

ABSTRACT
A mutation in herpes simplex virus 1 dUTPase (vdUTPase), which precluded its phosphorylation at Ser-187, decreased viral neurovirulence and increased mutation frequency in progeny virus genomes in the brains of mice where endogenous cellular dUTPase activity was relatively low, and overexpression of cellular dUTPase restored viral neurovirulence and mutation frequency altered by the mutation. Thus, phosphorylation of vdUTPase appeared to regulate viral virulence and genome integrity by compensating for low cellular dUTPase activity in vivo.
IMPORTANCE Many DNA viruses encode a homolog of host cell dUTPases, which are known to function in accurate replication of cellular DNA genomes. The viral dUTPase activity has long been assumed to play a role in viral replication by preventing mutations in progeny virus genomes if cellular dUTPase activity was not sufficient. Here, we showed that a mutation in herpes simplex virus 1 dUTPase, which precluded its phosphorylation at Ser-187 and reduced its activity, decreased viral neurovirulence and increased mutation frequency in progeny virus genomes in the brains of mice where endogenous cellular dUTPase activity was relatively low. In contrast, overexpression of cellular dUTPase restored viral neurovirulence and mutation frequency altered by the mutation in the brains of mice. This is the first report, to our knowledge, directly showing that viral dUTPase activity regulates viral genome integrity and pathogenicity by compensating for insufficient cellular dUTPase activity in vivo.
INTRODUCTION
Preserving the integrity of their genetic information during genome replication is of vital importance for all organisms. Noncanonical nucleotides are constantly generated during nucleotide metabolism, and their misincorporation into the genome during replication may result in increased mutagenesis and overload of the DNA excision repair system, leading to multiple DNA strand breaks and cell death (1, 2). The most common noncanonical nucleoside triphosphate is dUTP, which is continuously produced in the pyrimidine biosynthesis pathway by phosphorylation of dUDP or deamination of dCTP (1, 3). dUTPase catalyzes dUTP cleavage to dUMP and pyrophosphate, thereby reducing misincorporation of dUTP into the genome (4, 5). In addition, dUTPase also plays a role in providing a substrate for thymidylate synthase, which converts dUMP to TMP, a major biosynthetic pathway for TTP (6–8). Interestingly, a number of viruses, including herpesviruses, poxviruses, adenoviruses, D-type retroviruses, and African swine fever virus (ASFV), encode their own dUTPases, suggesting the importance of dUTPases in the life cycles of these viruses (4, 9–11).
In this study, we focused on the dUTPase encoded by herpes simplex virus 1 (HSV-1), one of the best-studied members of the Herpesviridae family, which causes a variety of diseases such as mucocutaneous diseases, keratitis, skin diseases, and encephalitis (12). HSV-1 dUTPase (vdUTPase) is encoded by the UL50 gene and is conserved throughout the Herpesviridae family (13, 14). We recently reported that Us3, an HSV-1-encoded serine/threonine protein kinase, phosphorylated vdUTPase at serine 187 (Ser-187), which upregulated its enzymatic activity in HSV-1-infected cells (15). We also showed that this phosphorylation promoted viral replication in human neuroblastoma SK-N-SH cells and viral replication and virulence in the central nervous system (CNS) of mice (15, 16). Thus, regulation of vdUTPase activity by Us3 phosphorylation of vdUTPase Ser-187 appeared to be important for HSV-1 replication and pathogenicity. Although the mechanism by which dUTPases encoded by HSV-1 and other viruses act in viral replication and pathogenicity in vitro and in vivo has not completely been elucidated, it has long been assumed that dUTPase activity is critical in the replication of viruses encoding a dUTPase. Such viral dUTPase activity may compensate if there is not sufficient host cell dUTPase activity for efficient viral replication, e.g., in resting and differentiated cells, such as neurons and macrophages, where cellular dUTPase activity has been suggested to be low (12, 17). In agreement with this hypothesis, replication of recombinant ASFV and D-type retroviruses with mutations in their vdUTPases was significantly reduced in nondividing cells in vitro, whereas replication in actively dividing cells was only minimally decreased (17–21).
