

Abstract
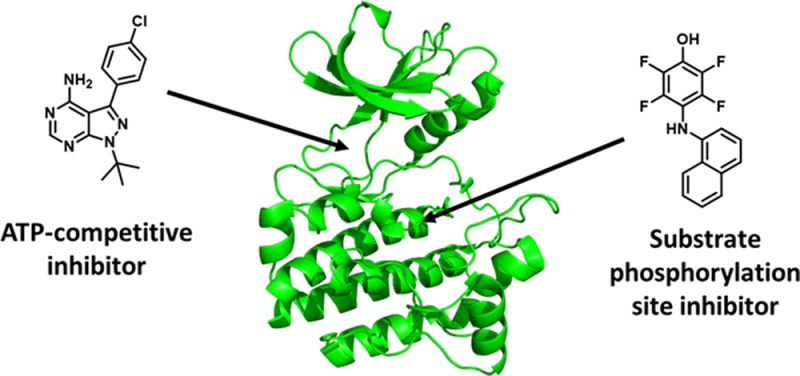
Protein kinases are important mediators of cellular communication and attractive drug targets for many diseases. Although success has been achieved with developing ATP-competitive kinase inhibitors, the disadvantages of ATP-competitive inhibitors have led to increased interest in targeting sites outside of the ATP binding pocket. Kinase inhibitors with substrate-competitive, ATP-noncompetitive binding modes are promising due to the possibility of increased selectivity and better agreement between biochemical and in vitro potency. However, the difficulty of identifying these types of inhibitors has resulted in significantly fewer small molecule substrate phosphorylation site inhibitors being reported compared to ATP-competitive inhibitors. This review surveys reported substrate phosphorylation site inhibitors and methods that can be applied to the discovery of such inhibitors, including a discussion of the challenges inherent to these screening methods.
Protein kinases catalyze the transfer of the gamma-phosphate of ATP to a serine, threonine, or tyrosine residue of a substrate protein or peptide. The human kinome includes 518 kinases and accounts for nearly 2% of the human genome.1 It is estimated that collectively the 518 human kinases can phosphorylate up to one-third of intracellular proteins to generate up to 20 000 distinct phosphoproteins.2 Phosphorylation of a substrate protein by a protein kinase is an important signal transduction mechanism within the cell and can yield diverse responses, including activation or deactivation of an enzyme, recruitment of adaptor proteins, and changes in cellular localization.3−6 Through their involvement in many critical signaling pathways, kinases control processes such as cell growth, apoptosis, motility, angiogenesis, metabolism, and inflammation.7−12
Illustrated in Figure 1 is the conserved structure of the kinase catalytic domain which consists of N-terminal and C-terminal lobes connected by a short loop termed the hinge region.14,15 The smaller N-terminal lobe is composed of five antiparallel β strands and one α helix, and the larger C-terminal lobe is composed of eight α helices and four β strands. The region between the N-terminal and C-terminal lobes and the hinge region forms a deep hydrophobic cleft that contains the ATP-binding site. ATP makes several key hydrogen bonds to the backbone of the hinge region which facilitate binding within the pocket. Additionally, the phosphate binding loop forms the ceiling of the ATP binding site and clamps down over the phosphate groups to orient them for catalysis. The protein substrate binding site is located within the C-terminal lobe. Also located in the C-terminal lobe is the activation loop. Many kinases are phosphorylated within this loop, which then undergoes a conformational change to activate the kinase and allow access to the substrate binding site. In addition to the catalytic domain, kinases may contain other regulatory domains which vary across the kinome and have diverse roles including modulating catalytic activity, recruiting substrates, controlling localization, and serving as scaffolding sites for other proteins.16−18
Figure 1.
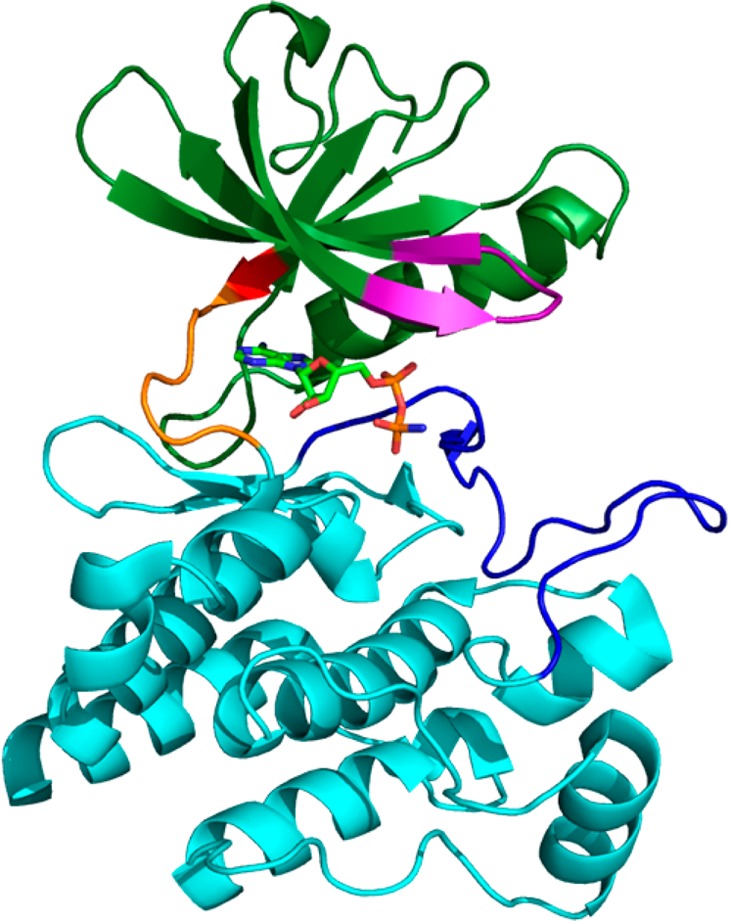
Crystal structure of the catalytic domain of Lck (PDB 1QPC).13 Highlighted are the N-terminal lobe (green), the C-terminal lobe (light blue), the hinge region (orange), the phosphate binding loop (purple), the activation loop (dark blue), and the gatekeeper residue (red). ATP is shown in stick depiction within the ATP binding site.
Due to the key roles of kinases in critical signaling pathways, the disregulation of kinase activity has been linked to over 400 diseases including many cancers, autoimmune disorders, inflammation, and diabetes.19−21 As a result, kinases are highly studied drug targets and constitute the largest drug target class after GPCRs.22 The first kinase inhibitor received FDA approval in 2001, and currently over 20 kinase inhibitors have been approved, mostly for use in oncology. Greater than 99% of reported kinase inhibitors, including all of the currently approved kinase-targeting drugs for oncology, inhibit kinase activity via competition for the ATP binding site.23 The heavy focus on ATP-competitive inhibitors can be largely attributed to the generality of this approach since all kinases contain an ATP binding site. Additionally, ATP-competitive inhibitors have been discovered with relative ease, initially through the design of adenosine analogs and later using techniques such as high throughput screening (HTS) and structure based drug design, due to the ATP binding site being a well formed pocket designed to bind small molecules.
Although many ATP-competitive kinase inhibitors have been described and several have proved successful in the clinic, there are drawbacks to these inhibitors that should be considered. First, the kinase ATP pocket is highly conserved across the kinome, leading to poor selectivity of most ATP-competitive kinase inhibitors.24−26 Off-target binding can result in additional toxicity of these compounds in the clinic and also prevents the use of most ATP-competitive inhibitors as biological probes. In addition to selectivity concerns, ATP-competitive inhibitors must contend with intracellular ATP levels that are typically in the millimolar range, while the ATP KM values for most kinases are in the low micromolar range. As a result of this, there is usually poor agreement between biochemical and cellular potency for ATP-competitive inhibitors, and a high affinity compound (typically nanomolar to picomolar) is required in order to see potent kinase inhibition in vivo.24,27 Finally, the rapid and common development of ATP pocket mutations, such as mutations of the “gatekeeper” residue that regulates access to a back hydrophobic pocket within the ATP site, both increases catalytic activity and confers resistance to many ATP-competitive inhibitors.28,29
As a result of these disadvantages, increased attention has been placed on developing small molecule inhibitors that instead target the protein substrate binding site. Like the ATP binding site, all protein kinases contain a protein substrate binding site; however, unlike the ATP binding site, the protein substrate binding site is less conserved between different kinases. Thus, similar to targeting the ATP binding site, targeting the protein substrate binding site is a strategy that can be applied to any protein kinase but offers the chance for improved selectivity compared to targeting the ATP binding site. Additionally, because kinase substrates are typically present at or below their KM value in vitro, a high biochemical affinity is not always required to yield in vitro activity.30 These features make the discovery of small molecule substrate phosphorylation site inhibitors highly desirable.
It should be noted that small molecule inhibitors have also been developed that target binding sites outside the ATP and substrate phosphorylation site, such as sites of autoinhibitory interactions, regulatory partner binding sites, or substrate docking interaction sites.31−35 While this will certainly increase the selectivity of these inhibitors and likely result in useful biological probes, this does not constitute a general targeting strategy that can be applied to any kinase like targeting the ATP or protein substrate binding site because these binding sites are not present in all kinases. It should also be noted that although some inhibitors targeting substrate docking sites show competition with peptide or protein substrates, they are considered outside the scope of this review because they do not bind at the protein substrate phosphorylation site. For clarity, we use the term “substrate phosphorylation site inhibitor” to refer to a substrate-competitive inhibitor that binds at the protein substrate binding site in a location analogous to that of the residue being phosphorylated.
Despite the potential benefits and the considerable effort put toward identifying small molecule substrate phosphorylation site inhibitors, their development has seen only limited success. This is directly related to the structure of the two binding sites. The relative ease of identifying small molecule ATP-competitive inhibitors is a result of targeting a well-defined pocket designed to bind a small molecule.14 Conversely, the substrate binding site is a shallow, open surface in order to facilitate the kinase–substrate protein–protein interaction.36 The differences between these two binding sites are demonstrated in Figure 2, which shows the structures of the insulin receptor tyrosine kinase (IRK) and the serine/threonine kinase Akt crystallized with ATP mimics bound to the ATP binding pocket and peptidic ligands bound to the substrate site. In both structures, the ATP mimic nestles deeply into the ATP cleft; in contrast, the peptide substrate mimic sits in a much shallower, solvent exposed cleft.
Figure 2.

Comparison of the ATP and substrate binding sites for (A) the tyrosine kinase IRK (PDB 1IR3)37 and (B) the serine/threonine kinase Akt (PBD 1O6K).38 ATP-competitive ligands are shown in green, and substrate-competitive ligands are shown in orange. The substrate binding site is less defined and more solvent exposed than the ATP binding site.
