

Abstract
Upon oxidation of the heterocyclic ring in 2′-deoxyguanosine (dG), the initial electrophilic intermediate displays a wide range of reactivities with nucleophiles leading to many downstream products. In the present study, the product profiles were mapped when aqueous solutions of dG were allowed to react with NH4Cl in the presence of the photooxidants riboflavin and Rose Bengal as well as the diffusible one-electron oxidant Na2IrCl6. Product characterization identified the 2′-deoxyribonucleosides of spiroiminodihydantoin, 5-guanidinohydantoin, and oxazolone resulting from H2O as the nucleophile. When NH3 was the nucleophile, a set of constitutional isomers that are diastereotopic were also observed, giving characteristic masses of dG + 31. ESI+-MS/MS of these NH3 adducts identified them to be spirocycles with substitution of either the C5 or C8 carbonyl with an amine. The NH3 adducts exhibit acid-catalyzed hydrolysis to spiroiminodihydantoin. Quantification of the NH3 and H2O adducts resulting from oxidation of dG in the nucleoside, single-stranded, and duplex oligodeoxynucleotide contexts were monitored allowing mechanisms for product formation to be proposed. These data also provide a cautionary note to those who purify their oligonucleotide samples with ammonium salts before oxidation because this will lead to unwanted side reactions in which ammonia participates in product formation.
Introduction
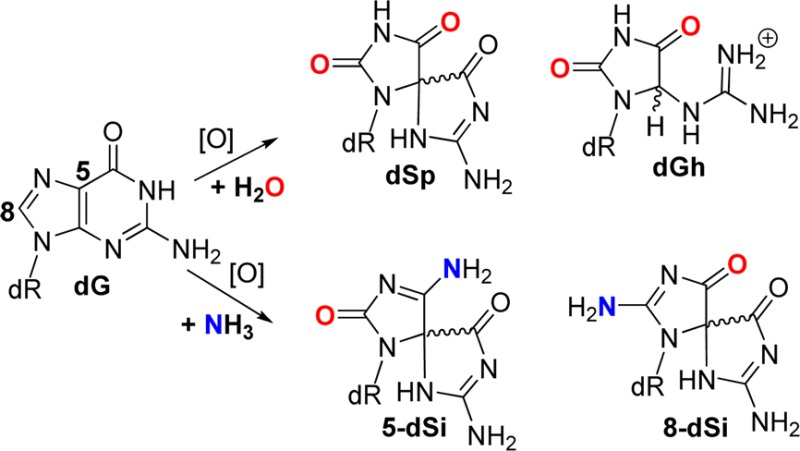
The guanine heterocyclic ring is rich in chemical reactivity toward oxidants and adduct-forming species.1,2 In DNA, 2′-deoxyguanosine (dG, mass = M) is the chief site for base oxidation due to its low redox potential leading to many products that are characterized by their mass signatures.3,4 The principal products characterized with one-electron oxidants in aqueous solutions can be grouped based on the site of reaction on the radical intermediate resulting from one-electron oxidation of dG.5−7 The 2′-deoxyribonucleoside products arising from initial reactivity at C5 of dG include a four-electron oxidation product, an imidazolone (dIz, M-39) that hydrolyzes to an oxazolone (dZ, M-21),8 and the two-electron oxidation product 5-carboxamido-5-formamido-2-iminohydantoin (d2Ih, M+34; Scheme 1).9−13 When the initial reaction occurs at C8 of dG, either 2,6-diamino-4-hydroxy-5-formamidopyrimidine (Fapy-dG, M + 18) is observed under reducing conditions or 8-oxo-7,8-dihydroguanine (dOG, M+16) is observed under oxidizing conditions, in which the latter compound is a key marker for monitoring oxidative stress in cells (Scheme 1).1,5 The two-electron oxidation product dOG is stable but is highly susceptible to further oxidation leading to two hydantoin compounds. The yield for the thermodynamically preferred product spiroiminodihydantoin (dSp, M + 32) is greatest in nucleoside reactions at pH > 6 or in unencumbered reaction contexts (i.e., single-stranded DNA or G-quadruplexes), while the yield of 5-guanidinohydantoin (dGh, M + 6) is greatest in nucleoside reactions at pH < 6 or in sterically demanding duplex contexts (Scheme 1).14−19 The hydantoins are also readily formed from direct four-electron oxidation with 1O2.20 This list of products represents those that are consistently observed from many oxidant systems conducted by several laboratories; however, other compounds have been reported in lower yield.21
Scheme 1. Major Oxidation Reaction Pathways and Products Observed for dG along with Their Mass Differences.

In the cellular context, oxidation reactions with dG can have other nucleophilic participants such as amines or phenols. In most cases, when amines or phenols participate in the reaction, products are observed with similar backbone structures as characterized with water serving as the nucleophile. For example, when dG is oxidized in the presence of lysine, products retaining the dG heterocycle and those with a core similar to dGh or dSp have been observed.22−25 The adducts characterized by our laboratory and others show lysine competing with water for sites of covalent bond formation, where a single lysine is observed at C8 analogous to dOG, at either C5 or C8 of a spirocyclic core similar to dSp, and last a bis-adduct at C5 and C8 with a spirocyclic ring structure has been identified (Figure 1).22 All products involve covalent attachment via the ε-amine group of lysine. The lysine adducts are all stable allowing their quantification and characterization. As a last example, the polyamine spermine has been adducted to dG and dOG under oxidative conditions. In the dG oxidation studies, a spermine adduct at C8 is observed retaining the dG heterocyclic core,26 while dOG oxidations yield an adduct at C5 that does not undergo acyl migration to create a spirocycle, but instead the polyamine can generate an unstable hemiaminal intermediate that decomposes leaving a ribosylurea lesion at the adduct formation site.27 Moreover, the facile formation of spermine adducts to dOG oxidation intermediates has been harnessed for quantification of dOG from DNA samples.28 In contrast to amines, phenols (i.e., tyrosine) are more redox active than amines and better able to compete with dG for oxidant during reactions. The products observed when phenols participate with dG during oxidation have more variability in their structures compared to amine adducts.25,29,30 These model studies aid in understanding the chemical nature of DNA–protein cross-links that are detrimental to cellular processes.5,31,32 In the current work, oxidations that allowed dG to react with NH3 were conducted while the full product and diastereomer distributions were monitored. After careful characterization of the nucleoside reactions, similar oxidations were conducted in the single-stranded and duplex oligodeoxynucleotide (ODN) contexts. The purified ammonia adducts were also studied with respect to their decomposition pathways leading to end products that have not been previously determined. Lastly, polymerase insertion studies were conducted to test the hypothesis that amine adducts might have altered base-pairing preferences; every substitution of an oxo group on a base for an amino group converts a hydrogen bond acceptor into a potential hydrogen bond donor. These results provide a fundamental understanding of the stability and base-pairing properties of amine adducts to dG that result during oxidation, which may occur in vivo due to the high concentration of nucleophilic amines in the vicinity of the genome.
Figure 1.

Adducts observed when dG and lysine are allowed to react in the presence of oxidant.
Results and Discussion
The unencumbered nucleoside dG (1 mM) was chosen for the initial oxidation reactions where NH4Cl (20 mM) provided the source of nucleophilic NH3. All reactions were conducted with 75 mM NaPi buffer at pH 7.4 at 22 °C. The oxidants chosen include the photooxidants riboflavin and Rose Bengal and the one-electron oxidant Na2IrCl6. Reactions were conducted in triplicate to achieve ∼70% conversion to products in each reaction, and the products (Scheme 2) were analyzed by a dual HPLC method. The first round of HPLC analysis utilized a reversed-phase column to identify dOG (M+16) and 8-amino-dG (M+15), while all other products eluted in the void volume. The void volume from the previous run was collected and analyzed on a Hypercarb HPLC column that allowed analysis of the hydantoins, ammonia adducts, and dZ. The precursor dIz was not detected by this analytical method as previously described.9 However, dIz hydrolyzes to dZ that was detected on the Hypercarb column; thus, formation of dIz is inferred from quantification of dZ. Moreover, the Hypercarb HPLC column also allowed separation and quantification of all spirocyclic diastereomeric pairs of products. The dGh diastereomers are interconvertible,20 and thus, their diastereomer ratios are not reported (see the Supporting Information for complete experimental details). Lastly, a test reaction with Na2IrCl6 was conducted in which half was directly analyzed by the Hypercarb HPLC column and the other half was analyzed by the dual HPLC method outlined above. The product distributions observed from these comparative studies were within ∼3% of one another. This observation is critical for the discussion that follows.
