

ABSTRACT
We have previously shown that ORF45, an immediate-early and tegument protein of Kaposi's sarcoma-associated herpesvirus (KSHV), causes sustained activation of p90 ribosomal S6 kinases (RSKs) and extracellular regulated kinase (ERK) (E. Kuang, Q. Tang, G. G. Maul, and F. Zhu, J Virol 82:1838–1850, 2008, http://dx.doi.org/10.1128/JVI.02119-07). We now have identified the critical region of ORF45 that is involved in RSK interaction and activation. Alanine scanning mutagenesis of this region revealed that a single F66A point mutation abolished binding of ORF45 to RSK or ERK and, consequently, its ability to activate the kinases. We introduced the F66A mutation into BAC16 (a bacterial artificial chromosome clone containing the entire infectious KSHV genome), producing BAC16-45F66A. In parallel, we also repaired the mutation and obtained a revertant, BAC16-45A66F. The reconstitution of these mutants in iSLK cells demonstrated that the ORF45-F66A mutant failed to cause sustained ERK and RSK activation during lytic reactivation, resulting in dramatic differences in the phosphoproteomic profile between the wild-type virus-infected cells and the mutant virus-infected cells. ORF45 mutation or deletion also was accompanied by a noticeable decreased in viral gene expression during lytic reactivation. Consequently, the ORF45-F66A mutant produced significantly fewer infectious progeny virions than the wild type or the revertant. These results suggest a critical role for ORF45-mediated RSK activation in KSHV lytic replication.
IMPORTANCE KSHV is the causative agent of three human malignancies. KSHV pathogenesis is intimately linked to its ability to modulate the host cell microenvironment and to facilitate efficient production of progeny viral particles. We previously described the mechanism by which the KSHV lytic protein ORF45 activates the cellular kinases ERK and RSK. We now have mapped the critical region of ORF45 responsible for binding and activation of ERK/RSK to a single residue, F66. We mutated this amino acid of ORF45 (F66A) and introduced the mutation into a newly developed bacterial artificial chromosome containing the KSHV genome (BAC16). This system has provided us with a useful tool to characterize the functions of ORF45-activated RSK upon KSHV lytic reactivation. We show that viral gene expression and virion production are significantly reduced by F66A mutation, indicating a critical role for ORF45-activated RSK during KSHV lytic replication.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) is the causative agent of Kaposi's sarcoma (KS), the most common malignancy in HIV-infected individuals (1, 2). Besides KS, KSHV is associated with two lymphoproliferative diseases, primary effusion lymphoma and multicentric Castleman's disease (3, 4). KSHV belongs to the Gammaherpesvirinae subfamily in the Herpesviridae family and is closely related to rhesus rhadinovirus (RRV), herpesvirus saimiri (HVS), and murine gammaherpesvirus 68 (MHV-68) in the Rhadinovirus genus (γ2). Its closest relative in humans is Epstein-Barr virus (EBV), which belongs to the Lymphocryptovirus genus (γ1) in the same subfamily (5). Like other herpesviruses, KSHV exhibits two alternative life cycles: latent and lytic. KSHV primarily establishes latent infection both in vitro and in vivo, during which only a few genes are expressed and no progeny are produced. Under appropriate conditions, latent viral genomes can be reactivated to express the full panel of viral genes in a temporal cascade, beginning with immediate-early genes, followed by early genes, and then late genes (6–9). The successful completion of lytic replication ultimately results in the production and release of progeny viruses. Although lytic replication ultimately can lead to cell death, there is considerable clinical and laboratory evidence suggesting crucial roles of lytic replication in KS development (10, 11).
Open reading frame 45 (ORF45) of KSHV is a multifunctional protein with important roles throughout the lytic viral life cycle (12). Its expression begins at the immediate-early phase but increases over time, and the protein accumulates until the late phase, when it is abundantly assembled into the tegument layer of virions (6, 13, 14). Although ORF45 is conserved among gammaherpesviruses (no homologue in either alpha- or betaherpesviruses), the homology is limited and lies mostly at the termini of the protein. The ORF45 homologues also differ in their protein lengths. KSHV ORF45 (407 amino acids [aa]) is much bigger than its counterpart in RRV (353 aa) and almost double the size of those in MHV-68 (206 aa) and EBV (217 aa). KSHV ORF45 is involved in the evasion of the host innate antiviral immune responses by inhibiting interferon regulatory factor 7 (IRF7) (15–17). It also plays a role in the intracellular transport of newly formed viral particles by association with the kinesin-2 motor protein KIF3A (18). More recently, we found that ORF45 persistently activates the cellular kinases ERK (extracellular regulated kinase) and its substrate p90 ribosomal S6 kinases (RSKs) by forming complexes with them and shielding them from dephosphorylation (19, 20). We have shown that the activation of RSK is required for optimal lytic replication of KSHV, because either knockdown of RSK expression by siRNAs or inhibition of RSK kinase activity by chemical inhibitors reduced the level of lytic replication (19). We also demonstrated that ORF45 is responsible for the sustained activation of ERK and RSK signaling pathways during KSHV lytic replication and is required for optimal progeny virion production (19, 20). Although our previous studies have suggested that both RSK and ORF45 are required for optimal KSHV lytic replication, the specific roles of the ORF45-mediated activation of RSK remained to be defined precisely.
In the present study, we have now mapped the critical region of ORF45 that is involved in RSK interaction and activation. We further identified a single site mutation (F66A) in ORF45 that abolished its binding to RSK or ERK and its ability to activate the kinases. This mutant provided us with a unique tool for defining the role of ERK/RSK activation by ORF45 specifically during KSHV lytic replication. Using the recently developed BAC16 and iSLK cells, we demonstrated a critical role of ORF45-mediated RSK activation in KSHV lytic infection.
MATERIALS AND METHODS
Antibodies, chemicals, and plasmids.
Anti-RxxS*/T* antibody was ordered from Cell Signaling Technology (110B7E). Anti-RTA (ORF50) monoclonal mouse antibody, anti-PF8 (ORF59) rabbit polyclonal antibody, and anti-vIL6 rabbit polyclonal antibodies were given by Ke Lan, Robert Ricciardi, and John Nicholas, respectively. Anti-ORF26, anti-ORF62, anti-ORF33, anti-ORF52, and 8B8 anti-ORF45 monoclonal antibodies were generated by the Florida State University hybridoma facility. All other antibodies and chemicals used in this study have been described previously (12, 19–21). Plasmids pKH3-RSK2, pKH3-RSK1, pCR3.1-ORF45, full-length pCMV-ORF45, and derivatives have been described previously (19, 20, 22). Site-directed mutagenesis of ORF45 was performed by using a QuikChange mutagenesis kit (Stratagene, La Jolla, CA). Primer sequences used for cloning and mutagenesis are available upon request. All clones were verified by DNA sequencing.
Expression and preparation of GST-fusion proteins.
Escherichia coli BL21 cultures transformed with plasmids encoding glutathione S-transferase (GST) or GST-fusion proteins were induced with 100 μM isopropyl-β-d-thiogalactopyranoside (IPTG) for 3 h at room temperature. Cells were pelleted and resuspended in phosphate-buffered saline (PBS) plus lysozyme and protease inhibitors. The cell suspension was sonicated, and Triton X-100 was added to a final concentration of 1%. After 30 min of incubation at 4°C with gentle agitation, cell debris was removed by centrifugation at 10,000 × g for 10 min. The supernatant was incubated with glutathione agarose beads at 4°C overnight. After five washes with PBS, GST proteins were eluted with 10 mM glutathione in 50 mM Tris-HCl, pH 8.5. The eluates were dialyzed in buffer A150 containing 25 mM Tris, pH 7.5, 1 mM EDTA, 150 mM NaCl, 0.1% NP-40, and 10% glycerol. The protein concentration was determined with a bicinchoninic acid protein assay kit (Pierce Biotechnology, Inc., Rockford, IL). The purified GST proteins were divided into aliquots and stored at −80°C until use.