As a result of the protein substrate site being designed for protein–protein interactions, the majority of reported substrate-competitive inhibitors are peptides which were either rationally designed from peptidic substrates or discovered from screens of combinatorial libraries generated using one-bead–one-compound techniques or phage display.39−41 These inhibitors often have modest affinity for their target (midmicromolar to millimolar) in addition to poor cellular permeability and stability due to their peptidic nature, and these features make them undesirable for use as biological probes or therapeutics. While the development of small molecule substrate phosphorylation site inhibitors could address the permeability and stability problems associated with the peptidic inhibitors, the discovery of such inhibitors has proved incredibly challenging. As discussed, traditional HTS approaches have rarely yielded substrate-competitive inhibitors due to the lack of a well-defined pocket in the protein substrate binding site. Additionally, HTS libraries are often highly biased toward small, flat, heterocyclic molecules that are more likely to function as ATP-mimics than as peptidomimetics.42
In spite of the challenging nature of targeting the substrate phosphorylation site with small molecules, a small number of these inhibitors have been reported. This review will survey some reported small molecule substrate-phosphorylation site inhibitors, focusing on the difficulty of ascertaining their binding modes and the limitations preventing their implementation as biological tools. Additionally, screening approaches that are likely to identify substrate phosphorylation site inhibitors will be discussed, including the benefits and challenges inherent to each of these methods.
Substrate Phosphorylation Site Inhibitors
Substrate phosphorylation site inhibitors remain under-reported, with the majority being peptidic inhibitors. Additionally, many small molecules initially reported as phosphorylation site inhibitors have later been shown to be ATP-competitive inhibitors or mixed competitive with respect to both peptide substrate and ATP. In this section of our review, we survey several small molecules reported as substrate phosphorylation site inhibitors. As seen below, many of the inhibitors reported remain ambiguous in their binding mode (and even whether they are actually inhibitors of the target kinase). These vagaries are likely due to the weak affinity that small molecule substrate phosphorylation site inhibitors possess.
Erbstatin Analogs
Many of the first reported kinase inhibitors were inspired by natural products. Erbstatin (Chart 1), a phenolic natural product isolated from the culture filtrate of Streptomyces, was originally reported to be a substrate phosphorylation site inhibitor of the tyrosine kinase EGFR (Ki = 5.6 μM). On the basis of its structural resemblance to tyrosine and Lineweaver–Burk analysis showing competition with peptide substrate but not ATP, erbstatin was initially reported as a substrate phosphorylation site inhibitor.43 The structure of erbstatin also resembled the styryl pharmacophore found in peptide inhibitors incorporating dehydrophenylalanine in place of tyrosine, further supporting the hypothesis that it bound at the substrate phosphorylation site.44
Chart 1. Chemical Structures of Erbstatin and Tyrphostins.

The Levitzki group has extensively studied benzene malonitriles based on erbstatin. We have highlighted here a few of these compounds, termed “tyrphostins,” but for a more comprehensive analysis of tyrphostins we direct the reader to several reviews on the subject.45−47 In their initial reports, the group disclosed several tyrphostins, exemplified by AG18 and AG99 (Chart 1), which inhibited EGFR with low micromolar potency and good selectivity over the highly similar IRK (AG18 EGFR Ki = 11 μM and IRK Ki = 1.2 mM; AG99Ki = 3.5 μM).48,49 The most potent inhibitors blocked EGFR autophosphorylation (AG18 IC50 = 40 μM; AG99 IC50 = 4 μM) and the EGF-dependent growth of A431/clone 15 cells but had minimal effects on EGF-independent growth. The hypothesis that tyrphostins would bind at the substrate phosphorylation site was supported by Dixon plots demonstrating competition with a peptide substrate for several compounds including AG18 and AG99; however, no evidence was provided to demonstrate that the compounds were noncompetitive with respect to ATP.
A second library of tyrphostins with substitution of a ketone or amide derivative at the α position was also developed by the Levitzki group as inhibitors of EGFR.50 Relative to AG99, many of these compounds showed improved affinity for EGFR, exemplified by AG538 (IC50 = 0.37 μM, Chart 1). However, most did not significantly improve the inhibition of EGFR autophosphorylation or EGF dependent proliferation, and importantly, competition with peptide substrate or ATP was not examined. AG538 was later shown to also inhibit other tyrosine kinases including IGF-1, IRK, and Src with low nanomolar to low micromolar potency.51 To explore the binding mode of AG538, modeling utilizing a crystal structure of IRK was used. In the autoinhibited structure of IRK, Tyr1162 within the activation loop binds in the active site in a position similar to the tyrosine of a peptide substrate (Figure 3).37,52 Modeling showed that the catechol moieties in AG538 could be superimposed over Tyr1162 and another tyrosine in the activation loop (Tyr1158), suggesting that AG538 could bind at the substrate phosphorylation site. This was supported by assay data showing that the IC50 value is sensitive to the concentration of substrate peptide but not the concentration of ATP, and Lineweaver–Burk analysis demonstrating competition with peptide substrate was also shown.
Figure 3.
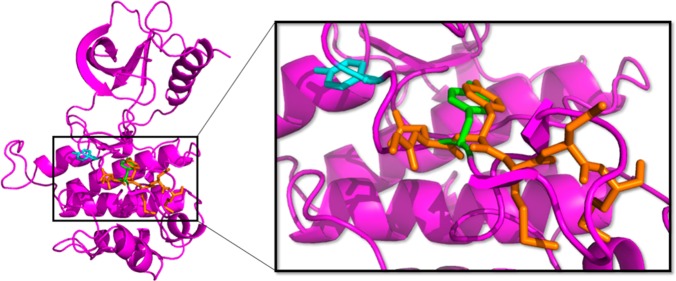
Crystal structure of the inactive form of IRK (PDB 1IRK)52 with Tyr1162 shown in green and Tyr1158 shown in cyan. Tyr1162 occupies the same binding site as the tyrosine residue in a substrate peptide, shown in orange (overlaid from PDB 1IR3).37
At the same time the Levitzki group was developing tyrphostins, the Watanabe group was also exploring benzene malononitrile erbstatin analogs, including analogs such as ST638 (Chart 1) which were substituted with thioethers.53,54ST638 was shown to inhibit EGFR (IC50 = 1 μM) and several other tyrosine kinases including Src family kinases but did not inhibit serine/threonine kinases. Using Lineweaver–Burk analysis, it was demonstrated that ST638 is competitive with respect to the EGFR substrate α-casein and noncompetitive with respect to ATP. The Levitzki group also examined substituted aryl thioether analogs such as AG824 (Chart 1).55AG824 has a high degree of similarity to ST638 and similar potency against EGFR (IC50 = 0.94 μM), but competition versus peptide substrate and ATP was not initially examined.
Although tyrphostins were designed to act as substrate phosphorylation site inhibitors and several were demonstrated to compete with a peptide substrate, it was disclosed in some initial reports that several compounds were found to have mixed-competitive binding modes. In hindsight, this is not surprising because although erbstatin was initially reported as a substrate phosphorylation site inhibitor, it has since been shown to be competitive with both ATP and peptide substrate for EGFR and to be ATP-competitive, peptide substrate-noncompetitive with other kinases.56−58 Upon additional analysis, it was demonstrated that AG18, AG99, AG824, and several other benzene malononitrile tyrphostins were competitive with both peptide substrate and ATP, but the substituted benzene malononitrile AG538 was demonstrated to be a substrate-competitive, ATP-noncompetitive inhibitor of EGFR.58 Although this analysis supports earlier biochemical data and the model suggesting AG538 binds at the substrate phosphorylation site, other tyrphostins with similar structures were shown to be competitive with both peptide substrate and ATP. More recently, a crystal structure was solved of the serine/threonine kinase CK2 with AG99 bound in the ATP site.59
As a whole, while there is evidence to support that a few tyrphostins such as AG538 may be substrate phosphorylation site inhibitors, this appears to be serendipity rather than rational design. Like erbstatin, most tyrphostins likely bind at the ATP site. ATP-competitive inhibitors that bind an inactive kinase conformation called the αC-helix out conformation have previously been demonstrated to be competitive with peptide substrate while not binding within the substrate phosphorylation site.60 In addition, it is worth noting that many tyrphostins have functionality (e.g., catechol) consistent with these compounds acting as PAINS (pan assay interference compounds), which complicates their evaluation in biochemical assays.61
Piceatannol Analogs
Piceatannol (Chart 2) is a plant secondary metabolite originally isolated from the seeds of Euphorbia lagascae and reported to have antileukemic properties.62 It was later shown by Geahlen and McLaughlin that piceatannol inhibits the activity of the tyrosine kinase Lck in a manner that is competitive with a peptide substrate and noncompetitive with ATP.63 Piceatannol also was structurally similar to tyrosine and contained the styryl pharmacophore known from peptide inhibitors. On the basis of this evidence, piceatannol was believed to be a substrate phosphorylation site inhibitor.
Chart 2. Chemical Structures of Piceatannol and Piceatannol Analogs.

Cushman and co-workers have examined two series of piceatannol derivatives.64,65 From a series of phenylhyrazones, the best inhibitor (Chart 2) was found to have similar potency against Lck (IC50 = 70 μM) as piceatannol (IC50 = 66 μM). The phenylhydrazone was also shown to be competitive with respect to a peptide substrate of Lck and noncompetitive with ATP, suggesting that like piceatannol it may be a substrate phosphorylation site inhibitor. A series of pyridine containing stilbene analogs of piceatannol was also examined. This series showed less potent inhibition of Lck compared to piceatannol, with the best compound having IC50 = 178 μM (Chart 2). Interestingly, the stilbene is competitive with ATP and uncompetitive with peptide substrate, suggesting that, similar to tyrphostins, small changes in structure can result in piceatannol analogs having variable binding modes.
