low catalyst concentration of 1.0 mM, hydrazone formation under catalysis of these novel compounds represents a useful alternative to other bioorthogonal reactions, yielding similar rates in comparison to common reactions such as Cu-catalyzed azide–alkyne cycloaddtiton (“CuAAC”) and maleimide–thiol coupling reactions, while outperforming reported Staudinger ligations and strain-promoted azide–alkyne cycloadditions.1b,1d,26
Acknowledgments
D.L. is thankful for travel grants for this work by the Danish Chemical Society and the Lundbeck, Oticon, and Augustinus foundations. We thank the Lundbeck Foundation for a Young Group Leader fellowship (M.P.) and a PhD fellowship (D.L.). E.T.K. acknowledges support from the U.S. National Institutes of Health (GM068122 and GM110050).
Supporting Information Available
Full list of tested aminophenol catalysts, supporting figures and procedures, equations, and examples of data fits used for kinetics. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Hackenberger C. P. R.; Schwarzer D. Angew. Chem., Int. Ed. 2008, 47, 10030. [DOI] [PubMed] [Google Scholar]; b Patterson D. M.; Nazarova L. A.; Prescher J. A. ACS Chem. Biol. 2014, 9, 592. [DOI] [PubMed] [Google Scholar]; c King M.; Wagner A. Bioconjugate Chem. 2014, 25, 825. [DOI] [PubMed] [Google Scholar]; d Sletten E. M.; Bertozzi C. R. Angew. Chem., Int. Ed. 2009, 48, 6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chang P. V.; Bertozzi C. R. Chem. Commun. 2012, 48, 8864. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Giepmans B. N. G.; Adams S. R.; Ellisman M. H.; Tsien R. Y. Science 2006, 312, 217. [DOI] [PubMed] [Google Scholar]
- a Jencks W. P. J. Am. Chem. Soc. 1959, 81, 475. [Google Scholar]; b Barrett E.; Lapworth A. J. Chem. Soc., Trans. 1908, 93, 85. [Google Scholar]; c Conant J. B.; Bartlett P. D. J. Am. Chem. Soc. 1932, 54, 2881. [Google Scholar]; d Kalia J.; Raines R. T. Angew. Chem., Int. Ed. 2008, 47, 7523. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Hine J.; Cholod M. S.; Chess W. K. Jr. J. Am. Chem. Soc. 1973, 95, 4270. [Google Scholar]
- a Dirksen A.; Dirksen S.; Hackeng T. M.; Dawson P. E. J. Am. Chem. Soc. 2006, 128, 15602. [DOI] [PubMed] [Google Scholar]; b Dirksen A.; Dawson P. E. Bioconjugate Chem. 2008, 19, 2543. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Byeon J.-Y.; Limpoco F. T.; Bailey R. C. Langmuir 2010, 26, 15430. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zeng Y.; Ramya T. N. C.; Dirksen A.; Dawson P. E.; Paulson J. C. Nat. Methods 2009, 6, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wendeler M.; Grinberg L.; Wang X.; Dawson P. E.; Baca M. Bioconjugate Chem. 2014, 25, 93. [DOI] [PubMed] [Google Scholar]; b Rashidian M.; Mahmoodi M. M.; Shah R.; ADozier J. K.; Wagner C. R.; Distefano M. D. Bioconjugate Chem. 2013, 24, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Blanden A. R.; Mukherjee K.; Dilek O.; Loew M.; Bane S. L. Bioconjugate Chem. 2011, 22, 1954. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lee J. H.; Domaille D. W.; Noh H.; Oh T.; Choi C.; Jin S.; Cha J. N. Langmuir 2014, 30, 8452. [DOI] [PubMed] [Google Scholar]; e Loskot S. A.; Zhang J.; Langenhan J. M. J. Org. Chem. 2013, 78, 12189. [DOI] [PubMed] [Google Scholar]; f Thygesen M. B.; Munch H.; Sauer J.; Cló E.; Jørgensen M. R.; Hindsgaul O.; Jensen K. J. J. Org. Chem. 2010, 75, 1752. [DOI] [PubMed] [Google Scholar]; g Dirksen A.; Hackeng T. M.; Dawson P. E. Angew. Chem., Int. Ed. 2006, 45, 7581. [DOI] [PubMed] [Google Scholar]