Scheme 2. One-Electron Oxidation Pathways Leading to Possible Products (Those in Boxes Were Detected in These Studies).

The photooxidant riboflavin led to the largest number of different products and was used to obtain suitable amounts of material for characterization. Initial product characterization was achieved by LC–ESI+-MS (Figure S1, Supporting Information), and the adducts observed that did not involve participation of NH3 during product formation included dZ (M - 21), dOG (M + 16), dGh (M + 6), and dSp (M + 32, Scheme 2). Confirmation of their structures was achieved by ESI+-MS/MS fragmentation of the free bases while monitoring the daughter fragments. The ESI+-MS/MS experiments were conducted on HPLC purified nucleosides in which the N-glycosyl bond was cleaved in the ionization source to liberate free bases that were further fragmented in the CID chamber to generate the daughter fragments used in identification of the structures. The MS/MS fragmentation spectra were compared to literature values for the two dSp diastereomers (Figure 2) and dZ to further confirm their structural assignments (Figures S2 and S3, Supporting Information). The structure for dSp has been further established by X-ray crystallography33 and NMR.34 Ammonia adducts observed included two pairs of chromatographic peaks for the diastereomers of spirodi(iminohydantoin)-2′-deoxyribonucleosides (dSi), whose names are based on the site of NH3 attachment to guanine, thus 5-dSi (M + 31) and 8-dSi (M + 31) (Scheme 2, see reference cited for the correct dSi IUPAC nomenclature).35 To confirm the identities of the 5- and 8-dSi constitutional isomers and their respective diastereomers, ESI+-MS/MS of the free bases were conducted (Figure 2). Comparison of the ESI+-MS/MS spectrum for 5-dSi with dSp gave a pair of peaks for 5-dSi that established C5 as the site of NH3 attachment (m/z [M + H]+ = 140 and 96, Figure 2). As for 8-dSi, comparison of its ESI+-MS/MS spectrum with those obtained from dSp and 5-dSi identified new masses that are best explained by NH3 attachment at C8 (m/z [M + H]+ = 141 and 97, Figure 2). On the basis of the proposed mechanism (Scheme 2), a product that includes bis-addition of NH3 at C5 and C8 of an oxidized guanine to yield a spirocycle (5,8-dSi, M+30)35 is possible; however, this adduct was not observed for reasons that will be elaborated on below. Additionally, NH3 adducts with a dGh core were not observed most likely due to the fact that the reactions were performed at pH 7.4 where yields of dGh are minimal. Lastly, the yields of dOG (M + 16) and 8-amino-dG (M + 15) were very low (<1%) as determined by LC-ESI+-MS (Figure S1, Supporting Information); hence, their quantities are not reported. The nucleosides dOG and C8 amine-adducted dG have redox potentials that are ∼600 mV below the parent nucleoside dG causing them to be much more susceptible to further oxidation.36,37 Further, one-electron oxidized dOG was observed to have an even lower redox potential than the parent compound dOG,38 which means that once oxidation of dOG, and likely 8-amino-dG, occurs product formation is inevitable due to the ∼70% conversion to product that yielded the spirocyclic compounds dSp, 5-dSi, or 8-dSi.
Figure 2.

ESI+-MS/MS spectra for dSp (top), 5-dSi (middle), and 8-dSi (bottom). The data provided was collected on the first eluting diastereomer of each spirocycle from a Hypercarb HPLC column. Data for the second eluting diastereomer for dSp, 5-dSi, and 8-dSi can be found in the Supporting Information (Figures S2, S4, and S5). The heavy lines represent the fragment observed and the thin lines represent the portion of the molecule lost upon fragmentation.
Relative product distributions observed with each oxidant system were determined by integration of the HPLC peak areas measured at 240 nm followed by normalization via each molecule’s unique extinction coefficient (ε240nm).9 Extinction coefficients for 5-dSi and 8-dSi are not known but were determined from experiments that are reported below. In the riboflavin-mediated oxidations, the major product was dZ (43%, Figure 3). This result was anticipated because riboflavin is a type I photooxidant and effects oxidation by electron transfer from dG yielding O2•– under aerobic reaction conditions.39 Oxidation of dG by one-electron and proton transfer yields an intermediate radical (dG•) that only couples with O2•– to initially yield dIz that is prone to hydration leading to dZ (Scheme 2),40,41 the species quantified. The adducts derived from the nucleophile trapping of electrophilic dG oxidation intermediates show NH3 participation through the products 5-dSi (29%) that was the highest yielding NH3 adduct followed by nearly 3-fold less 8-dSi (11%). The participation of H2O as the trapping nucleophile was identified by the lower yields of dSp (11%) and dGh (4%). The nucleophilicity of NH3 is much greater than H2O; therefore, as expected, adducts resulting from NH3 participation dominated over those derived from H2O.
Figure 3.
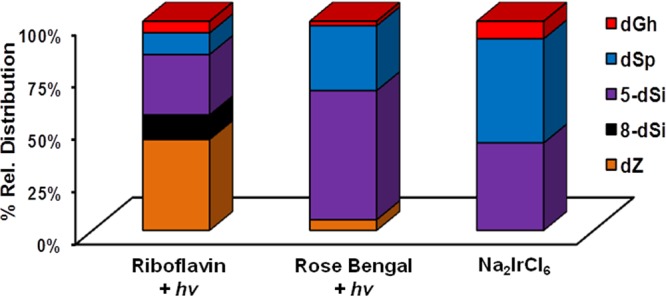
Relative product distributions observed when dG reacted with nucleophilic H2O or NH3 in oxidation reactions. The oxidants include the photooxidants riboflavin and Rose Bengal, as well as Na2IrCl6. Reactions were conducted with 1 mM dG, 20 mM NH4Cl, in 75 mM NaPi buffer (pH 7.4) at 22 °C. (1) Photoactivation of riboflavin (200 μM) was achieved with 350 nm light for 3 h, (2) photoactivation of Rose Bengal (100 μM) was achieved with 350 nm light for 3 h, and (3) oxidation with Na2IrCl6 (10 mM) was achieved by bolus addition of the salt. Results represent the average of triplicate trials and the error is ∼10% of each reported value.
