

Abstract

Recent advances in mass spectrometry (MS)-based proteomics allow the identification and quantitation of thousands of posttranslational modification (PTM) sites in a single experiment. This follows from the development of more effective class enrichment strategies, new high performance instrumentation and bioinformatic algorithms with rigorous scoring strategies. More widespread use of these combined capabilities have led to a vast expansion in our knowledge of the complexity of biological processes mediated by PTMs. The classes most actively pursued include phosphorylation, ubiquitination, O-GlcNAcylation, methylation, and acetylation. Very recently succinylation, SUMOylation, and citrullination have emerged. Among the some 260 000 PTM sites that have been identified in the human proteome thus far, only a few have been assigned to key regulatory and/or other biological roles. Here, we provide an update of MS-based PTM analyses, with a focus on current enrichment strategies coupled with revolutionary advances in high performance MS. Furthermore, we discuss examples of the discovery of recently described biological roles of PTMs and address the challenges of defining site-specific functions.
The human genome project revealed only approximately 20 000 protein-coding genes.1 The proteome, however, is far more complex and diverse because of post-translational modifications (PTMs) and to some extent isoform variations.2 While RNA sequencing detects the expression and sequence variations of the entire transcriptome,3 mass spectrometry (MS)-based proteomics has the advantage of being able to detect and structurally define any covalent changes in a protein after translation. A daunting number of such changes confer altered physiological activity, and many are reversible. There is a growing need to carry out accurate measurements of site-specific dynamics due to the lack of immunoaffinity reagents for the large numbers of newly identified proteins and their PTM analogs in rewired signaling networks, for example. Thus, the field is seeing an increase in use and further optimization of multiplexed targeted, selected-component quantitation by spectral acquisition in millisecond time frames. In fact, studies of large scale PTM dynamics will be driven by mass spectral-based quantitation—the methodology of choice. PTMs increase the functional diversity of proteins by adding covalent modifications such as phosphorylation, ubiquitination, glycosylation, methylation, and acetylation. Beside single PTMs, proteins are often modified through a combination of post-translational hydrolytic cleavages and the addition of functional groups through a stepwise processes leading to protein maturation or activation. Protein modifications influence and many times even define a large variety of normal and pathogenic cell biology functions. Therefore, identifying and understanding PTMs is critical for gaining a comprehensive understanding of cell biology, the detection and delineation of molecular defects underlying human and other diseases, drug target discovery and validation, and the eventual treatment and prevention of diseases.
A comprehensive treatment of our earlier level of knowledge of over 300 types of PTMs, which are known to occur physiologically, can be found in the Walsh monograph.4 Since then, revolutionary advances in enrichment strategies and improved performances of capillary liquid chromatography (LC) and new MS instrumentation have driven our growing knowledge of many PTMs. In fact, the delineation of the actual complexity of many PTMs has emerged mostly through the past decade. Thus, by significant enrichment of classes of modified peptides before MS-analysis, thousands of precise sites can now be identified with high confidence.5−13
In high-resolution tandem MS, two stages of mass analysis are used in a single experiment. The MS1 scan refers to the m/z of the precursor ion (peptide or protein), whereas the MS2 scans refer to the m/z values recorded for their fragmented ionic products. Major developments have led to new instrumentation that provides both high resolution MS and high mass measurement accuracies for both MS1 and MS2 levels simultaneously. The selection of appropriate energy deposition methods, however, is necessary to ensure generation of sequence ion series required for unambiguous site assignments. Having high mass spectral measurement quality has increased the reliability and efficiency of PTM identification at the peptide level and, in addition, has permitted the precise localization of modified sites for some intact protein sequences.14 In particular, MS fragmentation strategies that generate sufficient peptide fragmentation information are essential for precise PTM identification and localization, by definition. Among the different fragmentation strategies commonly employed, electron capture and transfer-based fragmentation techniques have proven to be essential for the localization of labile modifications as well as in dramatically expanding our experimental capability to carry out sequence analyses on large peptides or even medium size intact proteins. Labile structures include many serine or threonine phosphorylation sites as well as very labile modifications such as O-linked N-acetylglucosamine (O-GlcNAc), γ-carboxyglutamic acid, and others.15−18
In this review, we first describe how enrichment strategies and the revolutionary advances in mass spectrometry have contributed to high confidence PTM identification as well as site-specific localization. We then emphasize the discovery of previously unknown biological roles of PTMs including the signaling pathway activation role of arginine methylation and multiple cross-talk forms between PTMs. Finally, we address the biological challenges of defining individual site-specific functions. The complex fields of extracellular glycosylation and protein lipidation are not covered in this review, but the interested reader is referred to recently published reviews on these topics.19,20
PTM Enrichment Strategies
Since regulatory PTMs feature substoichiometric site occupancies in many cases, specific enrichment techniques are essential to achieve detection and characterization of low relative abundance components in digests of cell lysates (Table 1). Common PTM enrichment strategies use affinity purification based on charge properties or antibody recognition. They are applied at the peptide level usually ensuring higher accessibility of PTMs and allowing quite specific binding. Prior to enrichment, proteins are digested into peptides. The most commonly used enzyme for peptide level PTM analysis is trypsin, due to its high cleavage specificity after lysine and arginine.21 In some cases, however, the resulting tryptic peptides might be too long or too short for effective MS analysis. Instead of using trypsin alone, other proteases such as chymotrypsin, AspN, Lys-N, or endoproteinase Glu-C may be employed to increase the PTM coverage.22
Table 1. PTM Enrichment Strategies.