- a Dirksen A.; Yegneswaran S.; Dawson P. E. Angew. Chem., Int. Ed. 2010, 49, 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Beeren S. R.; Pittelkow M.; Sanders J. K. M. Chem. Commun. 2011, 47, 7359. [DOI] [PubMed] [Google Scholar]; c Nguyen R.; Huc I. Chem. Commun. 2003, 942. [DOI] [PubMed] [Google Scholar]; d Ciaccia M.; Cacciapaglia R.; Mencarelli P.; Mandolini L.; Di Stefano S. Chem. Sci. 2013, 4, 2253. [Google Scholar]
- a McKinnon D. D.; Domaille D. W.; Cha J. N.; Anseth K. S. Adv. Mater. 2014, 26, 865. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lin F.; Yu J.; Tang W.; Zheng J.; Defante A.; Guo K.; Wesdemiotis C.; Becker M. L. Biomacromolecules 2013, 14, 3749. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gauthier M. A.; Klok H.-A. Chem. Commun. 2008, 2591. [DOI] [PubMed] [Google Scholar]; d Alconcel S. N. S.; Kim S. H.; Tao L.; Maynard H. D. Macromol. Rapid Commun. 2013, 34, 983. [DOI] [PubMed] [Google Scholar]
- a Thakar D.; Migliorini E.; Coche-Guerente L.; Sadir R.; Lortat-Jacob H.; Boturyn D.; Renaudet O.; Labbe P.; Richter R. P. Chem. Commun. 2014, 50, 15148. [DOI] [PubMed] [Google Scholar]; b Zhang Y.; Yu M.; Zhang C.; Ma W.; Zhang Y.; Wang C.; Lu H. Anal. Chem. 2014, 86, 7920. [DOI] [PubMed] [Google Scholar]
- a Kool E. T.; Park D.-H.; Crisalli P. J. Am. Chem. Soc. 2013, 135, 17663. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kool E. T.; Crisalli P.; Chan K. M. Org. Lett. 2014, 16, 1454. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Crisalli P.; Kool E. T. J. Org. Chem. 2013, 78, 1184. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Crisalli P.; Kool E. T. Org. Lett. 2013, 15, 1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Curvall M.; Enzell C. R.; Pettersson B. Cell Biol. Toxicol. 1984, 1, 173. [DOI] [PubMed] [Google Scholar]; b Sauer U. G.; Vogel S.; Hess A.; Kolle S. N.; Ma-Hock L.; van Ravenzwaay B.; Landsiedel R. Toxicol. in Vitro 2013, 27, 174. [DOI] [PubMed] [Google Scholar]
- a Cordes E. H.; Jencks W. P. J. Am. Chem. Soc. 1962, 84, 826. [Google Scholar]; b Cordes E. H.; Jencks W. P. Biochemistry 1962, 1, 773. [DOI] [PubMed] [Google Scholar]
- Cordes E. H.; Jencks W. P. J. Am. Chem. Soc. 1962, 84, 832. [Google Scholar]
- Results show first-order dependence on hydrazine under the conditions applied here. We hypothesize that this can be explained by a quasi-steady-state approximation (see SI).
- Anslyn E. V.; Dougherty D. A.. Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, 2006. [Google Scholar]
- Galli C.; Mandolini L. Eur. J. Org. Chem. 2000, 3117. [Google Scholar]
- Vandenbelt J. M.; Henrich C.; Vanden Berg S. G. Anal. Chem. 1954, 26, 726. [Google Scholar]
- Hine J.; Li W.-S. J. Org. Chem. 1975, 40, 2622. [Google Scholar]
- Stahl N.; Jencks W. P. J. Am. Chem. Soc. 1986, 108, 4196. [Google Scholar]
- Brotzel F.; Mayr H. Org. Biomol. Chem. 2007, 5, 3814. [DOI] [PubMed] [Google Scholar]
- Li N. C.; Doody B. E.; White J. M. J. Am. Chem. Soc. 1957, 79, 5859. [Google Scholar]
- Dewick P. M.Essentials of Organic Chemistry; John Wiley & Sons: Chichester, U.K., 2006. [Google Scholar]
- Mayer J. M.; Testa B. Helv. Chim. Acta 1982, 65, 1868. [Google Scholar]
- El-Sherif A. A. J. Solution Chem. 2010, 39, 1562. [Google Scholar]
- Reference (5b) reports an increased rate of oxime formation on 2-pentanone with 1,3-diaminobenzene as catalyst, but using catalyst concentrations beyond the buffer capacity. Reference (5d) reports enhanced yields of hydrazone formation on an immobilized acetophenone with the same catalyst, but at pH 4.5.
- a Datta D.; Wang P.; Carrico I. S.; Mayo S. L.; Tirrell D. A. J. Am. Chem. Soc. 2002, 124, 5652. [DOI] [PubMed] [Google Scholar]; b Lang K.; Chin J. W. Chem. Rev. 2014, 114, 4764. [DOI] [PubMed] [Google Scholar]
- Schelté P.; Boeckler C.; Frisch B.; Schuber F. Bioconjugate Chem. 2000, 11, 118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.