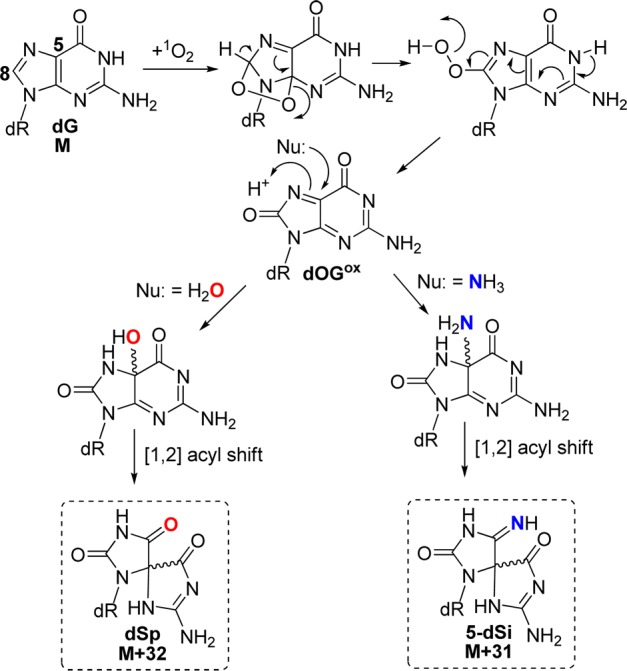
The second oxidant studied was Rose Bengal, a type II photooxidant (1O2) that furnished 5-dSi as the major product (62%) followed by a 3-fold lower amount of dSp (31%), and low yields of dZ (5%) and dGh (2%) were observed to complete the mass balance (Figure 3). As expected, this oxidant did not yield 8-dSi based on its mechanism of oxidation. Oxidation of dG by 1O2 proceeds by 4 + 2 cycloaddition to the imidazole ring, followed by ring opening to yield 8-HOO-G that eliminates water giving the proposed electrophile dOGox.20 Next, dOGox is trapped by nucleophiles at C5 leading to dSp with H2O or 5-dSi with NH3 (Scheme 3). Support for exclusive nucleophilic attack at C5 under 1O2 oxidations was derived from H218O studies followed by mapping the labeled site by ESI+-MS/MS;20 the current observation that the only NH3 adducts were the 5-dSi diastereomers further support this previously proposed mechanism, and further confirms our structural assignments for these peaks. Comparisons of the product distributions from the photooxidants riboflavin and Rose Bengal show a dramatic difference with respect to the major product observed (Figure 3). In the riboflavin oxidation, dZ was the major product while in the Rose Bengal oxidation, 5-dSi was the major product, and these observations can be ascribed to each oxidant’s unique mechanism of oxidation (Schemes 2 and 3).
Scheme 3. Proposed Mechanism for Product Formation Resulting from 1O2 Oxidation of dG.
The last oxidant studied was the one-electron oxidant Na2IrCl6. Product distributions from this oxidant included nearly equivalent distributions of dSp (50%) and 5-dSi (42%) with the mass balance completed by dGh (8%, Figure 3). In this oxidation reaction, the distribution did not yield NH3 adducts as the major products, which was unexpected on the basis of NH3 being the better nucleophile compared to H2O. Moreover, 8-dSi was not observed in this reaction.
Comparison of the product distributions observed with the one-electron oxidants riboflavin and Na2IrCl6 identified both NH3- and H2O-adducted compounds. The current results differ from those previously reported by our laboratory when lysine was adducted to dG in analogous oxidation reactions.22 Previously, in riboflavin oxidations spirodihydantoins with lysine at C8 were 2-fold greater than those with lysine at C5 (Figure 1), and in Na2IrCl6 oxidations a C8 lysine adduct was observed. In contrast to these results, riboflavin oxidations in the presence of NH4Cl gave more C5 adducts (29%) than C8 adducts (11%), and Na2IrCl6 oxidations did not yield detectable amounts of C8 adducts. We propose the difference in the current ammonia results compared to the lysine data can be attributed to a difference in the mechanism of product formation for the C8 adduct in the nucleoside context. The former work from our laboratory proposed that C8 amine adducts result from oxidation of the amine to the aminyl radical that adds at the C8 carbon of dG followed by oxidation leading to product formation (Scheme 4).22 In nucleoside dG, this proposed mechanism best supports the observation of ammonia adducts at C8, because upon one-electron oxidation of dG the initial radical cation (dG•+) formed is very acidic (pKa ∼ 3.9)40 and rapidly deprotonates to the neutral radical (dG•) that is not susceptible to nucleophilic attack.42 Thus, dG• reacts with O2•– yielding dIz/dZ and not with amines or H2O. This is consistent with dZ being the major oxidation product. Formation of amine adducts at C8 must result from a difference in the amine reactivity. The key difference between lysine and ammonia resides in their standard reduction potentials. In general, primary amines (1.0 V vs NHE, pH 10)43 have a lower redox potential than ammonia (>1.3 V vs NHE, pH 9);44 this trend should scale down to pH 7, in which the oxidations were conducted. Thus, oxidation of lysine to an aminyl radical that adds to C8 of dG is possible with Na2IrCl6 (0.9 V vs NHE, pH 7) and riboflavin (1.7 V vs NHE, pH 7);45 in contrast, the analogous reaction does not readily occur for NH3 because at pH 7 dG is the dominant site of oxidation due to its lower redox potential leading to products other than 8-dSi. In summary, one-electron oxidant driven oxidations of dG in the presence of NH3 lead to spirocyclic adducts at C5 and C8 that are in competition with H2O adducts of the same core structure.
Scheme 4. Proposed Radical Coupling Mechanism Leading to Amine Adducts at C8 of dG.

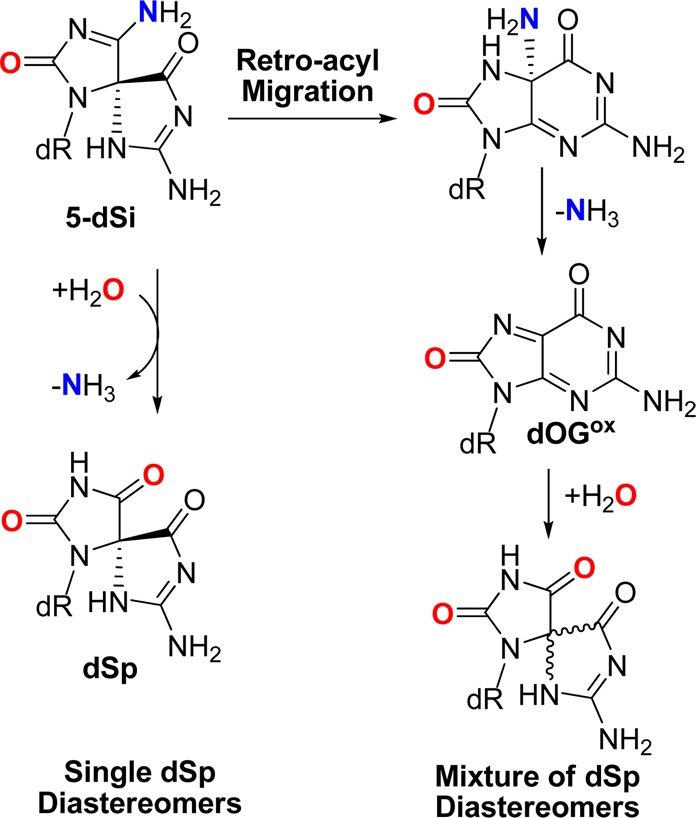
As the next step, we set out to determine the decomposition products and pathways through which the ammonia adducts proceed. HPLC was used to provide diastereomerically pure 5-dSi and 8-dSi samples that were subjected to conditions of pH 3 (0.1% formic acid) or pH 10 (20 mM NaPi) at 22 °C for 30 min or 10 h. Not only could the decomposition products be determined, but having diastereomerically pure starting material also allowed us to probe the mechanism of decomposition, specifically for 5-dSi. First, both 5- and 8-dSi were stable at pH 10 during this time frame, while 5-dSi readily hydrolyzed to dSp at pH 3 in 30 min and 8-dSi hydrolyzed to dSp after 10 h. For 5-dSi, deamination of the amine group can occur by two possible mechanisms: (1) A retro-acyl migration can occur followed by loss of NH3 to yield an electrophilic intermediate (dOGox) susceptible to H2O attack followed by a second acyl migration back to dSp or (2) acid-catalyzed deamination can occur directly to furnish dSp (Scheme 5). If the retro-acyl migration proceeds on the diastereomerically pure sample, it is expected that a mixture of dSp diastereomers would be observed, and if direct deamination occurs, then only one dSp diastereomer would be predicted. Upon incubation of one diastereomer of 5-dSi at pH 3, only one diastereomer of dSp was observed, supporting the direct deamination mechanism. Moreover, the early eluting 5- and 8-dSi NH3 adducts decomposed to the early eluting dSp isomer and the late eluting adducts deaminated to the later eluting dSp isomer (Figure S6, Supporting Information). These results aid in establishing the absolute configuration of the 5-dSi and 8-dSi diastereomers that will be discussed below. Moreover, these results also allude to an explanation as to why the bis-ammonium adduct was not observed in any of the reactions. Based on these data, the bis-ammonium adduct, if formed, could rapidly deaminate to yield either 5-dSi or 8-dSi, although this hypothesis could not be further validated.