We recently reported data supporting this hypothesis more directly (22). First, YK751 (vdUTPaseS187A), an HSV-1 mutant with an alanine substitution for vdUTPase Ser-187 (S187A) (Fig. 1), which blocked Us3 phosphorylation of vdUTPase Ser-187 and reduced vdUTPase activity in HSV-1-infected cells (15), replicated less efficiently than wild-type virus in SK-N-SK cells that had lower endogenous cellular dTUPase activity than in human carcinoma HEp-2 cells, in which YK751 (vdUTPaseS187A) replicated as well as did wild-type virus (22). Second, knockdown of cellular dUTPase in HEp-2 cells reduced replication of YK751 (vdUTPaseS187A) but not of wild-type virus (22). Third, overexpression of cellular dUTPase in SK-N-SH cells and in cellular dUTPase knockdown HEp-2 cells restored the replication of an HSV-1 mutant virus with the vdUTPase S187A mutation to that of the wild-type virus in both cell types (22). These results suggested that Us3 phosphorylation of vdUTPase Ser-187 upregulated its enzymatic activity and that this upregulation compensated for low cellular dUTPase activity for efficient HSV-1 replication, at least in cell cultures. This also appeared to be the case in vivo, based on our previous report that Us3 phosphorylation of vdUTPase Ser-187 promoted viral replication and virulence in the CNS of mice, in which most cells are not dividing, but played no obvious role in viral replication and pathogenicity in the eyes and vaginas of mice, in which the HSV-1 target epithelial cells are actively dividing (16, 23, 24). However, experimental data to directly prove that the dUTPase of HSV-1 and other viruses is needed to compensate for insufficient cellular dUTPase activity for viral replication and pathogenicity in some cells in vivo have not been reported thus far. Furthermore, there is a lack of information on the effect of regulation of vdUTPase activity on viral genome integrity during HSV-1 replication in vitro and in vivo.
FIG 1.
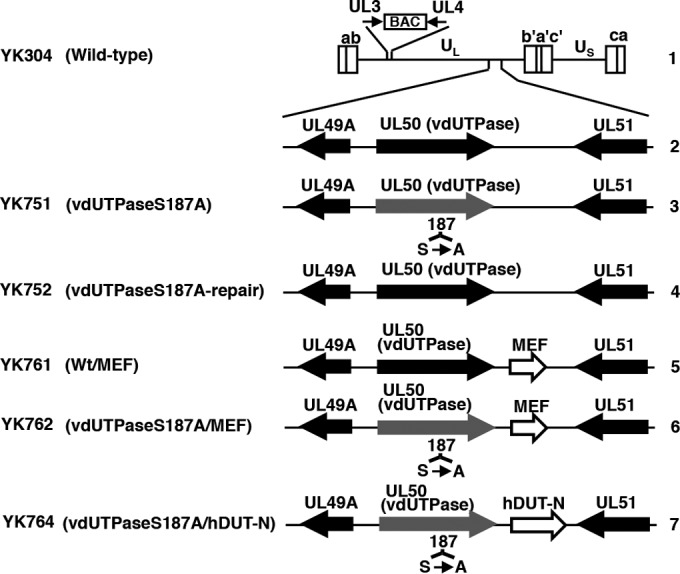
Schematic diagrams of the genome structure of the wild-type and recombinant HSV-1 viruses used in this study. Line 1, wild-type HSV-1 (YK304) genome carrying a bacmid (BAC) in the intergenic region between UL3 and UL4. Line 2, domain with the UL49A, UL50 (vdUTPase), and UL51 open reading frames. Line 3, recombinant virus YK751 with an S187A mutation in the UL50 (vdUTPase) gene. Line 4, recombinant virus YK752 with the repaired vdUTPase S187A mutation. Line 5, recombinant virus YK761 carrying the MEF foreign gene expression cassette (an MEF tag with Myc and Flag epitopes and a TEV protease cleavage site) inserted into the intergenic region between UL50 (vdUTPase) and UL51. Line 6, recombinant virus YK762 with the vdUTPase S187A mutation and carrying MEF. Line 7, recombinant virus YK764 with the vdUTPase S187A mutation and carrying the hDUT-N foreign gene expression cassette (the nuclear isoform of human dUTPase) inserted into the intergenic region between UL50 (vdUTPase) and UL51.
In the present study, we compared the levels of cellular dUTPase activity in mouse brains, eyes, and vaginas and examined whether overexpression of cellular dUTPase restored HSV-1 replication and virulence impaired by the vdUTPase S187A mutation in the CNS of mice, to address questions about the function of vdUTPase in HSV-1 infections. We also investigated the effect of the vdUTPase S187A mutation on the mutation frequency in progeny HSV-1 genomes in cellular dUTPase knockdown and control HEp-2 cells and in the CNS of mice. In addition, we studied the effect of overexpression of cellular dUTPase in the CNS of mice on the mutation frequency in progeny virus genomes of an HSV-1 strain with the vdUTPase S187A mutation.
MATERIALS AND METHODS
Cells and viruses.