Cell culture and transfection.
HEK293 and HEK293T cells were cultured under 5% CO2 at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics. iSLK-puro cells were cultured in DMEM containing 10% FBS, 450 μg/ml G418, and 1 μg/ml puromycin as previously described (22, 23). Transient transfections were performed in 6-well plates with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) or 100-mm dishes with calcium phosphate methods.
Immunoprecipitation and Western blot analysis.
Immunoprecipitation and Western blot analysis were performed as previously described (19, 20, 22). For immunoprecipitation with anti-FLAG or anti-hemagglutinin (HA) antibodies, the cell lysates were incubated with EZview red anti-Flag M2 or anti-HA affinity resin for 4 h or overnight at 4°C. After washing with the lysis buffer and Tris-buffered saline (TBS), proteins were eluted by incubation with 150 μg/ml 3× Flag or HA peptide for 1 h at 4°C. For immunoprecipitation of ORF45 from iSLK.BAC16 cells, we used a monoclonal anti-ORF45 antibody (8B8) conjugated to CNBr-activated Sepharose 4B (GE Life Sciences). Clarified lysates were bound to the beads for 2 h at 4°C and washed three times each with lysis buffer and TBS, and bound complexes were eluted by boiling. For Western blotting, about 20 μg of proteins was resolved by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked in 5% dried milk in 1× phosphate-buffered saline plus 0.2% Tween 20 and then incubated with diluted primary antibodies for 2 h at room temperature or overnight at 4°C. Anti-rabbit, anti-rat, and anti-mouse IgG antibodies conjugated to horseradish peroxidase (Pierce) were used as the secondary antibodies. SuperSignal chemiluminescence reagents (Pierce) were used for detection.
Genetic manipulation of KSHV BAC16 genome.
The mutagenesis of BAC16 (24) was performed using a recombineering system as described by Tischer et al. (25, 26). In brief, the Kan/I-SceI cassettes were amplified from plasmid pEPKan-S by PCR with primers KS45-F66A-5′ (5′-GGACCCACGGTGATAGATATGTCTGCCCCAGACGACGTCGCCGCCGAGGACACGCCATCGCAGGATGACGACGATAAGTAGGG-3′) and KS45-F66A-3′ (5′-ATCCAGAGGGGTTGCTGGCGGCGATGGCGTGTCCTCGGCGGCGACGTCGTCTGGGGCAGACAGCCAGTGTTACAACCAATTAACC-3′) for the F66A mutant and KS45-3STOP-5′ (5′-GTCAACCCCGTACAAGGCCATGGCGATGTTTGTGAGGACCAAGCTTTGATTAATTGATCGTCTAGCACACACGATGAAGGATGACGACGATAAGTAGGG-3′) and KS45-3STOP-3′ (5′-CAATTGGAAGCATTCTCTCTTCATCGTGTGTGCTAGACGATCAATTAATCAAAGCTTGGTCCTCACAAACATCGCCAGCCAGTGTTACAACCAATTAACC-3′) for the Stop45 mutant. The purified PCR fragments were electroporated into BAC16-containing GS1783 cells (26) that had been induced at 42°C for 15 min. The recombinant clones were selected at 32°C on LB plates containing 34 μg/ml chloramphenicol and 50 μg/ml kanamycin and then analyzed by restriction enzyme digestion. Positive clones were cultured with 1% l-arabinose, induced at 42°C again, and plated on LB plates containing 1% l-arabinose for secondary recombination. Colonies which survived on the l-arabinose plates were replicated on plates with 34 μg/ml chloramphenicol alone and on plates with both 34 μg/ml chloramphenicol and 50 μg/ml kanamycin. The kanamycin-sensitive clones were analyzed by restriction enzyme digestion, and proper mutations were further confirmed by DNA sequencing.
To generate a revertant mutant, we replaced the mutant ORF45 with the wild-type sequence by a strategy similar to that described above. The Kan/I-SceI cassettes were amplified from plasmid pEPKan-S by PCR with primers KS45-A66F-5′ (5′-GGACCCACGGTGATAGATATGTCTGCCCCAGACGACGTCTTCGCCGAGGACACGCCATCGCAGGATGACGACGATAAGTAGGG-3′) and KS45-A66F-3′ (5′-ATCCAGAGGGGTTGCTGGCGGCGATGGCGTGTCCTCGGCGAAGACGTCGTCTGGGGCAGACAGCCAGTGTTACAACCAATTAACC-3′) for the A66F mutant and KS45-3STOP-R-5′ (5′-GTCAACCCCGTACAAGGCCATGGCGATGTTTGTGAGGACCTCGTCTAGCACACACGATGAAGGATGACGACGATAAGTAGGG-3′) and KS45-3STOP-R-3′ (5′-CAATTGGAAGCATTCTCTCTTCATCGTGTGTGCTAGACGAGGTCCTCACAAACATCGCCAGCCAGTGTTACAACCAATTAACC-3′) for the Stop45-rev mutant.
Reconstitution of recombinant KSHVs.
Briefly, iSLK cells seeded in a 24-well plate were transfected with 1 μg of BAC DNAs by Effectene (Qiagen). One day after transfection, cells were subcultured into a T150 flask with fresh medium containing 450 μg/ml G418 and 1 μg/ml puromycin. The next day, hygromycin was added to a final concentration of 500 μg/ml for selection. After 12 days of selection, hygromycin-resistant colonies were trypsinized, pooled, and subcultured at a 1:9 dilution every 3 days. To induce viral lytic replication, BAC-containing iSLK cells were seeded into 6-well plates or a T150 flask, and 1 day later (when cells reached ∼90% confluence) medium was replaced with fresh medium containing 2 μg/ml doxycycline and 1 mM butyrate.
Kinase inhibitor assays.
Two kinase inhibitors, the MEK inhibitor U0126 and the RSK inhibitor BI-D1870, were used in this study. iSLK.BAC16 cells were pretreated for 1 h with 10 μM the indicated inhibitor or dimethyl sulfoxide (DMSO) control and then induced as described previously in the presence of the inhibitor. Lysates were collected at 2 days postinduction (dpi) and analyzed by Western blotting. Media were collected at 4 days postinduction, and the viral genome copy number was quantified as described below.
qRT-PCR analysis of virion DNA.
The virion DNA was prepared as previously described (12, 21). The medium from induced BAC-iSLK cells was collected, centrifuged, and passed through a 0.45-μm filter to clear cell debris. Treatment of 200 μl of the cleared supernatant with 10 U of Turbo DNase (Ambion, Austin, TX) at 37°C for 1 h degraded extravirion DNA. The reaction was stopped by the addition of EDTA to a final concentration of 5 mM, followed by heat inactivation at 70°C. Twenty microliters of proteinase K solution and 200 μl of buffer AL from a DNeasy kit (Qiagen, Valencia, CA) then were added. The mixture was kept at 70°C for 15 min and then extracted with phenol-chloroform. The DNA was precipitated by the addition of 2 volumes of ethanol with glycogen as a carrier, and the DNA pellet was dissolved in 40 μl of Tris-EDTA buffer. Two microliters of DNA was used in SYBR green real-time quantitative PCR (qRT-PCR) analyses using the Bio-Rad CFX96 real-time detection system and C1000 thermal cycler. Thermal amplification was performed with the following parameters: 3 min at 95°C, and then 45 amplification cycles, each with denaturation (95°C for 10 s), annealing (60°C for 20 s), and extension (72°C for 15 s) periods. The cycle threshold (CT) value was determined as the point (cycle) at which the amplification plot crossed the threshold line. The threshold line was automatically set at 10 times the standard deviation of the baseline by the program. Viral DNA copy numbers were calculated with external standards of known concentrations of serially diluted BAC16 DNA.