| enrichment strategy | PTM |
|---|---|
| antibody-based | tyrosine phosphorylation31,32 |
| arginine/lysine methylation35 | |
| lysine acetylation10 | |
| ubiquitin-like12,34 | |
| ionic interaction-based | serine/threonine/arginine phosphorylation24−926 |
| IMAC, TiO2, SIMAC | |
| metabolic tagging | |
| lectin | N-glycosylation/O-GlcNAc11 |
| His10 SUMO2 | SUMO13 |
| IodoTMT | S-nitrosylation40 |
| glyoxal derivate | citrullination941 |
| enzymatic-based | |
| subtiligase | proteolysis37 |
| PNGase | N-glycosylation11 |
| phospholipase | GPI-anchor942 |
Ionic Interaction-Based PTM Enrichment Strategies
Among all PTMs phosphorylation is the most extensively studied and approximately 197 000 human phosphorylation sites have been reported.23 Global analysis of serine- and threonine-phosphorylation is commonly achieved by metal ion-based enrichment methods such as immobilized metal affinity chromatography (IMAC) and titanium dioxide (TiO2).24,25 Both approaches use metal cations to bind the negatively charged phosphopeptides, and protocols designed to achieve higher enrichment efficiencies have been constantly improved over the past decade. Space does not permit discussion of the extensive literature here, but the interested reader is referred to the recent Engholm-Keller and Larsen review.26
Multiply phosphorylated peptides, however, remain challenging to detect and assign unambiguously. The combination of both IMAC and TiO2 enrichment methods, which is termed sequential elution from IMAC (SIMAC) enables efficient separations of monophosphorylated from multiply phosphorylated peptides and thus higher numbers of monophosphorylation site identifications.27 The use of graphite powder and titanium(IV) has been reported to increase the identification of multiply phosphorylated peptides.28,29 An alternative approach that holds promise to achieve higher phosphopeptide enrichment levels involve gallium complexes that stabilize the weak interaction between phosphoryl moiety and serine or threonine residues.30
Antibody-Based PTM Enrichment Methods
Global phosphorylation analysis revealed that 86%, 12%, and 2% of phosphorylation events occur on serine, threonine, and tyrosine, respectively.31 Although phosphotyrosine residues represent only a small percentage of all phosphosites the signal-initiating role of receptor tyrosine kinase (RTK) phosphorylation initiates key signaling cascades and plays a driving role in multiple diseases including cancer. To achieve higher identification coverage of phosphotyrosine-containing peptides, antibody-based enrichments in combination with LC-MS/MS analyses have been applied yielding quantitative profiling of hundreds of phosphorylated tyrosine residues.32,33
Antibody-based enrichment applications have been extended to quantitative profiling of ubiquitination, methylation, and acetylation. In particular, the development of antidiglycine-K antibodies led to the detection of more than 19 000 ubiquitination sites in a single proteomic workflow.12,34 Furthermore, antibodies targeting mono-, di-, and trimethylated lysine moieties and mono- and dimethyl arginine side chains have been applied to map the human methylome in depth.35
Enzymatic-Based PTM Enrichment
In addition to ionic interaction- or antibody-based enrichment strategies, enzymatic-based enrichments such as that, based upon application of the protein ligase, subtiligase, have been developed to study cellular substrates formed during intracellular proteolysis. During proteolytic cleavage, new free N-termini are generated, which are specifically biotinylated by the subtiligase, while native acetylated N-termini (present on almost 90% of human proteins) are not recognized. Subtiligase does not recognize ε-amino functions on lysine side chains. Upon labeling, avidin beads capture the biotinylated N-terminal amino functions of newly cleaved proteins. These labeled truncated proteins are digested with trypsin. The N-terminal peptide of the substrate is subsequently released from the avidin beads at the built in cleavage site by Tobacco etch virus (TEV) and analyzed by LC-MS/MS.36 This enrichment method allowed the identification of over 8000 proteolytic sites including more than 1700 caspase cleavages sites in human cells.37
Very recently, the wild type α-lytic protease was shown to cleave small ubiquitin-like modifier (SUMO) modified proteins to form a GG-K remnant, thus providing a new strategy for mapping of SUMO-modified proteins without the need for introduction of mutations.38 In addition, it has been reported that over 4000 SUMO sites have been uncovered using the decahistidine (His10)-tagged SUMO-2 strategy prior to MS measurement.13
Sequential Enrichment
The development of sequential enrichment strategies, which consist of using the flow-through of a first PTM enrichment step for a second or third enrichment step has become particularly desirable to reveal potential regulatory relationships from cross-talk among multiple PTMs from the same biological sample.8,39
In summary, enrichment strategies have been successfully established for proteome-wide identification of phosphorylation, ubiquitination, acetylation, methylation, proteolytic cleavages, SUMOylation, as well as lectin-based enrichment for O- and N-linked glycopeptides11 and iodoTMT-based S-nitrosylation enrichment.40 It is noted that neither analytical nor enrichment strategies exist yet for the vast majority of the other known PTMs.4
PTM Enrichment at the Protein Level
In contrast to peptide level approaches, direct PTM analysis of intact proteins preserves the intact structure of the protein that is mostly destroyed in peptide level approaches. Prior to MS analysis, intact proteins are separated from complex protein mixtures using a variety of enrichment techniques.41,42 These include gel-eluted liquid fraction entrapment electrophoresis, which separates proteins on the basis of their molecular weights, and LC techniques such as affinity, ion-exchange chromatography, size-exclusion chromatography, reverse phase chromatography, and online reversed phase LC tandem MS approach.14,43,44 Immunoaffinity methods can also be used as effective and specific protein purification protocols.45 Intact MS is a powerful technique to reveal global purity and relative stoichiometries and localize PTMs in highly modified but small proteins (10 to 50 kDa) without prior knowledge of targeted PTMs (Figure 1).41−43
Figure 1.

Overview of PTM analysis at the protein and peptide level. The principal steps for protein level PTM enrichment and MS analysis are represented on the left. Peptide level phosphorylation enrichment strategies are illustrated on the right. Isomeric isobars are proteins or peptides that may reveal the same amino acid sequence with equal numbers of PTMs but with different PTM configurations. The development of algorithms that search for non-redundant ions representing all possible PTM configurations enable the identification, quantification, and localization of all PTMs.
MS Fragmentation Strategies
In MS-based PTM analysis, it is essential to generate enough peptide fragmentation information (MS2 scans) for high confidence sequence identification and site localization of PTMs. Several fragmentation strategies including collision induced dissociation (CID), higher energy collisional dissociation (HCD), electron capture dissociation (ECD), and electron transfer dissociation (ETD) have been employed over the past decade for this purpose.
Collision Induced Dissociation
CID is the most common and widely applied unimolecular dissociation technique for MS-based proteome identification and quantification analysis. Under CID, the peptide or protein precursor ions are positively charged due to protonation of basic amino-acid residues and undergo collisions by interactions with neutral gas molecules (vibronic activation). In accordance with the mobile proton model,46 peptide bonds dissociate generating primary N-terminal b- and C-terminal y-type ions.47 The CID process is generally more effective for small and low-charge state peptides but is strongly influenced by the amino acid sequence and the distribution of the positive charges across the peptide backbone.
Higher Energy Collisional Dissociation
A similar strategy of fragmentation is the beam-type CID, also called HCD. HCD fragmentation is characterized by higher activation energy compared to CID. The higher fragmentation efficiency for HCD produces predominantly y-type fragment ions compared to b-type ions.48,49 In the HCD mode, Fourier transform detection in the Orbitrap analyzer results in better quality of MS2 mass spectra, but spectral acquisition times are longer compared to the greater acquisition speed recording CID spectra (MS/MS data collected in the ion trap).50,51 Both collisionally activated dissociation (CAD) methods, CID and HCD, are almost universal for analyses of stable PTMs and provide a high probability to generate and detect the modification-specific peptide sequence ion series.