Scheme 5. Proposed Decomposition Pathways for 5-dSi Leading to dSp.
Utilization of the Hypercarb HPLC column for analyzing the product distributions of these spirocycles allowed determination of the diastereomer ratios. It has previously been determined that (R)-dSp elutes first from this column and (S)-dSp elutes second.46−48 The absolute stereochemistry for the diastereomers of the ammonia adducts had not yet been determined. The results of these studies identified (R)-dSp and (S)-dSp yields to be nearly equal. Also, for the 5-dSi and 8-dSi ammonia adducts nearly equal yields for the diastereomers were observed. On the basis of these results, the defining point of the reaction to determine product stereochemistry must not be very sterically impeded in order to lead to such a small diastereomer preference.
As stated above, the diastereomerically pure ammonia adduct samples were determined to decompose to give a single diastereomer of dSp; therefore, on the basis of HPLC elution order, the absolute configuration for the diastereomers of the two dSi constitutional isomers can be determined. The first-eluting 5-dSi and 8-dSi diastereomers decomposed to the first dSp diastereomer (R), and the late-eluting diastereomer decomposed to the late-eluting dSp diastereomer (S); therefore, for 5-dSi the R diastereomer elutes first and the S diastereomer elutes second, because the R and S assignments are the same between dSp and 5-dSi. In contrast, the R and S assignments for the 8-dSi diastereomers are opposite those of dSp; thus, (S)-8-dSi elutes first and (R)-8-dSi elutes second and from the Hypercarb HPLC column (Figure 4). These examples provide a fascinating case outlining how the movement of ring substituents affects the R and S stereochemical assignments.
Figure 4.

Assignment of absolute configurations for the diastereomers of dSp, 5-dSi, and 8-dSi based on their elution profile from a Hypercarb HPLC column and their ECD spectra. The ECD spectra were recorded in ddH2O at 20 °C. The R and S assignments for the diastereomers of 8-dSi are the opposite of those for the dSp and 5-dSi isomers with the same geometric configuration of the spirocyclic ring, due to a change in the Cahn–Ingold–Prelog priorities.
The diastereotopic ammonia adducts were then probed by electronic circular dichroism spectroscopy (ECD). Previously, ECD was used in tandem with vibrational circular dichrosim spectroscopy, NMR, and X-ray crystallography to establish the absolute configuration for dSp.33,47,49 The ECD spectra for (R)- and (S)-dSp isomers gave three lobes that were mirror images of one another, as expected (Figure 4). Critical for assigning the absolute configuration for dSp was the low energy lobe, in which the S isomer gave a positive rotation at 258 nm, and the R isomer gave a negative rotation at 259 nm.49 In comparison to these results, the 5-dSi diastereomers gave ECD spectra that were different than dSp but showed a similarity in the low energy lobe (Figure 4). For (R)-5-dSp, the lobe at 258 nm gave a negative rotation and (S)-5-dSi gave a positive rotation at 258 nm. This observation was similar to that observed for the dSp diastereomers. The higher energy lobes observed in the ECD for the 5-dSi diastereomers were different than those measured for dSp (Figure 4). As a last comparison, the 8-dSi diastereomer ECD spectra were compared to those recorded for dSp (note: 8-dSi and dSp isomers with the same geometric configuration of the spirocyclic ring give opposite R and S assignments due to a difference in the Cahn–Ingold–Prelog priorities when the ring substituents are moved). For (S)-8-dSi, the low energy lobe gave a negative rotation at 262 nm similar to (R)-dSp, and (R)-8-dSi gave a positive rotation similar to (S)-dSp. Furthermore, the 8-dSi diastereomers also gave similar rotations and energies as their analogous dSp diastereomers at the ∼238 nm lobe, and 8-dSi and dSp gave similar rotations at the highest energy lobe, but the absolute energy was different (i.e., ∼202 nm for 8-dSi and ∼211 nm for dSp; Figure 4). The similarity in the critical low energy lobe further supports the absolute configuration assignments that were made from the deamination studies monitored by HPLC. Future computational studies to model these ECD spectra will help solidify these conclusions, and may address some of the challenges that occurred during modeling of the ECD spectra for the dSp diastereomers.48,49
The ability to deaminate the dSi adducts to dSp allowed the determination of the extinction coefficients for 5- and 8-dSi relative to dSp, in which these were the values used to determine the relative yields. Peak areas for identically pure 5-dSi samples were measured before and after incubation in formic acid and the change in peak area measured was used to determine the relative ε240 nm compared to the value established for dSp. A similar experiment was conducted for the 8-dSi diastereomers. From these experiments, the ε240 nm for the 5-dSi and 8-dSi diastereomers were determined to be 3800 and 3500 L·mol–1·cm–1, respectively, which are both slightly greater than that determined for the dSp diastereomers (3300 L·mol–1·cm–1).
The next step from the nucleoside studies
was to explore the context
dependence of NH3 adduct formation in single- and double-stranded
ODNs (ssODN and dsODN). The photooxidant riboflavin was chosen for
the studies because it gave the most diverse distribution of products
in the nucleoside reactions and might provide the most insight into
context effects. For these studies, the ssODNs selected for analysis
were the 18-mers ODN-1 and ODN-2, and the
dsODN context was studied via the duplex formed from these two single
strands (ODN-12). After oxidation of these ODN systems,
they were digested with a suite of nucleases and phosphatase to the
nucleosides followed by analysis using the previously described HPLC
methods. Previously, our laboratory demonstrated that the digestion
method used provides complete degradation of dSp-containing
ODNs to nucleosides;50 it is assumed that
the dSi adducts are equally digested to completion. The
nuclease digestion conditions were modified to include ammonium salts
for buffer to prevent loss of 5-dSi via deamination to dSp (Scheme 5), controls conducted
with sodium salts for buffers did not detect 5-dSi due
to deamination to dSp during the nuclease digestion (18
h at pH 5.4, Scheme 5).
A comparison between the contexts studied when dG was allowed to react with NH3 in the presence of the photooxidant riboflavin is provided in Figure 5. The first observation from these data was with respect to dZ distributions that dramatically decreased when proceeding from the nucleoside to ODN contexts; specifically, the dZ distribution in the dsODN context (2%) was 10-fold less than the ssODN context (∼20%) and 20-fold less than that observed in the nucleoside context (∼40%). This observation has already been reported in the literature and is proposed to result from the ODN context increasing the lifetime of the dG•+ that reacts with nucleophilic H2O to give the C8 product dOG while decreasing the lifetime of dG• that reacts with O2•– to yield dZ (Schemes 2 and 6).41 With respect to the yields of the hydantoins, more dSp relative to dGh was observed in the nucleoside context (11% vs 4%, respectively) versus the dsODN context (9% vs 21%, respectively). Again, this trend follows those in the literature in which the duplex context favors the less sterically demanding product dGh.17,51 The NH3 adducts 5-dSi and 8-dSi provided some interesting context-dependent product distributions. The yield of 5-dSi was greatest for the nucleoside (29%) and decreased by half in all ODN contexts studied (∼14%). In contrast, the yield of 8-dSi was smallest for the nucleoside (11%) and increased by more than 3-fold in the ssODN context (∼39%), and over half the products observed in the dsODN context were 8-dSi (54%). As previously stated, the lifetime of the electrophilic dG•+ is longer in the duplex context and the superior nucleophilicity of NH3 compared to H2O greatly increased the yield of 8-dSi in dsODN oxidations (Scheme 6). Furthermore, the increase in 8-dSi yield from the dsODN oxidations further supports this product resulting from nucleophilic addition of NH3 at C8 leading to 8-dSi, and not the NH3 aminyl radical adding at C8 to yield the same product. These results clearly demonstrate a dependence on the reaction context in formation of dG oxidation products and NH3 adducts. A look at the distribution of diastereomers resulting from oxidation of the ODN contexts gave nearly equal amounts of both R and S diastereomers. This observation parallels our previous work looking at dSp diastereomer formation in single-stranded and duplex contexts.51 Coupled together, these observations support a state of disorder in the duplex at the time of nucleophilic attack at C5 of oxidized dOG or 8-amino-dG (likely intermediates leading to the spirocycles) that leads to nearly equal covalent bond formation from both the re and si faces of these electrophiles.