KSHV real-time PCR array.
Whole-genomic KSHV real-time PCR arrays were performed as described previously (27, 28). Briefly, total RNA were reverse transcribed into cDNA with oligo(dT)20 primer using the Superscript III first-strand synthesis system (Invitrogen). The cDNA was used for SYBR green real-time PCR analyses under the conditions described above. The delta CT values were calculated for each sample and are represented as log2(fold change) in gene expression relative to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) internal control. The heat map of this array was generated using the pheatmap function in R (29, 30).
Virus stock preparation and infection.
Six T150 flasks of cells were induced for 5 days, and then medium was collected and centrifuged to remove cell debris. Virions were pelleted at 100,000 × g for 1 h on a 25% sucrose cushion with a Beckman SW28 rotor. The virus pellets were dissolved in 1/100 of the original volume of TNE buffer (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, and 1 mM EDTA) and stored at −80°C. The viral genome copy number was quantified by qRT-PCR. Infection was carried out as previously described (12, 21). Briefly, HEK293 cells plated in 24-well plates were incubated with 2-fold serial dilutions of concentrated virus plus Polybrene (4 μg/ml) and spun at 800 × g for 1 h at room temperature. The plates were incubated at 37°C for 2 h, and the inocula then were removed and replaced with fresh medium with 5% FBS. At 24 h postinduction, cells were washed twice and resuspended in PBS, and then green fluorescent protein (GFP) expression was measured using a BD FACSCanto analyzer.
RESULTS
Identification of aa 56 to 70 of KSHV ORF45 as a region critical for binding and activation of RSK.
We have shown previously that the N-terminal aa 1 to 115 of KSHV ORF45 is sufficient for binding and activation of RSK (19). To narrow down the RSK-binding domain further, we divided this region into three fragments (aa 1 to 50, aa 51 to 90, and aa 78 to 115) and fused each with GST to produce fusion proteins. Using pulldown assays, we found that only the fragment of aa 50 to 90 interacted with RSK, while the aa 1 to 50 and aa 77 to 115 fragments did not (Fig. 1A). To identify the critical residues, we generated a series of 5-aa internal deletion mutants of Flag-ORF45 (1-115) and examined their abilities to bind to HA-RSK1. Coimmunoprecipitation assays revealed that deletion of the aa 56 to 60, aa 61 to 65, or aa 66 to 70 region of ORF45 completely abolished its ability to bind to RSK (Fig. 1B, top). Furthermore, in vitro kinase assays of the immunoprecipitated HA-RSK revealed that these three mutants were unable to stimulate RSK kinase activity in cells (Fig. 1B, bottom). These results demonstrate that the region of aa 55 to 70 of KSHV ORF45 is critical for its binding and activation of RSK.
FIG 1.

Mapping of critical residues in KSHV ORF45 for its binding and activation of RSK. (A) The region between aa 50 and 90 of KSHV ORF45 is sufficient for binding to RSK. GST-fused ORF45 protein fragments were mixed with lysates of HEK293 cells transfected with HA-RSK1. After incubation for 2 h, equal portions of the mixtures were subjected to immunoaffinity purification with either glutathione beads or anti-HA affinity resins. The eluted samples were analyzed by immunoblotting with the designated antibodies. (B) The region between aa 56 and 70 of KSHV ORF45 is essential for binding to RSK. HEK293 cells were cotransfected with constructs expressing HA-RSK1 and Flag-ORF45 1-115 or a series of 5-aa deletion mutants in the region of aa 50 to 95. Half of the lysates of the transfected cells were immunoprecipitated (IP) with anti-Flag affinity resin and then analyzed by immunoblotting with anti-Flag and anti-HA antibodies. HA-RSK1 was immunoprecipitated from another half of the lysates with anti-HA affinity resins and used for in vitro kinase assays using GST-S6 as the substrate. (C) Alignment of sequences at aa 51 to 75 of KSHV ORF45 and corresponding homologues in RRV (AAD21372), HVS (CAA45668), and MHV-68 (murine herpesvirus 68; AAB66440). MHV-68 contains two similar motifs. (D) F66 (phenylalanine) in KSHV ORF45 is critical for binding to and activation of RSK. Residues in the conserved motif region of aa 51 to 76 of Flag-ORF45 (1-115) were mutated to alanine as indicated. The point mutation constructs were cotransfected with HA-RSK into HEK293 cells. After serum starvation, cell lysates were prepared and immunoprecipitated with anti-HA or anti-Flag resins. The whole-cell lysates (WCL) and immunocomplexes were analyzed by Western blotting (WB) with antibodies as indicated. (E) The wild type and F66A mutant in the full-length ORF45 expression constructs were analyzed by immunoprecipitation and Western blotting as described for panel D.
Crucial role of F66 of KSHV ORF45 in binding and activation of RSK.
Alignment of ORF45 sequences of gammaherpesviruses revealed limited homology in the aa 56 to 70 region (Fig. 1C). Interestingly, the homology seems to be shared by members of the genus Rhadinovirus (γ2) but not Lymphocryptovirus (γ1). For example, the homologous core is not found in BKRF4 of EBV, which belongs to γ1. We changed the conserved residues in this region individually to alanine and found that a change of phenylalanine 66 (F66) to alanine completely abolished the binding of ORF45 to RSK or ERK and ORF45's ability to activate the kinases (Fig. 1D). Changes of other residues had little effect, except D63A mutation, which reduced the level of RSK activation slightly (Fig. 1D). When the F66A mutation was introduced into the full-length ORF45, the mutation also abolished ORF45's interaction with RSK and ERK (Fig. 1E, lane 3, top and middle), as well as its ability to activate RSK and ERK (Fig. 1E, lane 3, bottom). In contrast, a D69A mutation in full-length ORF45 had no apparent effect on its binding or activation of RSK. These results suggest that the F66 residue of KSHV ORF45 is critical for binding and activation of RSK.
Generation of ORF45-F66A and ORF45-null mutants in BAC16, a newly developed and infectious bacterial artificial chromosomal clone of KSHV genome.
Because the F66A mutation prevented ORF45 from binding and activating RSK, it provided us an excellent tool for defining the roles of ORF45-mediated activation of RSK in the KSHV life cycle. We introduced this mutation into BAC16, a highly infectious BAC clone derived from KSHV.219 (24, 31), using two-step lambda Red-mediated seamless recombination (25, 26) (Fig. 2A). We also generated an ORF45-null mutant in which premature triple stop codon mutations were created after the eighth codon (Fig. 2A). To ensure any phenotypic changes indeed were caused by designed mutations rather than unintended ones, we restored the F66A and Stop45 mutations to the original wild-type sequence, producing revertant mutations (Fig. 2A). Putative mutant BAC clones were analyzed by digestion with restriction enzymes KpnI and HindIII. Representative results are shown in Fig. 2B. Because a HindIII site was introduced with the premature triple stop codons, digestion of the Stop45 mutant with HindIII yielded predicted fragments of 2.5 and 3.3 kb rather than 5.8 kb in the wild type (Fig. 2B, marked by arrows). Digestion of Stop45-rev yielded a pattern identical to that of the wild type, indicating successful repair of the mutation to the wild-type sequence. As expected, F66A mutation caused no change in digestion pattern of either KpnI or HindIII. Sequencing of the genomic region further confirmed all mutants had the desired sequences (Fig. 2C).
