

Abstract

Autophagy is a conserved cellular process that involves the degradation of cellular components for energy maintenance and cytoplasmic quality control that has recently gained interest as a novel target for a variety of human diseases, including cancer. A prime candidate to determine the potential therapeutic benefit of targeting autophagy is the kinase ULK1, whose activation initiates autophagy. Here, we report the first structures of ULK1, in complex with multiple potent inhibitors. These structures show features unique to the enzyme and will provide a path for the rational design of selective compounds as cellular probes and potential therapeutics.
Maintaining a homesostatic energy state during times of varying nutrient availability is an essential feature for the survival of cells. A key mechanism that allows eukaryotic cells to survive nutrient starvation is the catabolic process known as autophagy. Through this process, cellular components are sequestered within a double-membrane bound structure called the autophagosome, which is then fused with a lysosome to degrade the contents. By breaking down proteins and even whole organelles, cells can recover energy and building blocks to maintain essential functions during starvation.1
Autophagy was initially characterized in Saccharomyces cerevisiae through the discovery of 31 autophagy-related (Atg) genes,2 which included only one protein kinase, Atg1.3−5 Humans have four Atg1 orthologs, named ULK1 to ULK4, with ULK1 appearing to be the most indispensable kinase for autophagy.6 The enzyme is activated under nutrient deprivation by several upstream signals and then initiates autophagy7 through a poorly understood mechanism. ULK1 is a 112-kDa protein that consists of an N-terminal kinase domain, a serine-proline rich region, and a C-terminal interacting domain. Recent work has begun to shed light on the function of these domains and how they impact the role of ULK1 in autophagy.8 For example, the serine–proline-rich region has been shown to be the site of numerous regulatory phosphorylations by both mTORC1 and AMPK, which act as negative and positive regulators of ULK1 activity, respectively.9,10 The C-terminal interacting domain has been shown to scaffold the ULK1–ATG13–FIP200 complex,11 which is a key component of the autophagy initiation process. In contrast to these well-described functions, the kinase domain of ULK1 has been less well-characterized despite being one of the most attractive targets in the autophagy pathway.
In the past few years, autophagy has been linked to neurodegeneration,12 Crohn’s disease,13 and cancer.14 It must be noted that the role of autophagy in cancer is complex, with its effect changing as tumors develop and progress. For example, Beclin-1, a key regulator in autophagy, is found to be monoallelically deleted in 40–75% of breast, ovarian, and prostate cancers, indicating that impaired autophagy may aid in tumorigenesis.15−17 In contrast to this, established tumors seem to rely on autophagy to preserve cellular viability against both environmental18 and therapeutic stressors.19 To further complicate the potential impact of therapeutic autophagy inhibition, the currently available tool compounds lack the ability to specifically inhibit autophagy itself, which can lead to conflicting results concerning the potential beneficial effects of inhibiting autophagy20,21 and leave open the possibility that the observed effects are not specific to autophagy inhibition. The ability to fully assess the role of autophagy in cancer, and ULK1 in autophagy, has been hampered by a lack of structural information and chemical tools to modulate ULK1. Here, we report the first structure of ULK1 and present two high-resolution crystal structures of the kinase bound to potent inhibitors. The structures will help guide our understanding of ULK1 biology through rational mutagenesis studies and facilitate structure-based design of improved inhibitors to aid in the study of autophagy.
In order to study the kinase and obtain structural information, we developed a bacterial expression system for purifying the kinase domain of human ULK1. Using an N-terminal SUMO tag, we initially obtained no transformants of the kinase domain in an expression strain, suggesting that the kinase was toxic. Therefore, we coexpressed the kinase with bacteriophage lambda protein phosphatase and obtained colonies that grew overnight. This expression system yielded soluble protein that we could purify (Supporting Figure 1 and Supporting Methods). However, we were still unable to obtain any crystals of the kinase. We reasoned that a small molecule inhibitor of ULK1 could increase the stability of the kinase domain and facilitate its crystallization.