Figure 5.
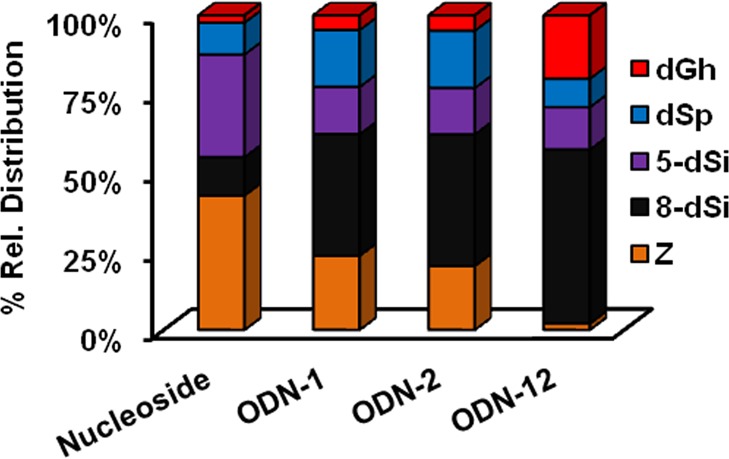
Relative product distributions measured when dG was allowed to react with NH3 in the presence of photoexcited riboflavin in various contexts. Reactions were conducted in 75 mM NaPi buffer (pH 7.4) at 22 °C, and 20 mM NH4Cl. In the nucleoside studies, 1 mM dG was studied, and in the ODN contexts 20 μM ssODN or 10 μM dsODN was studied. (1) Photoactivation of riboflavin (200 μM) was achieved with 350 nm light for 3 h in the nucleoside studies and 30 min in the ODN studies. These conditions achieved ∼70% conversion to product in the nucleoside reactions and ∼50% in the ODN studies. The ODNs were digested with nucleases and phosphatase prior to HPLC analysis. The data represent triplicate trials that gave ∼10% error on each reported value.
Scheme 6. Base Pair Dependent Reactivity of One Electron Oxidized dG.

It must first be noted that the bis-ammonium adduct could not be quantified due to its instability, but if formed, the adduct is most likely deaminated to either 5-dSi or 8-dSi. Keeping this limitation in mind, the following mechanistic proposal is drawn. The oxidation of dG by riboflavin initially yields dG•+ that rapidly deprotonates to dG• in the nucleoside contexts (pKa ∼ 3.9),40 dominating the product-forming step of the reaction. In contrast, dG•+ in the dsODN context retains more cationic character because the acidic proton is H-bonded with 2′-deoxycytosine in a base pair; therefore, the product-forming steps of the reaction are determined by the radical cation intermediate. As shown in Scheme 2, dG• could couple with radicals (e.g., O2•–) to ultimately yield dZ, while dG•+ reacts with nucleophiles (e.g., H2O and NH3) at C8, ultimately yielding a spirocyclic product after completion of the four-electron oxidation. Thus, the yield of 8-dSi was anticipated to be greatest in the duplex context, as indeed it is. Furthermore, these results support the original hypothesis by Steenken that the duplex context would favor the radical cation nature of one-electron oxidized dG,40 which is still the focus of many current research efforts.41,52 Because of the limitation imposed by the inability to detect the bis-ammonium adduct, or to understand its decomposition pathway, any comparison between the ammonia adduct distributions would be based on poorly defined assumptions; consequently, further discussion of these results is not provided.
For the last set of studies, we were inspired by the observation that the A ring of dSp mimics the H-bonding pattern of thymidine, while the A ring of 5-dSi mimics the H-bonding pattern of 2′-deoxycytidine (Figure 6). On the basis of this observation, polymerase insertion assays opposite dSp vs 5-dSi were conducted to determine if these H-bonding schemes applied during the selection of a nucleotide to insert opposite these spirocycles.
Figure 6.
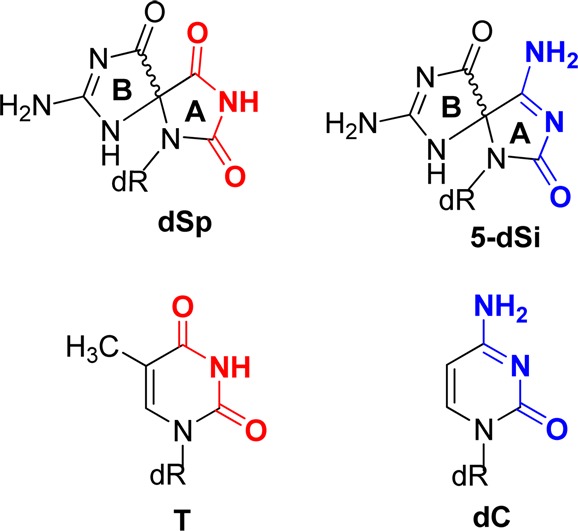
Comparison of H-bonding patterns between dSp vs T and 5-dSi vs dC.
Site-specific synthesis of dSp or 5-dSi was achieved by synthesizing dOG into an ODN by solid-phase synthesis and then oxidizing this strand with or without NH4Cl present with the one-electron oxidant Na2IrCl6 to furnish the desired products. Upon ion-exchange HPLC purification, diastereomerically pure samples of dSp and 5-dSi were obtained for the polymerase studies. The absolute stereochemical assignments for the dSp isomers have been established for the ODNs,46 and those for 5-dSi are assigned in this work to be analogous based on the mechanism of deamination discussed above (see Figure S7, Supporting Information, for more details). Standing start polymerase studies were conducted while providing the enzyme only one type of nucleotide per reaction. On the basis of previous studies, insertion of dTTP opposite dSp was not observed;53 therefore, to ensure that only one nucleotide was inserted opposite the lesion, and no extension past the lesions occurs, the sequence had an A placed 5′ to the spirocycle (Figure 7). Klenow fragment exo- was selected as the polymerase, and after the extension reactions, polyacrylamide gel electrophoresis (PAGE) was conducted to determine the amount of dATP, dTTP, dGTP, or dCTP incorporated opposite the dSp or 5-dSi diastereomers. The polymerase did not select either pyrimidine for insertion opposite the two lesions (Figure S7, Supporting Information); thus, only data for purine insertion opposite the lesions is provided in Figure 7. With respect to insertion opposite the dSp diastereomers, (R)-dSp was observed to give a slight preference for insertion of dATP opposite, and (S)-dSp gave nearly equal insertion of dATP and dGTP. In comparison to the dSp results, the 5-dSi isomers showed that (S)-5-dSi gives similar amounts of dATP and dGTP insertion, while (R)-5-dSi yields a slight preference for dATP insertion. Unfortunately, these results do not support the hypothesis that H-bonding in the A ring of the spirocycle is the key parameter for nucleotide selection, but do show that stereochemistry may be important for selecting a base pairing partner by this polymerase.
Figure 7.