FIG 2.

Construction and analysis of KSHV BAC16 mutants. (A) Schematic diagram of ORF45 and surrounding ORFs in the wild-type BAC or mutants. The nucleotide sequences refer to GenBank accession number U75698. For the F66A mutant, the 66th codon, TTC for phenylalanine (F), was changed to GGC for alanine. For the Stop45 mutant, a triple stop codon was inserted after the eighth codon. For easy identification, a HindIII site also was introduced. (B) Gel electrophoresis of KpnI- or HindIII-digested wild-type and mutant BAC DNA. F66A, Stop45, and their revertants were constructed as described in Materials and Methods. The red arrows indicate the predicted changes of restriction patterns caused by the introduced HindIII site in the Stop45 mutant. (C) Sequences of the wild-type and mutant BACs at the ORF45 locus. The sequence chromatogram and deduced amino acids are shown. The designed mutations are boxed or underlined.
Binding to and sustained activation of RSK and ERK is abolished by ORF45-F66A mutation.
F66A, Stop45, their revertants, and wild-type BAC16 DNA were transfected into iSLK cells which express RTA under a doxycycline-inducible promoter (23). After hygromycin selection of BAC DNA-transfected cells, comparable levels of GFP expression and viral DNA were found in iSLK cells harboring the different KSHV-BACs (data not shown). Upon doxycycline and butyrate treatment, RTA is expressed to initiate the lytic replication program that results in expression of lytic genes and ultimately the release of infectious viral particles (23). To verify that the ORF45 wild type, but not the F66A mutant, binds to ERK and RSK in the context of KSHV lytic replication, we induced iSLK.BAC16 F66A or the revertant (F66A-rev), harvested cells at 2 dpi, and immunoprecipitated lysates with a monoclonal anti-ORF45 antibody. As expected, both ERK and RSK interacted with wild-type ORF45 during lytic reactivation, and their binding was almost completely abrogated by F66A mutation (Fig. 3A; compare lanes 5 and 6). Significantly, the majority of total cellular RSK1 appears to form complexes with ORF45, as evidenced by the reduction of RSK1 signal in the flowthrough. We next investigated the effect of ORF45 mutation or deletion on the activation of ERK and RSK throughout a time course of lytic reactivation. The levels of pRSK and pERK increased dramatically and reached a maximum at 2 dpi in the wild-type- and revertant-infected cells. In contrast, the levels of pRSK and pERK barely increased upon lytic reactivation in the BAC16-45F66A or ORF45-null BAC16-Stop45 mutant, confirming critical roles of ORF45 and the F66 residue in sustained activation of RSK and ERK during KSHV lytic replication (Fig. 3B).
FIG 3.

Binding and sustained activation of RSK and ERK is abolished by ORF45-F66A mutation. (A) Wild-type ORF45 but not the F66A mutant binds with ERK and RSK during KSHV lytic replication. Lysates from uninduced (U) or induced (I) iSLK cells carrying BAC16 F66A or the revertant were immunoprecipitated using an anti-ORF45 antibody. Input, eluate (IP), and flowthrough (FT) fractions were immunoblotted with the indicated antibodies. (B) Sustained activation of RSK and ERK is abolished by F66A or ORF45 null mutation. Stable iSLK cells carrying wild-type BAC16, F66A, Stop45, or revertant viral genomes were induced with doxycycline and sodium butyrate. Lysates of cells collected at different times after induction were analyzed with antibodies as indicated. (C) The ORF45-F66A mutation resulted in significant changes in the phosphoproteomic profile of infected cells. The lysates shown in panel B were immunoblotted with an antibody that recognizes the RxxS*/T* motif (Cell Signaling Technology). (D) Pharmacological inhibition of RSK reduces phosphorylation at the RxxS*/T* motif. iSLK.16 F66A-rev cells were left untreated or were pretreated for 1 h with the indicated kinase inhibitor. Cells were induced as described previously, and lysates were collected at 2 dpi and probed with the same antibody as that used for panel C.
RSKs phosphorylate protein substrates at serine or threonine residues within the motif RxRxxS/T (R, arginine; S, serine; T, threonine; x, any amino acid), identical to the well-characterized AKT phosphorylation motif (32–34). Therefore, we used a commercially available motif-specific antibody to determine the impact of ORF45-F66A mutation on gross phosphorylation of putative RSK substrates in KSHV-infected cells. As shown in Fig. 3C, phosphorylation detected by this antibody dramatically increased over time, peaking at 2 dpi, but was essentially abolished by the ORF45-F66A or Stop45 mutation (Fig. 3C). Furthermore, pharmacological inhibition of RSK (BI-D1870) but not the upstream MEK (U0126) reduced phosphorylation at the putative RSK phosphorylation motif in a manner similar to that observed due to ORF45 mutation/deletion (Fig. 3D). These results not only confirmed critical roles of ORF45-activated RSK during KSHV lytic replication but also indicated widespread downstream effects of this activation on viral/cellular protein phosphorylation.
Effect of ORF45-F66A mutation on viral gene expression.
We first examined the expression of several viral proteins over time by Western blotting (Fig. 4). No significant differences were noticed for expression of RTA (ORF50), PF8 (ORF59), and vIL6 by the late stage of lytic replication, although small differences were apparent at 24 h. However, the expression of some late lytic genes, such as ORF65 (capsid), ORF26 (capsid), ORF52 (tegument), and ORF33 (tegument), obviously was lower in F66A and Stop45 mutants than in revertant mutants.
FIG 4.
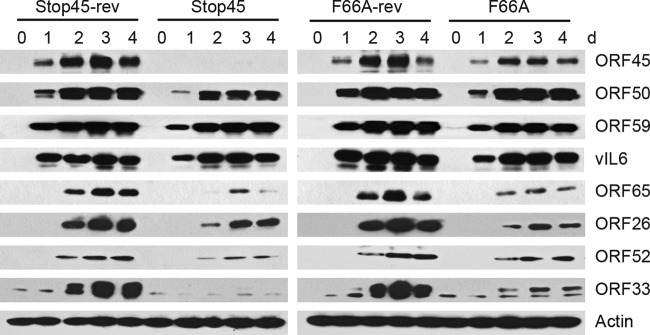
Viral protein expression of wild-type BAC16 and ORF45 mutants in iSLK cells. The lysates described in the legend to Fig. 3 were analyzed by Western blotting using anti-ORF50, anti-ORF45, anti-ORF59, anti-vIL6, anti-ORF52, anti-ORF26, anti-ORF65, and anti-ORF33 antibodies as indicated.
To assess the impact of ORF45-F66A mutation on KSHV gene expression at the genomic level, we analyzed the transcription of all KSHV ORFs by qRT-PCR array (27, 28, 35). The results suggest that the expression of a subset of KSHV genes are affected by ORF45-F66A or Stop45 mutation (Fig. 5). Many of the most affected ones appear to be late genes involved in virion assembly (i.e., tegument and capsid proteins), indicating roles for the ORF45/RSK axis in the regulation of viral late gene expression. Interestingly, some of these genes appear to be clustered in the region between ORF19 and ORF44, which has been shown to exhibit features of heterochromatin and to be bound by repressive histone marks during latency (36, 37). For a subset of genes, qRT-PCR analysis was repeated with samples collected in triplicate from the indicated cell lines at 48 hpi (Fig. 6). These data are consistent with the qRT-PCR array, in that while ORF45 mutation or deletion does not significantly affect the expression of latent or early lytic genes, it does dramatically reduce the expression of several late genes, including ORF25, ORF26, and ORF33.