Electron Capture and Electron Transfer Dissociations
In certain cases, however, CAD methods do not provide site-specific modification ions for long amino acid sequences. Moreover, the localization of the phosphorylation event within identified peptides that harbor more than one serine, threonine, or tyrosine is sometimes ambiguous. In addition, it has proven to be very challenging to decipher the complex histone patterns of modifications or assign PTMs that are labile in the gas phase using CAD energy deposition methods. For these cases, electron-based fragmentation methods such as ECD and ETD can be applied, achieving fragmentation through neutralization of backbone protonation sites with thermal electrons (ECD) or radical anions (ETD).14,17,52 The resulting nonergodic cleavages of N–Cα backbone bonds generate c- and z- type fragment ions without losing the PTM localization information.14,53 While ECD can only be implemented on Fourier transform ion cyclotron resonance (FTICR) MS instruments, ETD can be implemented on high resolution tandem MS instruments and is able to achieve higher detection sensitivity of labile PTM sites as well as complex PTM occupancies than with ECD-based approaches.17,54−56 ECD and ETD are complementary to CAD because they perform better with highly charged state analytes, whereas CAD is more efficient with low-charge state peptides.53,57 ECD and ETD, however, have major advantages over CAD for detecting unstable PTMs because peptide backbone fragmentation is virtually independent of the amino acid sequence, neutral losses such as phosphate groups are reduced, and O-GlcNAc elimination does not occur.8,17,57,58
HCF-1 is a transcriptional coregulator of cell proliferation and has been previously described as one of the most highly O-GlcNAcylated proteins. Nearly 30 HCF-1 O-GlcNAc sites have previously been reported.17 Using ETD and HCD, the site-specific localization of nearly 20 additional O-GlcNAc on HCF-1 could be identified with high confidence, allowing better understanding of the transcriptional regulating role of O-GlcNAc modification of HCF-1.17 In contrast, αB-crystalline has been found to be O-GlcNAcylated at only one serine residue.59O-GlcNActransferase (OGT) was found to be phosphorylated by Glycogen Synthase Kinase 3β (GSK3β), and the phosphorylation site has been localized to serine 3 or 4.60
Intact Protein Isoforms Profiling
Protein isoforms share a high percentage of amino acid sequence homology but often dramatically differ in their cellular concentration and biological roles.61 While MS on a digest mixture may not detect the peptide carrying a given particular isoform modification, intact protein level analysis has the advantage of mapping the complete amino acid sequence. In intact MS/MS analysis, precursor ions are conventionally selected by quadrupole or ion trap device before fragmentation. These methods, however, suffer from low mass selectivity. The stored waveform inverse Fourier transform (SWIFT) method enables high mass selectivity and achieves better isoform detection but can only be implemented on an FTICR instrument.62 Although FTICR mass spectrometers are expensive and less easily accessible than other mass spectrometers, traditionally, intact MS analysis has been most successful with FTICR-MS, since it provides the highest possible resolution for intact protein sample analysis.63,64 Early work combining ECD and the SWIFT technology on an FTICR-MS instrument has revealed the PTM occupancies of intact histone variants H2B.1 and H2B.2 from tetrahymena as well as novel H3 protein isoforms in rat.65,66
Dynamics-Quantitation
New MS designs have proven to provide powerful tools to quantify selected components of protein and PTM networks that provide new insight into cellular dynamics. The dynamics of histone modifications have been recently quantitatively established. Metabolic labeling of human cells using 13C glucose has been shown to enable monitoring of the dynamics of 13C-labeled acetyl groups’ incorporation on specific histone lysine. In this work, the turnover of acetylation was determined to be generally faster than methylation but slower than phosphorylation. Moreover, the modification rate varied depending on the histone type, targeted residues, as well as neighboring modifications.67
Caspase cleavage dynamics have also been quantitatively assessed by MS. Applying the N-termini subtiligase enrichment strategy, MS experiments across three human cell lines have revealed that the cellular cleavage kinetics of over 500 caspase substrates vary strongly between cell types and cytotoxic drug treatments. Furthermore, common caspase cleavage substrates that can be used to monitor the pro-apoptotic effects of cancer drug treatment have been identified.68
Uncovering PTM-Mediated Biological Processes
Arginine Methylation Initiates Smad Signaling
Phosphorylation is arguably the most common modification and a central mechanism for cell transduction. Indeed many transmembrane receptors are kinases or act through cytoplasmic kinase domains. In some cases, however, the low kinetics of substrate phosphorylation suggests a preceding step in the signal initiation. Recently, arginine methylation has been shown to activate the bone morphogenetic proteins (BMP) induced Smad-signaling pathway. Upon BMP binding inhibitory Smad6 is methylated by PRMT1. Smad6 in its unmethylated form dissociates from the BMP kinase receptor complex allowing activation of regulatory Smads through phosphorylation (Figure 2A).69 This observation raises the possibility that ligand-induced methylation may play a role in the activation of other signaling pathways.
Figure 2.
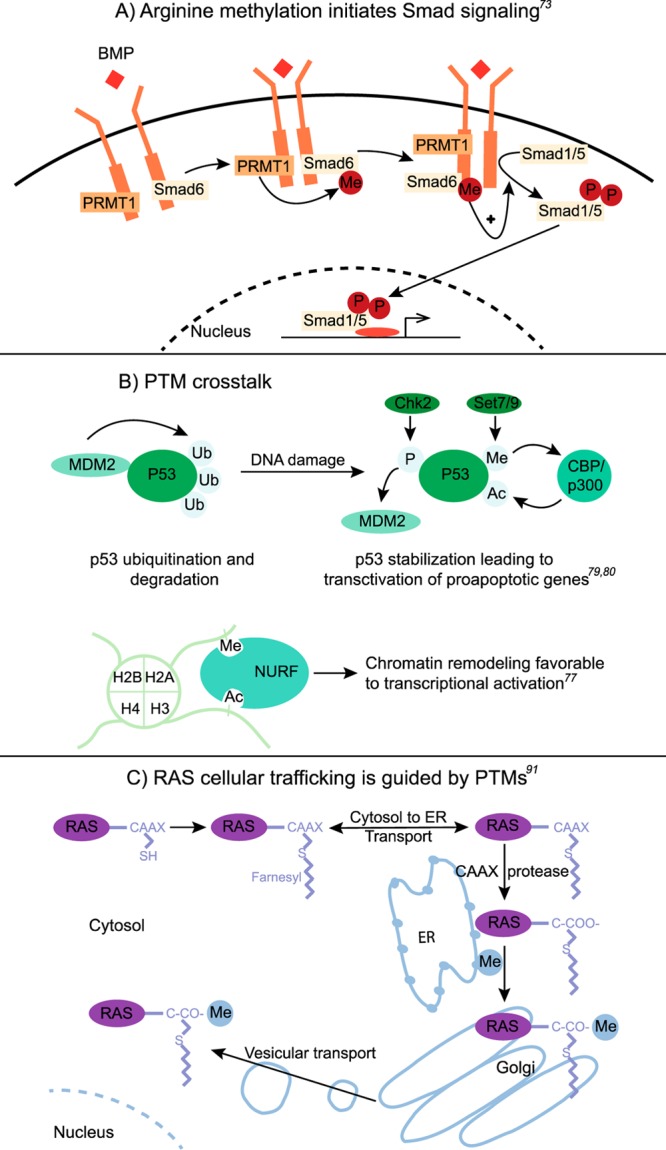
Examples of PTM-mediated biological processes. (A) Arginine methylation-mediated Smad signaling pathway. (B) p53 interaction with the CBP/p300 complex through methylation and acetylation exchange after DNA damage. P53 is subsequently stabilized and transactivates pro-apoptotic genes. The lower portion of panel B represents a protein-mediated histone crosstalk between methylation and acetylation, leading to chromatin remodeling favorable to transcriptional activation. (C) PTM-mediated cellular trafficking of RAS.