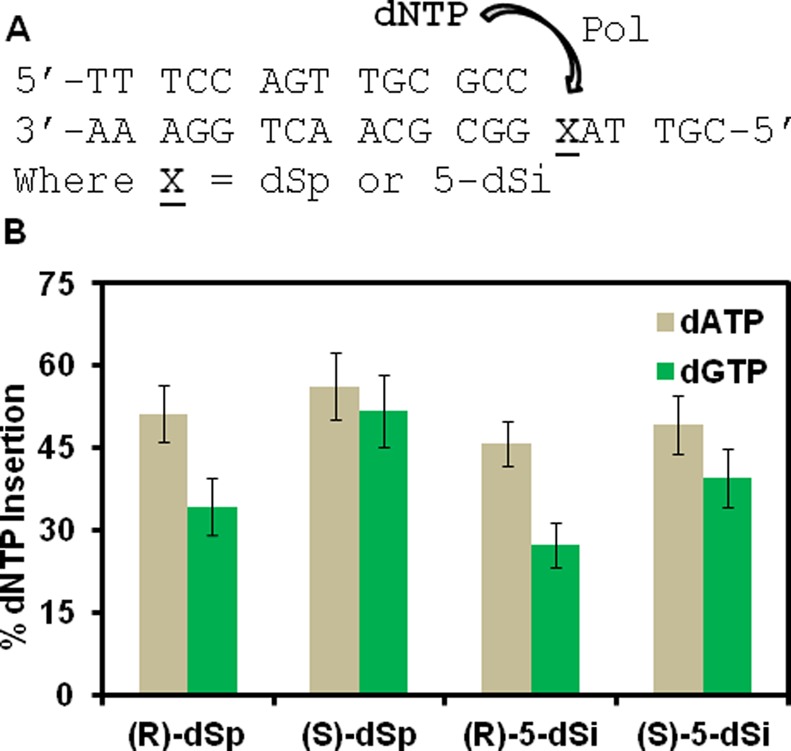
Sequence studied for polymerase dNTP insertion studies and the percent dNTP insertions opposite the diastereomers of dSp and 5-dSi. (A) Sequence for the dsODN construct used in the polymerase insertion assays and (B) comparison of the percent purine dNTP insertion opposite the lesions dSp and 5-dSi by the DNA polymerase Klenow fragment exo-.
Conclusion
The present studies mapped the pathways and quantified the products observed when dG was allowed to react with NH4Cl in the presence of the photooxidants riboflavin and Rose Bengal, as well as the one-electron oxidant Na2IrCl6. On the basis of the analysis of the products in the nucleoside context, the major products were oxidant dependent. The major product for riboflavin was dZ, Rose Bengal yielded the 5-dSi diastereomers, and Na2IrCl6 gave the dSp diastereomers as the major products (Figure 3). Mechanisms were proposed to explain these results (Schemes 2–4). Further analyses determined that 5-dSi and 8-dSi decompose via an acid-catalyzed deamination pathway leading to dSp as an end product (Scheme 5). Oxidations of dG in the nucleoside, ss- and dsODN contexts with photochemically activated riboflavin demonstrated three major context effects: (1) The yield of dZ was highest in the nucleoside studies and decreased dramatically in dsODN contexts; (2) when NH3 participated as the nucleophile, 5-dSi was obtained in the highest yield in nucleoside studies, while the yield of 8-dSi was highest in dsODN contexts; and (3) when H2O was the nucleophile in nucleoside reactions, dSp presented in the highest yield, while in dsODN contexts dGh was obtained in the highest yield (Figure 4). Furthermore, a combination of mapping the decomposition pathways of the dSi compounds to dSp, in which the absolute configuration has been assigned, allowed the determination of the absolute configurations for the diastereomers of 5- and 8-dSi. These assignments were further supported by preliminary ECD spectra (Figure 4). These studies provide fundamental chemical insight into formation of amine adducts to dG and their stability. Further, these studies insert a cautionary note to researchers who purify their ODNs using ammonium salts and then conduct oxidation reactions after purification. Ammonia is a better nucleophile and out-competes water for the electrophilic intermediates derived from dG or dOG oxidation, resulting in a new mass of dG + 31 or dOG + 15, respectively.54 A similar observation has been highlighted for oxidations occurring in Tris buffer that generate Tris adducts to dG.55
Experimental Section
Nucleoside Oxidation Studies
Oxidations were conducted with 2′-deoxyguanosine (dG) at 1 mM concentration in 75 mM NaPi buffer (pH 7.4) and 22 °C. Reactions with 20 mM NH4Cl and without this salt allowed product profile comparisons. The oxidants and specific reaction conditions were achieved as follows: (1) Riboflavin oxidations were initiated by adding 200 μM riboflavin while exposing the samples to 350 nm light for 3 h. (2) Rose Bengal oxidations were achieved by adding 100 μM Rose Bengal and exposing these samples to 350 nm for 3 h. The light source for the riboflavin and Rose Bengal reactions came from a sun lamp that was placed ∼7 cm above the reaction Eppendorf tubes. The tube lids were left open to allow all wavelengths of light to pass into the reaction samples. (3) The Na2IrCl6 oxidations were initiated by a bolus addition of the oxidizing salt with a final concentration of 10 mM, after a 30 min reaction the samples were quenched with 50 mM EDTA (pH 8). The reaction products and their distributions were determined by a dual HPLC method following a previously reported set of protocols (specific details can be found in the Supporting Information file).9 The first RP-HPLC run allowed analysis of dOG and 8-amino-dG (both observed in <1% yield), while all other products eluted in the void volume of this run. The void volume was collected, dried, and reinjected on a Hypercarb HPLC column to analyze the diastereomers of dGh, dSp, 5-dSi, and 8-dSi, as well as the product dZ, while monitoring their absorbance at 240 nm. To determine product distributions the peak areas were integrated and normalized by each compound’s ε240nm (values provided below).
Product Identification
Products were characterized initially by LC–ESI+-MS, and then each compound was HPLC purified for further structural analysis. The dGh diastereomers were previously characterized by NMR,20dZ was also previously characterized by NMR,39 and the dSp diastereomers have been characterized by X-ray crystallography.33 Structural characterization of 5-dSi and 8-dSi by NMR was not conducted because of the instability of these compounds toward deaminating to dSp, and the lack of nonexchangeable protons on the ring of either ammonia adducts makes them very challenging for structural analysis. Furthermore, if deamination of either 5-dSi or 8-dSi to dSp occurs in the NMR tube during analysis, the peaks observed would be similar and challenging to interpret; therefore, the best method for obtaining structural data on the diastereomers of 5-dSi and 8-dSi is via the ESI+-MS/MS that was conducted (Figure 2), and provided satisfactory results to determine their structures. Characterization for each molecule is as follows, the mixture of the dGh diastereomers (not resolvable) gave tR = 6 min; LC–ESI+-MS m/z [M + H]+ calcd 274.3, found 274.1; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C9H15N5O5Na 296.0971, found 296.0979; UV–vis ε240 = 2400 