FIG 5.

Transcriptional profile of wild-type BAC16 and ORF45 mutants in iSLK cells. Stable iSLK cells carrying wild-type BAC16, F66A, F66A-rev, or Stop45 viral genomes were induced with doxycycline and sodium butyrate for 48 h. Total RNA was extracted, reverse-transcribed into cDNA, and used for KSHV whole-genome qPCR array analysis. The delta CT values for each primer set were calculated and converted to a heat map using R. ORFK1 was omitted because the primers failed to amplify significant product. IE, immediate early; E, early; L, late; −, unknown expression kinetics.
FIG 6.
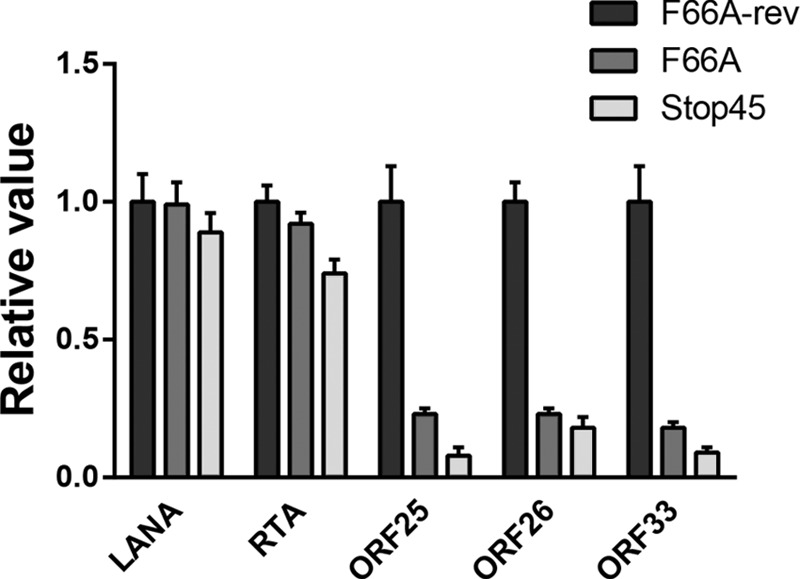
Late gene expression is compromised by ORF45 mutation or deletion. For a subset of genes, qRT-PCR of the indicated sample at 48 hpi was repeated with 3 biological replicates, and the fold change induction of gene expression was calculated, normalized to GAPDH, and represented as gene expression relative to that of the F66A-rev mutant.
Several KSHV genes which have been shown to activate the ERK mitogen-activated protein kinase (MAPK) pathway, including gB (ORF8) (38), K8.1 (38), and K15 (39), exhibit reduced expression in the F66A or Stop45 mutant to varied extents (Fig. 5). Therefore, we need to be cautious not to overemphasize the functional significance of ORF45 as a direct critical factor for RSK activation. Although our data suggest a major role for ORF45 in the activation of ERK and RSK, there could be indirect mechanisms contributing to this activation via the ORF45-dependent expression of other KSHV genes.
ORF45 F66A mutation impairs progeny virion production.
The extracellular viruses were collected daily from the culture media of induced iSLK.BAC16 cells (WT and mutants), and viral genomic copy in the medium was determined by real-time qPCR as described previously (12, 21). As shown in Fig. 7A, the F66A mutant produced about 10-fold fewer viruses and the ORF45-null mutant produced 30- to 50-fold fewer viruses than the wild-type or revertant viruses. Notably, MEK or RSK inhibition also significantly impaired progeny virion production, confirming a role for the MAPK signaling pathway in efficient lytic replication (Fig. 7B). The extracellular wild-type and ORF45 mutant viruses were concentrated by ultracentrifugation and normalized based on the viral genomic DNA copies. The equality of viral particles was confirmed by similar levels of capsid protein ORF26 and tegument proteins ORF33 and ORF52 (Fig. 7C). We infected HEK293 cells with a series of 2-fold dilutions of these progeny viruses. The GFP marker on BAC16 facilitated the counting of infected cells by flow cytometry analysis (Fig. 7D). At equivalent dilutions, the infection rate by ORF45-null virus was at least an order of magnitude lower than those of the wild-type and revertant viruses, while infectivity of the F66A mutant was only slightly reduced (Fig. 7D). Interestingly, although similar percentages of cells were scored as GFP positive, the intensity of GFP was lower in BAC16-45-F66A-infected cells than in the wild type and revertants at equivalent dilutions (Fig. 7E and F). The effect became more dramatic at higher dilutions (lower MOI), and GFP intensity became close to that of ORF45-null mutant-infected cells. These results suggested that the expression of genes from latent episomes is reduced by the loss of ORF45 and its ability to activate RSK, particularly at lower MOI. Collectively, these results suggest important functions of ORF45 and its activation of RSK in the production of infectious virions upon KSHV lytic reactivation.
FIG 7.

Production of infectious viruses is compromised by ORF45 mutation or deletion. (A) Growth curves for recombinant KSHV. Stable iSLK cells carrying wild-type BAC16, F66A, Stop45, or revertant viral genomes were induced with doxycycline and sodium butyrate. Supernatants of induced medium were collected at different time points as indicated. Viral genome copies in the medium were determined by real-time PCR and were plotted against the time since induction. (B) iSLK.BAC16 cells were pretreated for 1 h with the indicated kinase inhibitor, followed by induction of lytic reactivation in the presence of inhibitor. Viral genome copies were measured at 96 hpi as described for panel A. (C) Extracellular virions were collected and concentrated from the media of induced iSLK.BAC16 wild-type and ORF45 mutant cell lines as outlined in Materials and Methods. After normalizing by viral genome copy number, virion composition was analyzed by Western blotting with the indicated antibodies. (D to F) 293 cells were infected with 2-fold serial dilutions of wild-type and ORF45 mutant KSHV. Twenty-four h postinduction, GFP-positive cells were counted by fluorescence-activated cell sorting (FACS). The percentages of GFP-positive cells (D) and GFP median values (E) were calculated for each dilution. (F) Representative histograms of the FACS data.
DISCUSSION
Crucial roles of ORF45-mediated activation of RSK during KSHV lytic replication.
In the work reported here, we have precisely mapped the domain involved in RSK interaction and activation to a small region (∼15 aa) near the N terminus of KSHV ORF45. We further identified a single F66A point mutation that abolished ORF45's binding to RSK and ERK and, consequently, its ability to activate the kinases. We engineered a mutant virus carrying this mutation in the genome of KSHV and performed functional analyses of this mutant in iSLK cells. Our studies took advantage of recent technological advances in the field, including (i) BAC16, an infectious bacterial artificial chromosome clone of the KSHV genome, which yields much higher titer of viruses than the previously used BAC36 (24); (ii) iSLK, a cell line in which RTA is expressed under an inducible promoter that permits efficient induction of KSHV lytic replication by doxycycline (23), thereby avoiding use of TPA, a pleotropic agent known to induce ERK signaling, which would complicate our analyses; and (iii) a two-step RED-mediated recombination technique that allows seamless manipulation of the cloned KSHV BAC genome in E. coli (25, 26). These tools permitted us to introduce the ORF45-F66A mutation into the KSHV genome and produce a recombinant virus.