To identify such an inhibitor, we screened a collection of 764 compounds against ULK1 using a standard 32P-ATP radioactive assay with MBP as the substrate. Among the top hits we identified were numerous pyrazole aminoquinazolines (Figure 1a) exemplified by compound 1, which, when retested in a dose-dependent assay, inhibited ULK1 with an IC50 of 160 nM. Differential scanning fluorimetry confirmed that the inhibitor dramatically stabilized the enzyme (Supporting Figure 2).22 Crystallization trials with 1 yielded small crystals that we were unable to obtain with the apo protein or with ADP. However, the crystals showed poor diffraction. Therefore, we attempted to improve the packing by mutating two residues (Glu37 and Lys38) at the surface of the N-terminal lobe, which were predicted to be of high entropy,23 to alanine. This mutation had a negligible effect on the activity of the enzyme (Supporting Figure 3) but resulted in large crystals of the ULK1/1 complex in several conditions. The best crystals diffracted to better than 1.6 Å. We then solved the structure by molecular replacement using the structure of polo-like kinase 3 as the search model, which has 34% identity to the ULK1 kinase domain.
Figure 1.

Structure of ULK1 bound to a small-molecule inhibitor. (a) (Left) Structure of the chemotype identified in our screen. (Right) Structure of the lead compound 1 from the initial screen. (b) Overall structure of ULK1. The key features indicated are the phosphorylated threonine in the activation loop, the inhibitor, and the interlobe loop. (c) Electrostatic surface rendering of ULK1. The front view is shown on the left. The back view, which contains the basic patch, is shown on the right. (d) Closeup view of 1 bound to ULK1. The electron density is from an fo–fc difference map contoured at 3σ. Key residues that contact the inhibitor are labeled and shown in yellow. The inhibitor is shown in cyan.
The overall structure of ULK1 kinase is shown in Figure 1b. ULK1 possesses the standard kinase fold with several unique features, most notably a large loop between the N- and C-terminal lobes (Figure 1b). Likely stabilized through crystal contacts, the positively charged loop is ordered and extends out from the kinase domain from the active site. Among ULKs, ULK1, -2, and -4 have this feature (Supporting Figure 4); however, it is rare in the remainder of the kinome. Only five other families of kinases have a similarly sized insertion: Sgk, Wee1, Clk, Mlk, and JAK. The poorly understood ULK4 has an even larger insertion here, as does the yeast ortholog Atg1. Moreover, the loop is one of the most nonconserved features between the highly similar ULK1 and ULK2, suggesting a role in ULK1-specific biology. Unexpectedly, we also observed strong density for a phosphothreonine in the activation loop at Thr180. This modification is an autophosphorylation event, since ULK1 was expressed in bacteria. The phosphothreonine appears to have strong interactions with Arg137 and Arg170, which might explain why the modification survived the presence of phosphatase used for expression. Interestingly, when we mutated this threonine in the activation loop to alanine, we found that it dramatically reduced the activity of the kinase, suggesting that autophosphorylation plays an important role in the activation of ULK1 (Supporting Figure 3). Last, when we looked at an electrostatic surface rendering of the kinase, we observed a large basic patch on the opposite side of the ATP binding site (Figure 1c). This positively charged surface might play a role in the binding of the enzyme to membrane structures in the process of assembling autophagophores. Alternatively, it could be involved in interacting with its binding partner Atg13 or the C-terminal domain of ULK1 itself. One intriguing observation is that the recent structure of the C-terminal domain of Atg1 (the ULK1 yeast ortholog) bound to Atg13 showed a large area of negative charge on the surface (Supporting Figure 5).24 We also note that Lys162 is adjacent to this patch and is a known regulatory site of ULK1 through acetylation by TIP60.25 This residue makes several important contacts within the kinase, so acetylation of it could alter the conformation of the protein.
Although ULK1 crystallized as a monomer in the asymmetric unit, there are numerous interfaces through crystallographic symmetry. The most striking one is a dimer between two kinases with their active sites facing each other (Supporting Figure 6), which has a large buried surface area of 2142.3 Å2. While this is often indicative of physiological interactions, we suspect that this is not a physiologically relevant dimer based on several lines of evidence. First, the protein was a stable monomer on gel filtration. Second, the dimer observed in our crystal structure does not resemble the weak dimerization interfaces reported in other kinase crystal structures that are stabilized by crystallization but are otherwise undetectable by gel filtration.26 Last, the calculated thermodynamics of the interface is not suggestive of a strong interaction.27 There are several other dimer interfaces in the crystal structure, but they are of much smaller surface area.