L ·mol–1·cm–1.9(R)-dSp: tR = 11 min; LC–ESI+-MS m/z [M + H]+ calcd 300.3, found 300.3; HRMS (ESI-TOF) m/z [M + Na]+ for C10H13N5O6Na calcd 322.0764, found 322.0761; ESI+-MS/MS m/z [M + H]+ lit.15 184, 156, 141, 114, 113, 99, 86, found 184, 156, 141, 114, 113, 99, and 86; UV–vis ε240 = 3,300 L·mol–1·cm–1;9 CD λ (c 1.24 × 10–5 M in ddH2O) nm 259 (Δε −8.7), 236 (+35.1), and 211 (−37.4). (S)-dSp: tR = 18 min; LC–ESI+-MS m/z [M + H]+ calcd 300.3, found 300.3; HRMS (ESI-TOF) m/z [M + Na]+ for C10H13N5O6Na calcd 322.0764, found 322.0761; ESI+-MS/MS m/z [M + H]+ lit.15 184, 156, 141, 114, 113, 99, 86, found 184, 156, 141, 114, 113, 99, and 86; UV–vis ε240 = 3300 L·mol –1·cm–1;9 CD λ (c 1.30 × 10–5 M in ddH2O) nm 258 (Δε +8.7), 234 (−32.9), and 212 (+40.5). (R)-5dSi: tR = 9 min; LC–ESI+-MS m/z [M + H]+ calcd 299.3, found 299.3; HRMS (ESI-TOF) m/z [M + Na]+ for C10H14N6O5Na calcd 321.0923, found 321.0920; ESI+-MS/MS m/z [M + H]+ found values 183, 166, 140, 123, 113, 96, and 86; UV–vis ε240 = 3800 L·mol–1·cm–1; CD λ (c 1.50 × 10–5 M in ddH2O) nm 258 (Δε −8.2), 242 (+20.8), and 216 (+61.8). (S)-5dSi: tR = 12 min; LC-ESI+-MS m/z [M + H]+ calcd 299.3, found 299.3; HRMS (ESI-TOF) m/z [M + Na]+ for C10H14N6O5Na calcd 321.0923, found 321.0916; ESI+-MS/MS m/z [M + H]+ found 183, 166, 140, 123, 113, 96, and 86; UV–vis ε240 = 3800 L·mol–1·cm–1; CD λ (c 1.45 × 10–5 M in ddH2O) nm 258 (Δε +8.2), 242 (−20.1), and 216 (−62.2). (R)-8dSi: tR = 7 min; LC–ESI+-MS m/z [M + H]+ calcd 299.3, expt 299.3; HRMS (ESI-TOF) m/z [M + Na]+ for C10H14N6O5Na calcd 321.0923, found 321.0924; ESI+-MS/MS m/z [M + H]+ found 183, 165, 155, 138, 113, 98, and 86; UV–vis ε240 = 3500 L·mol–1·cm–1; CD λ (c 1.30 × 10–5 M in ddH2O) nm 261 (Δε +20.0), 237 (−43.2), and 203 (+18.6). (S)-8dSi: tR = 14 min; LC–ESI+-MS m/z [M + H]+ calcd 299.3, expt 299.3; HRMS (ESI-TOF) m/z [M + Na]+ for C10H14N6O5Na calcd 321.0923, found 321.0926; ESI+-MS/MS m/z [M + H]+ found 183, 165, 155, 138, 113, 98, and 86; UV–vis ε240 = 3500 L·mol–1·cm–1; CD λ (c 1.24 × 10–5 M in ddH2O) nm 262 (Δε −13.7), 239 (+46.0), and 201 (−33.1). dZ: tR = 27 min; LC–ESI+-MS m/z [M + H]+ calcd 247.3, found 247.3; HRMS (ESI-TOF) m/z [M + Na]+ for C8H14N4O5Na calcd 269.0862, found 269.0870; ESI+-MS/MS m/z [M + H]+ lit.56 131 and 117, found 131 and 117; UV–vis ε240 = 1800 L·mol–1·cm–1.9
ODN Oxidation Reactions
The ODN samples were synthesized by standard solid-phase synthesis methods. The ODNs were HPLC purified using an ion-exchange HPLC column, and the purification salts (NaOAc) were removed by dialysis prior to their oxidation following previously reported methods.9 The riboflavin oxidations were conducted similarly to those reported for the nucleoside studies, with the following exceptions: the ssODN oxidations were conducted on 20 μM samples while the dsODN oxidations were conducted on 10 μM samples. In addition, the reaction times were decreased for the ODN reactions to 30 min. After the oxidations, the ODNs were digested with a suite of nucleases and phosphatases to nucleoside samples following a previously established protocol,50 with the exception that all buffers during the digestion process were comprised of ammonium salts. Next, the digested mixture was analyzed by the HPLC method used in the nucleoside studies. The complete details can be found in the Supporting Information.
Synthesis of ODNs for the Polymerase Studies
The polymerase insertion assays were conducted on duplex ODN samples that had site specific incorporation of dSp or 5-dSi in the template strand. The site-specific synthesis was commenced on ODNs that had a dOG phosphoramidite synthesized at the desired site of modification within the sequence 5′-CGT TAX GGC GCA ACT GGA AA-3′ where X = dOG. The modifications were synthesized by taking 1 nmol of the dOG-containing ODN and placing it in 100 μL of reaction buffer (75 mM NaPi, pH 7.4) with or without 2 mM NH4Cl. The reaction without NH4Cl gave the dSp diastereomers and the reaction with NH4Cl gave the 5-dSi diastereomers. The individual diastereomers were purified using an ion-exchange HPLC column running NaOAc as the resolving salt and characterized via digestion of the ODN to nucleosides followed by HPLC analysis (purification details can be found in the Supporting Information).
Polymerase Insertion Studies
The primer template duplex for the insertion studies was made by annealing 125 nM primer (5′-TT TCC AGT TGC GCC-3′) with 156 nM lesion-containing template (5′-CGT TAX GGC GCA ACT GGA AA-3′ where X = (R)-dSp, (S)-dSp, (R)-5-dSi, or (S)-5-dSi to obtain 100 nM duplex in Klenow fragment exo- buffer (50 mM Tris, 50 mM NaCl, 5 mM MgCl2, 1 mM DTT at pH 8). To a 25 μL reaction 20 μL of the annealed duplex was added, 1 μL of Klenow fragment exo- (0.2 units/μL), 0.5 μL of dNTP (500 μM stock solution), and 8.5 μL of Klenow buffer to obtain a 100 nM duplex solution with 10 μM dNTP and 0.2 U of polymerase. The dNTPs were studied individually. The reaction was incubated at 37 °C for 30 min, after which loading dye (95% DMF plus 0.025% bromophenol blue, and 0.025% xylene cylanol) was added to the samples and they were heated at 95 °C for 20 min to quench the reaction and denature the DNA–protein complex. The denatured samples were loaded on a 20% PAGE gel and electrophoresed for 2 h at 45 W. Upon completion of the electrophoresis, the gel was placed in a phosphor screen overnight and imaged by storage phosphor autoradiography. The band intensities were quantified using ImageQuant software.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (CA090689).