PTM Crosstalk
Since recent studies have provided a global view of the widespread occurrence of many PTMs, the next challenge is to understand the interplay among different PTM classes and specific sites of regulation. PTM-mediated crosstalk has been classified as positive or negative.70 In the case of positive crosstalk, one PTM serves as an active signal for the addition or removal of another PTM (e.g., phosphorylation dependent ubiquitination71 and SUMOylation72) or as a trigger for binding proteins that carry out a second modification (e.g., histone mark binding proteins73). In contrast, negative crosstalk can include direct competition of different modifications targeting the same amino acid or one modification can mask the recognition site for another PTM (e.g., the acetylation-mediated inhibition of kinase phosphorylation74).
Crosstalk between phosphorylation and ubiquitination is one of the most studied relationships. For example, phosphorylation can either promote or inhibit ubiquitination by regulating the activity of the E3 ubiquitin ligase controlling proteasomal degradation. For example, following DNA damage, p53 becomes phosphorylated, thus decreasing the interaction affinity with E3 ligase MDM2. P53 in its deubiquitinated form further interacts with the CBP/p300 complex through methylation and acetylation exchange. P53 is consequently stabilized and transactivates pro-apoptotic genes (Figure 2B).75,76 Crosstalk between phosphorylation and ubiquitination is also essential for the cancer-implicated epidermal growth factor (EGF)-mediated extracellular-signal-regulated kinases signaling pathway.71 Here, E3 ligase Cbl binds phosphotyrosine residues of the activated EGF receptor (EGFR). Subsequent proteasome-independent ubiquitination of EGFR and endocytic adaptor proteins mediate the EGFR internalization. Interestingly, deubiquitination directs EGFR through the recycling pathway back to the cell membrane.77
The histone code presents one of the most important examples for extensive interplay between different PTMs. ETD-based MS revealed several H3 lysine residues including lysine 9 (H3K9) that can be exclusively methylated or acetylated with different biological outcomes. While H3K9 methylation correlates with transcriptional repression, H3K9 acetylation induces transcriptional activation. H3K9 can be mono-, di-, or trimethylated with varying biological output depending on the degree of methylation.78 Other histone 3 crosstalk forms include the recently described acetylation-dependent SUMOylation.13 This new type of crosstalk is of particular interest, since histone SUMOylation has been previously associated with transcriptional repression, whereas histone acetylation is linked to transcriptional activation. Another form of crosstalk describes proteins that bind to different types of PTMs. For example, the nucleosome remodeling factor (NURF) contains two domains that bind the H3K4 di- or trimethylation and H4K16 acetylation sites, leading to the transcriptional activation of homeotic genes (Figure 2B).73 An additional form of histone crosstalk involves the phosphorylation of H3 (H3S10), which leads to the acetylation of H4. H3S10 creates a binding site for 14–3–3, a phosphoserine binding protein. 14–3–3 recruits a histone acetyltransferase MOF, which subsequently acetylates H4 on lysine 16. Acetylated H4K16 in turn forms a binding site for a kinase that phosphorylates RNA Polymerase II to facilitate transcriptional elongation of FOSL1, a gene activated in response to serum.79 Histone modifications can also prevent the recruitment of binding proteins. For example, the heterochromatin protein 1 (HP1) is not able to bind H3K9 when the adjacent serine 10 is phosphorylated during mitosis or gene activation.80 In general, the interplay among PTMs on histones appears to be context and time specific, increasing the challenge of understanding the regulated changes.
A few examples have shown that O-GlcNAcylation interacts with phosphorylation, acetylation, methylation, and ubiquitination.60,81−84 While the crosstalk between O-GlcNAcylation and phosphorylation has been hypothesized to be particularly important for multiple cellular processes, including the regulation of enzyme activity, cell division, and cytoskeletal functions,17,81,85 large-scale experiments revealed that only 7% of all O-GlcNAcylation sites were found to be phosphorylated also and the frequency of negative crosstalk between these two PTMs is virtually equal to the frequency expected by chance alone.8 Over the past decade, however, many particular examples of crosstalk have been described, showing that PTMs can work in concert to determine the final biological read-out.60,70,86
PTMs Regulate the Cellular Ras Trafficking
While the activation status of Ras proteins is regulated by the exchange of GTP, for GDP their cellular localization is mediated by PTMs. Ras traffics between various subcellular compartments guided by modifications such as farnesylation, proteolysis, methylation, palmitoylation, and phosphorylation. While farnesylation of Ras proteins increases its affinity toward the endoplasmic reticulum (ER), proteolysis and subsequent methylation and palmitoylation trap the two Ras isoforms HRas and NRas in the Golgi apparatus. Ras proteins are subsequently transported to the plasma membrane via vesicles (Figure 2C).87
These PTMs present potential therapeutic targets for the development of small molecule Ras inhibitors in cancer.