The simian kidney epithelial Vero line was described previously (25). sh-hDUT-HEp-2 and sh-Luc-HEp-2 cells were a cellular dUTPase knockdown HEp-2 cell line stably expressing short hairpin RNA (shRNA) against the 3′ untranscribed region (UTR) in cellular dUTPase mRNA and a control cell line expressing shRNA against the open reading frame (ORF) in firefly luciferase mRNA, respectively, and were described previously (22). HSV-1 wild-type strain HSV-1(F); recombinant virus YK751 with a vdUTPase S187A mutation (vdUTPaseS187A); recombinant virus YK752, in which the vdUTPase S187A mutation in YK751 was repaired (vdUTPaseS187A-repair); recombinant virus YK761 carrying an expression cassette consisting of the Egr-1 promoter, an MEF tag consisting of Myc and Flag epitopes, and a tobacco etch virus (TEV) protease cleavage site and bidirectional polyadenylation signals of HSV-1 UL21 and UL22 genes (EGRp-MEF-polyA) in the intergenic region between UL50 and UL51 genes (Wt/MEF); recombinant virus YK762 with an S187A mutation in vdUTPase and the EGRp-MEF-polyA expression cassette (vdUTPaseS187A/MEF); and recombinant virus YK764 with an S187A mutation in vdUTPase and an expression cassette consisting of the Egr-1 promoter, the human hDUT-N ORF encoding the dominant nuclear isoform of human dUTPase, and bidirectional polyadenylation signals of HSV-1 UL21 and UL22 genes (vdUTPaseS187A/hDUT-N) were described previously (15, 22, 26) (Fig. 1).
Animal studies.
Female ICR mice were purchased from Charles River. For all animal studies, after choosing the mean weight ± 4 g of mice, mice were randomly allocated into each group. For intracranial infection, 3-week-old mice were infected intracranially with 102 PFU of each of the indicated viruses as described previously (25, 27). Mice were monitored daily, and mortality from 1 to 21 days postinfection was attributed to the inoculated virus. To determine viral titers in the brains, mice were inoculated intracranially with 102 PFU of the indicated virus as described previously (27, 28). Briefly, at 3 days postinfection, the mice were sacrificed, and the whole brain was removed, sonicated in 500 μl of 199 medium containing 1% fetal calf serum and antibiotics, and frozen at −80°C. Frozen samples were later thawed, and viral titers in the supernatants obtained after centrifugation of the samples were determined by standard plaque assays on Vero cells. All animal experiments were carried out in accordance with the Guidelines for Proper Conduct of Animal Experiments, Science Council of Japan. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the Institute of Medical Science, The University of Tokyo (IACUC protocol approval number 19-26).
dUTPase enzyme assay.
Brains, eyes, or vaginas were removed from 3-week-old female mice, 5-week-old female mice, or 5-week-old female mice pretreated with 1.67 mg of medroxyprogesterone (Depo-M; Vesco), respectively; washed with 10 ml phosphate-buffered saline (PBS); and sonicated in 500 μl Nonidet P-40 (NP-40) buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1.0% NP-40). Protein concentrations in the supernatants obtained after a brief centrifugation were determined using a Bio-Rad protein assay kit. A 60-ng sample of each supernatant was then assayed for dUTPase enzymatic activity as described previously (22, 29, 30). Briefly, sample from HSV-1-infected mouse tissues was mixed with 200 μl reaction buffer (50 mM Tris-HCl [pH 8.0], 2 mM β-mercaptoethanol, 1 mM MgCl2, 0.1% bovine serum albumin, 2 mM p-nitrophenylphosphate, 0.24 nM [3H]dUTP [28.8 Ci/mmol; PerkinElmer]). The reaction was allowed to proceed for 30 min at 37°C and then terminated by spotting the reaction mixture onto DE81 circle discs (Whatman). The discs were washed three times for 5 min each with washing solution (1 mM ammonium formate, 4 M formic acid), followed by one wash with 95% ethanol for 3 min. The discs were air dried and counted for radioactivity using an LS3801 scintillation counter (Beckman).
HSV mutation frequency in cell cultures and mouse CNS.
To measure the mutation frequency in the progeny virus genomes in cell cultures, sh-hDUT-HEp-2 and sh-Luc-HEp-2 cells were infected with the indicated virus at a multiplicity of infection (MOI) of 5, harvested 36 h postinfection, and lysed in 500 μl lysis buffer (10 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1.5 mM MgCl2, 0.1% Nonidet P-40 [NP-40]). After a brief centrifugation, β-mercaptoethanol and EDTA were added to 400 μl of each supernatant to final concentrations of 50 mM and 1 mM, respectively. DNA was extracted with phenol-chloroform and precipitated with ethanol. HSV-1 UL54 nucleotides 114425 to 115153 were then amplified by PCR from the isolated viral genomes with Tks Gflex DNA polymerase (TaKaRa) and cloned into pBluescript II KS(+) (Stratagene). DH5α cells were transformed with the cloned DNA, and 192 or 96 white colonies containing replicated viral genomes from sh-hDUT-HEp-2 or sh-Luc-HEp-2 cells, respectively, infected with each of the indicated viruses were picked. Sequencing was performed using a BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems). To measure the mutation frequency in the progeny virus genomes in the CNS of infected mice, a 3-week-old female ICR mouse was infected intracranially with 102 PFU of each of the indicated viruses and sacrificed at 3 days postinfection. To isolate viral DNAs from the brains of infected mice, whole brains were then removed, washed in PBS, frozen at −80°C, later thawed, lysed in urea lysis buffer (4.7 M urea, 1.3% [wt/vol] SDS, 0.23 M NaCl, 0.67 mM EDTA [pH 8.0], 6.7 mM Tris-HCl [pH 8.0]), extracted with phenol-chloroform, and ethanol precipitated as described previously (31). UL54 nucleotides 114425 to 115153 were then amplified, cloned, and sequenced as described above, except using a pCR-Blunt II-TOPO vector (Invitrogen) and XL1-Blue cells.
