

ABSTRACT
The HIV-1 capsid plays multiple roles in infection and is an emerging therapeutic target. The small-molecule HIV-1 inhibitor PF-3450074 (PF74) blocks HIV-1 at an early postentry stage by binding the viral capsid and interfering with its function. Selection for resistance resulted in accumulation of five amino acid changes in the viral CA protein, which collectively reduced binding of the compound to HIV-1 particles. In the present study, we dissected the individual and combinatorial contributions of each of the five substitutions Q67H, K70R, H87P, T107N, and L111I to PF74 resistance, PF74 binding, and HIV-1 infectivity. Q67H, K70R, and T107N each conferred low-level resistance to PF74 and collectively conferred strong resistance. The substitutions K70R and L111I impaired HIV-1 infectivity, which was partially restored by the other substitutions at positions 67 and 107. PF74 binding to HIV-1 particles was reduced by the Q67H, K70R, and T107N substitutions, consistent with the location of these positions in the inhibitor-binding pocket. Replication of the 5Mut virus was markedly impaired in cultured macrophages, reminiscent of the previously reported N74D CA mutant. 5Mut substitutions also reduced the binding of the host protein CPSF6 to assembled CA complexes in vitro and permitted infection of cells expressing the inhibitory protein CPSF6-358. Our results demonstrate that strong resistance to PF74 requires accumulation of multiple substitutions in CA to inhibit PF74 binding and compensate for fitness impairments associated with some of the sequence changes.
IMPORTANCE The HIV-1 capsid is an emerging drug target, and several small-molecule compounds have been reported to inhibit HIV-1 infection by targeting the capsid. Here we show that resistance to the capsid-targeting inhibitor PF74 requires multiple amino acid substitutions in the binding pocket of the CA protein. Three changes in CA were necessary to inhibit binding of PF74 while maintaining viral infectivity. Replication of the PF74-resistant HIV-1 mutant was impaired in macrophages, likely owing to altered interactions with host cell factors. Our results suggest that HIV-1 resistance to capsid-targeting inhibitors will be limited by functional constraints on the viral capsid protein. Therefore, this work enhances the attractiveness of the HIV-1 capsid as a therapeutic target.
INTRODUCTION
Retrovirus particles contain a capsid shell that is critical for completion of the early postentry steps in HIV-1 infection. The capsid surrounds the genome and associated proteins and plays a critical role in reverse transcription and entry into the nucleus of target cells. In HIV-1, the viral capsid consists of a lattice of approximately 250 hexamers of the CA protein, with an additional 12 pentamers that create the curvature at both ends of the capsid that allow for a closed volume (reviewed in reference 1). While retroviral capsids are diverse with respect to shape, the capsids of lentiviruses such as HIV-1 typically adopt a conical morphology. Lentiviruses also exhibit the ability to efficiently infect nondividing cells, a feature that has been linked genetically to the capsid (2, 3). Mutations in the HIV-1 CA protein can result in impairments in several early steps of infection, including reverse transcription, nuclear entry, and integration. Impaired activity of HIV-1 CA mutants has been associated with changes in the structure of the capsid as well as its stability, suggesting that the process of disassembly of the capsid in target cells is important for infection (4–6). Many mutations that either increase or decrease the intrinsic stability of the capsid, or the rate at which it is dissociated from the genome following entry into the cell, reduce HIV-1 infectivity (6–8). Additionally, the capsid represents the functional viral determinant for several host factors on which HIV-1 infection depends, including cyclophilin A, CPSF6, TNPO3, Nup153, and Nup358/RanBP2 (9–15), as well as the target of host-intrinsic antiviral defenses (16–21). Because the viral capsid plays a critical role in HIV-1 infection, the CA protein has recently attracted interest as a target for antiviral therapy (22).
The small molecule PF-3450074 (PF74) inhibits HIV-1 replication by targeting the viral capsid (23, 24). PF74 binds to a pocket in the N-terminal domain of CA, at a location near the interface between adjacent N-terminal and C-terminal domains (NTD and CTD, respectively) of adjacent CA subunits within the hexamer (23). Paradoxically, while binding of PF74 promotes self-assembly of the viral CA protein in vitro, addition of PF74 to purified HIV-1 cores results in their accelerated disassembly. These observations suggested that the compound acts by destabilizing the viral capsid in target cells (23, 24). Consistent with this notion, a substitution in CA (E45A) that stabilizes the capsid renders the virus resistant to PF74 in single-round infection assays without reducing binding of the inhibitor (24).
Previously, Blair and coworkers isolated a PF74-resistant HIV-1 mutant by prolonged serial passage of HIV-1 in the presence of escalating doses of a closely related compound (23). The resulting virus, named “5Mut,” encodes five amino acid substitutions in the CA NTD. Three of the substitutions (Q67H, K70R, and T107N) reside within the PF74 binding site, while the other two changes (H87P and L111I) are at distal positions. 5Mut virions exhibit reduced binding to PF74, suggesting that its resistance to the inhibitor results from decreased affinity for the compound. However, the strong resistance exhibited by the E45A CA mutant implied that PF74 resistance may also occur through other mechanisms, such as increased capsid stability. Additionally, the conserved nature of the PF74 binding pocket and the presence of two amino acid substitutions (H87P and L111I) outside the pocket suggested that some of the changes might be important for viral fitness. To test these hypotheses, we deconstructed the substitutions in the 5Mut virus and studied their individual and combined contributions to HIV-1 infectivity, PF74 sensitivity, and PF74 binding.
MATERIALS AND METHODS
Cells and viruses.
The HIV-1 viruses used in this study were generated from the full-length infectious proviral clone R9. R9-5Mut contains the following substitutions in CA: Q67H, K70R, H87P, T107N, and L111I. The 30 mutant R9 constructs containing all possible subsets of the 5 mutations were generated by overlap PCR, and the corresponding restriction fragments were replaced into the R9 clone. The sequences of the PCR-amplified regions were confirmed. Virus stocks were produced by transfection of 293T cells cultured in 100-mm dishes in Dulbecco's modified Eagle's medium (DMEM) supplemented with fetal bovine serum (10%), penicillin, and streptomycin (D10 medium). Culture supernatants were harvested 36 to 48 h after transfection, clarified by passage through 0.45-μm-pore syringe filters, frozen in aliquots, and stored at −80°C. Aliquots were thawed once prior to use in infection assays or to inoculate cultures for replication studies. CEM, a human T cell line, was cultured in RPMI 1640 supplemented with FBS (10%), penicillin, and streptomycin. HeLa cells expressing CPSF6-358 (the kind gift of Vineet Kewalramani) and TZM-bl cells were cultured in Dulbecco's modified Eagle's medium supplemented with FBS (10%), penicillin, and streptomycin (D10 medium).