Analyses of this mutant revealed its significant deficiency in RSK activation and in production of infectious progeny viruses upon KSHV lytic reactivation. Therefore, our results demonstrate the importance of ORF45-mediated RSK activation during KSHV lytic infection. Although other KSHV lytic proteins, such as gB, K8.1, and vGPCR, as well as latent proteins kaposin and LANA, have been reported to activate the ERK MAPK pathway (38, 40–42), most of them were only assessed under ectopically overexpressed conditions. Moreover, none of them have been studied in the context of viral infection, let alone evaluated by loss-of-function reverse genetic analysis. Because some of these genes appear to be differentially expressed upon ORF45 mutation or deletion (Fig. 5), we cannot exclude the possibility that their activation of the ERK MAPK pathway also is impaired, thereby amplifying the defect in RSK activation associated with the F66A/Stop45 phenotype. However, our results argue for a dominant role of ORF45 in sustained activation of RSK during KSHV lytic replication.
Rewiring of kinase signaling by ORF45 during KSHV lytic replication.
As obligate intracellular parasites, viruses must modulate the host intracellular environment. Although ORF45 has no enzymatic activity, its association with RSK and ERK causes a significant increase of their enzymatic activity and duration, resulting in drastic changes of protein phosphorylation profiles in cells. Phosphorylation of proteins can regulate diverse functions in cells. By rewiring the RSK and ERK signaling pathways to phosphorylate specific protein substrates, ORF45 may exert distinct functions during KSHV lytic replication by altering the repertoire of proteins phosphorylated by these kinases.
Although we have previously demonstrated that KSHV ORF45 increases RSK and ERK activities by protecting the active kinases from dephosphorylation, the detailed mechanism remained to be elucidated. We now have identified the critical region of ORF45 involved in RSK binding and activation. Although this small region is conserved among γ2 herpesviruses, ORF45 homologues seem to differ in their abilities to activate RSK and ERK (E. Kuang and F. Zhu, unpublished data), suggesting additional regions are involved. Recent studies by other groups have confirmed critical roles of the ERK pathway in MHV-68 and RRV lytic infection cycles that are at least partially dependent on ORF45 (43–45). More studies are needed to elucidate the molecular mechanisms by which ORF45 causes persistent activation of RSK and ERK. Another interesting question is how ORF45 homologues have evolved to acquire their abilities to rewire cellular kinase signaling. The significant expansion of protein length and distinct subcellular localizations of KSHV ORF45 may increase its capacity to recruit specific substrates to these kinases; therefore, they could allow it to attain unique functions not shared by other gammaherpesviruses. The identification of the ORF45 interactome and specific substrates of ORF45-activated kinases will facilitate a better understanding of these functions.
Multiple functions of ORF45 and its mediated RSK activation in KSHV life cycle.
Our previous reverse genetic studies using ORF45-null BAC36 in 293T cells suggested roles for ORF45 in both early and late stages of KSHV lytic replication, although the detailed functions remained to be determined (12). Indeed, KSHV ORF45 has been known to be involved in the evasion of innate immune responses and in the intracellular transportation of newly assembled virions (13, 18, 46). Rewiring of the ERK/RSK signaling pathway enables ORF45 to modulate the host cellular environment to a great extent through phosphorylation of a distinct repertoire of protein substrates. We recently identified a prominent cytoplasmic substrate, eIF4B, a translation initiation factor, as a specific substrate of ORF45-activated RSK, revealing a role for ORF45 in translational regulation (22). Here, we show evidence that ORF45 also is involved in transcriptional regulation, because the transcription of a cluster of late genes surrounding ORF25 was depressed in BAC16-45F66A- and BAC16-Stop45-infected cells. Interestingly, this region appeared to be bound by repressive histone modifications during latency, but the epigenetic landscape changes significantly during KSHV lytic replication (36, 37). These results suggest that ORF45 and its mediated activation of the ERK/RSK signaling pathway affect the expression of a subset of viral genes through the regulation of epigenetic modifications. ERK and RSK have been shown to be involved in transcriptional regulation by phosphorylation of transcription factors, histones, and histone-modifying enzymes (32). Furthermore, a role for ORF45 in transcriptional regulation is supported by the recent report from Glaunsinger's group that ORF45 potently increases HIV long terminal repeat (LTR) transcription activity in an RSK2-dependent manner (47). Because of its role in transcriptional and translational regulation, ORF45 could have a broad impact on the expression of not only viral but also cellular genes. The biological consequences of such regulations on cellular genes remain to be assessed. Comprehensive identification of substrates specified by ORF45 will be crucial for understanding its roles throughout KSHV lytic replication.
In summary, KSHV ORF45 possesses regulatory functions at multiple levels: epigenetics, transcription, translation, and posttranslational modification. Further studies should employ systems biology approaches to examine the epigenome, transcriptome, proteome, and phosphoproteome by both gain-of-function and loss-of-function analyses. The F66A mutant will be a great tool for these studies.
The lower infection rate of HEK293 cells by the ORF45-null BAC16-Stop45 mutant indicates that ORF45 plays a critical role during primary infection. This result is in agreement with what we have previously reported (12). Interestingly, although the infectivity of the F66A mutant is only slightly impaired, the GFP intensity is significantly reduced compared to those of the revertant and wild type. The effect is dependent on the multiplicity of infection (MOI), and the deficiency is more severe at lower MOI, where the GFP intensity of F66A-infected and Stop45-infected cells became similar. These results suggest that the activation of RSK by virion-contained ORF45 is required for optimal expression of genes from viral episomes during de novo infection, particularly at lower MOI, which is more common under physiological conditions. Although we cannot exclude the potential involvement of ORF45 in early processes of infection (such as entry, virion transport, uncoating, and nuclear import of viral genomes), the effect is in agreement with ORF45 having a role in the regulation of transcription and translation.
Several tegument proteins likely are involved in viral assembly and maturation during late stages of herpesvirus replication, but it is challenging to dissect the roles of ORF45 in these processes, since its absence affects the expression of some late viral structural proteins. Interestingly, although some viral structural proteins were reduced by the loss of ORF45, the packaging of these proteins into virions appeared not to be greatly affected by the ORF45-null mutation. Additionally, the assembly of ORF45 itself into the virion was not affected by the F66A mutation, suggesting that interaction with RSK is not directly involved in the packaging of ORF45 into virions.
Potential significance of sustained RSK activation by ORF45 in pathogenesis of KSHV-associated diseases.