The inhibitor binds in the ATP binding site making hinge contacts with its amino pyrazole core (Figure 1d). In addition, the aniline forms a contact with Asp165 of the DFG motif, while the cyclopropyl substitutent fits into a pocket adjacent to the gatekeeper methionine. The quinazoline is hemmed in at the 6 position by the backbone of the kinase and at the 7 position by a number of side chains from the crystallographic symmetry-mate. In accordance with this steric obstruction, no compounds with substituents at these positions showed any activity against ULK1 in our original inhibitor screen.
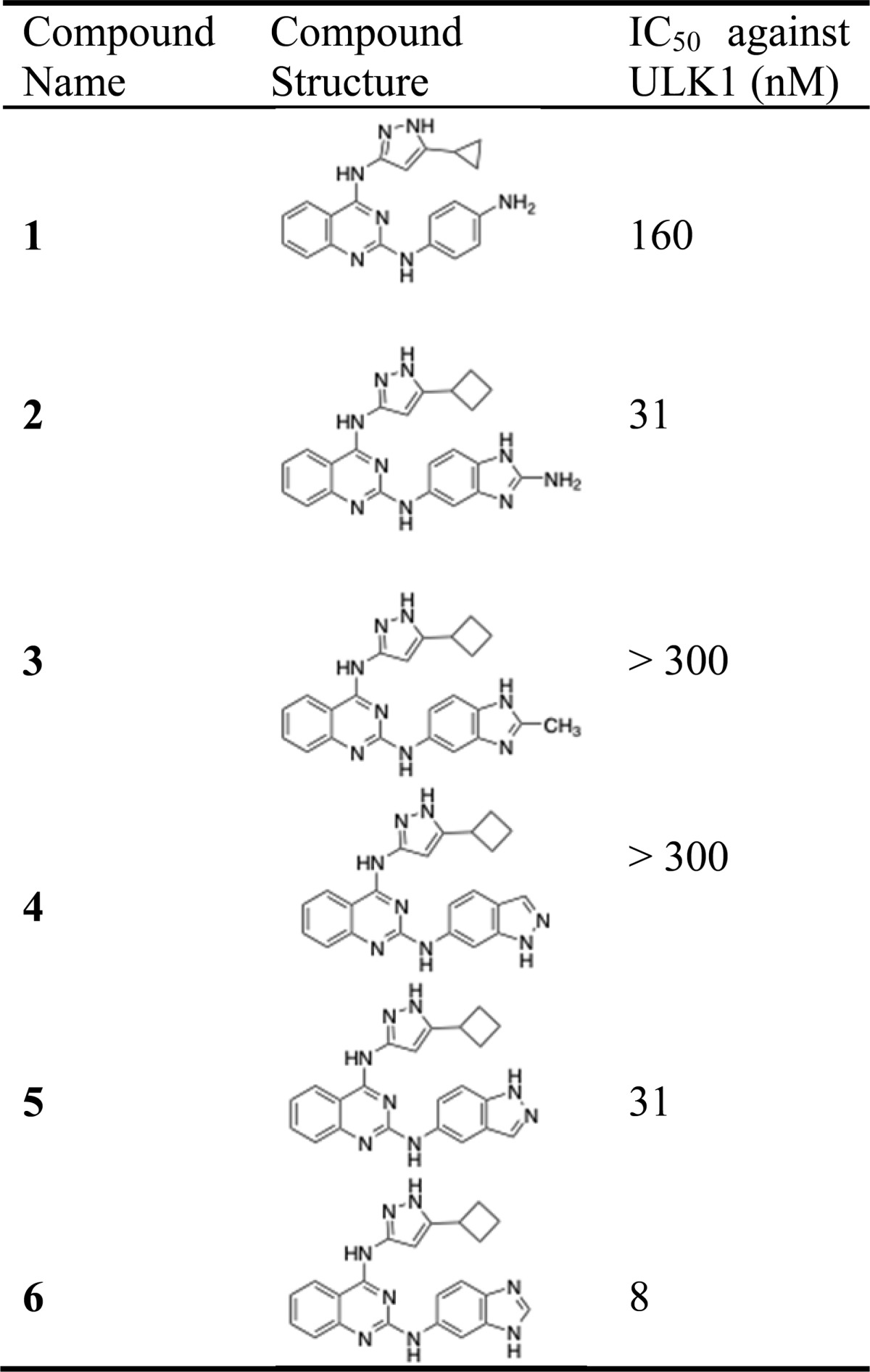
Using the crystal structure as a guide, we reasoned that building off the R1 and R2 (Figure 1a) positions would be accommodated and could potentially increase the potency and selectivity of the inhibitor. In our initial screening data, we noticed that compounds with a cyclopropyl or cyclobutyl ring at the R1 position were tolerated while any cyclopentyl derivative showed no inhibition. Therefore, we used the cyclobutyl group at the R1 position to maximize the bulkiness and take advantage of our relatively flexible methionine gatekeeper residue. Keeping R1 constant as a cyclobutyl ring, we then synthesized a series of analogs varying at the R2 position. A number of compounds showed significant improvement in potency, leading to our best inhibitor, compound 6 with an IC50 of 8 nM (Table 1 and Supporting Figure 7). Strikingly, a single methyl group at the 2 position of the benzimidazole (compound 3) showed almost no inhibition while a closely related 2-amino benzimidazole (compound 2) maintained potent inhibition of the enzyme. Additionally, the positional isomers 4 and 5 showed an equally dramatic difference in their ability to inhibit ULK1.