Mononuclear cell-enriched blood was purchased from Biological Specialty Corp., Colmar, PA. Monocytes were purified by CD14+ selection using the EasySep human CD14 selection kit (STEMCELL Technologies) and were seeded in 48-well plates at 250,000 cells per well. Purified monocytes were >95% CD14+ CD3− as assessed by flow cytometry. Monocytes were differentiated into macrophages by being cultured for 1 week in D10 medium supplemented with granulocyte-macrophage colony-stimulating factor (GM-CSF) (500 U/ml [Peprotech]), with fresh GM-CSF added after 3 days prior to inoculation with HIV-1.
Chemicals.
PF74 and triiodo-PF74 were synthesized and purified in the Chemical Synthesis Core, Vanderbilt Institute for Chemical Biology. PF74 was dissolved at 10 mM in dimethyl sulfoxide (DMSO) and added to cultures to achieve final concentrations of up to 20 μM. [3H]PF74 was produced by 3H-I exchange reaction using triiodo-PF74 by Moravek Biochemicals and Radiochemicals (La Brea, CA) to a specific radioactivity of 52 Ci/mmol and purified by high-performance liquid chromatography (HPLC). Radiochemical purity was confirmed by chromatography with unlabeled PF74 as a reference compound, and the identity was confirmed by mass spectrometry. The antiviral potency of [3H]PF74 was quantified in parallel with unlabeled PF74 and found to be equivalent.
Single-cycle infection and replication assays.
For single-cycle assays of HIV-1 infection, dilutions of HIV-1 stocks were inoculated onto cultures of TZM-bl cells (10,000 cells per well seeded 1 day prior), cultured in D10 medium in 96-well black wall plates (Costar). PF74 was added to the cultures at the time of virus inoculation. Cultures were maintained for 48 h prior to quantification of cellular luciferase activity. Supernatants were aspirated, and cells were washed with 50 μl phosphate-buffered saline (PBS) and lysed in situ by addition of 30 μl of Reportasol extraction buffer (Novagen). Luminescence was quantified in a Molecular Devices Lmax microplate luminometer (15 s of integration) upon injection of 150 μl of luciferase assay cocktail (75 mM Tris-HCl, 15 mM magnesium acetate, 4 mM ATP [pH 7.8]) and 100 μl of d-luciferin (1 mM solution). HIV-1 titration experiments demonstrated that the assay was linear up to 1,000 relative luminescence units (RLU); the assay exhibited a background of approximately 2 RLU, so virus dilutions producing signals within the linear range of the assay (between 20 and 400 RLU) were employed for the wild-type (WT) and mutant virus stocks. To calculate relative infectivity, the luminescence values were normalized by the reverse transcriptase activity values of the corresponding virus stocks. PF74 was employed at concentrations up to 20 μM; higher concentrations produced significant cytotoxicity and thus were avoided. Fifty percent inhibitory concentration (IC50) values were computed by fitting the data to a least-squares model using Graphpad Prism software.
To assess HIV-1 replication in CEM cells, cultures of 40,000 cells in 200 μl of medium were inoculated with virus doses corresponding to 10 ng of p24, and 100 μl of the culture supernatants was removed periodically and assayed for reverse transcriptase activity. Medium was replenished with an equal volume containing the appropriate concentrations of PF74 for the cultures. Reverse transcriptase activity was quantified by determining the extent of DNA synthesis in reaction mixtures containing [3H]TTP and oligo(dT)-poly(rA) primer template, as previously described (25).
To assay HIV-1 replication in macrophages, cultures of human monocyte-derived macrophages were inoculated with the R5-tropic HIV-1 isolate R9-BaL, and HIV-1 replication was assessed by periodically quantifying the accumulation of reverse transcriptase activity in culture supernatants.
Analysis of sensitivity to CPSF6-358 restriction.
HIV-green fluorescent protein (GFP) reporter viruses pseudotyped by vesicular stomatitis virus glycoprotein G (VSV-G) were produced by transfection of 293T cells with the Env-defective reporter virus construct HIV-GFP (20 μg) and corresponding CA mutants with plasmid pHCMV-G encoding VSV-G. Virus stocks were quantified by p24 enzyme-linked immunosorbent assay (ELISA) and titrated on cultures of HeLa cells stably expressing CPSF6-358 or the control empty LPCX retroviral vector. (These cell lines were the generous gift of Vineet Kewalramani, National Cancer Institute, Frederick, MD.) Cells were harvested by trypsinization 2 days after inoculation, fixed with paraformaldehyde, and analyzed for GFP expression by flow cytometry.
Assay of [3H]PF74 binding to HIV-1 particles.
Wild-type and CA mutant HIV-1 particles were produced by transfection of 293T cells, pelleted through a layer of 20% sucrose, and resuspended in D10 medium to a concentration of 10 μg p24 per ml. [3H]PF74 was added to a concentration of 0.38 μM, and the samples were incubated for 2 h at room temperature. Unbound PF74 was removed by pelleting the particles through a cushion of 20% sucrose in STE buffer (10 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 mM EDTA), the pellets were rinsed with 0.2 ml of STE buffer and recentrifuged, and the supernatants were removed. The pelleted particles were dissolved in SDS sample buffer, and samples were subjected to 3H quantification by liquid scintillation counting and assayed for p24 by ELISA. Results were expressed as the molar ratio of [3H]PF74 to p24 in the samples and were normalized to the values determined in parallel for wild-type HIV-1 particles.
Determination of PF74 affinity for recombinant CA hexamers.
HIV-1 CA hexamers, stabilized by engineered intersubunit disulfide bonds, were produced by assembly of recombinant CA containing four amino acid substitutions, as previously reported (26). Equilibrium dialysis was performed by adding 500 μl of various concentrations of [3H]PF74 into the buffer chamber and 300 μl of CA hexameric protein (1 μM) into the sample chamber of a Thermo Scientific rapid equilibrium dialysis plate. Each compound concentration was analyzed in duplicate. Plates were sealed and incubated at 37°C for 24 h with rotation at 100 rpm. Samples were removed from each side of the chamber, and the PF74 concentrations were determined by liquid scintillation counting for 3H and comparison to a reference consisting of the purified radiochemical. Kd (dissociation constant) values were calculated by fitting the data to a one-site binding model with GraphPad Prism software.