Bological Challenge of Defining Site-Specific Functions
The revolutionary development of PTM enrichment methods and new MS strategies has enabled the identification and quantification of more than 260 000 PTMs.23 Only a small proportion of PTM sites, however, have been associated with a particular biological function. One major challenge for the discovery of site-specific functions is to select a small number of sites from a large-scale data set for follow-up experiments. In general, any given PTMs are selected depending on the identification accuracy, reasonable stoichiometry, and their potential regulatory role in the process of interest. Evolutionary conservation usually points to important functionality and can therefore be used as a guide in selection criteria.88 Quantitative large scale MS is an additional powerful tool that can be employed to initially screen a subset of modified proteins that are regulated by the pathway of interest. For example, large-scale MS revealed that less than 15% of phosphorylated sites are modulated by EGF treatment.31
Functional follow-up experiments often include in vitro enzyme assays to determine specific enzyme substrates or lysine point mutations to investigate the functional role of a particular PTM. The core components of the Clustered Regulatory Interspaced Palindromic Repeats (CRISPR) system includes the Cas9 nuclease, which is able to create double-strand breaks in DNA and guide RNA (gRNA), which directs the CRISPR complex to a target sequence complementary to gRNA.89 Using CRISPR, point mutations have been generated in the genome of mice, which led to single amino acid substitutions in proteins of interest to probe site-specific PTM functions under in vivo conditions.90 Furthermore, the combination of CRISPR and Chromatin Affinity Purification (ChAP)-MS provides a new tool to study epigenetic regulation. Applying CRISPR-ChAP-MS, a specific section of chromatin can be purified for subsequent identification of associated histone PTMs and proteins by high resolution MS. Unlike Chromatin Immunoprecipitation (ChIP), this proteomic approach does not depend on prior knowledge of the targeted protein or PTM.91 Other useful MS follow up experiments rely on short hairpin RNA (shRNA) knockdown of specific regulators to investigate protein network interactions.92
MS-based proteomic measurements with additional conditions, such as targeted kinase or receptor inhibition, perturbing the PTMs of interest can also be used as functional follow-up experiments. For instance, large-scale proteomics experiments have elucidated the mTOR-regulated phosphoproteome93 and cellular outputs of the fibroblast growth factor receptor.94 In addition, MS analyses revealed new SUMOylated protein sites uncovering the regulatory role of SUMOylation in all nuclear processes.13 Despite these technological advances, follow up experiments are often limited to selected PTMs and/or sites on particular proteins and therefore would not reveal potential functionality for the majority of PTMs that can be characterized. Hence, it is reasonable to call into question the biological relevance of at least some fraction of the 260 000 reported human PTM sites.95
In summary, enrichment strategies and revolutionary advances in mass spectrometry have enabled rigorous identification and quantification of large numbers of PTMs, which certainly constitute the most complex regulatory networks in eukaryote cells. Future challenges for PTM proteomics include the optimization of PTM enrichment strategies and the development of PTM screens that are faster, more sensitive, and reproducible. Finally, the emerging importance of PTM multisite occupancies and potential modes of PTM crosstalk in proteins require the development of novel methods for full-spectrum PTM identification not only at the peptide level but also at the intact protein level.
Acknowledgments
Financial support was provided by National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS) grant 8P41GM103481 and the Adelson Medical Research Foundation. We thank R. J. Chalkley and F. Gnad for helpful input.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Consortium I. H. G. S. (2004) Finishing the euchromatic sequence of the human genome. Nature 431, 931–945. [DOI] [PubMed] [Google Scholar]
- Jensen O. N. (2004) Modification-specific proteomics: Characterization of post-translational modifications by mass spectrometry. Curr. Opin. Chem. Biol. 8, 33–41. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Gerstein M.; Snyder M. (2009) RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. T. (2006) Posttranslational Modification of Proteins: Expanding Nature’s Inventory, 1st ed., pp 1–490, Roberts and Company, Publishers, Greenwood Village, CO. [Google Scholar]
- Hebert A. S.; Richards A. L.; Bailey D. J.; Ulbrich A.; Coughlin E. E.; Westphall M. S.; Coon J. J. (2014) The one hour yeast proteome. Mol. Cell. Proteomics 13, 339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura H.; Sugiyama N.; Wakabayashi M.; Ishihama Y. (2014) Large-scale identification of phosphorylation sites for profiling protein kinase selectivity. J. Proteome Res. 13, 3410–3419. [DOI] [PubMed] [Google Scholar]
- Masuda T.; Sugiyama N.; Tomita M.; Ishihama Y. (2011) Microscale phosphoproteome analysis of 10 000 cells from human cancer cell lines. Anal. Chem. 83, 7698–7703. [DOI] [PubMed] [Google Scholar]
- Trinidad J. C.; Barkan D. T.; Gulledge B. F.; Thalhammer A.; Sali A.; Schoepfer R.; Burlingame A. L. (2012) Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol. Cell. Proteomics 11, 215–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J. V.; Vermeulen M.; Santamaria A.; Kumar C.; Miller M. L.; Jensen L. J.; Gnad F.; Cox J.; Jensen T. S.; Nigg E. A.; Brunak S.; Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3. 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- Choudhary C.; Kumar C.; Gnad F.; Nielsen M. L.; Rehman M.; Walther T. C.; Olsen J. V.; Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. [DOI] [PubMed] [Google Scholar]
- Zielinska D. F.; Gnad F.; Schropp K.; Wiśniewski J. R.; Mann M. (2012) Mapping N-glycosylation sites across seven evolutionarily distant species reveals a divergent substrate proteome despite a common core machinery. Mol. Cell 46, 542–548. [DOI] [PubMed] [Google Scholar]
- Kim W.; Bennett E. J.; Huttlin E. L.; Guo A.; Li J.; Possemato A.; Sowa M. E.; Rad R.; Rush J.; Comb M. J.; Harper J. W.; Gygi S. P. (2011) Systematic and quantitative assessment of the ubiquitin modified proteome. Mol. Cell 44, 325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks I. A.; D’Souza R. C. J.; Yang B.; Verlaan-de Vries M.; Mann M.; Vertegaal A. C. O. (2014) Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 21, 927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliuk S. M.; Maltby D.; Panning B.; Burlingame A. L. (2010) High resolution electron transfer dissociation studies of unfractionated intact histones from murine embryonic stem cells using on-line capillary LC separation. Mol. Cell. Proteomics 9, 824–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syka J. E. P.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. (2004) Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 101, 9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jünger M. A.; Aebersold R. (2014) Mass spectrometry-driven phosphoproteomics: Patterning the systems biology mosaic. Wiley Interdiscip. Rev. Dev. Biol. 3, 83–112. [DOI] [PubMed] [Google Scholar]
- Myers S. A.; Daou S.; Affar E. B.; Burlingame A. (2013) Electron transfer dissociation (ETD): The mass spectrometric breakthrough essential for O-GlcNAc protein site assignments—a study of the O-GlcNAcylated protein host cell factor C1. Proteomics 13, 982–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramström M.; Sandberg H. (2011) Characterization of γ-carboxylated tryptic peptides by collision-induced dissociation and electron transfer dissociation mass spectrometry. Eur. J. Mass Spectrom. 17, 497–506. [DOI] [PubMed] [Google Scholar]
- Moremen K. W.; Tiemeyer M.; Nairn A. V. (2012) Vertebrate protein glycosylation: Diversity, synthesis, and function. Nat. Rev. Mol. Cell Biol. 13, 448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X.; Yang K.; Gross R. W. (2012) Multi-dimensional mass spectrometry-based shotgun lipidomics and novel strategies for lipidomic analyses. Mass Spectrom. Rev. 31, 134–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J. V.; Ong S.-E.; Mann M. (2004) Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol. Cell. Proteomics 3, 608–614. [DOI] [PubMed] [Google Scholar]
- Gilmore J. M.; Kettenbach A. N.; Gerber S. A. (2012) Increasing phosphoproteomic coverage through sequential digestion by complementary proteases. Anal. Bioanal. Chem. 402, 711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- www.phosphosite.org.