Statistical analysis.
Differences in relative dUTPase activity and virus titers were statistically analyzed using the two-tailed Student t test. Differences in frequency of the mutated clones were statistically analyzed using the χ2 test. Differences in mortality of infected mice were statistically analyzed by the log rank test. For the three comparison analyses, P values of <0.0167 (0.05/3), 0.025 (0.05/2), or 0.05 (0.05/1) were sequentially considered significant after Holm's sequentially rejective Bonferroni multiple-comparison adjustment.
RESULTS
Endogenous cellular dUTPase activity in mice.
We reported previously that YK751 (vdUTPaseS187A) replication and virulence in the brains of mice following intracranial inoculation were impaired compared to the YK752 (vdUTPaseS187A-repair) repaired virus (Fig. 1), but YK751 (vdUTPaseS187A) and YK752 (vdUTPaseS187A-repair) replication and pathogenicity in the eyes and vaginas of mice following ocular and vaginal inoculation, respectively, were almost identical (16). To examine endogenous cellular dUTPase activity in mouse tissues, the brains, eyes, and vaginas were removed from 3-week-old female mice, 5-week-old female mice, and 5-week-old female mice pretreated with Depo-M, respectively, which were the same conditions as those in the mouse experimental models that we used previously (16). As shown in Fig. 2, endogenous dUTPase activity in the brains of 3-week-old female mice was significantly lower than in the eyes of 5-week-old female mice or in the vaginas of 5-week-old female mice pretreated with Depo-M. Thus, low endogenous cellular dUTPase activity in viral host tissues may be linked to the lower virulence and replication of YK751 (vdUTPaseS187A) described above, which had reduced vdUTPase activity due to its mutation (15).
FIG 2.
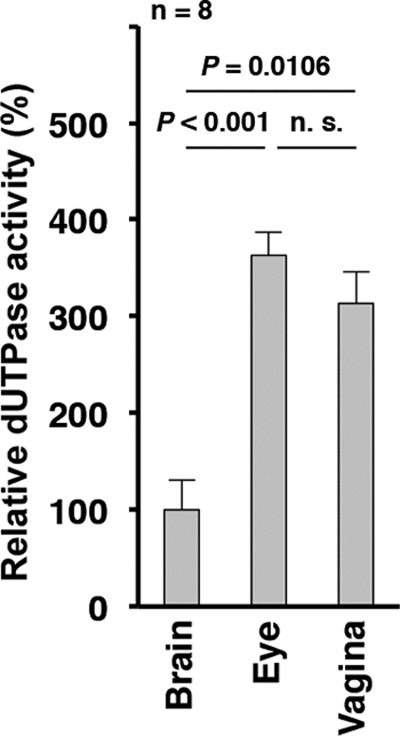
Endogenous cellular dUTPase activity in mouse brains, eyes, and vaginas. Using the same conditions as in the mouse experimental models described previously (16), the brains, eyes, and vaginas from eight 3-week-old female mice, 5-week-old female mice, and 5-week-old female mice pretreated with Depo-M for a week, respectively, were solubilized and 60-ng samples were assayed for dUTPase activity. Each value is the mean ± standard error and is expressed relative to the mean dUTPase activity from brains, which was normalized to 100%. P represents the statistical significance value according to the two-tailed Student t test. n.s., not significant.
Effect of overexpression of cellular endogenous dUTPase on replication and virulence of HSV-1 with the vdUTPase S187A mutation.
To investigate a direct linkage between low cellular dUTPase activity and HSV-1 replication and virulence, we studied the effect of cellular dUTPase overexpression in the CNS of mice on the replication and virulence of HSV-1 with reduced vdUTPase activity due to the vdUTPase S187A mutation (15). Three previously reported (22) recombinant viruses were used for these experiments (Fig. 1): (i) YK761 (Wt/MEF), which carried an expression cassette for the MEF tag in the intergenic region between UL50 and UL51; (ii) YK762 (vdUTPaseS187A/MEF), which had the vdUTPase S187A mutation and carried the MEF tag expression cassette in the intergenic region between UL50 and UL51; and (iii) YK764 (vdUTPaseS187A/hDUT-N), which had the vdUTPase S187A mutation and carried the hDUT-N expression cassette. The MEF tag gene was used as a control foreign gene unrelated to the cellular hDUT-N gene. We previously reported that insertion of foreign genes into the intergenic region between UL50 and UL51 had no effect on viral replication in cell cultures or on viral virulence in mice following intracranial inoculation (32). We also reported that (i) YK764 (vdUTPaseS187A/hDUT-N) overexpressed cellular dUTPase in cell cultures, (ii) the growth curves of the three recombinant viruses (YK761, YK762, and YK764) were almost identical to that of wild-type HSV-1(F) in Vero cells infected at multiplicities of infection (MOIs) of 5 and 0.01, and (iii) insertion of the foreign genes into the intergenic region between UL50 and UL51 in the three recombinant viruses had no effect on expression of UL50 and UL51 (22).