Analysis of CPSF6 association with assembled CA tubes.
Binding of CPSF6 protein in HeLa cell extracts was performed as previously described (9). Briefly, disulfide-cross-linked tubular CA assemblies were prepared from recombinant 14C/45C CA proteins (with the indicated additional mutations) expressed in Escherichia coli. Protein extracts prepared from cultured HeLa cells were incubated with assembled CA tubes, and the complexes were pelleted and subjected to SDS-PAGE and immunoblotting using rabbit antiserum to CPSF6 (Novus Biologicals). Bands were detected following probing with IR dye-conjugated secondary antibody and detected with a LI-COR Odyssey imaging system. Band intensities were quantified with the instrument software.
RESULTS
Effects of the 5Mut substitutions on HIV-1 fitness.
The PF74-resistant virus 5Mut encodes five changes in CA (Q67H, K70R, H87P, T107N, and L111I) and exhibits up to 40-fold resistance to the inhibitor in single-cycle infection assays (23, 24). The original selection experiment in which 5Mut was recovered involved a dose-escalation strategy. Sequencing of the CA region of Gag throughout the course of the experiment identified a progressive accumulation of the mutations in the order H87P T107N, Q67H, K70R, and L111I (Fig. 1A). In this selection experiment, the changes at positions 87 and 107 emerged first, followed by Q67H, K70R, and finally L111I. To evaluate the contribution of each change to PF74 resistance, we generated HIV-1 clones representing all 31 possible combinations of the five substitutions. To determine whether any of the mutations affect particle assembly, each mutant clone was transfected into 293T cells, and the resulting culture supernatants were assayed for HIV-1 particles by p24 ELISA and reverse transcriptase activity. The wild-type HIV-1 clone was included as a control. The results showed that the mutations had no major effects on particle production or particle-associated reverse transcriptase levels (data not shown).
FIG 1.

Combinations of mutations in the CA N-terminal domain confer resistance to PF74. (A) Selection for resistance by dose escalation in CEM cells. The related compound PF-3759857 was used in this experiment, in which the virus accumulated 5 mutations. Shown is the time of first detection of each mutation. The ordinate shows the dose-escalation selection protocol. This panel shows the original drug selection experiment reported in reference 23. (B) Single-cycle infectivity assays of all viruses containing all possible combinations of the 5 mutations. Viruses were titrated on TZM-bl cells, and the relative extent of infection was determined by luciferase activity assay. Shown are the mean infectivity values (normalized by reverse transcriptase [RT] activity) obtained from three or four independent determinations.
Many amino acid changes in CA have been shown to result in impaired HIV-1 infectivity. To determine whether any substitutions in the 5Mut virus alter HIV-1 infectivity, we titrated the virus stocks onto TZM-bl cells and quantified the expression of luciferase resulting from transactivation of the cellular reporter gene upon expression of Tat from the integrated HIV-1 provirus. Assays of the panel of 32 viruses revealed marked differences in the extent of infection (Fig. 1B). Among the single mutants, K70R and L111I reduced HIV-1 infectivity by approximately 90% and 50%, respectively, and the virus containing both substitutions (designated 70/111) was also markedly impaired. The T107N substitution slightly reduced HIV-1 infectivity, although the extent of the reduction varied between experiments. In contrast, the Q67H and H87P mutants exhibited infectivity levels slightly higher than that of the wild type.
The infectivity of the 5Mut virus was approximately two-thirds that of the wild type, suggesting that the strong infectivity impairment induced by the K70R substitution was mitigated by one or more of the other changes in 5Mut. Accordingly, we observed that the Q67H K70R mutant (designated 67/70) was nearly as infectious as the wild type, and the infectivity of the Q67H K70R L111I triple mutant (67/70/111) was much greater than that of the K70R L111I mutant (70/111). Moreover, addition of Q67H markedly enhanced the infectivity of the K70R H87P L111I mutant. Similarly, addition of the T107N substitution relieved the infectivity impairment exhibited by mutants containing K70R but lacking Q67H. These results indicate that Q67H and T107N substitutions can relieve the infectivity impairment associated with K70R. In contrast, the H87P substitution did not have a strong effect on HIV-1 infectivity alone or in combination with the other substitutions. Nonetheless, reversion of this substitution to a His codon slightly reduced the infectivity of the 5Mut virus, suggesting that H87P plays a minor role in maintaining viral fitness.
To examine the effects of the mutations on continuous HIV-1 replication in T cells, cultures of the CEM human T cell line were inoculated with equivalent quantities of the mutant virions, and HIV-1 replication was periodically assessed by reverse transcriptase assay of culture supernatants. The replication assays were performed in duplicate to determine the extent of variation in the cultures, which was minimal. The wild-type virus reached a plateau at day 10 postinoculation (Fig. 2). Under these conditions, the replication of the 5Mut virus was similar to that of the wild type. Several of the mutants were strongly impaired for replication; these were the K70R single mutant and mutants 67/111, 70/111, and 70/87/111. Mutants that replicated with attenuated kinetics (∼8-day delay) included the L111I single mutant, the double mutants 70/87, 87/111, and 107/111, and the triple mutants 67/70/111 and 87/107/111. The double mutant 87/107 was delayed by 3 to 4 days relative to the wild type. All of the quadruple mutants replicated well, with only a slight delay exhibited by 67/87/107/111. For the mutants exhibiting delayed replication, the duplicate cultures yielded consistent results, suggesting that stochastic differences did not contribute to the emergence of the viruses. However, the sequences of the viruses that emerged in these cultures were not analyzed, and it is possible that accumulation of additional mutations contributed to their replication. Together, the results indicate that the replication defect of double mutant viruses containing K70R was markedly relieved by the Q67H, H87P, and T107N substitutions.
FIG 2.

Replication of the panel of CA mutants in human T cells. Cultures of the CEM T cell line were inoculated with normalized quantities of the viruses (20 ng p24), and replication was monitored for up to 3 weeks by assaying reverse transcriptase activity in the culture supernatants. Shown are the mean values of duplicate parallel cultures, which typically varied by less than 20%. The results are representative of two independent experiments. The single, double, triple, and quadruple mutants are shown in panels A, B, C, and D, respectively.