Piceatannol has also been shown to have other activities in addition to Lck inhibition. These include inhibition of other kinases, tyrosine kinases such as Syk and JAK1, the serine/threonine kinase IκB, and the lipid kinase PI3K, as well as nonkinase activity such as binding to and activating estrogen receptors.66 Its effects on diverse families of kinases suggest that piceatannol is likely functioning as an ATP-competitive inhibitor and not a substrate phosphorylation site inhibitor, and, in fact, the inhibition of PI3K by piceatannol has been shown to be ATP-competitive.67 As a result of its effects on multiple signaling pathways, including nonkinase targets, and its probable ATP-competitive binding mode, the use of piceatannol (and likely analogs) as a biological probe likely offers no advantages over other more potent and moderately selective ATP-competitive probes. As with the tyrphostins, the presence of PAINS functionalities likely explains their complicated biochemical assay results.61
ON012380
In 2005, Gumireddy and co-workers reported the discovery of ON012380 (Chart 3).68 A library of styryl benzylsulfones previously shown by the authors to have potent antitumor activity was screened against purified BCR-Abl and identified ON012380 as a potent BCR-Abl inhibitor (IC50 = 9 nM).69−71ON012380 also inhibited BCR-Abl with the T315I gatekeeper mutation (IC50 = 1.5 nM), a mutation which renders the kinase resistant to most ATP-competitive inhibitors. Lineweaver–Burk analysis indicated that ON012380 was competitive with a protein substrate of Abl and was noncompetitive with ATP. Together these data suggested that ON012380 was a substrate phosphorylation site inhibitor. ON012380 was also shown to induce apoptosis in 32Dcl3 and K562 leukemia cells (LD50 = 10–15 nM), including cells expressing BCR-Abl mutants resistant to the ATP-competitive inhibitor imatinib. Additionally, treatment of K562 cells with ON012380 decreased BCR-Abl autophosphorylation and phosphorylation of two substrates, Crk and STAT5.
Chart 3. Chemical Structure of ON012380.

Wu and colleagues later studied the effects of ON012380 in more depth both in intact cells and in samples from chronic myeloid leukemia (CML) patients.72 Similar to the results of Gumireddy and co-workers, it was found that ON012380 reduced the survival of K562 and BV-173 leukemia cells expressing both wild-type and T315I BCR-Abl, as well as three samples derived from leukemia patients who had developed the T315I BCR-Abl mutation after treatment with imatinib. However, the authors found that these effects were not due to inhibition of BCR-Abl. BaF3 is an interleukin (IL)-3 dependent murine hematopoietic cell line that when transfected with a constitutively active kinase becomes kinase-dependent, IL-3 independent.73 When the authors evaluated the ATP-competitive Abl inhibitors imatinib and dasatinib in IL-3-dependent BaF3 cells and both BCR-Abl and T315I BCR-Abl transformed BaF3 cells, only the proliferation of the BCR-Abl transformed cells was inhibited. In contrast, ON012380 reduced the viability of both wild-type and T315I BCR-Abl transformed cells; however, it also inhibited the growth of the IL-3-dependent cells, indicating that the growth inhibition was independent of Abl.
Wu and co-workers also examined changes in tyrosine phosphorylation in BaF3, BV-173, and K562 cells in the presence of imatinib, dasatinib, and ON012380.72 They observed that while imatinib and dasatinib treatment reduced total phosphorylation levels, including the levels of BCR-Abl substrates, ON012380 had no effect on phosphorylation levels, which indicates that ON012380 does not inhibit Abl kinase activity and supports the results from the BaF3 cell study. Furthermore, when the activation of caspase cascades was examined, it was found that as expected apoptosis induced by treatment with imatinib was dependent on BCR-Abl transformation and inhibition of Abl. In contrast, treatment with ON012380 induced apoptosis independent of BCR-Abl transformation and occurred in the absence of kinase inhibition. The authors note that the discrepancies between their data and Gumireddy’s may be due to differences in assays and cell models used; however, these data support that ON012380 is not a direct inhibitor of Abl kinase, and it induces cellular apoptosis through a kinase-independent mechanism.
KX-01
The Hangauer lab has developed ATP-noncompetitive inhibitors of c-Src by employing qualitative molecular modeling.74,75 As a crystal structure of c-Src with a ligand bound to the protein substrate binding site is not currently available, the authors used a crystal structure of autoinhibited IRK to guide compound design. In the autoinhibited conformation, a tyrosine residue in the activation loop (Tyr1162) binds in the IRK active site in a position similar to the tyrosine of a substrate peptide (Figure 3).37,52 Inhibitor core scaffolds were superimposed on Tyr1162, and from this a series of hydroxynaphthalene and hydroxyindole methyl esters and amides predicted to bind at the substrate phosphorylation site were designed. Several inhibitors with low micromolar IC50 values against c-Src were identified, such as compounds 2f and 2k (Chart 4; IC50 = 16 μM and 38 μM, respectively), and the IC50 values of 2f and 2k were not susceptible to changes in ATP concentration, supporting an ATP-noncompetitive binding mode. However, no direct evidence was presented for competition with peptide substrate or binding at the substrate phosphorylation site.
Chart 4. Chemical Structures of KX-01, Hydroxynaphthalene Amide 2f, and Hydroxyindole Amide 2k.

Using a similar modeling approach, Hangauer and colleagues developed KX-01 (also called KX2–391; Chart 4) as a c-Src substrate phosphorylation site inhibitor.76,77 Initially, no experimental data were presented to support this claim, and while a recent a NMR study using a paramagnetic ATP-competitive probe has provided experimental evidence to support that KX-01 binds outside of the ATP binding site, its exact binding location is still unconfirmed.78 Although KX-01 has micromolar potency when evaluated with isolated enzyme (c-Src IC50 = 46 μM), it was a low nanomolar inhibitor of cell proliferation in HT-29 colon cancer (GI50 = 13 nM) and c-Src transformed 3T3 cells (GI50 = 23 nM). The authors originally proposed that this discrepancy may be due to the binding site of KX-01 not being formed outside of the cellular environment, and thus, when in the cellular environment where the binding site is formed the inhibitor shows greater potency. However, it is more likely that this is due to a secondary mechanism of action, and indeed it was later shown through photoaffinity labeling that KX-01 interacts with a novel binding site on heterodimeric tubulin and prevents tubulin polymerization.79,80
The dual mechanism of action of KX-01 was confirmed by Tu and co-workers using a proteomics strategy and Western blotting.81 When PC3-LN4 prostate cancer cells were treated with either KX-01 or vinblastine, a microtubule inhibitor, downregulation of expression of tubulin isotypes was observed, which is hypothesized to be due to microtubule polymerization inhibitors increasing the pool of tubulin, thereby leading to a decrease in tubulin synthesis. However, the total level of Src and Src autophosphorylation was decreased only after exposure to KX-01 and not after exposure to vinblastine. The authors also performed photoaffinity labeling studies which confirmed that KX-01 binds to tubulin.
The effects of KX-01 in multiple cell lines have also been studied, further supporting that KX-01 inhibits both c-Src and tubulin polymerization in cellulo. Anbalagan and co-workers confirmed that exposure to KX-01 decreased c-Src autophosphorylation and phosphorylation of c-Src substrates in MDA-MB-231, MDA-MB-157, and MDA-MB-468 breast cancer cells treated with KX-01 or combinations of KX-01 and paclitaxel.82 Additionally, the authors demonstrated that KX-01 and combinations of KX-01 and paclitaxel disrupted microtubules in MDA-MB-231 cells and tumor xenografts. Similar effects on Src activity were observed in MCF-7 breast cancer cell and tumor xenographs, and it was also shown that treatment of tamoxifen resistant MCF-7 cells with KX-01 restored tamoxifen sensitivity and resulted in synergistic growth inhibition.83 Finally, Liu and colleagues demonstrated inhibition of Src activity and microtubule polymerization in RMUG-S ovarian cancer cells and that combinations of KX-01 and the cytotoxic drug oxaliplatin resulted in synergistic inhibition of tumor growth.84
KX-01 has been evaluated in phase I/II clinical trials for multiple cancers including solid tumors, acute myeloid leukemia, and lymphoma, and a trial for a combination of KX-01 with paclitaxel for the treatment of solids tumors is also currently recruiting subjects.85 However, while current data suggest that the dual mechanism of action of KX-01 may prove beneficial in the clinical setting, this will hinder its use as a biological probe to study c-Src signaling. Furthermore, the large increase in potency observed going from evaluation with purified enzyme to evaluation in vitro suggests that the in vitro effects are due to the inhibition of tubulin polymerization and not inhibition of c-Src.
MEB-SCI
We recently reported the discovery of substrate-competitive c-Src inhibitors using a substrate activity screening (SAS)-based approach (vide infra).86 The lead inhibitor, MEB-SCI (see Scheme 2), had low micromolar affinity for c-Src (Ki = 16 μM), and Lineweaver–Burk analysis demonstrated that MEB-SCI was competitive with a peptide substrate and noncompetitive with ATP. Induced fit docking also suggests that MEB-SCI binds at the protein substrate binding site. Our computational model was supported by biochemical data showing a decrease in affinity for a Src mutant with changes in the protein substrate binding site and synergistic inhibition when combined with ATP-competitive inhibitors. MEB-SCI inhibited the growth of SK-BR-3 breast cancer cells (GI50 = 14 μM) with potency equivalent to some of the most potent ATP-competitive Src inhibitors. Additionally, analysis of Src-dependent and independent signaling pathways in SK-BR-3 cells showed that only Src-dependent pathways were inhibited, suggesting that MEB-SCI has good selectivity for Src in cellulo. Currently, MEB-SCI has only been evaluated for inhibition of isolated kinases with Src family kinases and c-Abl, and while selectivity against Src family kinases was modest (3–20 fold), it does show good selectivity against the highly similar c-Abl (Ki > 1 mM).
Scheme 2.

Screening Methods to Identify Substrate-Phosphorylation Site Inhibitors
As demonstrated above, many reported small molecule substrate phosphorylation site inhibitors were designed using the structure of natural products thought to be substrate phosphorylation site inhibitors or through the use of molecular modeling approaches. However, as evidenced by both the small number of reported inhibitors and the number of inhibitors later shown to have alternate binding modes or targets, this approach has not been particularly successful. As such, the development of new methods for the identification of substrate phosphorylation site inhibitors remains an important goal. Here we highlight screening methods that can be used for the discovery of substrate phosphorylation site inhibitors, with a focus on benefits and potential pitfalls of each method.
Biased Activity-Based Biochemical Screens
Activity based assays have long been the first choice for kinase inhibitor HTS.87 These approaches have traditionally been more likely to discover ATP competitive inhibitors, but several groups have recently reported activity-based screens in which the assay conditions were modified to promote the identification of ATP-noncompetitive inhibitors. This was generally accomplished by encouraging formation of the enzyme–ATP complex, which was predicted to favor the binding of ATP-noncompetitive ligands and discourage the binding of weak and modest ATP-competitive inhibitors (Scheme 1). However, while this approach aims to reduce the number of ATP-competitive hits, it can also bias toward the identification of highly potent ATP-competitive inhibitors.
Scheme 1.

Liu and colleagues reported a biased activity assay for the identification of ATP-noncompetitive inhibitors of LRRK2.88 Knowledge of the kinetic mechanism and the kinetic parameters for LRRK2, coupled with mechanistic simulations, enabled determination of initial concentrations of ATP and PLK-peptide substrate which would bias toward formation of the enzyme–ATP complex. A time-resolved Forster resonance energy transfer (TR-FRET) assay performed under these conditions was used to quantify phosphorylation of the PLK-peptide substrate in the presence of potential inhibitors. From a screen of 63 400 compounds, 21 hits with IC50 <10 μM were identified. The lead compound from the screen is an allosteric inhibitor, as analysis of its effects on the ATP and substrate kinetic parameters demonstrated that it is noncompetitive with both ATP and peptide substrate. This suggests that the lead compound is not a substrate phosphorylation site inhibitor.