- Gruhler A.; Olsen J. V.; Mohammed S.; Mortensen P.; Faergeman N. J.; Mann M.; Jensen O. N. (2005) Quantitative phosphoproteomics applied to the yeast pheromone signaling pathway. Mol. Cell. Proteomics 4, 310–327. [DOI] [PubMed] [Google Scholar]
- Pinkse M. W. H.; Uitto P. M.; Hilhorst M. J.; Ooms B.; Heck A. J. R. (2004) Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide precolumns. Anal. Chem. 76, 3935–3943. [DOI] [PubMed] [Google Scholar]
- Schmidt A.; Trentini D. B.; Spiess S.; Fuhrmann J.; Ammerer G.; Mechtler K.; Clausen T. (2014) Quantitative phosphoproteomics reveals the role of protein arginine phosphorylation in the bacterial stress response. Mol. Cell. Proteomics 13, 537–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engholm-Keller K.; Larsen M. R. (2013) Technologies and challenges in large-scale phosphoproteomics. Proteomics 13, 910–931. [DOI] [PubMed] [Google Scholar]
- Thingholm T. E.; Jensen O. N.; Robinson P. J.; Larsen M. R. (2008) SIMAC (Sequential Elution from IMAC), a phosphoproteomics strategy for the rapid separation of monophosphorylated from multiply phosphorylated peptides. Mol. Cell. Proteomics 7, 661–671. [DOI] [PubMed] [Google Scholar]
- Larsen M. R.; Graham M. E.; Robinson P. J.; Roepstorff P. (2004) Improved detection of hydrophilic phosphopeptides using graphite powder microcolumns and mass spectrometry: Evidence for in vivo doubly phosphorylated dynamin I and dynamin III. Mol. Cell. Proteomics 3, 456–465. [DOI] [PubMed] [Google Scholar]
- Zhou H.; Low T. Y.; Hennrich M. L.; van der Toorn H.; Schwend T.; Zou H.; Mohammed S.; Heck A. J. R. (2011) Enhancing the identification of phosphopeptides from putative basophilic kinase substrates using Ti (IV)-based IMAC enrichment. Mol. Cell. Proteomics 10, M110–006452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svane S.; Kryuchkov F.; Lennartson A.; McKenzie C. J.; Kjeldsen F. (2012) Overcoming the instability of gaseous peptide phosphate ester groups by dimetal protection. Angew. Chem., Int. Ed. Engl. 51, 3216–3219. [DOI] [PubMed] [Google Scholar]
- Olsen J. V.; Blagoev B.; Gnad F.; Macek B.; Kumar C.; Mortensen P.; Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648. [DOI] [PubMed] [Google Scholar]
- Rush J.; Moritz A.; Lee K. A.; Guo A.; Goss V. L.; Spek E. J.; Zhang H.; Zha X.-M.; Polakiewicz R. D.; Comb M. J. (2005) Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 23, 94–101. [DOI] [PubMed] [Google Scholar]
- Pighi C.; Gu T.-L.; Dalai I.; Barbi S.; Parolini C.; Bertolaso A.; Pedron S.; Parisi A.; Ren J.; Cecconi D.; Chilosi M.; Menestrina F.; Zamo A. (2011) Phospho-proteomic analysis of mantle cell lymphoma cells suggests a pro-survival role of B-cell receptor signaling. Cell. Oncol. (Dordr.) 34, 141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udeshi N. D.; Svinkina T.; Mertins P.; Kuhn E.; Mani D. R.; Qiao J. W.; Carr S. A. (2013) Refined preparation and use of anti-diglycine remnant (K-ε-GG) antibody enables routine quantification of 10 000s of ubiquitination sites in single proteomics experiments. Mol. Cell. Proteomics 12, 825–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson S. M.; Gozani O. (2014) Emerging technologies to map the protein methylome. J. Mol. Biol. 426, 3350–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara H. A. I.; Mahrus S.; Wells J. A. (2008) Tags for labeling protein N-termini with subtiligase for proteomics. Bioorg. Med. Chem. Lett. 18, 6000–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiita A. P.; Seaman J. E.; Wells J. A. (2014) Global analysis of cellular proteolysis by selective enzymatic labeling of protein N-termini. Methods Enzymol. 544, 327–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer J. G.; Yang B.; Bennett E.; Komives E. A. (2014) A novel comprehensive discovery approach for SUMO modified proteins. Mol. Cell. Proteomics 13, S41–S43. [Google Scholar]
- Mertins P.; Qiao J. W.; Patel J.; Udeshi N. D.; Clauser K. R.; Mani D. R.; Burgess M. W.; Gillette M. A.; Jaffe J. D.; Carr S. A. (2013) Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 10, 634–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christophorou M. A.; Castelo-Branco G.; Halley-Stott R. P.; Oliveria C. S.; Loos R.; Radzisheuskaya A.; Mowen K. A.; Bertone P.; Silva J. C. R.; Zernicka-Goetz M.; Nielsen M. L.; Gurdon J. B.; Kouzarides T. (2014) Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature 507, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeg M. A.; Davitz M. A. (1995) Glycosylphosphatidylinositol-phospholipase D: A tool for glycosylphosphatidylinositol structural analysis. Methods Enzymol. 250, 630–640. [DOI] [PubMed] [Google Scholar]
- Qu Z.; Meng F.; Bomgarden R. D.; Viner R. I.; Li J.; Rogers J. C.; Cheng J.; Greenlief C. M.; Cui J.; Lubahn D. B.; Sun G. Y.; Gu Z. (2014) Proteomic quantification and site-mapping of S-nitrosylated proteins using isobaric iodoTMT reagents. J. Proteome Res. 13, 3200–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y.; Rybakova I. N.; Xu Q.; Moss R. L. (2009) Top-down high-resolution mass spectrometry of cardiac myosin binding protein C revealed that truncation alters protein phosphorylation state. Proc. Natl. Acad. Sci. U.S.A. 106, 12658–12663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyne M. T.; Pesavento J. J.; Mizzen C. A.; Kelleher N. L. (2006) Precise characterization of human histones in the H2A gene family by top down mass spectrometry. J. Proteome Res. 5, 248–253. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Ge Y. (2011) Comprehensive analysis of protein modifications by top-down mass spectrometry. Circ.: Cardiovasc. Genet. 4, 711–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan S.; Burlingame A. L. (2010) Data processing algorithms for analysis of high resolution MSMS spectra of peptides with complex patterns of posttranslational modifications. Mol. Cell. Proteomics 9, 804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk J. C.; Li J.; Wang H.; Aletta J. M.; Qu J.; Read L. K. (2013) Proteomic analysis reveals diverse classes of arginine methylproteins in mitochondria of trypanosomes. Mol. Cell. Proteomics 12, 302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Dong X.; Hacker T. A.; Ge Y. (2010) Deciphering modifications in swine cardiac troponin I by top-down high-resolution tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 21, 940–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paizs B.; Suhai S. (2005) Fragmentation pathways of protonated peptides. Mass Spectrom. Rev. 24, 508–548. [DOI] [PubMed] [Google Scholar]