Supporting Information Available
Complete experimental methods, ESI+-MS, ESI+-MS/MS, HPLC, and PAGE analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Dedication
Dedicated to Professor Iwao Ojima on the occasion of his 70th birthday and his 42nd year of chemical biology and drug discovery research at Stony Brook University.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cadet J.; Douki T.; Ravanat J.-L. Free Radical Biol. Med. 2010, 49, 9–21. [DOI] [PubMed] [Google Scholar]
- Gates K. S. Chem. Res. Toxicol. 2009, 22, 1747–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney S.; Jarem D. A.; Volle C. B.; Yennie C. J. Free Radical Res. 2012, 46, 420–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimisis T.; Cismas C. Eur. J. Org. Chem. 2006, 2006, 1351–1378. [Google Scholar]
- Cadet J.; Wagner J. R.; Shafirovich V.; Geacintov N. E. Int. J. Radiat. Biol. 2014, 90, 423–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratviel G.; Meunier B. Chem.—Eur. J. 2006, 12, 6018–6030. [DOI] [PubMed] [Google Scholar]
- Burrows C. J.; Muller J. G. Chem. Rev. 1998, 98, 1109–1152. [DOI] [PubMed] [Google Scholar]
- Pouget J. P.; Frelon S.; Ravanat J. L.; Testard I.; Odin F.; Cadet J. Radiat. Res. 2002, 157, 589–595. [DOI] [PubMed] [Google Scholar]
- Fleming A. M.; Muller J. G.; Ji I.; Burrows C. J. Org. Biomol. Chem. 2011, 9, 3338–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokhlenko Y.; Geacintov N. E.; Shafirovich V. J. Am. Chem. Soc. 2012, 134, 4955–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialas C.; Claparols C.; Pratviel G.; Meunier B. J. Am. Chem. Soc. 2000, 122, 2157–2167. [Google Scholar]
- Ye W.; Sangaiah R.; Degen D. E.; Gold A.; Jayaraj K.; Koshlap K. M.; Boysen G.; Williams J.; Tomer K. B.; Mocanu V.; Dicheva N.; Parker C. E.; Schaaper R. M.; Ball L. M. J. Am. Chem. Soc. 2009, 131, 6114–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banu L.; Blagojevic V.; Bohme D. K. J. Phys. Chem. B 2012, 116, 11791–11797. [DOI] [PubMed] [Google Scholar]
- Luo W.; Muller J. G.; Rachlin E. M.; Burrows C. J. Chem. Res. Toxicol. 2001, 14, 927–938. [DOI] [PubMed] [Google Scholar]
- Luo W.; Muller J. G.; Rachlin E. M.; Burrows C. J. Org. Lett. 2000, 2, 613–616. [DOI] [PubMed] [Google Scholar]
- Fleming A. M.; Burrows C. J. Chem. Res. Toxicol. 2013, 26, 593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremaud J. N.; Martin B. D.; Sugden K. D. Chem. Res. Toxicol. 2010, 23, 379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niles J. C.; Wishnok J. S.; Tannenbaum S. R. Chem. Res. Toxicol. 2004, 17, 1510–1519. [DOI] [PubMed] [Google Scholar]
- Crean C.; Geacintov N. E.; Shafirovich G. V. Angew. Chem., Int. Ed. 2005, 44, 5057–5060. [DOI] [PubMed] [Google Scholar]
- Ye Y.; Muller J. G.; Luo W.; Mayne C. L.; Shallop A. J.; Jones R. A.; Burrows C. J. J. Am. Chem. Soc. 2003, 125, 13926–13927. [DOI] [PubMed] [Google Scholar]
- Neeley W. L.; Essigmann J. M. Chem. Res. Toxicol. 2006, 19, 491–505. [DOI] [PubMed] [Google Scholar]
- Xu X.; Muller J. G.; Ye Y.; Burrows C. J. J. Am. Chem. Soc. 2008, 130, 703–709. [DOI] [PubMed] [Google Scholar]
- Perrier S.; Hau J.; Gasparutto D.; Cadet J.; Favier A.; Ravanat J. L. J. Am. Chem. Soc. 2006, 128, 5703–10. [DOI] [PubMed] [Google Scholar]
- Solivio M. J.; Joy T. J.; Sallans L.; Merino E. J. J. Inorg. Biochem. 2010, 104, 1000–5. [PubMed] [Google Scholar]
- Solivio M. J.; Nemera D. B.; Sallans L.; Merino E. J. Chem. Res. Toxicol. 2012, 25, 326–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silerme S.; Bobyk L.; Taverna-Porro M.; Cuier C.; Saint-Pierre C.; Ravanat J. L. Chem. Res. Toxicol. 2014, 27, 1011–8. [DOI] [PubMed] [Google Scholar]
- Hosford M. E.; Muller J. G.; Burrows C. J. J. Am. Chem. Soc. 2004, 126, 9540–1. [DOI] [PubMed] [Google Scholar]
- Bajacan J. E. V.; Hong I. S.; Penning T. W.; Greenberg M. M. Chem. Res. Toxicol. 2014, 27, 1227–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Fleming A. M.; Muller J. G.; Burrows C. J. J. Am. Chem. Soc. 2008, 130, 10080–10081. [DOI] [PubMed] [Google Scholar]
- Dai J.; Sloat A. L.; Wright M. W.; Manderville R. A. Chem. Res. Toxicol. 2005, 18, 771–779. [DOI] [PubMed] [Google Scholar]
- Johansen M. E.; Muller J. G.; Xu X.; Burrows C. J. Biochemistry 2005, 44, 5660–71. [DOI] [PubMed] [Google Scholar]
- Gherezghiher T. B.; Ming X.; Villalta P. W.; Campbell C.; Tretyakova N. Y. J. Proteome Res. 2013, 12, 2151–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckenroth B. E.; Fleming A. M.; Sweasy J. B.; Burrows C. J.; Doublie S. Biochemistry 2014, 53, 2075–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam W.; Arnold M. A.; Grune M.; Nau W. M.; Pischel U.; Saha-Möller C. R. Org. Lett. 2002, 4, 537–540. [DOI] [PubMed] [Google Scholar]
- The numbering for the spirodi(iminohydantoin) structures is abbreviated here in such a way as to represent the carbon of dG at which ammonia attacks. The correct IUPAC nomenclature for 5-dSi is 1-(β-D-2′-deoxyribose)-4,7-diamino-1,3,6,8-tetraazaspiro[4.4]nona-3,7-diene-2,9-dione, 8-dSi is 1-(β-D-2′-deoxyribose)-2,7-diamino-1,3,6,8-tetraazaspiro[4.4]nona-2,7-diene-4,9-dione, and 5,8-dSi is 1-(β-D-2′-deoxyribose)-2,7-diamino-9-imino-1,3,6,8-tetraazaspiro[4.4]nona-2,7-diene-4-one.
- Stover J. S.; Ciobanu M.; Cliffel D. E.; Rizzo C. J. J. Am. Chem. Soc. 2007, 129, 2074–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenken S.; Jovanovic S. V.; Bietti M.; Bernhard K. J. Am. Chem. Soc. 2000, 122, 2373–2374. [Google Scholar]
- Shukla L. I.; Adhikary A.; Pazdro R.; Becker D.; Sevilla M. D. Nucleic Acids Res. 2004, 32, 6565–6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet J.; Berger M.; Buchko G. W.; Joshi P. C.; Raoul S.; Ravanat J.-L. J. Am. Chem. Soc. 1994, 116, 7403–7404. [Google Scholar]
- Steenken S. Chem. Rev. 1989, 89, 503–520. [Google Scholar]
- Rokhlenko Y.; Cadet J.; Geacintov N. E.; Shafirovich V. J. Am. Chem. Soc. 2014, 136, 5956–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynisson J.; Steenken S. Phys. Chem. Chem. Phys. 2002, 4, 527–532. [Google Scholar]
- Koppang M. D.; Witek M. g.; Blau J.; Swain G. M. Anal. Chem. 1999, 71, 1188–1195. [Google Scholar]
- Poskrebyshev G. A.; Huie R. E.; Neta P. J. Phys. Chem. A 2003, 107, 7423–7428. [Google Scholar]
- Lu C.; Lin W.; Wang W.; Han Z.; Yao S.; Lin N. Phys. Chem. Chem. Phys. 2000, 2, 329–334. [Google Scholar]
- Fleming A. M.; Orendt A. M.; He Y.; Zhu J.; Dukor R. K.; Burrows C. J. J. Am. Chem. Soc. 2013, 135, 18191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwowski B.; Dupeyrat F.; Bardet M.; Ravanat J. L.; Krajewski P.; Cadet J. Chem. Res. Toxicol. 2006, 19, 1357–1365. [DOI] [PubMed] [Google Scholar]
- Ding S.; Jia L.; Durandin A.; Crean C.; Kolbanovskiy A.; Shafirovich V.; Broyde S.; Geacintov N. E. Chem. Res. Toxicol. 2009, 22, 1189–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming A. M.; Orendt A. M.; He Y.; Zhu J.; Dukor R. K.; Burrows C. J. J. Am. Chem. Soc. 2013, 135, 18191–18204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Fleming A. M.; Muller J. G.; Burrows C. J. New J. Chem. 2013, 37, 3440–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming A. M.; Muller J. G.; Dlouhy A. C.; Burrows C. J. J. Am. Chem. Soc. 2012, 134, 15091–15102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanduri D.; Adhikary A.; Sevilla M. D. J. Am. Chem. Soc. 2011, 133, 4527–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornyushyna O.; Burrows C. J. Biochemistry 2003, 42, 13008–13018. [DOI] [PubMed] [Google Scholar]
- Delaney S.; Delaney J. C.; Essigmann J. M. Chem. Res. Toxicol. 2007, 20, 1718–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupan A.; Sauliere A.; Broussy S.; Seguy C.; Pratviel G.; Meunier B. ChemBioChem 2006, 7, 125–33. [DOI] [PubMed] [Google Scholar]
- Matter B.; Malejka-Giganti D.; Csallany A. S.; Tretyakova N. Nucleic Acids Res. 2006, 34, 5449–5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.