In general, the mutants exhibited replication kinetics consistent with the single-cycle infectivity measurements, with viruses exhibiting less than 10% of wild-type infectivity failing to replicate within the 15-day culture period and those with 25 to 75% infectivity exhibiting delayed replication. However, some exceptions were noted. The 67/111 virus failed to replicate, although it exhibited 28% of wild-type infectivity. Moreover, among the mutants with intermediate infectivity impairments, the replication kinetics were somewhat inconsistent. For example, replication of 67/87/107/111, which exhibited 30% of wild-type infectivity, was only slightly delayed, while the 87/111 mutant, which was 70% as infectious as the wild type, was markedly delayed in its replication.
The growth curves shown in Fig. 2 were performed by inoculation of the cultures with 20 ng of p24, resulting in a peak of wild-type virus replication after 10 days. In experiments with lower initial virus inputs, we observed that replication of the 5Mut virus was strongly dependent on the multiplicity of infection: the virus replicated with kinetics similar to those of the wild type at the highest inoculum, but reduction of the virus input by 2- and 4-fold led to markedly delayed replication relative to that in the corresponding wild-type cultures (Fig. 3). Several other debilitated mutants, including the L111I and 87/107/111 mutants, were not as strongly dependent on the initial multiplicity of infection (MOI) as 5Mut. In sum, the results indicate that the replicative efficiency of the 5Mut virus is dependent on a minimum of three amino acid substitutions and is strongly affected by the quantity of the inoculum.
FIG 3.

Replication of 5Mut is strongly dependent on initial multiplicity of infection. CEM cultures were inoculated with doses of wild-type and 5Mut particles corresponding to 5, 10, and 20 ng of p24. Replication was assessed as described in the legend to Fig. 2. Shown for comparison is the analysis of the wild-type and the L111I and 87/107/111 mutants.
Multiple mutations contribute to PF74 resistance of 5Mut.
To assess the contribution of each of the 5Mut substitutions and combinations thereof to resistance to PF74, we performed single-cycle infection assays of the panel of mutants in the presence of a range of PF74 concentrations and used the data to calculate the corresponding IC50 values. PF74 was employed at concentrations up to 20 μM; higher concentrations were toxic to cells and thus were avoided. As previously reported, wild-type HIV-1 was inhibited by PF74 with an IC50 of approximately 0.5 μM (23, 24). Assays of the single mutants revealed that three of the viruses exhibited dose-response curves that were shifted to higher concentrations, indicating that the mutations Q67H, K70R, and T107N rendered HIV-1 slightly less susceptible to inhibition by PF74 (Fig. 4A). The increase in IC50 for each of these mutants was statistically significant (P < 0.05). In contrast, the H87P and L111I mutants were approximately as sensitive to PF74 as the wild type.
FIG 4.

Analysis of the effects of the 5Mut substitutions on PF74 resistance and PF74 binding to virions. (A) Quantification of IC50 values for viral inhibition. Shown are the mean values, determined from three or four independent experiments, depending on the mutant. Error bars represent 1 standard deviation. (B) Extent of binding of PF74 to virions. Concentrated virus particles were incubated with [3H]PF74 at room temperature and pelleted by ultracentrifugation. Pellets were assayed for 3H by scintillation counting and for viral antigen by p24 ELISA. The values shown represent the ratio of 3H to p24, normalized to wild-type HIV-1. Shown are the mean values from three independent determinations, with error bars representing 1 standard deviation.
Because no single mutation conferred the level of resistance to PF74 exhibited by 5Mut, and because H87P and L111I lie outside the PF74 binding site, we sought to determine which mutations act in concert to confer resistance. To this end, the remaining mutants were tested in parallel for sensitivity to PF74 in the single-cycle infection assay (Fig. 4A). We observed that the 67/107 mutant was less sensitive to PF74 than any of the single mutants. Of the 10 double mutants, 67/107 was the least sensitive, exhibiting an IC50 of 4.3 μM. The 70/107, 87/107, and 107/111 mutants were comparable to the T107N single mutant (IC50, ∼2 μM). However, addition of the H87P or L111I substitution did not appear to enhance the resistance of any of the single mutants. Collectively, these results support the major conclusion drawn from analysis of the single mutants: mutations Q67H, K70R, and T107N contribute significantly to PF74 resistance.
Assays of the triple mutants revealed that two exhibited strong resistance (IC50, 8 to 10 μM): these were 67/70/107 and 67/70/111. Intermediate resistance (IC50, ∼4 μM) was observed for 67/87/107 and 67/107/111; the remaining triple mutants were only slightly resistant (IC50, 1 to 2 μM). Collectively, these results indicate that minimally three substitutions, including Q67H plus K70R and either T107N or L111I, are required for strong resistance to PF74. Similar results were observed with the quadruple mutants, of which only 67/70/87/107 and 67/70/107/111 exhibited strong resistance to PF74. Thus, mutants containing all three of the position 67, 70, and 107 substitutions exhibited maximal PF74 resistance in single-cycle infection assays.
5Mut substitutions act in combination to reduce PF74 binding.
In a previous study, we showed that PF74 binds to wild-type HIV-1 particles, while binding of the compound to 5Mut virions was essentially undetectable (24). To further examine the relationship between PF74 antiviral activity and capsid binding, we quantified the extent of binding of [3H]PF74 to wild-type and mutant HIV-1 particles (Fig. 4B). The results revealed that all of the mutants except the H87P mutant and 87/111 exhibited reduced binding to PF74. Consistent with our previous report, binding to 5Mut was at or just above the limit of detection. Similarly, mutants 67/70, 70/107, 67/70/87, 67/70/107, 67/70/111, 67/107/111, 70/87/107, 70/87/111, 70/107/111, 67/70/87/111, and 67/70/107/111 exhibited very low binding to the compound. Thus, the combination of Q67H and K70R substitutions strongly inhibited HIV-1 binding to PF74. Similarly, mutants containing both K70R and T107N were markedly impaired for binding, with the exception of 67/70/87/107 and 70/87/107/111, for which binding was approximately 10% of the wild type. Thus, while H87P generally did not inhibit binding of PF74, it did appear to slightly enhance binding to the 67/70/107 and 67/107/111 mutants. PF74 also bound the 70/87/111 mutant to a very low extent, indicating that the H87P substitution cooperates with the K70R and L111I substitutions to inhibit binding.