- Lau K. W.; Hart S. R.; Lynch J. A.; Wong S. C. C.; Hubbard S. J.; Gaskell S. J. (2009) Observations on the detection of b- and y-type ions in the collisionally activated decomposition spectra of protonated peptides. Rapid Commun. Mass Spectrom. 23, 1508–1514. [DOI] [PubMed] [Google Scholar]
- Frese C. K.; Altelaar A. F. M.; Hennrich M. L.; Nolting D.; Zeller M.; Griep-Raming J.; Heck A. J. R.; Mohammed S. (2011) Improved peptide identification by targeted fragmentation using CID, HCD, and ETD on an LTQ-Orbitrap Velos. J. Proteome Res. 10, 2377–2388. [DOI] [PubMed] [Google Scholar]
- Cui W.; Thompson M. S.; Reilly J. P. (2005) Pathways of peptide ion fragmentation induced by vacuum ultraviolet light. J. Am. Soc. Mass Spectrom. 16, 1384–1398. [DOI] [PubMed] [Google Scholar]
- Nagaraj N.; D’Souza R. C. J.; Cox J.; Olsen J. V.; Mann M. (2010) Feasibility of large-scale phosphoproteomics with higher energy collisional dissociation fragmentation. J. Proteome Res. 9, 6786–6794. [DOI] [PubMed] [Google Scholar]
- Jedrychowski M. P.; Huttlin E. L.; Haas W.; Sowa M. E.; Rad R.; Gygi S. P. (2011) Evaluation of HCD- and CID-type fragmentation within their respective detection platforms for murine phosphoproteomics. Mol. Cell. Proteomics 10(12), M111–009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young N. L.; DiMaggio P. A.; Plazas-Mayorca M. D.; Baliban R. C.; Floudas C. A.; Garcia B. A. (2009) High throughput characterization of combinatorial histone codes. Mol. Cell. Proteomic 8, 2266–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T.-Y.; McLuckey S. A. (2010) Gas-phase chemistry of multiply charged bioions in analytical mass spectrometry. Annu. Rev. Anal. Chem. 3, 365–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister G. C.; Berggren W. T.; Griep-Raming J.; Horning S.; Makarov A.; Phanstiel D.; Stafford G.; Swaney D. L.; Syka J. E. P.; Zabrouskov V.; Coon J. J. (2008) A proteomics grade electron transfer dissociation-enabled hybrid linear ion trap-orbitrap mass spectrometer. J. Proteome Res. 7, 3127–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobott F.; Watt S. J.; Smith J.; Edelmann M. J.; Kramer H. B.; Kessler B. M. (2009) Comparison of CID versus ETD-based MS/MS fragmentation for the analysis of protein ubiquitination. J. Am. Soc. Mass Spectrom. 20, 1652–1659. [DOI] [PubMed] [Google Scholar]
- Scott N. E.; Parker B. L.; Connolly A. M.; Paulech J.; Edwards A. V. G.; Crossett B.; Falconer L.; Kolarich D.; Djordjevic S. P.; Hojrup P.; Packer N. H.; Larsen M. R.; Cordwell S. J. (2011) Simultaneous glycan-peptide characterization using hydrophilic interaction chromatography and parallel fragmentation by CID, higher energy collisional dissociation, and electron transfer dissociation MS applied to the N-linked glycoproteome of Campylobacter jejuni. Mol. Cell. Proteomics 10(2), M000031–MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubarev R. A.; Horn D. M.; Fridriksson E. K.; Kelleher N. L.; Kruger N. A.; Lewis M. A.; Carpenter B. K.; McLafferty F. W. (2000) Electron capture dissociation for structural characterization of multiply charged protein cations. Anal. Chem. 72, 563–573. [DOI] [PubMed] [Google Scholar]
- Witze E. S.; Old W. M.; Resing K. A.; Ahn N. G. (2007) Mapping protein post-translational modifications with mass spectrometry. Nat. Methods 4, 798–806. [DOI] [PubMed] [Google Scholar]
- Chalkley R. J.; Burlingame A. L. (2001) Identification of GlcNAcylation sites of peptides and α-crystallin using Q-TOF mass spectrometry. J. Am. Soc. Mass Spectrom. 12, 1106–1113. [DOI] [PubMed] [Google Scholar]
- Kaasik K.; Kivimäe S.; Allen J. J.; Chalkley R. J.; Huang Y.; Baer K.; Kissel H.; Burlingame A. L.; Shokat K. M.; Ptáček L. J.; Fu Y.-H. (2013) Glucose sensor O-GlcNAcylation coordinates with phosphorylation to regulate circadian clock. Cell Metab. 17, 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stastna M.; Van Eyk J. E. (2012) Analysis of protein isoforms: Can we do it better?. Proteomics 12, 2937–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan S.; Burlingame A. L. (2010) High mass selectivity for top-down proteomics by application of SWIFT technology. J. Am. Soc. Mass Spectrom. 21, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall A. G.; Hendrickson C. L.; Jackson G. S. (1998) Fourier transform ion cyclotron resonance mass spectrometry: A primer. Mass Spectrom. Rev. 17, 1–35. [DOI] [PubMed] [Google Scholar]
- Tipton J. D.; Tran J. C.; Catherman A. D.; Ahlf D. R.; Durbin K. R.; Kelleher N. L. (2011) Analysis of intact protein isoforms by mass spectrometry. J. Biol. Chem. 286, 25451–25458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzihradszky K. F.; Zhang X.; Chalkley R. J.; Guan S.; McFarland M. A.; Chalmers M. J.; Marshall A. G.; Diaz R. L.; Allis C. D.; Burlingame A. L. (2004) Characterization of tetrahymena histone H2B variants and posttranslational populations by electron capture dissociation (ECD) Fourier transform ion cyclotron mass spectrometry (FT-ICR MS). Mol. Cell. Proteomics 3, 872–886. [DOI] [PubMed] [Google Scholar]