A similar approach was taken by Lo and co-workers to screen for ATP-noncompetitive inhibitors of CDK4.89 It was predicted that increasing the concentration of ATP in the assay to 12-fold above its apparent KM value would bias toward the enzyme–ATP complex. As mentioned earlier, because the assay format also would allow for highly potent ATP-competitive inhibitors to be identified, the IC50 values for initial hits were determined at ATP concentrations equal to KM and 12-fold greater than KM. From a screen of 250 000 compounds, three compounds were identified with potencies that were relatively insensitive to ATP concentration, suggesting that they are ATP-noncompetitive inhibitors. Additional analysis of the most potent hit (IC50 = 2.4 μM) in the presence of increasing concentrations of substrate demonstrated that potency was also insensitive to peptide substrate concentration. Surprisingly, although the lead compound appears to be an allosteric inhibitor of CDK4, it was found to be an ATP competitor of the tyrosine kinase Lck.
Our lab has also modified the conditions of an activity-based assay to favor the discovery of ATP-noncompetitive inhibitors of c-Src by increasing the concentration of ATP.90 In one screen, the ATP concentration was increased to 10-fold above its KM value, and fragment libraries were screened. Although several hits were identified, no substrate-competitive, ATP-noncompetitive inhibitors were found. We then tried to further bias the assay conditions by increasing the ATP concentration to 50-fold higher than KM and screening against the c-Src T338 M gatekeeper mutant. Because gatekeeper mutations are known to cause resistance to ATP-competitive inhibitors, we hypothesized that this would also favor the discovery of ATP-noncompetitive inhibitors. Several hits were identified from this screen, and Lineweaver–Burk analysis suggests that the lead inhibitor is noncompetitive with both ATP and peptide substrate.
These examples highlight that although activity based assays can be biased toward identifying ATP-noncompetitive inhibitors, the discovery of substrate phosphorylation site inhibitors from such assays remains elusive. Assay conditions that favor the identification of substrate-competitive inhibitors will also favor the identification of allosteric inhibitors that bind at neither the ATP nor the substrate sites. Additionally, while these modifications discourage ligands with weak to moderate affinity for the ATP site, they can also promote the identification of potent ATP-competitive inhibitors. As a result of this, further analysis of each hit will be required in order to determine the binding mode. Overall, it appears that assays monitoring enzyme inhibition will continue to be a poor choice for the identification of substrate phosphorylation site inhibitors.
In contrast to assays that monitor inhibition of enzymatic activity, the Ellman lab has developed a screening approach termed substrate activity screening (SAS) that instead identifies molecules that serve as substrates of an enzyme.91−96 The identified substrates can then be optimized and later converted into inhibitors by replacement of the reactive functionality. Because SAS identifies substrates of an enzyme, hits that are converted into inhibitors should inherently be substrate-competitive inhibitors. This approach is similar to that used for the development of peptidic substrate phosphorylation site inhibitors from peptidic kinase substrates.97−101
The Ellman lab has previously described SAS methodology for the discovery of small molecule inhibitors of several proteases and phosphatases, and our group recently reported the development of a SAS method for the identification of small molecule substrate phosphorylation site inhibitors of tyrosine kinases.86 In the SAS method for tyrosine kinases, diverse phenolic fragments are screened using an assay that monitors ADP generation to identify substrates of the kinase. The phosphorylatable phenol is then modified to prevent the phosphotransfer reaction, thereby converting the substrate into an inhibitor (Scheme 2). Using this method, we reported the first small molecule substrates of any protein kinase. Building on previous work by Graves and co-workers which demonstrated that fluorination of a substrate tyrosine yielded peptide inhibitors of IRK, we fluorinated the phenol of a small molecule substrate to yield a substrate phosphorylation site inhibitor that was not phosphorylated by c-Src.98,102 Further optimization yielded the lead inhibitor MEB-SCI, a substrate phosphorylation site inhibitor of c-Src (Ki = 16 μM) with activity similar to ATP-competitive inhibitors in cellulo and promising selectivity (vide supra).
Competitive Binding Screens
Although activity-based screens generally have not identified substrate phosphorylation site inhibitors, binding assays appear better poised for success. Binding assays can be used to either directly detect binding of a ligand to the target or indirectly detect binding through competitive displacement of a probe. Direct binding assays using surface plasmon resonanace (SPR) and affinity selection mass spectrometry (ASMS) have been used to discover ATP-noncompetitive ligands for kinase targets; however, these methods are nonbiased, and ligands can bind to all exposed sites on the protein.103−105 As such, these screens are similar to activity based screens in that they will be far more likely to identify ATP-competitive ligands, and extensive additional analysis is required to determine the binding mode of each hit. In contrast, using a competitive-binding assay will allow for identifying ligands that bind to a specific site on the target. A general scheme demonstrating how competitive binding assays can be used to identify substrate phosphorylation site inhibitors is shown in Scheme 3. These assays rely on the net displacement of a probe to measure ligand binding, and therefore using a probe that binds at the substrate phosphorylation site should enable the exclusive identification of substrate phosphorylation site inhibitors. The net displacement of the probe from the target can be evaluated by multiple methods, including fluorescence-based techniques such as fluorescence polarization (FP) and Forster resonance energy transfer (FRET) and biophysical techniques such as SPR.
Scheme 3.
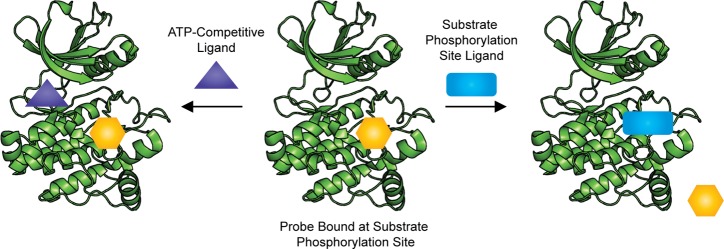
Despite the promise of competitive binding assays for the discovery of substrate phosphorylation site inhibitors, these screens have not yet been used for the identification of substrate phosphorylation site inhibitors. Interestingly, fluorescently labeled substrate phosphorylation site inhibitor peptides that could be used in competitive binding assays to identify small molecule substrate phosphorylation site inhibitors have been developed but were not used for this purpose. For example, Saldanha and co-workers used a peptidic substrate phosphorylation site probe (Kd = 4.4 nM) in the development of a ligand-regulated competition (LiReC) screen to identify compounds that modulate the interactions between the catalytic and regulatory domains of PKA, and Tsuganezawa and colleagues used a peptidic substrate phosphorylation site probe (Kd = 5 μM) in a fluorescent correlation spectroscopy (FCS) assay to identify ATP-competitive inhibitors of Pim-1 that also make interactions with residues known to be important for substrate binding.106,107 While these probes could be used in competitive binding screens for substrate phosphorylation site inhibitors of PKA or Pim-1, this application has not been reported. Stebbins and colleagues have also reported using a competitive binding assay for the identification of inhibitors targeting a scaffolding site in JNK, but although these inhibitors were shown to compete with a substrate protein, they do not bind at the substrate phosphorylation site.108
The limited development of competitive-binding screens using substrate phosphorylation site probes is likely a direct result of the assay design requirements.109 Ideally, the probe should have high affinity for the target in order to ensure that a high fraction of the probe is bound without requiring large quantities of enzyme. Most reported substrate phosphorylation site ligands are peptides with modest affinities (low to high micromolar) and do not fulfill this requirement. One way to address the modest potency of many substrate phosphorylation site ligands is to develop bisubstrate ligands.110 These compounds contain a substrate phosphorylation site ligand (usually a peptide) covalently linked to a ligand for the ATP binding site. The bisubstrate compound usually has greatly increased potency relative to the peptide alone, which makes them more amenable for use as probes. As illustrated in Scheme 4, these bisubstrate probes will be displaced by both ligands for the substrate phosphorylation site and ligand for the ATP site. There have been several reports of the use of bisubstrate probes in the development of competitive-binding assays for kinase targets that can identify and characterize both ATP-competitive inhibitors and substrate phosphorylation site inhibitors.
Scheme 4.
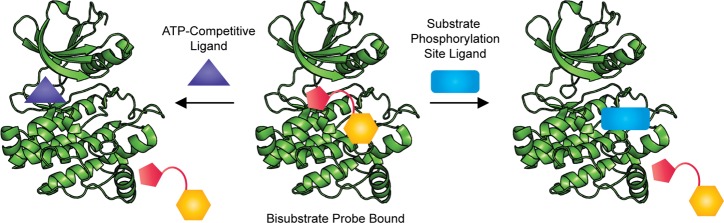
The Uri group has developed several bisubstrate inhibitors, termed ARCs, by linking adenosine to arginine rich peptides, and they have begun using these ARCs as probes to develop competitive binding assays. A SPR competitive-binding assay for the determination of the affinities of both ATP-competitive ligands and substrate phosphorylation site ligands of PKA was developed by immobilizing an ARC via a streptavidin–biotin complex.111 The immobilized probe ARC-704 (Chart 5) had excellent affinity for PKA (Kd = 16 nM), and the SPR assay was able to detect displacement of the bisubstrate probe by the binding of known ATP-competitive inhibitors, other ARCs, and protein substrates of PKA. The Kd values for the known inhibitors characterized with this assay were in good agreement with reported values.
Chart 5. Chemical Structures of Bisubstrate Probes for Competitive Binding Assays.

The same group has also developed an FP method utilizing a bisubstrate probe based on an ARC for the characterization of ligands of PKA and ROCK.112 The FP probe ARC-538 (Chart 5) was generated by labeling the N-terminus of the peptidic portion of the ARC with the fluorescent tag TAMRA. The probe had excellent affinity for PKA (Kd = 480 pM), and displacement of the probe from PKA was observed with ATP-competitive inhibitors, other ARCs, and protein substrates of PKA. The probe is also reported to be a ligand for ROCK (Kd = 3.6 nM), but displacement of the probe by a substrate phosphorylation site ligand for ROCK was not evaluated. In both cases, the Kd values for known inhibitors obtained using the FP assay were in good agreement with literature reports.
Our lab has also developed a bisubstrate TR-FRET tracer that can identify substrate-competitive inhibitors.113 A bisubstrate inhibitor of c-Src was fluorescently labeled with Cy5 to generate a TR-FRET tracer. Similar to what was seen with the ARC probes, this probe has excellent affinity for c-Src (Kd = 6 nM). In a TR-FRET assay with c-Src, displacement of the tracer by known ATP-competitive and substrate-competitive ligands could be detected, and the Kd values obtained for the ligands using this assay were in good agreement with literature values.