The virus binding data indicated that the substitutions at positions 67, 70, 107, and, to a lesser extent, 111 act in concert to inhibit binding of PF74. Combinations of the substitutions at positions 67, 70, and 107 markedly inhibited binding. For several mutants, including 67/70, 70/107, 70/87/111, 70/107/111, and 67/70/87/111, the virions bound low levels of PF74 yet remained relatively sensitive to inhibition by the compound. Thus, the antiviral activity was not strictly correlated with the extent of binding to virus particles.
In an effort to more accurately quantify the effects of mutations on PF74 binding, we determined the affinity of disulfide-stabilized recombinant CA hexamers for PF74 by equilibrium dialysis. CA hexamer protein was placed on one side of the dialysis membrane, and various concentrations of [3H]PF74 were allowed to equilibrate across the membrane. The concentration of the compound in each chamber was then quantified by scintillation counting, and the Kd was determined by analysis of the binding isotherm. The results are shown in Fig. 5. PF74 binding to the control cross-linked hexamer exhibited a Kd of 0.105 ± 0.034 μM. The affinity for hexamers bearing individual Q67H, K70R, or T107N substitutions was reduced by 2- to 3-fold. The H87P and L111I substitutions did not markedly alter the PF74 affinity, consistent with the minimal effects of these two substitutions on PF74 antiviral activity. We also observed that the combination of the Q67H and K70R substitutions additively increased the Kd for PF74, as did inclusion of the combination of the substitutions at positions 67, 70, and 107. However, the latter increase was variable in repeated experiments. We also observed that the 70/87/111 and 70/107/111 mutant CA hexamers exhibited higher affinity for PF74 than did the 5Mut hexamers, thereby accounting for the intermediate sensitivity of these HIV-1 mutants to inhibition by PF74. Nonetheless, 67/70/87/111 hexamers exhibited low affinity for PF74 that was indistinguishable from that of 5Mut. Thus, while most of the results are consistent with the conclusion that reduction in PF74 affinity imposed by the substitutions in the PF74 binding pocket provides the major contribution to the resistance exhibited by the 5Mut virus, alterations in PF74 affinity may not fully account for HIV-1 resistance to the inhibitor.
FIG 5.

Effects of CA substitutions on PF74 affinity for disulfide-stabilized recombinant CA hexamers. Affinities of [3H]PF74 for recombinant proteins were determined by equilibrium dialysis using 6 concentrations of labeled compound in duplicate. (A) Representative isotherms are shown for each protein. (B) Mean Kd values and standard deviations were computed from the results from multiple assays with each protein.
PF74 resistance is associated with decreased binding of CPSF6.
Previous studies have shown that amino acid substitutions in the region of the PF74 binding site, particularly N74D, confer resistance to restriction by the CPSF6-358 protein by preventing its binding to the capsid (10, 27). To examine the relationship between PF74 resistance and capsid-host factor interactions, we analyzed the sensitivity of viruses containing the 5Mut substitutions by titrating the corresponding GFP reporter viruses on control cells and cells expressing CPSF6-358. The results showed that 5Mut is highly resistant to inhibition by the protein (Fig. 6). In contrast, the Q67H and H87P mutant viruses were inhibited to an extent similar to that of the wild type. K70R, T107N, and L111I mutant viruses were partially resistant to restriction by CPSF6-358. These results indicate that the 5Mut virus is highly resistant to CPSF6-358, likely owing to reduced binding of the restriction factor resulting from amino acid substitutions in the CPSF6 binding site.
FIG 6.

PF74 resistance mutations reduce the inhibition of HIV-1 infection by CPSF6-358. VSV-G-pseudotyped HIV-GFP reporter viruses were titrated on control HeLa cells or cells expressing CPSF6-358. Infection was quantified by flow cytometry for GFP expression. Shown are the results from an experiment in which all of the viruses were analyzed in parallel.
To directly test the effects of 5Mut substitutions on CPSF6 binding affinity, we generated disulfide-stabilized CA tubes by assembly of recombinant 5Mut CA protein containing the A14C and E45C substitutions allowing spontaneous cross-linking following assembly in vitro. HeLa cell extracts were incubated with the assemblies, which were subsequently pelleted by low-speed centrifugation and analyzed by immunoblotting with a CPSF6-specific antibody. Titration of the cell extracts showed that 5Mut tubes bound less CPSF6 than the corresponding control CA tubes, particularly at the lower input levels (Fig. 7). As a negative control, N74D mutant CA tubes were analyzed in parallel and exhibited low CPSF6 binding, as previously reported (9). We conclude that the 5Mut CA assemblies exhibit reduced binding to CPSF6, likely accounting for the decreased sensitivity to CPSF6-358.
FIG 7.
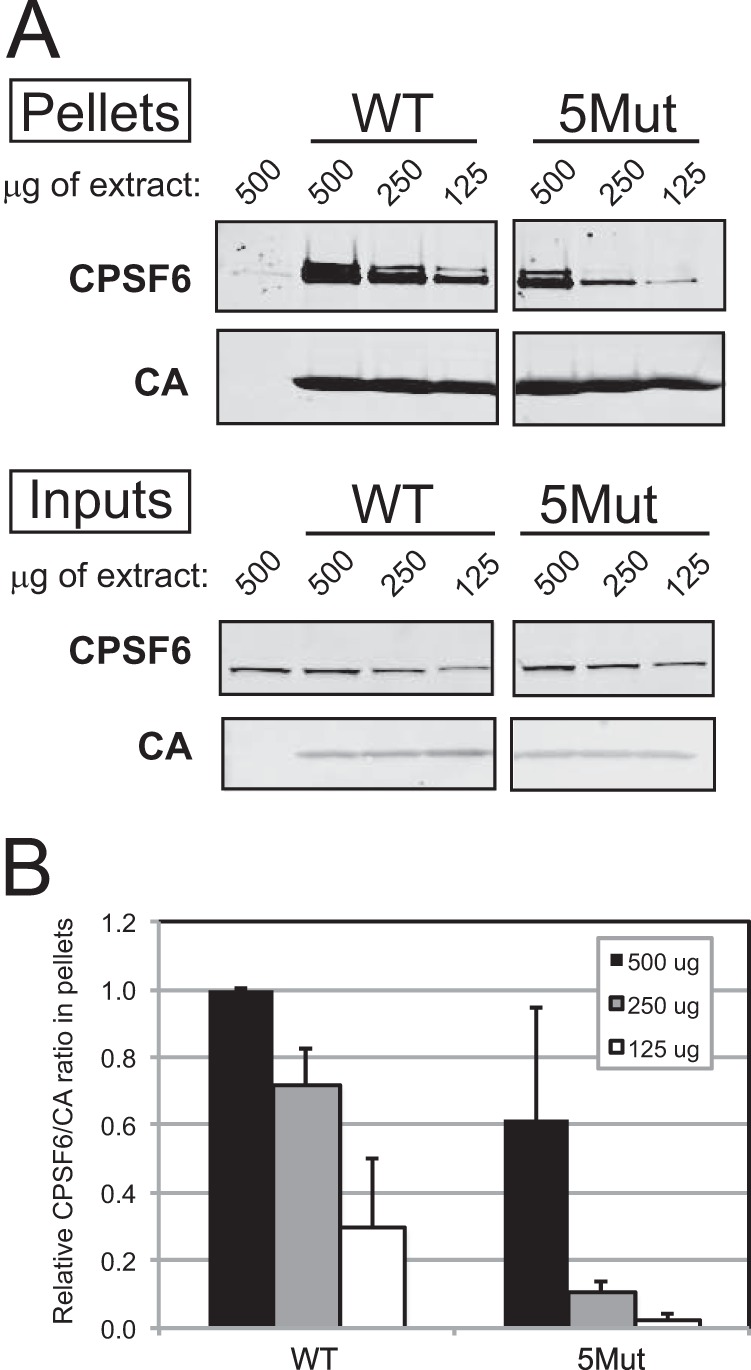