To investigate the effect of overexpression of cellular dUTPase on HSV-1 replication and virulence in the brains of mice, 3-week-old female ICR mice were infected intracranially with 102 PFU of YK761 (Wt/MEF), YK762 (vdUTPaseS187A/MEF), or YK764 (vdUTPaseS187A/hDUT-N) (Fig. 1); virus titers were assayed in the brains of infected mice at 3 days postinfection; and mouse survival was monitored for 21 days postinfection. In agreement with our previous results that the vdUTPase S187A mutation significantly reduced HSV-1 replication and virulence in the CNS of mice following intracranial inoculation (16), there was significantly less progeny virus in the brains of mice infected intracranially with YK762 (vdUTPaseS187A/MEF) than in the brains of mice infected with YK761 (Wt/MEF) (Fig. 3A) and greater survival in mice infected with YK762 (vdUTPaseS187A/MEF) than in mice infected with YK761 (Wt/MEF) (Fig. 3B). In contrast, the survival of mice infected with YK764 (vdUTPaseS187A/hDUT-N) was significantly less than that of mice infected with YK762 (vdUTPaseS187A/MEF), although it was not as low as that of mice infected with YK761 (Wt/MEF) (Fig. 3B). Furthermore, the level of replication of YK764 (vdUTPaseS187A/hDUT-N) in the brains of mice was similar to that of YK761 (Wt/MEF) (Fig. 3A). These results indicated that overexpression of cellular hDUT-N significantly compensated for the reduction in HSV-1 replication and virulence due to the vdUTPase S187A mutation in the CNS of mice.
FIG 3.
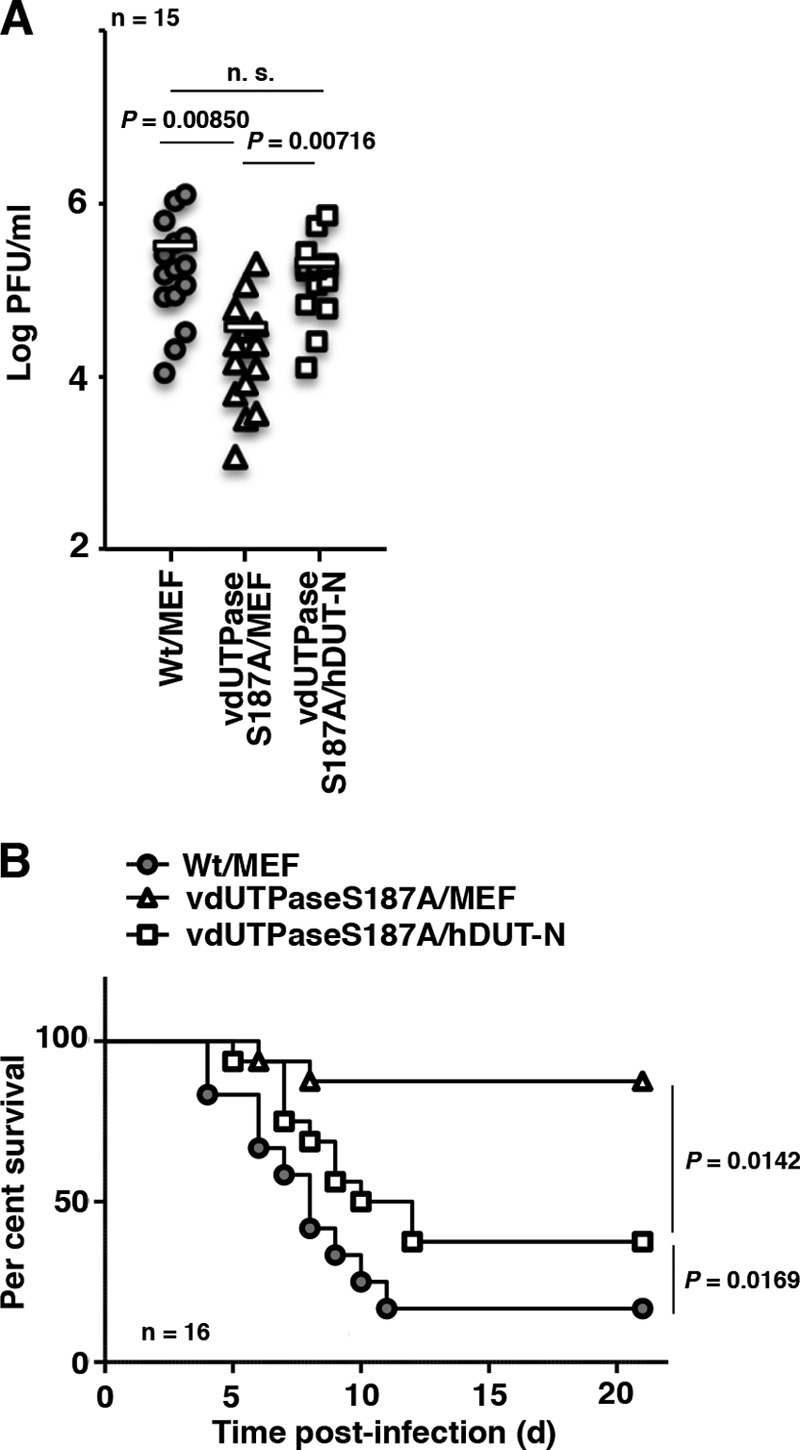
Effect of cellular dUTPase overexpression on HSV-1 replication in the CNS of mice. (A) Fifteen 3-week-old female mice were infected intracranially with the indicated viruses. At 3 days postinfection, the brains of infected mice were harvested, and virus titers were assayed. Each data point is the virus titer in the brain of one mouse. The horizontal bars indicate the mean for each group. (B) Effect of cellular dUTPase overexpression on neurovirulence in the CNS of mice. Sixteen 3-week-old female ICR mice were infected intracranially with the indicated viruses, and survival was monitored for 21 days. P represents the statistical significance value according to the two-tailed Student t test (A) or the log rank test (B). n.s., not significant.
Effect of the vdUTPase S187A mutation on the mutation frequency in progeny viral genomes in cell cultures and mouse CNS.