These examples demonstrate that a variety of competitive binding assay formats can be used with bisubstrate probes to recognize substrate phosphorylation site ligands, but thus far screens for substrate phosphorylation site inhibitors using these methods have not been reported. Although bisubstrate probes hold promise for the identification of substrate phosphorylation site inhibitors, a complication of using bisubstrate probes instead of probes that only target the substrate phosphorylation site is that ATP-competitive inhibitors will be identified as well. Therefore, a counter screen against an ATP-competitive probe should be performed to rule out compounds which displace the bisubstrate probe by competing for binding to the ATP site. A further complication that could arise during counter screening is that recent work by Lebakken and co-workers has shown that some ligands binding outside the ATP site, including substrate phosphorylation site inhibitors, can displace ATP-competitive TR-FRET tracers by causing perturbations within the ATP binding pocket.114 This raises the possibility that substrate phosphorylation site inhibitors identified from a competitive binding assay using a bisubstrate probe may be ruled out as ATP-competitive inhibitors during counter screening. Due to these issues, the use of a probe for the substrate phosphorylation site instead of a bisubstrate probe would be preferable when available.
A remaining issue with both substrate phosphorylation site and bisubstrate probes is that they are not likely to bind to a large number of kinases due to the less conserved nature of the substrate binding site. This means that while in general competitive binding assays could be developed with any kinase of interest, a single probe cannot be used for all (or even most) kinases, and new probes will need to be developed in order to access different subsets of targets. This will be most challenging for new kinase targets; however, while the development of substrate phosphorylation site probes may not be initially feasible for new targets since high potency substrate phosphorylation site ligands will likely not yet be known, a bisubstrate approach may be possible. Many services offering broad kinase inhibitor profiling screens include kinases whose functions are currently unknown in their panels, and published data sets of kinase inhibitor selectivity show that a potent ATP-competitive inhibitor can be identified for most kinases. These data could aid in the development of bisubstrate probes for competitive-binding assays with new targets.
NMR Screening
NMR screening has become a popular screening method due to its ability to detect even weakly binding fragments, but while NMR screens against kinase targets have been successful in identifying ATP-competitive fragments, the propensity of the ligands to bind within the better defined ATP-pocket has stalled the discovery of substrate phosphorylation site inhibitors.115 Recently, however, some success in identifying ATP-noncompetitive ligands has been achieved by utilizing paramagnetic spin-labeled ATP-competitive probes. In these experiments, NMR spectra of a compound with the kinase of interest are obtained both in the presence and in the absence of the spin-labeled probe. The spin-label will increase the relaxation time of nearby protons, and thus compounds which bind simultaneously near the probe can be identified by observing a paramagnetic relaxation enhancement (PRE) in the NMR spectrum.116 When using an ATP-competitive spin-labeled probe, other ATP-competitive ligands will not be identified since these compounds cannot bind at the same time as the probe. The probes are sensitive to ligands binding up to 25 Å away from the spin label, a distance which includes the protein substrate binding site (Figure 4).
Figure 4.
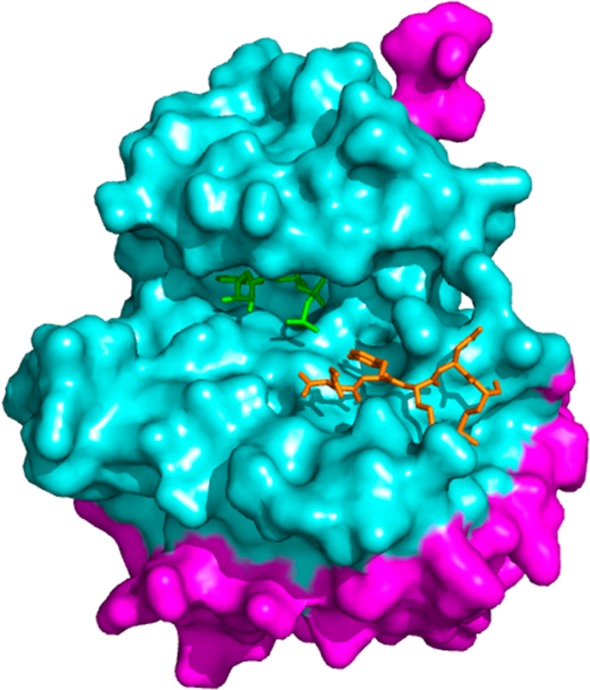
Crystal structure of IRK bound to an ATP analog (green) and a peptidic substrate mimic (orange), with residues within 20 Å of the ATP binding site highlighted in cyan (PDB 1IR3).37 The substrate binding site is located within this 20 Å radius.
In 2005, McCoy and co-workers reported the use of manganese-chelated ATP as a paramagnetic probe for identifying ATP-noncompetitive ligands.117 The authors demonstrated that the probe could detect the binding of a known ATP-noncompetitive inhibitor of MEK1, but no new ligands were reported. Although this probe should bind to any kinase of interest, ATP (and therefore the probe) has micromolar affinity for many kinases. Due to the modest affinity, a large excess of the probe may be required in order saturate the kinase and obtain the maximum signal, but this could also result in nonspecific binding of the probe. Furthermore, Mn2+ can also bind nonspecifically to proteins. To ensure that only ligands binding within 25 Å of the ATP binding site are identified, the authors noted that the probe should be used with kinases for which it has good affinity (low micromolar to nanomolar), and if the buffer used contains manganese, the concentration of Mn2+ should be less than 100 μM.
At the same time, Jahnke and co-workers reported the TEMPO-labeled adenine analog probe 1 (Chart 6).118 While an example NMR spectrum for the identification of an ATP-noncompetitive ligand (ligand and kinase not disclosed) and recommendations for confirming hits are outlined, no new ligands were reported. The close structural resemblance of probe 1 to ATP should allow it to bind to most kinases. However, similar to the ATP-manganese chelate probe, probe 1 will likely have micromolar affinity for many kinases, and a large quantity of the probe may be required. As mentioned previously, this can lead to nonspecific binding, and in fact, the authors report that the probe does bind nonspecifically to some kinases.
Chart 6. Chemical Structures of Paramagnetic Probes for NMR Screening.

To remedy the low affinity and nonspecific binding of previous probes, research groups have begun modifying potent ATP-competitive inhibitors with spin labels. Moy and co-workers have developed the spin-labeled probe 2 (Chart 6) based on an ATP-competitive inhibitor that was reported to bind potently to several kinases.78 Profiling of probe 2 against a panel of 19 kinases showed that it bound eight kinases with an IC50 value less than 40 nM. However, of the other 11 kinases examined, nine had IC50 > 50 μM, suggesting that probe 2 cannot be widely applied to any kinase of interest. The ability of probe 2 to identify compounds binding outside the ATP pocket was confirmed by detecting the binding of KX-01 (Chart 4) to c-Src. While a full scale screen was not reported using this probe, the authors were able to identify the binding of the fragment N-phenylanthranilic acid to Lck. The binding site for this fragment has not been conclusively determined, but the weak PRE signal and modeling of the probe in complex with Lck predicted that it binds in a pocket adjacent to the ATP binding site and the substrate binding site.
Large screens utilizing spin-labeled ATP-competitive probes have not yet been reported, and it is important to remember that these probes will identify any ligand that binds within 25 Å of the spin-label. As such, these probes will not exclusively identify substrate phosphorylation site inhibitors, and after a screen using one of these probes additional experiments may be required to determine if the ligand binds to the substrate phosphorylation site or an allosteric site. This potentially could be minimized by carefully designing the probe so that the spin label is placed close to the substrate phosphorylation site. Vasquez and co-workers have used a similar approach to bias toward the identification of inhibitors binding to a scaffold docking site in JNK.119 Ligands not binding at the desired site experience weak PRE due to binding far from the spin-label and thus can be easily discarded. However, this strategy has not been applied to screening for substrate phosphorylation site inhibitors.
Similar to competitive binding assays, the affinity and selectivity of the spin-labeled probes will also complicate their use. Probes based on ATP or adenine will bind to most if not all kinases, but micromolar affinity for most kinases and nonspecific binding will limit their use. Conversely, designing probes based on potent ATP-competitive inhibitors such as probe 2 will minimize nonspecific binding, but these probes will not bind to the full kinome. As a result, the development of new probes will be required for some targets of interest; however, as discussed previously, selectivity data sets show that potent ATP-competitive inhibitors can be found for most kinases.
Conclusions and Future Directions
Despite the interest in using small molecule substrate phosphorylation site kinase inhibitors as biological probes and therapeutics, a tiny fraction of reported kinase inhibitors falls into this category. The vast majority of small molecule kinase inhibitors are ATP-competitive inhibitors. This can largely be attributed to the surface of the ATP and protein substrate binding sites. While the ATP pocket is well formed and designed to bind a small molecule, the protein substrate site is a shallow, solvent exposed surface designed to facilitate protein–protein interactions instead of binding small molecules.
Many compounds initially reported as substrate phosphorylation site inhibitors have later been demonstrated to be ATP-competitive inhibitors or to have other nonkinase targets that are responsible for the observed effects in vitro. As such, the development of new methods for the discovery of a substrate phosphorylation site inhibitor remains a priority. Although “traditional” HTS using activity based assays generally do not identify substrate phosphorylation site inhibitors, more screening methods are being developed that increase the likelihood of discovering this class of inhibitors. These approaches include biased activity assays, competitive binding assays, and NMR screening using ATP-competitive probes. However, many of these methods still have considerable disadvantages, such as the likelihood of identifying compounds targeting other sites than the substrate phosphorylation site, the need to develop new probes for different kinases, or not being applicable to all classes of protein kinases.
Overall, while these methods will aid in the identification of new substrate phosphorylation site inhibitors, their shortcomings demonstrate that there is still a continuing need to improve current screening methods as well as develop new methods. Ideally, a screening method would exclusively identify substrate phosphorylation site inhibitors, would be applicable to any kinase of interest, and would not require structural knowledge of the target or the development of multiple probes for different targets. Such a screening method would be of great value for advancing the discovery of new substrate phosphorylation site inhibitors to serve as biological probes and potential therapeutics.