5Mut substitutions reduce the binding of CPSF6 in vitro. (A) Disulfide cross-link-stabilized CA tubes (5 μM) were incubated with 500, 250, or 125 μg of HeLa cell extracts for 1 h at room temperature with gentle mixing. (Upper panel [Pellets]) Reaction mixtures were subjected to low-speed centrifugation, and pelleted proteins were analyzed by SDS-PAGE and immunoblotting for CPSF6 and CA. (Lower panel [Inputs]) Prior to centrifugation, samples corresponding to 10% of each reaction mixture were withdrawn for analysis. (B) Quantification of CPSF6 association with CA tubes relative to amount of pelleted CA. The results are the mean values from 3 independent experiments, with error bars representing 1 standard deviation.
The 5Mut virus is impaired for replication in human macrophages.
Impaired binding of the HIV-1 capsid to CPSF6 is associated with decreased replication capacity in primary human cell targets of infection, particularly macrophages (9, 28, 29). The resistance to CPSF6-358 restriction exhibited by 5Mut suggested that, akin to the previously reported N74D mutant (10), 5Mut may replicate poorly in macrophages. To test this, we performed continuous replication assays in cultured macrophages derived by differentiation from monocytes purified from human blood. We observed that replication of the 5Mut virus encoding the envelope glycoproteins from the R5-tropic HIV-1 isolate R9-BaL was markedly attenuated (Fig. 8).
FIG 8.
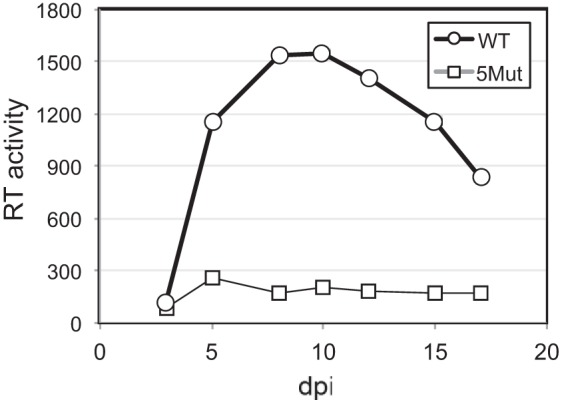
The HIV-1 5Mut virus is impaired for replication in macrophages. Growth of R9-BaL wild-type, and 5Mut viruses in cultures of macrophages derived by differentiation of purified human monocytes was assessed by quantification of accumulation of reverse transcriptase activity in culture supernatants. The results are representative of two independent experiments. dpi, day postinfection.
DISCUSSION
In this study, we examined the contributions of the five amino acid substitutions in the 5Mut virus to viral fitness and resistance to the capsid-targeting HIV-1 inhibitor PF74. 5Mut was originally identified by selection for resistance to a related compound in a 53-day dose-escalation study (23). During the selection process, the changes in CA accumulated in a stepwise fashion, resulting in efficient replication in the presence of high concentrations of PF74. In the present study, we observed that combinations of three substitutions in CA—Q67H plus K70R and either T107N or L111I—are sufficient for high-level resistance to PF74 and for efficient replication in CEM T cells. Therefore, a minimum of three amino acid substitutions is required for the strong PF74 resistance exhibited by 5Mut while maintaining viral fitness.
The fitness impairment associated with the K70R and L111I substitutions warrants further investigation. In the original resistance selection experiment, these changes were detected only after the virus acquired the T107N and Q67H substitutions, which partially rescue the infectivity impairment. Both K70R and L111I are conservative substitutions within or near the PF74 binding pocket in CA. In additional experiments, we observed that both of the single mutants contained viral cores that were moderately more stable than the wild type, as reflected by the level of core-associated CA recovered from a density gradient following removal of the virion envelope (V. Shah, unpublished observations). However, the cores from neither mutant were as stable as those of 5Mut. Thus, it is not clear whether the apparent stability changes associated with these mutants are the cause of their impaired infectivity. The PF74-binding pocket also overlaps the binding sites for the host proteins CPSF6 and Nup153, suggesting that inhibitors targeting this site may alter the interaction of the viral capsid with cellular proteins that are important for HIV-1 infection (10, 12, 27). Substitutions at Thr107, including T107N found in the 5Mut virus, decrease the sensitivity of HIV-1 to inhibition by expression of the truncated CPSF6 protein (CPSF6-358). The CPSF6 binding site overlaps a highly conserved region of HIV-1 CA (30), and CPSF6 binding has recently been linked to evasion of innate immune sensing of the viral DNA following reverse transcription (28). Moreover, CA mutations that arise in vivo to restore fitness to cytotoxic T lymphocyte (CTL) escape mutants appear to preserve CPSF6 binding, despite facile loss of the interaction during HIV-1 replication in vitro (9). Thus, constraints to avoid loss of CPSF6 binding via mutations in the PF74 binding pocket may impose a barrier to emergence of clinical resistance to inhibitors targeting this site. We observed that 5Mut CA tubes bind CPSF6 to an extent between those of the corresponding wild-type and N74D mutant tubes, suggesting that the combination of the mutations may partially preserve CPSF6 binding. Analogous to the phenotype of the N74D mutant (28), it is plausible that the impaired binding to CPSF6 contributes to the markedly impaired replication exhibited by 5Mut in cultured macrophages.