- Garcia B. A.; Thomas C. E.; Kelleher N. L.; Mizzen C. A. (2008) Tissue-specific expression and post-translational modification of histone H3 variants. J. Proteome Res. 7, 4225–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evertts A. G.; Zee B. M.; DiMaggio P. A.; Gonzales-Cope M.; Coller H. A.; Garcia B. A. (2013) Quantitative dynamics of the link between cellular metabolism and histone acetylation. J. Biol. Chem. 288, 12142–12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimbo K.; Hsu G. W.; Nguyen H.; Mahrus S.; Trinidad J. C.; Burlingame A. L.; Wells J. A. (2012) Quantitative profiling of caspase-cleaved substrates reveals different drug-induced and cell-type patterns in apoptosis. Proc. Natl. Acad. Sci. U.S.A. 109, 12432–12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Wang A. H.; Oses-Prieto J.; Makhijani K.; Katsuno Y.; Pei M.; Yan L.; Zheng Y. G.; Burlingame A.; Brückner K.; Derynck R. (2013) Arginine methylation initiates BMP-induced Smad signaling. Mol. Cell 51, 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T. (2007) The age of crosstalk: Phosphorylation, ubiquitination, and beyond. Mol. Cell 28, 730–738. [DOI] [PubMed] [Google Scholar]
- Nguyen L. K.; Kolch W.; Kholodenko B. N. (2013) When ubiquitination meets phosphorylation: A systems biology perspective of EGFR/MAPK signalling. Cell Commun. Signal. 11, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.; Kaneko S.; Li X. K.; Li X. (2014) The PI3K/Akt signal hyperactivates Eya1 via the SUMOylation pathway. Oncogene 1. 10.1038/onc.2014.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg A. J.; Li H.; Patel D. J.; David Allis C. (2007) Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell Biol. 8, 983–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Keitany G.; Li Y.; Wang Y.; Ball H. L.; Goldsmith E. J.; Orth K. (2006) Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312, 1211–1214. [DOI] [PubMed] [Google Scholar]
- Moll U. M.; Petrenko O. (2003) The MDM2-p53 interaction. Mol. Cancer Res. 1, 1001–1008. [PubMed] [Google Scholar]
- Coutts A. S.; Adams C. J.; La Thangue N. B. (2009) p53 ubiquitination by Mdm2: A never ending tail?. DNA Repair 8, 483–490. [DOI] [PubMed] [Google Scholar]
- Meijer I. M. J.; van Leeuwen J. E. M. (2011) ERBB2 is a target for USP8-mediated deubiquitination. Cell. Signal. 23, 458–467. [DOI] [PubMed] [Google Scholar]
- Latham J. A.; Dent S. Y. R. (2007) Cross-regulation of histone modifications. Nat. Struct. Mol. Biol. 14, 1017–1024. [DOI] [PubMed] [Google Scholar]
- Lee J.-S.; Smith E.; Shilatifard A. (2010) The language of histone crosstalk. Cell 142, 682–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W.; Tseng B. S.; Dormann H. L.; Ueberheide B. M.; Garcia B. A.; Shabanowitz J.; Hunt D. F.; Funabiki H.; Allis C. D. (2005) Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 438, 1116–1122. [DOI] [PubMed] [Google Scholar]
- Hart G. W.; Slawson C.; Ramirez-Correa G.; Lagerlof O. (2011) Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 80, 825–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison D. F.; Wamsley J. J.; Kumar M.; Li D.; Gray L. G.; Hart G. W.; Jones D. R.; Mayo M. W. (2012) Modification of RelA by O-linked N-acetylglucosamine links glucose metabolism to NF-KB acetylation and transcription. Proc. Natl. Acad. Sci. U.S.A. 109, 16888–16893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakabe K.; Hart G. W. (2010) O-GlcNAc transferase regulates mitotic chromatin dynamics. J. Biol. Chem. 285, 34460–34468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan H.-B.; Nie Y.; Yang X. (2013) Regulation of protein degradation by O-GlcNAcylation: Crosstalk with ubiquitination. Mol. Cell. Proteomics 12, 3489–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Hart G. W. (2014) O-GlcNAc profiling: From proteins to proteomes. Clin. Proteomics 11, 8. 10.1186/1559-0275-11-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venne A. S.; Kollipara L.; Zahedi R. P. (2014) The next level of complexity: Crosstalk of posttranslational modifications. Proteomics 14, 513–524. [DOI] [PubMed] [Google Scholar]
- Ahearn I. M.; Haigis K.; Bar-Sagi D.; Philips M. R. (2011) Regulating the regulator: Post-translational modification of Ras. Nat. Rev. Mol. Cell Biol. 13, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrao P.; Bork P.; Krogan N. J.; van Noort V. (2013) Evolution and functional cross-talk of protein post-translational modifications. Mol. Syst. Biol. 9, 714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P.; Esvelt K. M.; Church G. M. (2013) Cas9 as a versatile tool for engineering biology. Nat. Methods 10, 957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inui M.; Miyado M.; Igarashi M.; Tamano M.; Kubo A.; Yamashita S.; Asahara H.; Fukami M.; Takada S. (2014) Rapid generation of mouse models with defined point mutations by the CRISPR/Cas9 system. Sci. Rep. 4, 5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldrip Z. J.; Byrum S. D.; Storey A. J.; Gao J.; Byrd A. K.; Mackintosh S. G.; Wahls W. P.; Taverna S. D.; Raney K. D.; Tackett A. J. (2014) A CRISPR-based approach for proteomic analysis of a single genomic locus. Epigenetics 9, 1207–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y.; Zhang C.; Croucher D. R.; Soliman M. A.; St-Denis N.; Pasculescu A.; Taylor L.; Tate S. A.; Hardy W. R.; Colwill K.; Dai A. Y.; Bagshaw R.; Dennis J. W.; Gingras A.-C.; Daly R. J.; Pawson T. (2013) Temporal regulation of EGF signalling networks by the scaffold protein Shc1. Nature 499, 166–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P. P.; Kang S. A.; Rameseder J.; Zhang Y.; Ottina K. A.; Lim D.; Peterson T. R.; Choi Y.; Gray N. S.; Yaffe M. B.; Marto J. A.; Sabatini D. M. (2011) The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332, 1317–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francavilla C.; Rigbolt K. T. G.; Emdal K. B.; Carraro G.; Vernet E.; Bekker-Jensen D. B.; Streicher W.; Wikström M.; Sundström M.; Bellusci S.; Cavallaro U.; Blagoev B.; Olsen J. V. (2013) Functional proteomics defines the molecular switch underlying FGF receptor trafficking and cellular outputs. Mol. Cell 51, 707–722. [DOI] [PubMed] [Google Scholar]
- Lienhard G. E. (2008) Non-functional phosphorylations?. Trends Biochem. Sci. 33, 351–352. [DOI] [PubMed] [Google Scholar]