Acknowledgments
We thank the National Institutes of Health (R01GM088546 to M.B.S.) and the University of Michigan College of Pharmacy for financial support. M.E.B. was supported, in part, by a Pharmacological Sciences Training Program NIH training grant (GM007767). We thank R. Pricer for assistance with graphics.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Manning G.; Whyte D. B.; Martinez R.; Hunter T.; Sudarsanam S. (2002) The Protein Kinase Complement of the Human Genome. Science 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- Johnson S. A.; Hunter T. (2005) Kinomics: methods for deciphering the kinome. Nat. Methods 2, 17–25. [DOI] [PubMed] [Google Scholar]
- Ryšlavá H.; Doubnerová V.; Kavan D.; Vaněk O. (2013) Effect of posttranslational modifications on enzyme function and assembly. J. Proteomics 92, 80–109. [DOI] [PubMed] [Google Scholar]
- Pawson T.; Scott J. D. (2005) Protein phosphorylation in signaling – 50 years and counting. Trends Biochem. Sci. 30, 286–290. [DOI] [PubMed] [Google Scholar]
- Graves J. D.; Krebs E. G. (1999) Protein Phosphorylation and Signal Transduction. Pharmacol. Ther. 82, 111–121. [DOI] [PubMed] [Google Scholar]
- Hunter T. (1995) Protein kinases and phosphatases: The Yin and Yang of protein phosphorylation and signaling. Cell 80, 225–236. [DOI] [PubMed] [Google Scholar]
- Dhanasekaran D. N.; Reddy E. P. (2008) JNK signaling in apoptosis. Oncogene 27, 6245–6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning B. D.; Cantley L. C. (2007) AKT/PKB Signaling: Navigating Downstream. Cell 129, 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A.-K.; Dimberg A.; Kreuger J.; Claesson-Welsh L. (2006) VEGF receptor signalling: in control of vascular function. Nat. Rev. Mol. Cell Biol. 7, 359–371. [DOI] [PubMed] [Google Scholar]
- Mitra S. K.; Hanson D. A.; Schlaepfer D. D. (2005) Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56–68. [DOI] [PubMed] [Google Scholar]
- Zarubin T.; Han J. (2005) Activation and signaling of the p38 MAP kinase pathway. Cell Res. 15, 11–18. [DOI] [PubMed] [Google Scholar]
- Birchmeier C.; Birchmeier W.; Gherardi E.; Vande Woude G. F. (2003) Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 4, 915–925. [DOI] [PubMed] [Google Scholar]
- Zhu X.; Kim J. L.; Newcomb J. R.; Rose P. E.; Stover D. R.; Toledo L. M.; Zhao H.; Morgenstern K. A. (1999) Structural analysis of the lymphocyte-specific kinase Lck in complex with non-selective and Src family selective kinase inhibitors. Structure 7, 651–661. [DOI] [PubMed] [Google Scholar]
- Hubbard S. R.; Till J. H. (2000) Protein Tyrosine Kinase Structure and Function. Annu. Rev. Biochem. 69, 373–398. [DOI] [PubMed] [Google Scholar]
- Hanks S. K.; Hunter T. (1995) Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 9, 576–96. [PubMed] [Google Scholar]
- Deshmukh K.; Anamika K.; Srinivasan N. (2010) Evolution of domain combinations in protein kinases and its implications for functional diversity. Prog. Biophys. Mol. Biol. 102, 1–15. [DOI] [PubMed] [Google Scholar]
- Endicott J. A.; Noble M. E. M.; Johnson L. N. (2012) The Structural Basis for Control of Eukaryotic Protein Kinases. Annu. Rev. Biochem. 81, 587–613. [DOI] [PubMed] [Google Scholar]
- Dohlman H. G. (2008) A Scaffold Makes the Switch. Sci. Signal. 1, pe46-. [DOI] [PubMed] [Google Scholar]
- Blume-Jensen P.; Hunter T. (2001) Oncogenic kinase signalling. Nature 411, 355–365. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Boehm J.; Lee J. C. (2003) p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discovery 2, 717–726. [DOI] [PubMed] [Google Scholar]
- Evans J. L.; Goldfine I. D.; Maddux B. A.; Grodsky G. M. (2002) Oxidative Stress and Stress-Activated Signaling Pathways: A Unifying Hypothesis of Type 2 Diabetes. Endocr. Rev. 23, 599–622. [DOI] [PubMed] [Google Scholar]
- Cohen P. (2002) Protein kinases - the major drug targets of the twenty-first century?. Nat. Rev. Drug Discovery 1, 309–315. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Yang P. L.; Gray N. S. (2009) Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight Z. A.; Shokat K. M. (2005) Features of Selective Kinase Inhibitors. Chem. Biol. 12, 621–637. [DOI] [PubMed] [Google Scholar]
- Vulpetti A.; Bosotti R. (2004) Sequence and structural analysis of kinase ATP pocket residues. Farmaco 59, 759–765. [DOI] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–U124. [DOI] [PubMed] [Google Scholar]
- Scapin G. (2006) Protein Kinase Inhibition: Different Approaches to Selective Inhibitor Design. Curr. Drug Targets 7, 1443–1454. [DOI] [PubMed] [Google Scholar]
- Krishnamurty R.; Maly D. J. (2010) Biochemical Mechanisms of Resistance to Small-Molecule Protein Kinase Inhibitors. ACS Chem. Biol. 5, 121–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub H.; Specht K.; Ullrich A. (2004) Strategies to overcome resistance to targeted protein kinase inhibitors. Nat. Rev. Drug Discovery 3, 1001–1010. [DOI] [PubMed] [Google Scholar]
- Lawrence D. S.; Niu J. (1998) Protein Kinase Inhibitors: The Tyrosine-Specific Protein Kinases. Pharmacol. Ther. 77, 81–114. [DOI] [PubMed] [Google Scholar]
- Adrian F. J.; Ding Q.; Sim T.; Velentza A.; Sloan C.; Liu Y.; Zhang G.; Hur W.; Ding S.; Manley P.; Mestan J.; Fabbro D.; Gray N. S. (2006) Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat. Chem. Biol. 2, 95–102. [DOI] [PubMed] [Google Scholar]
- Mahadevan D.; Powis G.; Mash E. A.; George B.; Gokhale V. M.; Zhang S.; Shakalya K.; Du-Cuny L.; Berggren M.; Ali M. A.; Jana U.; Ihle N.; Moses S.; Franklin C.; Narayan S.; Shirahatti N.; Meuillet E. J. (2008) Discovery of a novel class of AKT pleckstrin homology domain inhibitors. Mol. Cancer Ther. 7, 2621–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzáez M.; Gortat A.; Mondragón L.; Bachs O.; Pérez-Payá E. (2009) ATP-Noncompetitive Inhibitors of CDK–Cyclin Complexes. ChemMedChem. 4, 19–24. [DOI] [PubMed] [Google Scholar]
- Licht-Murava A.; Eldar-Finkelman H. (2012) Exploiting Substrate Recognition for Selective Inhibition of Protein Kinases. Curr. Pharm. Des. 18, 2914–2920. [DOI] [PubMed] [Google Scholar]
- Schnieders M. J.; Kaoud T. S.; Yan C.; Dalby K. N.; Ren P. (2012) Computational Insights for the Discovery of Non-ATP Competitive Inhibitors of MAP Kinases. Curr. Pharm. Des. 18, 1173–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose R.; Holbert M. A.; Pickin K. A.; Cole P. A. (2006) Protein tyrosine kinase-substrate interactions. Curr. Opin. Struct. Biol. 16, 668–675. [DOI] [PubMed] [Google Scholar]
- Hubbard S. R. (1997) Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 16, 5572–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Cron P.; Good V. M.; Thompson V.; Hemmings B. A.; Barford D. (2002) Crystal structure of an activated Akt/Protein Kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat. Struct. Biol. 9, 940–944. [DOI] [PubMed] [Google Scholar]
- Bidwell G. L.; Raucher D. (2009) Therapeutic peptides for cancer therapy. Part I – peptide inhibitors of signal transduction cascades. Expert Opin. Drug Delivery 6, 1033–1047. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H.; Eisenstein M. (2009) Peptide Inhibitors Targeting Protein Kinases. Curr. Pharm. Des. 15, 2463–2470. [DOI] [PubMed] [Google Scholar]
- Bogoyevitch M. A.; Barr R. K.; Ketterman A. J. (2005) Peptide inhibitors of protein kinases—discovery, characterisation and use. Biochim. Biophys. Acta 1754, 79–99. [DOI] [PubMed] [Google Scholar]
- Akritopoulou-Zanze I.; Hajduk P. J. (2009) Kinase-targeted libraries: The design and synthesis of novel, potent, and selective kinase inhibitors. Drug Discovery Today 14, 291–297. [DOI] [PubMed] [Google Scholar]
- Imoto M.; Umezawa K.; Isshiki K.; Kunimoto S.; Sawa T.; Takeuchi T.; Umezawa H. (1987) Kinetic Studies of Tyrosine Kinase Inhibition by Erbstatin. J. Antibiot. 40, 1471–1473. [DOI] [PubMed] [Google Scholar]
- Wong T. W.; Goldberg A. R. (1984) Kinetics and mechanism of angiotensin phosphorylation by the transforming gene product of Rous sarcoma virus. J. Biol. Chem. 259, 3127–3131. [PubMed] [Google Scholar]
- Levitzki A. (2002) Tyrosine kinases as targets for cancer therapy. Eur. J. Cancer 38(Supplement 5), S11–S18. [DOI] [PubMed] [Google Scholar]
- Levitzki A. (1999) Protein Tyrosine Kinase Inhibitors as Novel Therapeutic Agents. Pharmacol. Ther. 82, 231–239. [DOI] [PubMed] [Google Scholar]
- Levitzki A.; Mishani E. (2006) Tyrphostins and Other Tyrosine Kinase Inhibitors. Annu. Rev. Biochem. 75, 93–109. [DOI] [PubMed] [Google Scholar]
- Yaish P.; Gazit A.; Gilon C.; Levitzki A. (1988) Blocking of EGF-Dependent Cell Proliferation by EGF Receptor Kinase Inhibitors. Science 242, 933–935. [DOI] [PubMed] [Google Scholar]