PF74 binds within a natural pocket formed between helices 3, 4, 5, and 7 in the CA N-terminal domain (23). 5Mut particles bind very low levels of PF74 relative to the wild type, likely accounting for most or all of the observed resistance. While several of the double and triple mutants were apparently as impaired for PF74 binding as 5Mut, some exhibited intermediate resistance to PF74. Thus, the resistance exhibited by 5Mut may not be completely due to decreased PF74 binding. We previously showed that the E45A CA mutant, which exhibits hyperstable cores, is highly resistant to PF74 despite binding normal levels of the inhibitor (24). Conversely, the quadruple mutant 67/70/87/111 bound PF74 very weakly, yet infection by the virus was fairly sensitive to PF74 inhibition. Thus, it remains possible that additional effects of the CA substitutions, such as changes in intrinsic capsid stability or interactions with host factors in target cells, contribute to PF74 resistance. Consistent with the latter interpretation, PF74 antiviral activity is potentiated by binding of the host protein cyclophilin A to the viral capsid in target cells (24), and depletion of the nuclear transport proteins TNPO3 or Nup153 from target cells reduces HIV-1 sensitivity to PF74 (12, 31). Additionally, the 5Mut virus was insensitive to inhibition by CPSF6-358, owing to changes in the CPSF6 binding site in CA. The impaired replication of 5Mut in macrophages is also suggestive of altered virus-host cell interactions, as is the pronounced dependence of 5Mut replication on the virus inoculum that we observed in T cells. Thus, it should be informative to examine the interaction of the 5Mut capsid with additional host factors—for example, Nup153, which also appears to interact with CA at the PF74 binding pocket (12). The inefficient binding of 5Mut to CPSF6 may also promote a cellular innate immune response, as previously reported for the N74D CA mutant (28).
Our results demonstrate that HIV-1 resistance to PF74 requires multiple amino acid substitutions in CA that reduce the binding of PF74 while relieving fitness defects resulting from the K70R and L111I substitutions. Many substitutions in CA result in replication defects, indicating that CA is under strict evolutionary constraints (5, 32). Moreover, CA epitopes are targets for the host cellular immune response, further restricting CA sequence variation in vivo (33–35). The requirement for multiple changes in CA for strong PF74 resistance suggests that clinical resistance to inhibitors targeting the PF74 binding pocket could be limited by evolutionary constraints, making it difficult for HIV-1 to evade effective antivirals targeting the viral capsid.
Several HIV-1 capsid-targeting small-molecule inhibitors have been reported, although none of the molecules exhibits the requisite potency for drug development. BI-2 is another CA-targeting inhibitor that has been described as binding CA in the PF74-binding pocket (36). BI-2 is less potent an HIV-1 inhibitor than PF74, and effective resistance to this compound can be conferred by a single amino acid substitution (T107N). BI-2 lacks the indole group of PF74 that extends out of the NTD pocket toward the NTD-CTD interface. Thus, the higher potency of PF74 and its more complex resistance pattern may be due to structural effects of the compound acting at the NTD-CTD interface. Accordingly, PF74 appears to destabilize the viral capsid, while BI-2 does not. These observations suggest that efforts to improve the potency of PF74 may benefit from a focus on modifications targeting its indole moiety.
ACKNOWLEDGMENTS
This work was supported by NIH grant R01 AI089401.
We thank Reeves Crabtree for technical assistance and Vineet Kewalramani for the CPSF6-358-expressing cell line. PF74 and triiodo-PF74 were provided by the Vanderbilt Institute of Chemical Biology Synthesis Core, Vanderbilt University, Nashville, TN. The following reagent was obtained through the NIH AIDS Research and Reference Reagent Program: hybridoma 183-H12-5C from Bruce Chesebro.
REFERENCES
- 1.Sundquist WI, Krausslich HG. 2012. HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med 2:a006924. doi: 10.1101/cshperspect.a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamashita M, Perez O, Hope TJ, Emerman M. 2007. Evidence for direct involvement of the capsid protein in HIV infection of nondividing cells. PLoS Pathog 3:1502–1510. doi: 10.1371/journal.ppat.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamashita M, Emerman M. 2005. The cell cycle independence of HIV infections is not determined by known karyophilic viral elements. PLoS Pathog 1:e18. doi: 10.1371/journal.ppat.0010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang S, Murakami T, Agresta BE, Campbell S, Freed EO, Levin JG. 2001. Human immunodeficiency virus type 1 N-terminal capsid mutants that exhibit aberrant core morphology and are blocked for initiation of reverse transcription in infected cells. J Virol 75:9357–9366. doi: 10.1128/JVI.77.23.12592-12602.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. 2003. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J Virol 77:5439–5450. doi: 10.1128/JVI.77.9.5439-5450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byeon IJ, Meng X, Jung J, Zhao G, Yang R, Ahn J, Shi J, Concel J, Aiken C, Zhang P, Gronenborn AM. 2009. Structural convergence between cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell 139:780–790. doi: 10.1016/j.cell.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forshey BM, von Schwedler U, Sundquist WI, Aiken C. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol 76:5667–5677. doi: 10.1128/JVI.76.11.5667-5677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, Ahn J, Gronenborn AM, Schulten K, Aiken C, Zhang P. 2013. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 497:643–646. doi: 10.1038/nature12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henning MS, Dubose BN, Burse MJ, Aiken C, Yamashita M. 2014. In vivo functions of CPSF6 for HIV-1 as revealed by HIV-1 capsid evolution in HLA-B27-positive subjects. PLoS Pathog 10:e1003868. doi: 10.1371/journal.ppat.1003868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, Takemura T, Shelton K, Taniuchi I, Li Y, Sodroski J, Littman DR, Coffin JM, Hughes SH, Unutmaz D, Engelman A, KewalRamani VN. 2010. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7:221–233. doi: 10.1016/j.chom.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matreyek KA, Engelman A. 2011. The requirement for nucleoporin NUP153 during human immunodeficiency virus type 1 infection is determined by the viral capsid. J Virol 85:7818–7827. doi: 10.1128/JVI.00325-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matreyek KA, Yucel SS, Li X, Engelman A. 2013. Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog 9:e1003693. doi: 10.1371/journal.ppat.1003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, Hue S, Fletcher AJ, Lee K, KewalRamani VN, Noursadeghi M, Jenner RG, James LC, Bushman FD, Towers GJ. 2011. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog 7:e1002439. doi: 10.1371/journal.ppat.1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meehan AM, Saenz DT, Guevera R, Morrison JH, Peretz M, Fadel HJ, Hamada M, J van Deursen Poeschla EM. 2014. A cyclophilin homology domain-independent role for Nup358 in HIV-1 infection. PLoS Pathog 10:e1003969. doi: 10.1371/journal.ppat.1003969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krishnan L, Matreyek KA, Oztop I, Lee K, Tipper CH, Li X, Dar MJ, Kewalramani VN, Engelman A. 2010. The requirement for cellular transportin 3 (TNPO3 or TRN-SR2) during infection maps to human immunodeficiency virus type 1 capsid and not integrase. J Virol 84:397–406. doi: 10.1128/JVI.01899-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sayah DM, Sokolskaja E, Berthoux L, Luban J. 2004. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430:569–573. doi: 10.1038/nature02777. [DOI] [PubMed] [Google Scholar]
- 17.Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. 2004. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 18.Goujon C, Moncorge O, Bauby H, Doyle T, Ward CC, Schaller T, Hue S, Barclay WS, Schulz R, Malim MH. 2013. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502:559–562. doi: 10.1038/nature12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Z, Pan Q, Ding S, Qian J, Xu F, Zhou J, Cen S, Guo F, Liang C. 2013. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 14:398–410. doi: 10.1016/j.chom.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 20.Kane M, Yadav SS, Bitzegeio J, Kutluay SB, Zang T, Wilson SJ, Schoggins JW, Rice CM, Yamashita M, Hatziioannou T, Bieniasz PD. 2013. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 502:563–566. doi: 10.1038/nature12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nisole S, Lynch C, Stoye JP, Yap MW. 2004. A Trim5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc Natl Acad Sci U S A 101:13324–13328. doi: 10.1073/pnas.0404640101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bocanegra R, Rodriguez-Huete A, Fuertes MA, Del Alamo M, Mateu MG. 2012. Molecular recognition in the human immunodeficiency virus capsid and antiviral design. Virus Res 169:388–410. doi: 10.1016/j.virusres.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 23.Blair WS, Pickford C, Irving SL, Brown DG, Anderson M, Bazin R, Cao J, Ciaramella G, Isaacson J, Jackson L, Hunt R, Kjerrstrom A, Nieman JA, Patick AK, Perros M, Scott AD, Whitby K, Wu H, Butler SL. 2010. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog 6:e1001220. doi: 10.1371/journal.ppat.1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi J, Zhou J, Shah VB, Aiken C, Whitby K. 2011. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J Virol 85:542–549. doi: 10.1128/JVI.01406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aiken C, Trono D. 1995. Nef stimulates human immunodeficiency virus type 1 proviral DNA synthesis. J Virol 69:5048–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pornillos O, Ganser-Pornillos BK, Banumathi S, Hua Y, Yeager M. 2010. Disulfide bond stabilization of the hexameric capsomer of human immunodeficiency virus. J Mol Biol 401:985–995. doi: 10.1016/j.jmb.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price AJ, Fletcher AJ, Schaller T, Elliott T, Lee K, KewalRamani VN, Chin JW, Towers GJ, James LC. 2012. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog 8:e1002896. doi: 10.1371/journal.ppat.1002896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, Jacques DA, Selwood DL, James LC, Noursadeghi M, Towers GJ. 2013. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 503:402–405. doi: 10.1038/nature12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ambrose Z, Lee K, Ndjomou J, Xu H, Oztop I, Matous J, Takemura T, Unutmaz D, Engelman A, Hughes SH, KewalRamani VN. 2012. Human immunodeficiency virus type 1 capsid mutation N74D alters cyclophilin A dependence and impairs macrophage infection. J Virol 86:4708–4714. doi: 10.1128/JVI.05887-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li G, Verheyen J, Rhee SY, Voet A, Vandamme AM, Theys K. 2013. Functional conservation of HIV-1 Gag: implications for rational drug design. Retrovirology 10:126. doi: 10.1186/1742-4690-10-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shah VB, Shi J, Hout DR, Oztop I, Krishnan L, Ahn J, Shotwell MS, Engelman A, Aiken C. 2013. The host proteins transportin SR2/TNPO3 and cyclophilin A exert opposing effects on HIV-1 uncoating. J Virol 87:422–432. doi: 10.1128/JVI.07177-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rihn SJ, Wilson SJ, Loman NJ, Alim M, Bakker SE, Bhella D, Gifford RJ, Rixon FJ, Bieniasz PD. 2013. Extreme genetic fragility of the HIV-1 capsid. PLoS Pathog 9:e1003461. doi: 10.1371/journal.ppat.1003461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinez-Picado J, Prado JG, Fry EE, Pfafferott K, Leslie A, Chetty S, Thobakgale C, Honeyborne I, Crawford H, Matthews P, Pillay T, Rousseau C, Mullins JI, Brander C, Walker BD, Stuart DI, Kiepiela P, Goulder P. 2006. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J Virol 80:3617–3623. doi: 10.1128/JVI.80.7.3617-3623.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Battivelli E, Migraine J, Lecossier D, Yeni P, Clavel F, Hance AJ. 2011. Gag cytotoxic T lymphocyte escape mutations can increase sensitivity of HIV-1 to human TRIM5alpha, linking intrinsic and acquired immunity. J Virol 85:11846–11854. doi: 10.1128/JVI.05201-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schneidewind A, Brockman MA, Yang R, Adam RI, Li B, Le Gall S, Rinaldo CR, Craggs SL, Allgaier RL, Power KA, Kuntzen T, Tung CS, LaBute MX, Mueller SM, Harrer T, McMichael AJ, Goulder PJ, Aiken C, Brander C, Kelleher AD, Allen TM. 2007. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J Virol 81:12382–12393. doi: 10.1128/JVI.01543-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lamorte L, Titolo S, Lemke CT, Goudreau N, Mercier JF, Wardrop E, Shah VB, von Schwedler UK, Langelier C, Banik SS, Aiken C, Sundquist WI, Mason SW. 2013. Discovery of novel small molecule HIV-1 replication inhibitors that stabilize capsid complexes. Antimicrob Agents Chemother 57:4622–4631. doi: 10.1128/AAC.00985-13. [DOI] [PMC free article] [PubMed] [Google Scholar]