The Ras/Raf/MEK/ERK pathway is one of the most important signaling cascades frequently dysregulated in human cancers, and components of this pathway have been extensively explored as therapeutic targets (48–50). The persistent activation of RSK and ERK caused by ORF45 could have important implications in KSHV pathogenesis. First, activation of RSKs by ORF45 is important for KSHV lytic replication, a life cycle now known to be crucial for KS development because of its roles in paracrine regulation of the inflammatory and angiogenic microenvironment of tumor cells and in providing a reservoir of viruses from which new latently infected cells can be derived (51, 52). The inhibition of lytic replication was found to reduce KS development in patients (53). Second, as mentioned above, ORF45-mediated activation of RSK is involved in the direct interaction between KSHV and HIV-1, which is well known to drastically increase both the incidence and severity of KS. ORF45 has been shown to work synergistically with the HIV-1-encoded protein Tat to potently increase LTR transcription activity in an RSK-dependent manner (47, 54). Conversely, Tat has been found to increase KSHV lytic gene expression and infectivity (55–57). Third, Chang and Ganem recently discovered a unique transcriptional program consisting of both latency and limited expression of lytic genes in KSHV-infected lymphatic endothelial cells (LECs) but not in the similarly infected blood endothelial cells (BECs) (58). The LECs are highly relevant to the spindle cells in KS lesions. ORF45 was shown to contribute to RSK-dependent but AKT-independent upregulation of mTOR signaling in KSHV-infected LECs, making the cells dependent on mTOR signaling. As a result, LECs but not BECs are sensitized to rapamycin-mediated killing. This finding provided a molecular mechanism to explain the observed phenomenon that rapamycin causes the regression of posttransplant KS lesions in renal transplant recipients. Lastly, because of its roles in transcriptional and translational regulation, ORF45 could affect the expression of cellular genes involved in a variety of physiological and pathological pathways. The involvement of ORF45-RSK in these processes suggests that both RSK and its interaction with ORF45 can be explored as therapeutic targets for KSHV-associated diseases. The natural and synthetic inhibitors of RSKs and small molecules that disrupt ORF45-RSK interaction could be pursued in future studies (48, 59, 60).
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant R01DE016680 to F.Z. D.A. was supported by National Institutes of Health grant F31CA183250-01. Z.T. was partially supported by the FSU Undergraduate Research Opportunity Program. A.V. was partially supported by the FSU Undergraduate Research and Creative Activity Award.
We thank Brian Washburn, Rani Dhanarajan, and Kristina Poduch for making hybridomas. We thank Ruth Didier at the Florida State University Flow Cytometry Facility for assistance with flow cytometry. We are grateful to Klaus Osterrieder for providing pEPKan-S plasmid, Gregory Smith for providing E. coli strain GS1783, Rolf Renne for providing E. coli strain GS1783 carrying BAC16, and Jinjong Myoung and Don Ganem for providing the iSLK cells. We thank Ke Lan for the anti-RTA monoclonal antibody, Robert Ricciardi for the anti-PF8 polyclonal antibody, and John Nicholas for the anti-vIL6 polyclonal antibody. We thank members of the Zhu laboratory for critical reading of the manuscript and for helpful discussions. We also thank Jen Kennedy at the Florida State University for excellent editorial assistance.
REFERENCES
- 1.Ganem D. 2007. Kaposi's sarcoma-associated herpesvirus, p 2847–2888. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 3.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 4.Dupin N, Gorin I, Deleuze J, Agut H, Huraux J-M, Escande J-P. 1995. Herpes-like DNA sequences, AIDS-related tumors, and Castleman' disease. N Engl J Med 333:797–798. doi: 10.1056/NEJM199509213331211. [DOI] [PubMed] [Google Scholar]
- 5.Davison AJ, Eberle R, Ehlers B, Hayward GS, McGeoch DJ, Minson AC, Pellett PE, Roizman B, Studdert MJ, Thiry E. 2009. The order Herpesvirales. Arch Virol 154:171–177. doi: 10.1007/s00705-008-0278-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu FX, Cusano T, Yuan Y. 1999. Identification of the immediate-early transcripts of Kaposi's sarcoma-associated herpesvirus. J Virol 73:5556–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun R, Lin SF, Staskus K, Gradoville L, Grogan E, Haase A, Miller G. 1999. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J Virol 73:2232–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med 2:342–346. doi: 10.1038/nm0396-342. [DOI] [PubMed] [Google Scholar]
- 9.Sarid R, Flore O, Bohenzky RA, Chang Y, Moore PS. 1998. Transcription mapping of the Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J Virol 72:1005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ganem D. 2010. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Invest 120:939–949. doi: 10.1172/JCI40567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mesri EA, Cesarman E, Boshoff C. 2010. Kaposi's sarcoma and its associated herpesvirus. Nat Rev Cancer 10:707–719. doi: 10.1038/nrc2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu FX, Li X, Zhou F, Gao SJ, Yuan Y. 2006. Functional characterization of Kaposi's sarcoma-associated herpesvirus ORF45 by bacterial artificial chromosome-based mutagenesis. J Virol 80:12187–12196. doi: 10.1128/JVI.01275-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu FX, Yuan Y. 2003. The ORF45 protein of Kaposi's sarcoma-associated herpesvirus is associated with purified virions. J Virol 77:4221–4230. doi: 10.1128/JVI.77.7.4221-4230.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu FX, Chong JM, Wu L, Yuan Y. 2005. Virion proteins of Kaposi's sarcoma-associated herpesvirus. J Virol 79:800–811. doi: 10.1128/JVI.79.2.800-811.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu FX, King SM, Smith EJ, Levy DE, Yuan Y. 2002. A Kaposi's sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc Natl Acad Sci U S A 99:5573–5578. doi: 10.1073/pnas.082420599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sathish N, Zhu FX, Golub EE, Liang Q, Yuan Y. 2011. Mechanisms of autoinhibition of IRF-7 and a probable model for inactivation of IRF-7 by Kaposi's sarcoma-associated herpesvirus protein ORF45. J Biol Chem 286:746–756. doi: 10.1074/jbc.M110.150920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang Q, Fu B, Wu F, Li X, Yuan Y, Zhu F. 2012. ORF45 of Kaposi's sarcoma-associated herpesvirus inhibits phosphorylation of interferon regulatory factor 7 by IKKepsilon and TBK1 as an alternative substrate. J Virol 86:10162–10172. doi: 10.1128/JVI.05224-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sathish N, Zhu FX, Yuan Y. 2009. Kaposi's sarcoma-associated herpesvirus ORF45 interacts with kinesin-2 transporting viral capsid-tegument complexes along microtubules. PLoS Pathog 5:e1000332. doi: 10.1371/journal.ppat.1000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuang E, Tang Q, Maul GG, Zhu F. 2008. Activation of p90 ribosomal S6 kinase by ORF45 of Kaposi's sarcoma-associated herpesvirus and its role in viral lytic replication. J Virol 82:1838–1850. doi: 10.1128/JVI.02119-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuang E, Wu F, Zhu F. 2009. Mechanism of sustained activation of ribosomal S6 kinase (RSK) and ERK by Kaposi's sarcoma-associated herpesvirus ORF45: multiprotein complexes retain active phosphorylated ERK AND RSK and protect them from dephosphorylation. J Biol Chem 284:13958–13968. doi: 10.1074/jbc.M900025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li X, Zhu F. 2009. Identification of the nuclear export and adjacent nuclear localization signals for ORF45 of Kaposi's sarcoma-associated herpesvirus. J Virol 83:2531–2539. doi: 10.1128/JVI.02209-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuang E, Fu B, Liang Q, Myoung J, Zhu F. 2011. Phosphorylation of eukaryotic translation initiation factor 4B (EIF4B) by open reading frame 45/p90 ribosomal S6 kinase (ORF45/RSK) signaling axis facilitates protein translation during Kaposi sarcoma-associated herpesvirus (KSHV) lytic replication. J Biol Chem 286:41171–41182. doi: 10.1074/jbc.M111.280982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J Virol Methods 174:12–21. doi: 10.1016/j.jviromet.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J Virol 86:9708–9720. doi: 10.1128/JVI.01019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 26.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 27.Yoo SM, Zhou FC, Ye FC, Pan HY, Gao SJ. 2005. Early and sustained expression of latent and host modulating genes in coordinated transcriptional program of KSHV productive primary infection of human primary endothelial cells. Virology 343:47–64. doi: 10.1016/j.virol.2005.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fakhari FD, Dittmer DP. 2002. Charting latency transcripts in Kaposi's sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J Virol 76:6213–6223. doi: 10.1128/JVI.76.12.6213-6223.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kolde R. 2013. pheatmap: pretty heatmaps. R package, version 0.7.7 http://CRAN.R-project.org/package=pheatmap.