- Gazit A.; Yaish P.; Gilon C.; Levitzki A.; Tyrphostins I. (1989) synthesis and biological activity of protein tyrosine kinase inhibitors. J. Med. Chem. 32, 2344–2352. [DOI] [PubMed] [Google Scholar]
- Gazit A.; Osherov N.; Posner I.; Yaish P.; Poradosu E.; Gilon C.; Levitzki A. (1991) Tyrphostins. II. Heterocyclic and.alpha.-substituted benzylidenemalononitrile tyrphostins as potent inhibitors of EGF receptor and ErbB2/neu tyrosine kinases. J. Med. Chem. 34, 1896–1907. [DOI] [PubMed] [Google Scholar]
- Blum G.; Gazit A.; Levitzki A. (2000) Substrate Competitive Inhibitors of IGF-1 Receptor Kinase. Biochemistry 39, 15705–15712. [DOI] [PubMed] [Google Scholar]
- Hubbard S. R.; Wei L.; Hendrickson W. A. (1994) Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 372, 746–754. [DOI] [PubMed] [Google Scholar]
- Shiraishi T.; Domoto T.; Imai N.; Shimada Y.; Watanabe K. (1987) Specific inhibitors of tyrosine-specific protein kinase, synthetic 4-hydroxycinnamamide derivatives. Biochem. Biophys. Res. Commun. 147, 322–328. [DOI] [PubMed] [Google Scholar]
- Shiraishi T.; Koji Owada M.; Tatsuka M.; Yamashita T.; Watanabe K.; Kakunaga T. (1989) Specific Inhibitors of Tyrosine-specific Protein Kinases: Properties of 4-Hydroxycinnamamide Derivatives in Vitro. Cancer Res. 49, 2374–2378. [PubMed] [Google Scholar]
- Gazit A.; Osherov N.; Posner I.; Bar-Sinai A.; Gilon C.; Levitzki A. (1993) Tyrphostins. 3. Structure-activity relationship studies of.alpha.-substituted benzylidenemalononitrile 5-S-aryltyrphostins. J. Med. Chem. 36, 3556–3564. [DOI] [PubMed] [Google Scholar]
- Bishop W. R.; Petrin J.; Wang L.; Ramesh U.; Doll R. J. (1990) Inhibition of protein kinase c by the tyrosine kinase inhibitor erbstatin. Biochem. Pharmacol. 40, 2129–2135. [DOI] [PubMed] [Google Scholar]
- Hsu C.-Y. J.; Jacoski M. V.; Maguire M. P.; Spada A. P.; Zilberstein A. (1992) Inhibition kinetics and selectivity of the tyrosine kinase inhibitor erbstatin and a pyridone-based analogue. Biochem. Pharmacol. 43, 2471–2477. [DOI] [PubMed] [Google Scholar]
- Posner I.; Engel M.; Gazit A.; Levitzki A. (1994) Kinetics of inhibition by tyrphostins of the tyrosine kinase activity of the epidermal growth factor receptor and analysis by a new computer program. Mol. Pharmacol. 45, 673–683. [PubMed] [Google Scholar]
- Lolli G.; Cozza G.; Mazzorana M.; Tibaldi E.; Cesaro L.; Donella-Deana A.; Meggio F.; Venerando A.; Franchin C.; Sarno S.; Battistutta R.; Pinna L. A. (2012) Inhibition of Protein Kinase CK2 by Flavonoids and Tyrphostins. A Structural Insight. Biochemistry 51, 6097–6107. [DOI] [PubMed] [Google Scholar]
- Georghiou G.; Kleiner R. E.; Pulkoski-Gross M.; Liu D. R.; Seeliger M. A. (2012) Highly specific, bisubstrate-competitive Src inhibitors from DNA-templated macrocycles. Nat. Chem. Biol. 8, 366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. (2010) New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- Ferrigni N. R.; McLaughlin J. L.; Powell R. G.; Smith C. R. (1984) Use of Potato Disc and Brine Shrimp Bioassays to Detect Activity and Isolate Piceatannol as the Antileukemic Principle from the Seeds of Euphorbia lagascae. J. Nat. Prod. 47, 347–352. [DOI] [PubMed] [Google Scholar]
- Geahlen R. L.; McLaughlin J. L. (1989) Piceatannol (3,4,3′,5′-tetrahydroxy-trans-stilbene) is a naturally occurring protein-tyrosine kinase inhibitor. Biochem. Biophys. Res. Commun. 165, 241–245. [DOI] [PubMed] [Google Scholar]
- Cushman M.; Nagarathnam D.; Gopal D.; Geahlen R. L. (1991) Synthesis and evaluation of new protein-tyrosine kinase inhibitors. Part 1. pyridine-containing stilbenes and amides. Bioorg. Med. Chem. Lett. 1, 211–214. [Google Scholar]
- Cushman M.; Nagarathnam D.; Gopal D.; Geahlen R. L. (1991) Synthesis and evaluation of new protein-tyrosine kinase inhibitors. Part 2. Phenylhydrazones. Bioorg. Med. Chem. Lett. 1, 215–218. [Google Scholar]
- Piotrowska H.; Kucinska M.; Murias M. (2012) Biological activity of piceatannol: Leaving the shadow of resveratrol. Mutat. Res. 750, 60–82. [DOI] [PubMed] [Google Scholar]
- Choi K. H.; Kim J.-E.; Song N. R.; Son J. E.; Hwang M. K.; Byun S.; Kim J. H.; Lee K. W.; Lee H. J. (2010) Phosphoinositide 3-kinase is a novel target of piceatannol for inhibiting PDGF-BB-induced proliferation and migration in human aortic smooth muscle cells. Cardiovasc. Res. 85, 836–844. [DOI] [PubMed] [Google Scholar]
- Gumireddy K.; Baker S. J.; Cosenza S. C.; John P.; Kang A. D.; Robell K. A.; Reddy M. V. R.; Reddy E. P.; non-ATP-competitive A. (2005) inhibitor of BCR-ABL overrides imatinib resistance. Proc. Natl. Acad. Sci. U.S.A. 102, 1992–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy E. P.; Reddy M. V. R.. Substituted styryl benzylsulfones for treating proliferative disorders. U.S. Patent 6,486,210, 2002.
- Reddy E. P.; Reddy M. V. R.. alpha., .beta.-unsaturated sulfones for treating proliferative disorders. U.S. Patent 6,541,475, 2003.
- Reddy E. P.; Reddy M. V. R.. Alpha, beta-unsaturated sulfones for treating proliferative disorders U.S. Patent 6,599,932, 2003.
- Wu J.; Meng F.; Ying Y.; Peng Z.; Daniels L.; Bornmann W. G.; Quintas-Cardama A.; Roulston D.; Talpaz M.; Peterson L. F.; Donato N. J. (2010) ON012380, a putative BCR-ABL kinase inhibitor with a unique mechanism of action in imatinib-resistant cells. Leukemia 24, 869–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmuth M.; Kim S.; Gu X.-j.; Xia G.; Adrian F. (2007) Ba/F3 cells and their use in kinase drug discovery. Curr. Opin. Oncol. 19, 55–60. [DOI] [PubMed] [Google Scholar]
- Marsilje T. H.; Milkiewicz K. L.; Hangauer D. G. (2000) The design, synthesis and activity of non-ATP competitive inhibitors of pp60c-src tyrosine kinase. Part 1: Hydroxynaphthalene derivatives. Bioorg. Med. Chem. Lett. 10, 477–481. [DOI] [PubMed] [Google Scholar]
- Milkiewicz K. L.; Marsilje T. H.; Woodworth R. P. Jr.; Bifulco N. Jr.; Hangauer M. J.; Hangauer D. G. (2000) The design, synthesis and activity of non-ATP competitive inhibitors of pp60c-src tyrosine kinase. Part 2: Hydroxyindole derivatives. Bioorg. Med. Chem. Lett. 10, 483–486. [DOI] [PubMed] [Google Scholar]
- Hangauer D. G.Compositions for treating cell proliferation disorders. U.S. Patent 7,300,931, 2007.
- Hangauer D. G.Biaryl compositions and methods for modulating a kinase cascade. U.S. Patent 7,968,574, 2011.
- Moy F. J.; Lee A.; Gavrin L. K.; Xu Z. B.; Sievers A.; Kieras E.; Stochaj W.; Mosyak L.; McKew J.; Tsao D. H. H. (2009) Novel Synthesis and Structural Characterization of a High-Affinity Paramagnetic Kinase Probe for the Identification of Non-ATP Site Binders by Nuclear Magnetic Resonance. J. Med. Chem. 53, 1238–1249. [DOI] [PubMed] [Google Scholar]
- Hangauer D. G.; Smolinski M.; Bu Y. H.; Kazim L.; Qu J., Photoaffinity labeling studies to better define the mechanism of action for Phase II oncology drug KX2–391. In Abstracts of Papers, 239th American Chemical Society National Meeting, San Francisco, CA, 2010.
- Yahao B.; Qu J.; Kazim L.; Dyster L.; Gelman I.; Hangauer D. G., Mechanism of action of KX01 (KX2-391), a novel tubulin polymerization and Src signaling inhibitor proceeding to Phase II clinical trials. In Abstracts of Papers, 101st AACR Annual Meeting, Washington, DC, 2010.
- Tu C.; Li J.; Bu Y.; Hangauer D.; Qu J. (2012) An ion-current-based, comprehensive and reproducible proteomic strategy for comparative characterization of the cellular responses to novel anti-cancer agents in a prostate cell model. J. Proteomics 77, 187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anbalagan M.; Ali A.; Jones R. K.; Marsden C. G.; Sheng M.; Carrier L.; Bu Y.; Hangauer D.; Rowan B. G. (2012) Peptidomimetic Src/Pretubulin Inhibitor KX-01 Alone and in Combination with Paclitaxel Suppresses Growth, Metastasis in Human ER/PR/HER2-Negative Tumor Xenografts. Mol. Cancer Ther. 11, 1936–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anbalagan M.; Carrier L.; Glodowski S.; Hangauer D.; Shan B.; Rowan B. (2012) KX-01, a novel Src kinase inhibitor directed toward the peptide substrate site, synergizes with tamoxifen in estrogen receptor α positive breast cancer. Breast Cancer Res. Treat. 132, 391–409. [DOI] [PubMed] [Google Scholar]
- Liu T.; Hu W.; Dalton H. J.; Choi H. J.; Huang J.; Kang Y.; Pradeep S.; Miyake T.; Song J. H.; Wen Y.; Lu C.; Pecot C. V.; Bottsford-Miller J.; Zand B.; Jennings N. B.; Ivan C.; Gallick G. E.; Baggerly K. A.; Hangauer D. G.; Coleman R. L.; Frumovitz M.; Sood A. K. (2013) Targeting Src and Tubulin in Mucinous Ovarian Carcinoma. Clin. Cancer Res. 19, 6532–6543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. National Institutes of Health. www.clinicaltrials.gov.