Table 1. Potencies of ULK1 Inhibitors.
To understand the activity differences observed between compounds 2 and 3 and between 4 and 5, as well as the greatly increased potency of 6, we crystallized the kinase with compound 6 (Figure 2a and Table 2). Interestingly, although the complex crystallized under the same conditions, it produced a different space group, suggesting that compound 6 induced conformational changes within the kinase domain. We solved the structure with molecular replacement using the first structure as a search model. The kinase crystallized with similar packing but with four protomers in the asymmetric unit. The kinase showed a similar overall confirmation with an RMSD of 0.67 Å. A major change was observed in the conformation of the interlobe loop, supporting the idea that this is a flexible region of the protein. Within the active site, the largest change involves Asp165 in the DFG (Figure 2a), whose side chain tucks in toward the kinase in order to accommodate the bulkier benzimidazole. A water molecule takes the place previously occupied by the aspartate carboxylic acid and forms a water-mediated hydrogen bond between the benzimidazole and Lys46. It also appears that the benzimidazole forms a likely hydrogen bond to the backbone carbonyl of Gln142. Last, the gatekeeper methionine is pushed slightly farther back into the back hydrophobic pocket by the now bulkier cyclobutyl ring.
Figure 2.

Structure of ULK1 bound to compound 6. (a) Closeup view of 6. Key interactions between the inhibitor (yellow) and the protein include an ordered water molecule (shown as a red sphere) that connects the benzimidazole to the catalytic lysine. (b) Surface view of 6 bound to ULK1. The compound is shown in yellow with sphere rendering.
Table 2. Data Collection and Refinement Statistics.
| compound 1/ULK1 complex | compound 6/ULK1 complex | |
|---|---|---|
| data collection | ||
| space group | P42212 | P2221 |
| cell dimensions | ||
| a, b, c (Å) | 66.78, 66.78, 116.23 | 100.41, 113.80, 100.63 |
| α, β, γ (deg) | 90, 90, 90 | 90, 90, 90 |
| resolution (Å) | 58.11–1.56 (1.59–1.56)a | 46.02–1.88 (1.98–1.88)a |
| Rsym or Rmerge | 0.070 (0.500) | 0.077 (0.801) |
| I/σI | 18.1 (2.0) | 10.4 (2.0) |
| completeness (%) | 100 (100) | 99.9 (100) |
| redundancy | 7.0 (3.6) | 4.2 (4.3) |
| refinement | ||
| resolution (Å) | 57.90–1.56 | 46.02–1.88 |
| no. reflections | 38221 | 94125 |
| Rwork/Rfree | 0.1784/0.2076 | 0.1922/0.2264 |
| no. atoms | ||
| protein | 2220 | 8839 |
| ligand/ion | 27 | 120 |
| water | 161 | 243 |
| B factors | ||
| protein | 20.82 | 43.56 |
| ligand/ion | 15.52 | 34.01 |
| water | 26.23 | 35.78 |
| RMS deviations | ||
| bond lengths (Å) | 0.007 | 0.008 |
| bond angles (deg) | 1.235 | 1.048 |
Values in parentheses are for the highest-resolution shell. Each data set was from a single crystal.