- 30.CoreTeam. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- 31.Vieira J, O'Hearn PM. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225–240. doi: 10.1016/j.virol.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 32.Anjum R, Blenis J. 2008. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol 9:747–758. doi: 10.1038/nrm2509. [DOI] [PubMed] [Google Scholar]
- 33.Romeo Y, Zhang X, Roux PP. 2012. Regulation and function of the RSK family of protein kinases. Biochem J 441:553–569. doi: 10.1042/BJ20110289. [DOI] [PubMed] [Google Scholar]
- 34.Cargnello M, Roux PP. 2011. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papin J, Vahrson W, Hines-Boykin R, Dittmer DP. 2005. Real-time quantitative PCR analysis of viral transcription. Methods Mol Biol 292:449–480. doi: 10.1385/1-59259-848-X:449. [DOI] [PubMed] [Google Scholar]
- 36.Toth Z, Maglinte DT, Lee SH, Lee HR, Wong LY, Brulois KF, Lee S, Buckley JD, Laird PW, Marquez VE, Jung JU. 2010. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog 6:e1001013. doi: 10.1371/journal.ppat.1001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gunther T, Grundhoff A. 2010. The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog 6:e1000935. doi: 10.1371/journal.ppat.1000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma-Walia N, Krishnan HH, Naranatt PP, Zeng L, Smith MS, Chandran B. 2005. ERK1/2 and MEK1/2 induced by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J Virol 79:10308–10329. doi: 10.1128/JVI.79.16.10308-10329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pietrek M, Brinkmann MM, Glowacka I, Enlund A, Havemeier A, Dittrich-Breiholz O, Kracht M, Lewitzky M, Saksela K, Feller SM, Schulz TF. 2010. Role of the Kaposi's sarcoma-associated herpesvirus K15 SH3 binding site in inflammatory signaling and B-cell activation. J Virol 84:8231–8240. doi: 10.1128/JVI.01696-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cannon M, Philpott NJ, Cesarman E. 2003. The Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor has broad signaling effects in primary effusion lymphoma cells. J Virol 77:57–67. doi: 10.1128/JVI.77.1.57-67.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kliche S, Nagel W, Kremmer E, Atzler C, Ege A, Knorr T, Koszinowski U, Kolanus W, Haas J. 2001. Signaling by human herpesvirus 8 kaposin A through direct membrane recruitment of cytohesin-1. Mol Cell 7:833–843. doi: 10.1016/S1097-2765(01)00227-1. [DOI] [PubMed] [Google Scholar]
- 42.Liu J, Martin HJ, Liao G, Hayward SD. 2007. The Kaposi's sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc. J Virol 81:10451–10459. doi: 10.1128/JVI.00804-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woodson EN, Anderson MS, Loftus MS, Kedes DH. 2014. Progressive accumulation of activated ERK2 within highly stable ORF45-containing nuclear complexes promotes lytic gammaherpesvirus infection. PLoS Pathog 10:e1004066. doi: 10.1371/journal.ppat.1004066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Woodson EN, Kedes DH. 2012. Distinct roles for extracellular signal-regulated kinase 1 (ERK1) and ERK2 in the structure and production of a primate gammaherpesvirus. J Virol 86:9721–9736. doi: 10.1128/JVI.00695-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stahl JA, Chavan SS, Sifford JM, Macleod V, Voth DE, Edmondson RD, Forrest JC. 2013. Phosphoproteomic analyses reveal signaling pathways that facilitate lytic gammaherpesvirus replication. PLoS Pathog 9:e1003583. doi: 10.1371/journal.ppat.1003583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu FX, Sathish N, Yuan Y. 2010. Antagonism of host antiviral responses by Kaposi's sarcoma-associated herpesvirus tegument protein ORF45. PLoS One 5:e10573. doi: 10.1371/journal.pone.0010573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karijolich J, Zhao Y, Peterson B, Zhou Q, Glaunsinger B. 2014. Kaposi's sarcoma-associated herpesvirus ORF45 mediates transcriptional activation of the HIV-1 long terminal repeat via RSK2. J Virol 88:7024–7035. doi: 10.1128/JVI.00931-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Romeo Y, Roux PP. 2011. Paving the way for targeting RSK in cancer. Expert Opin Ther Targets 15:5–9. doi: 10.1517/14728222.2010.531014. [DOI] [PubMed] [Google Scholar]
- 49.Dhillon AS, Hagan S, Rath O, Kolch W. 2007. MAP kinase signalling pathways in cancer. Oncogene 26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 50.Roberts PJ, Der CJ. 2007. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 51.Cesarman E, Mesri EA, Gershengorn MC. 2000. Viral G protein-coupled receptor and Kaposi's sarcoma: a model of paracrine neoplasia? J Exp Med 191:417–422. doi: 10.1084/jem.191.3.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grundhoff A, Ganem D. 2004. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J Clin Invest 113:124–136. doi: 10.1172/JCI200417803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martin DF, Kuppermann BD, Wolitz RA, Palestine AG, Li H, Robinson CA. 1999. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. N Engl J Med 340:1063–1070. doi: 10.1056/NEJM199904083401402. [DOI] [PubMed] [Google Scholar]
- 54.Huang LM, Chao MF, Chen MY, Shih H, Chiang YP, Chuang CY, Lee CY. 2001. Reciprocal regulatory interaction between human herpesvirus 8 and human immunodeficiency virus type 1. J Biol Chem 276:13427–13432. doi: 10.1074/jbc.M011314200. [DOI] [PubMed] [Google Scholar]
- 55.Zeng Y, Zhang X, Huang Z, Cheng L, Yao S, Qin D, Chen X, Tang Q, Lv Z, Zhang L, Lu C. 2007. Intracellular Tat of human immunodeficiency virus type 1 activates lytic cycle replication of Kaposi's sarcoma-associated herpesvirus: role of JAK/STAT signaling. J Virol 81:2401–2417. doi: 10.1128/JVI.02024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aoki Y, Tosato G. 2004. HIV-1 Tat enhances Kaposi sarcoma-associated herpesvirus (KSHV) infectivity. Blood 104:810–814. doi: 10.1182/blood-2003-07-2533. [DOI] [PubMed] [Google Scholar]
- 57.Ensoli B, Sturzl M. 1998. Kaposi's sarcoma: a result of the interplay among inflammatory cytokines, angiogenic factors and viral agents. Cytokine Growth Factor Rev 9:63–83. doi: 10.1016/S1359-6101(97)00037-3. [DOI] [PubMed] [Google Scholar]
- 58.Chang HH, Ganem D. A unique herpesviral transcriptional program in KSHV-infected lymphatic endothelial cells leads to mTORC1 activation and rapamycin sensitivity. Cell Host Microbe 13:429–440. doi: 10.1016/j.chom.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nguyen TL. 2008. Targeting RSK: an overview of small molecule inhibitors. Anticancer Agents Med Chem 8:710–716. doi: 10.2174/187152008785914770. [DOI] [PubMed] [Google Scholar]
- 60.Utepbergenov D, Derewenda ZS. 2013. The unusual mechanism of inhibition of the p90 ribosomal S6 kinase (RSK) by flavonol rhamnosides. Biochim Biophys Acta 1834:1285–1291. doi: 10.1016/j.bbapap.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]