- Breen M. E.; Steffey M. E.; Lachacz E. J.; Kwarcinski F. E.; Fox C. C.; Soellner M. B. (2014) Substrate Activity Screening with Kinases: Discovery of Small-Molecule Substrate-Competitive c-Src Inhibitors. Angew. Chem., Int. Ed. 53, 7010–7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Ahsen O.; Bömer U. (2005) High-Throughput Screening for Kinase Inhibitors. ChemBioChem. 6, 481–490. [DOI] [PubMed] [Google Scholar]
- Liu M.; Poulose S.; Schuman E.; Zaitsev A. D.; Dobson B.; Auerbach K.; Seyb K.; Cuny G. D.; Glicksman M. A.; Stein R. L.; Yue Z. (2010) Development of a mechanism-based high-throughput screen assay for leucine-rich repeat kinase 2—Discovery of LRRK2 inhibitors. Anal. Biochem. 404, 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo M.-C.; Ngo R.; Dai K.; Li C.; Liang L.; Lee J.; Emkey R.; Eksterowicz J.; Ventura M.; Young S. W.; Xiao S.-H. (2012) Development of a time-resolved fluorescence resonance energy transfer assay for cyclin-dependent kinase 4 and identification of its ATP-noncompetitive inhibitors. Anal. Biochem. 421, 368–377. [DOI] [PubMed] [Google Scholar]
- Steffey M. E.; Soellner M. B. Unpublished data, 2012.
- Baguley T. D.; Xu H.-C.; Chatterjee M.; Nairn A. C.; Lombroso P. J.; Ellman J. A. (2013) Substrate-Based Fragment Identification for the Development of Selective, Nonpeptidic Inhibitors of Striatal-Enriched Protein Tyrosine Phosphatase. J. Med. Chem. 56, 7636–7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyva M. J.; DeGiacomo F.; Kaltenbach L. S.; Holcomb J.; Zhang N.; Gafni J.; Park H.; Lo D. C.; Salvesen G. S.; Ellerby L. M.; Ellman J. A. (2010) Identification and Evaluation of Small Molecule Pan-Caspase Inhibitors in Huntington’s Disease Models. Chem. Biol. 17, 1189–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brak K.; Doyle P. S.; McKerrow J. H.; Ellman J. A. (2008) Identification of a New Class of Nonpeptidic Inhibitors of Cruzain. J. Am. Chem. Soc. 130, 6404–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soellner M. B.; Rawls K. A.; Grundner C.; Alber T.; Ellman J. A. (2007) Fragment-based substrate activity screening method for the identification of potent inhibitors of the Mycobacterium tuberculosis phosphatase PtpB. J. Am. Chem. Soc. 129, 9613–9615. [DOI] [PubMed] [Google Scholar]
- Patterson A. W.; Wood W. J. L.; Hornsby M.; Lesley S.; Spraggon G.; Ellman J. A. (2006) Identification of Selective, Nonpeptidic Nitrile Inhibitors of Cathepsin S Using the Substrate Activity Screening Method. J. Med. Chem. 49, 6298–6307. [DOI] [PubMed] [Google Scholar]
- Wood W. J. L.; Patterson A. W.; Tsuruoka H.; Jain R. K.; Ellman J. A. (2005) Substrate activity screening: A fragment-based method for the rapid identification of nonpeptidic protease inhibitors. J. Am. Chem. Soc. 127, 15521–15527. [DOI] [PubMed] [Google Scholar]
- Ye G. F.; Tiwari R.; Parang K. (2008) Development of Src tyrosine kinase substrate binding site inhibitors. Curr. Opin. Invest. Drugs (BioMed Cent.) 9, 605–613. [PubMed] [Google Scholar]
- Yuan C. J.; Jakes S.; Elliott S.; Graves D. J. (1990) A rationale for the design of an inhibitor of tyrosyl kinase. J. Biol. Chem. 265, 16205–16209. [PubMed] [Google Scholar]
- Al-Obeidi F. A.; Wu J. J.; Lam K. S. (1998) Protein tyrosine kinases: Structure, substrate specificity, and drug discovery. Biopolymers 47, 197–223. [DOI] [PubMed] [Google Scholar]
- Niu J.; Lawrence D. S. (1997) Nonphosphorylatable Tyrosine Surrogates: Implications for Protein Kinase Inhibitor Design. J. Biol. Chem. 272, 1493–1499. [DOI] [PubMed] [Google Scholar]
- Chapelat J.; Berst F.; Marzinzik A. L.; Moebitz H.; Drueckes P.; Trappe J.; Fabbro D.; Seebach D. (2012) The Substrate-Activity-Screening methodology applied to receptor tyrosine kinases: A proof-of-concept study. Eur. J. Med. Chem. 57, 1–9. [DOI] [PubMed] [Google Scholar]
- Martin B. L.; Wu D. L.; Jakes S.; Graves D. J. (1990) Chemical Influences on the Specificity of Tyrosine Phosphorylation. J. Biol. Chem. 265, 7108–7111. [PubMed] [Google Scholar]
- Comess K. M.; Sun C.; Abad-Zapatero C.; Goedken E. R.; Gum R. J.; Borhani D. W.; Argiriadi M.; Groebe D. R.; Jia Y.; Clampit J. E.; Haasch D. L.; Smith H. T.; Wang S.; Song D.; Coen M. L.; Cloutier T. E.; Tang H.; Cheng X.; Quinn C.; Liu B.; Xin Z.; Liu G.; Fry E. H.; Stoll V.; Ng T. I.; Banach D.; Marcotte D.; Burns D. J.; Calderwood D. J.; Hajduk P. J. (2010) Discovery and Characterization of Non-ATP Site Inhibitors of the Mitogen Activated Protein (MAP) Kinases. ACS Chem. Biol. 6, 234–244. [DOI] [PubMed] [Google Scholar]
- Ashwell M. A.; Lapierre J.-M.; Brassard C.; Bresciano K.; Bull C.; Cornell-Kennon S.; Eathiraj S.; France D. S.; Hall T.; Hill J.; Kelleher E.; Khanapurkar S.; Kizer D.; Koerner S.; Link J.; Liu Y.; Makhija S.; Moussa M.; Namdev N.; Nguyen K.; Nicewonger R.; Palma R.; Szwaya J.; Tandon M.; Uppalapati U.; Vensel D.; Volak L. P.; Volckova E.; Westlund N.; Wu H.; Yang R.-Y.; Chan T. C. K. (2012) Discovery and Optimization of a Series of 3-(3-Phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amines: Orally Bioavailable, Selective, and Potent ATP-Independent Akt Inhibitors. J. Med. Chem. 55, 5291–5310. [DOI] [PubMed] [Google Scholar]
- Navratilova I.; Macdonald G.; Robinson C.; Hughes S.; Mathias J.; Phillips C.; Cook A. (2012) Biosensor-Based Approach to the Identification of Protein Kinase Ligands with Dual-Site Modes of Action. J. Biomol. Screening 17, 183–193. [DOI] [PubMed] [Google Scholar]
- Saldanha S. A.; Kaler G.; Cottam H. B.; Abagyan R.; Taylor S. S. (2006) Assay Principle for Modulators of Protein–Protein Interactions and Its Application to Non-ATP-Competitive Ligands Targeting Protein Kinase A. Anal. Chem. 78, 8265–8272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuganezawa K.; Watanabe H.; Parker L.; Yuki H.; Taruya S.; Nakagawa Y.; Kamei D.; Mori M.; Ogawa N.; Tomabechi Y.; Handa N.; Honma T.; Yokoyama S.; Kojima H.; Okabe T.; Nagano T.; Tanaka A. (2012) A Novel Pim-1 Kinase Inhibitor Targeting Residues That Bind the Substrate Peptide. J. Mol. Biol. 417, 240–252. [DOI] [PubMed] [Google Scholar]
- Stebbins J. L.; De S. K.; Machleidt T.; Becattini B.; Vazquez J.; Kuntzen C.; Chen L.-H.; Cellitti J. F.; Riel-Mehan M.; Emdadi A.; Solinas G.; Karin M.; Pellecchia M. (2008) Identification of a new JNK inhibitor targeting the JNK-JIP interaction site. Proc. Natl. Acad. Sci. U.S.A. 105, 16809–16813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zettner A. (1973) Principles of Competitive Binding Assays (Saturation Analyses). I. Equilibrium Techniques. Clin. Chem. 19, 699–705. [PubMed] [Google Scholar]
- Gower C. M.; Chang M. E. K.; Maly D. J. (2014) Bivalent inhibitors of protein kinases. Crit. Rev. Biochem. Mol. Biol. 49, 102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viht K.; Schweinsberg S.; Lust M.; Vaasa A.; Raidaru G.; Lavogina D.; Uri A.; Herberg F. W. (2007) Surface-plasmon-resonance-based biosensor with immobilized bisubstrate analog inhibitor for the determination of affinities of ATP- and protein-competitive ligands of cAMP-dependent protein kinase. Anal. Biochem. 362, 268–277. [DOI] [PubMed] [Google Scholar]
- Vaasa A.; Viil I.; Enkvist E.; Viht K.; Raidaru G.; Lavogina D.; Uri A. (2009) High-affinity bisubstrate probe for fluorescence anisotropy binding/displacement assays with protein kinases PKA and ROCK. Anal. Biochem. 385, 85–93. [DOI] [PubMed] [Google Scholar]
- Brandvold K. R.; Santos S. M.; Breen M. E.; Lachacz E. J.; Steffey M. E.; Soellner M. B. Manuscript in preparation, 2014.
- Lebakken C. S.; Reichling L. J.; Ellefson J. M.; Riddle S. M. (2012) Detection of Allosteric Kinase Inhibitors by Displacement of Active Site Probes. J. Biomol. Screening 17, 813–821. [DOI] [PubMed] [Google Scholar]
- Pellecchia M.; Bertini I.; Cowburn D.; Dalvit C.; Giralt E.; Jahnke W.; James T. L.; Homans S. W.; Kessler H.; Luchinat C.; Meyer B.; Oschkinat H.; Peng J.; Schwalbe H.; Siegal G. (2008) Perspectives on NMR in drug discovery: a technique comes of age. Nat. Rev. Drug Discovery 7, 738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnke W.; Perez L. B.; Paris C. G.; Strauss A.; Fendrich G.; Nalin C. M. (2000) Second-Site NMR Screening with a Spin-Labeled First Ligand. J. Am. Chem. Soc. 122, 7394–7395. [Google Scholar]
- McCoy M. A.; Senior M. M.; Wyss D. F. (2005) Screening of Protein Kinases by ATP-STD NMR Spectroscopy. J. Am. Chem. Soc. 127, 7978–7979. [DOI] [PubMed] [Google Scholar]
- Jahnke W.; Blommers M. J. J.; Fernández C.; Zwingelstein C.; Amstutz R. (2005) Strategies for the NMR-Based Identification and Optimization of Allosteric Protein Kinase Inhibitors. ChemBioChem. 6, 1607–1610. [DOI] [PubMed] [Google Scholar]
- Vazquez J.; De S. K.; Chen L.-H.; Riel-Mehan M.; Emdadi A.; Cellitti J.; Stebbins J. L.; Rega M. F.; Pellecchia M. (2008) Development of Paramagnetic Probes for Molecular Recognition Studies in Protein Kinases. J. Med. Chem. 51, 3460–3465. [DOI] [PMC free article] [PubMed] [Google Scholar]