The most dramatic change in the inhibitor conformation is the twisting of the benzimidazole ring compared to the aniline in the previous structure (Supporting Figure 8). By rotating, the benzimidazole binds right up against the two lobes of the kinase, forming a tight binding surface (Figure 2b). The strong interactions made by the benzimidazole with both the N and C lobes cause the kinase to clamp down around the inhibitor, which causes the C-terminal lobe to move closer to the N-terminal lobe (Supporting Figure 9).
An overlay of the structures of compounds 1 and 6 revealed that both the aniline in 1 and benzimidazole in 6 make a potential hydrogen bond either directly with Lys46 or through a water molecule, which may help explain the differing activities of compounds 2 and 3. Given how tightly the benzimidazole fits into the pocket it is likely that the methyl group of 3 pushes the benzimidazole back and prevents it from making this hydrogen bond, potentially explaining its lack of binding potency. The amine substituent on compound 2 would still push the benzimidazole away from the kinase but could potentially maintain this hydrogen bond through the same extra amine. Similarly, compound 5 would be predicted to be able to both fit into the benzimidazole pocket and potentially form this hydrogen bond while compound 4 would be unable to both make the hydrogen bond and bind into the benzimidazole pocket. Compound 6 can make two hydrogen bonds, both to the critical water molecule and to the Gln142 backbone, explaining its improved potency over the other analogs.
To determine if 6 would be suitable for cellular experiments, we tested its selectivity against a small panel of representative kinases. Although built on a promiscuous scaffold, we anticipated that the larger substituents of our improved inhibitor (the cyclobutyl and benzimidazole moieties) would improve its kinome wide selectivity. However, the profiling revealed that the compound is not yet selective enough for cellular use (Supporting Table 1). Nevertheless, we have achieved sufficient potency, and further gains in selectivity should lead to a cellular probe of autophagy.
In summary, we have identified a series of potent ULK1 inhibitors that allowed for the crystallization of the human ULK1 kinase domain. The structures reveal several novel features of the enzyme and will enable the hypothesis driven mutagenesis of ULK1 to better elucidate its biological role in cells. In addition, our preliminary SAR has identified both a scaffold and a potentially critical hydrogen bond that are necessary for potent inhibition of ULK1. Further optimization of these inhibitors should lead to the first potent and selective modulators of autophagy in cells.
Methods
For details on experimental methods, including a description of protein purification, crystallography, chemical synthesis, and differential scanning fluorimetry, please see the Supporting Information.
Acknowledgments
We thank the staff at A.L.S. beamline 8.2.2. This work was supported by the Howard Hughes Medical Institute and R01 AI094098 to K.M.S. M.B.L. is a Merck fellow of the Helen Hay Whitney Foundation.
Supporting Information Available
Supporting Figures, Tables, and Methods are available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The structures of ULK1 bound to 1 and 6 have been deposited with the Protein Data Bank as entries 4WNO and 4WNP, respectively.
Author Contributions
† These authors contributed equally to this work.