To examine whether Us3 phosphorylation of vdUTPase Ser-187 was involved in regulation of viral genome integrity in cells with low cellular dUTPase activity, two sets of experiments were performed. In the first set of experiments, we used sh-hDUT-HEp-2 and sh-Luc-HEp-2 cells (22). It was also reported previously that cellular dUTPase was barely detectable in sh-hDUT-HEp-2 cells and that endogenous dUTPase activity in sh-hDUT-HEp-2 cells was significantly lower than that in sh-Luc-HEp-2 cells (22). Therefore, sh-hDUT-HEp-2 and sh-Luc-HEp-2 cells were infected at an MOI of 5 with wild-type HSV-1(F), YK751 (vdUTPaseS187A), or YK752 (vdUTPaseS187A-repair) and harvested 36 h postinfection, and the mutation frequency in the progeny virus genomes in the infected cells was measured. As shown in Table 1, in two experiments the relative mutation frequency in progeny virus genomes in sh-hDUT-HEp-2 cells infected with YK751 (vdUTPaseS187A) was 5.15-fold higher than that in sh-hDUT-HEp-2 cells infected with wild-type HSV-1(F) and was 5.42-fold higher than that in sh-hDUT-HEp-2 cells infected with YK752 (vdUTPaseS187A-repair). In contrast, the relative mutation frequency of progeny virus genomes in sh-Luc-HEp-2 cells infected with YK751 (vdUTPaseS187A) was similar to that in sh-hDUT-HEp-2 cells infected with wild-type HSV-1(F) or YK752 (vdUTPaseS187A-repair) (Tables 1 and 2). The S187A mutation in vdUTPase significantly elevated the frequency of the mutated clones from progeny virus genomes in sh-hDUT-HEp-2 cells (Table 1), whereas the mutation had no effect on that in sh-Luc-HEp-2 (Table 2). These results indicated that the level of endogenous cellular dUTPase activity in HEp-2 cells was sufficient for accurate replication of HSV-1 genomes with the vdUTPase S187A mutation and that wild-type vdUTPase enzymatic activity, which was downregulated by the vdUTPase S187A mutation (15), was required for suppression of incorporation of mutations into replicating HSV-1 genomes in cells with low endogenous dUTPase activity.
TABLE 1.
Sequence analysis of progeny HSV-1 genomes in sh-hDUT-HEp-2 cells
| Virus | No. of expt | Total no. of clones analyzed | No. of mutated clones | Frequency of mutated clonesa | Total no. of nucleotides analyzed | No. of mutated nucleotides | Mutation frequencyb | Fold enhancementc |
|---|---|---|---|---|---|---|---|---|
| HSV-1(F) (wild type) | 1 | 89 | 3 | 57,227 | 3 | 5.24 × 10−5 | ||
| 2 | 86 | 3 | 55,298 | 3 | 5.43 × 10−5 | |||
| Mean | 87.5 | 3 | 0.0343d | 56,263 | 3 | 5.33 × 10−5 | 1.00 | |
| vdUTPaseS187A | 1 | 88 | 12 | 56,584 | 15 | 26.5 × 10−5 | ||
| 2 | 93 | 15 | 59,799 | 17 | 28.4 × 10−5 | |||
| Mean | 90.5 | 13.5 | 0.149 | 58,192 | 16 | 27.5 × 10−5 | 5.15 | |
| vdUTPaseS187A-repair | 1 | 92 | 2 | 59,156 | 2 | 3.38 × 10−5 | ||
| 2 | 92 | 4 | 59,156 | 4 | 6.76 × 10−5 | |||
| Mean | 92 | 3 | 0.0326e,f | 59,156 | 3 | 5.07 × 10−5 | 0.945 |
Frequency of mutated clones was calculated as (mean of number of mutated clones)/(mean of total number of clones analyzed).
Mutation frequency was calculated as (number of mutated nucleotides)/(total number of nucleotides analyzed).
Fold enhancement was calculated as [mean of mutation frequency of HSV-1(F) genome], (mean of mutation frequency of vdUTPaseS187A genome), or (mean of mutation frequency of vdUTPaseS187A-repair genome)/[mean of mutation frequency of HSV-1(F) genome].
Statistically significant difference from vdUTPaseS187A (P < 0.001).
Statistically significant difference from vdUTPaseS187A (P = 0.00286).
Statistically nonsignificant difference from HSV-1(F).
TABLE 2.
Sequence analysis of progeny HSV-1 genomes in sh-Luc-HEp-2 cells
| Virus | No. of expt | Total no. of clones analyzed | No. of mutated clones | Frequency of mutated clonesa | Total no. of nucleotides analyzed | No. of mutated nucleotides | Mutation frequencyb | Fold enhancementc |
|---|---|---|---|---|---|---|---|---|
| HSV-1(F) (wild type) | 1 | 47 | 1 | 30,221 | 1 | 3.31 × 10−5 | ||
| 2 | 47 | 1 | 30,221 | 1 | 3.31 × 10−5 | |||
| Mean | 47 | 1 | 0.0213d | 30,221 | 1 | 3.31 × 10−5 | 1.00 | |
| vdUTPaseS187A | 1 | 46 | 0 | 29,578 | 0 | 0.00 × 10−5 | ||
| 2 | 48 | 1 | 30,864 | 1 | 3.24 × 10−5 | |||
| Mean | 47 | 0.5 | 0.0106 | 30,221 | 0.5 | 1.62 × 10−5 | 0.490 | |
| vdUTPaseS187A-repair | 1 | 47 | 1 | 30,221 | 1 | 3.31 × 10−5 | ||
| 2 | 48 | 1 | 30,864 | 1 | 3.24 × 10−5 | |||
| Mean | 47.5 | 1 | 0.0211d,e | 30,543 | 1 | 3.28 × 10−5 | 0.990 |
Frequency of mutated clones was calculated as (mean of number of mutated clones)/(mean of total number of clones analyzed).
Mutation frequency was calculated as (number of mutated nucleotides)/(total number of nucleotides analyzed).
Fold enhancement was calculated as [mean of mutation frequency of HSV-1(F) genome], (mean of mutation frequency of vdUTPaseS187A genome), or (mean of mutation frequency of vdUTPaseS187A-repair genome)/[mean of mutation frequency of HSV-1(F) genome].
Statistically nonsignificant difference from vdUTPaseS187A.
Statistically nonsignificant difference from HSV-1(F).
In the second set of experiments, 3-week-old female ICR mice were infected intracranially with 102 PFU of YK751 (vdUTPaseS187A), YK752 (vdUTPaseS187A-repair), YK761 (Wt/MEF), YK762 (vdUTPaseS187A/MEF), or YK764 (vdUTPaseS187A/hDUT-N), and the mutation frequency in progeny virus genomes in the brains of the infected mice was measured at 3 days postinfection. As shown in Table 3, the relative mutation frequency in progeny virus genomes in mice infected with YK751 (vdUTPaseS187A) was higher (3.79-fold) than that in mice infected with YK752 (vdUTPaseS187A-repair). In agreement with this result, the relative mutation frequency in progeny virus genomes in mice infected with YK762 (vdUTPaseS187A/MEF) was higher (4.61-fold) than that in mice infected with YK761 (Wt/MEF) (Table 4). The relative mutation frequency in progeny virus genomes in mice infected with YK764 (vdUTPaseS187A/hDUT-N) was lower than that in mice infected with YK762 (vdUTPaseS187A/MEF) in the two experiments (Table 4). The S187A mutation in vdUTPase significantly elevated the frequency of mutated clones from progeny virus genomes in mice and overexpression of cellular dUTPase in part but significantly restored the wild-type frequency of the mutated clones from progeny virus genomes in mice (Tables 3 and 4). These results indicated that, in the brains of infected mice, wild-type vdUTPase activity was required for suppression of incorporation of mutations into replicating HSV-1 genomes, and overexpression of cellular dUTPase (hDUT-N) reduced the increase in mutation frequency in progeny virus genomes due to the vdUTPase S187A mutation.