The authors declare no competing financial interest
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Mizushima N.; Levine B.; Cuervo A. M.; Klionsky D. J. (2008) Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N.; Yoshimori T.; Ohsumi Y. (2011) The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27, 107–132. [DOI] [PubMed] [Google Scholar]
- Tsukada M.; Ohsumi Y. (1993) Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 333, 169–174. [DOI] [PubMed] [Google Scholar]
- Harding T. M.; Morano K. A.; Scott S. V.; Klionsky D. J. (1995) Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J. Cell Biol. 131, 591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thumm M.; Egner R.; Koch B.; Schlumpberger M.; Straub M.; Veenhuis M.; Wolf D. H. (1994) Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 349, 275–280. [DOI] [PubMed] [Google Scholar]
- Chan E. Y.; Kir S.; Tooze S. A. (2007) siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 282, 25464–25474. [DOI] [PubMed] [Google Scholar]
- Wong P. M.; Puente C.; Ganley I. G.; Jiang X. (2013) The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 9, 124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stjepanovic G.; Davies C. W.; Stanley R. E.; Ragusa M. J.; Kim do J.; Hurley J. H. (2014) Assembly and dynamics of the autophagy-initiating Atg1 complex. Proc. Natl. Acad. Sci. U. S. A. 111, 12793–12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan D. F.; Shackelford D. B.; Mihaylova M. M.; Gelino S.; Kohnz R. A.; Mair W.; Vasquez D. S.; Joshi A.; Gwinn D. M.; Taylor R.; Asara J. M.; Fitzpatrick J.; Dillin A.; Viollet B.; Kundu M.; Hansen M.; Shaw R. J. (2011) Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Kundu M.; Viollet B.; Guan K. L. (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley I. G.; Lam du H.; Wang J.; Ding X.; Chen S.; Jiang X. (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 284, 12297–12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer B.; Potkar R.; Trejo M.; Rockenstein E.; Patrick C.; Gindi R.; Adame A.; Wyss-Coray T.; Masliah E. (2009) Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 29, 13578–13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell K.; Liu J. Y.; Brown S. L.; Miyoshi H.; Loh J.; Lennerz J. K.; Kishi C.; Kc W.; Carrero J. A.; Hunt S.; Stone C. D.; Brunt E. M.; Xavier R. J.; Sleckman B. P.; Li E.; Mizushima N.; Stappenbeck T. S.; Virgin H. W. t (2008) A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R.; Karantza-Wadsworth V.; White E. (2007) Role of autophagy in cancer. Nat. Rev. Cancer 7, 961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X. H.; Jackson S.; Seaman M.; Brown K.; Kempkes B.; Hibshoosh H.; Levine B. (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402, 672–676. [DOI] [PubMed] [Google Scholar]
- Karantza-Wadsworth V.; White E. (2007) Role of autophagy in breast cancer. Autophagy 3, 610–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z.; Jin S.; Yang C.; Levine A. J.; Heintz N. (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. U. S. A. 100, 15077–15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P.; Gonzalez-Polo R. A.; Casares N.; Perfettini J. L.; Dessen P.; Larochette N.; Metivier D.; Meley D.; Souquere S.; Yoshimori T.; Pierron G.; Codogno P.; Kroemer G. (2005) Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 25, 1025–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Q. W.; Cheng C.; Hackett C.; Feldman M.; Houseman B. T.; Nicolaides T.; Haas-Kogan D.; James C. D.; Oakes S. A.; Debnath J.; Shokat K. M.; Weiss W. A. (2010) Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci. Signaling 3, ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzawa T.; Germano I. M.; Komata T.; Ito H.; Kondo Y.; Kondo S. (2004) Role of autophagy in Temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 11, 448–457. [DOI] [PubMed] [Google Scholar]
- Paglin S.; Hollister T.; Delohery T.; Hackett N.; McMahill M.; Sphicas E.; Domingo D.; Yahalom J. (2001) A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 61, 439–444. [PubMed] [Google Scholar]
- Niesen F. H.; Berglund H.; Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221. [DOI] [PubMed] [Google Scholar]
- Goldschmidt L.; Cooper D. R.; Derewenda Z. S.; Eisenberg D. (2007) Toward rational protein crystallization: A Web server for the design of crystallizable protein variants. Protein Sci. 16, 1569–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka Y.; Suzuki S. W.; Yamamoto H.; Kondo-Kakuta C.; Kimura Y.; Hirano H.; Akada R.; Inagaki F.; Ohsumi Y.; Noda N. N. (2014) Structural basis of starvation-induced assembly of the autophagy initiation complex. Nat. Struct. Mol. Biol. 21, 513–521. [DOI] [PubMed] [Google Scholar]
- Lin S. Y.; Li T. Y.; Liu Q.; Zhang C.; Li X.; Chen Y.; Zhang S. M.; Lian G.; Liu Q.; Ruan K.; Wang Z.; Zhang C. S.; Chien K. Y.; Wu J.; Li Q.; Han J.; Lin S. C. (2012) GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 336, 477–481. [DOI] [PubMed] [Google Scholar]
- Pike A. C.; Rellos P.; Niesen F. H.; Turnbull A.; Oliver A. W.; Parker S. A.; Turk B. E.; Pearl L. H.; Knapp S. (2008) Activation segment dimerization: a mechanism for kinase autophosphorylation of non-consensus sites. EMBO J. 27, 704–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.