TABLE 3.
Sequence analysis of progeny HSV-1 genomes in the brains of mice infected with YK751 (vdUTPaseS187A) or YK752 (vdUTPaseS187A-repair)
| Virus | No. of expt | Total no. of clones analyzed | No. of mutated clones | Frequency of mutated clonesa | Total no. of nucleotides analyzed | No. of mutated nucleotides | Mutation frequencyb | Fold enhancementc |
|---|---|---|---|---|---|---|---|---|
| vdUTPaseS187A | 1 | 74 | 9 | 47,582 | 9 | 18.9 × 10−5 | ||
| 2 | 93 | 14 | 59,799 | 14 | 23.4 × 10−5 | |||
| Mean | 83.5 | 11.5 | 0.138 | 53,691 | 11.5 | 21.2 × 10−5 | 3.79 | |
| vdUTPaseS187A-repair | 1 | 88 | 2 | 56,584 | 2 | 3.54 × 10−5 | ||
| 2 | 74 | 5 | 47,582 | 5 | 10.5 × 10−5 | |||
| Mean | 81 | 3.5 | 0.0432d | 52,083 | 3.5 | 7.02 × 10−5 | 1.00 |
Frequency of mutated clones was calculated as (mean of number of mutated clones)/(mean of total number of clones analyzed).
Mutation frequency was calculated as (number of mutated nucleotides)/(total number of nucleotides analyzed).
Fold enhancement was calculated as (mean of mutation frequency of vdUTPaseS187A genome) or (mean of mutation frequency of vdUTPaseS187A-repair genome)/(mean of mutation frequency of vdUTPaseS187A-repair genome).
Statistically significant difference from vdUTPaseS187A (P < 0.001).
TABLE 4.
Sequence analysis of progeny HSV-1 genomes in the brains of mice infected with YK761 (Wt/MEF), YK762 (vdUTPaseS187A/MEF), or YK764 (vdUTPaseS187A/hDUT-N)
| Virus | No. of expt | Total no. of clones analyzed | No. of mutated clones | Proportion of mutated clonesa | Total no. of nucleotides analyzed | No. of mutated nucleotides | Mutation frequencyb | Fold enhancementc |
|---|---|---|---|---|---|---|---|---|
| Wt/MEF | 1 | 86 | 2 | 55,298 | 2 | 3.62 × 10−5 | ||
| 2 | 94 | 5 | 60,442 | 6 | 9.93 × 10−5 | |||
| Mean | 90 | 3.5 | 0.0389d | 57,870 | 4 | 6.77 × 10−5 | 1.00 | |
| vdUTPaseS187A/MEF | 1 | 78 | 11 | 50,154 | 12 | 23.9 × 10−5 | ||
| 2 | 78 | 13 | 50,154 | 13 | 25.9 × 10−5 | |||
| Mean | 78 | 12 | 0.154 | 50,154 | 12.5 | 24.9 × 10−5 | 4.61 | |
| vdUTPaseS187A/hDUT-N | 1 | 87 | 5 | 55,941 | 5 | 8.94 × 10−5 | ||
| 2 | 81 | 8 | 52,083 | 8 | 15.4 × 10−5 | |||
| Mean | 84 | 6.5 | 0.0774e,f | 54,012 | 6.5 | 12.0 × 10−5 | 2.01 |
Frequency of mutated clones was calculated as (mean of number of mutated clones)/(mean of total number of clones analyzed).
Mutation frequency was calculated as (number of mutated nucleotides)/(total number of nucleotides analyzed).
Fold enhancement was calculated as (mean of mutation frequency of Wt/MEF), (mean of mutation frequency of vdUTPaseS187A/MEF), or (mean of mutation frequency of vdUTPaseS187A/hDUT-N genome)/(mean of mutation frequency of Wt/MEF genome).
Statistically significant difference from vdUTPaseS187A/MEF (P < 0.001).
Statistically significant difference from vdUTPaseS187A/MEF (P = 0.0116).
Statistically nonsignificant difference from Wt/MEF.
DISCUSSION
In the present study, we showed that the endogenous dUTPase activity in the CNS of mice, in which the HSV-1 vdUTPase S187A mutation reduced viral replication and pathogenicity (16), was significantly lower than that in the eyes or vaginas of mice, in which the HSV-1 vdUTPase S187A mutation had no effect on viral replication and pathogenicity (16). These observations indicated that low endogenous cellular dUTPase activity in viral host tissues in vivo was linked to lower replication and pathogenicity of HSV-1 viruses with the vdUTPase S187A mutation. The more striking feature in this study was that overexpression of cellular dUTPase significantly restored the viral replication and virulence that were impaired by the vdUTPase S187A mutation in the CNS of mice. Taken together, the data presented here and in our previous studies suggested that a particular level of dUTPase activity was required for efficient HSV-1 replication and virulence in the CNS of mice and that Us3 phosphorylation of vdUTPase Ser-187 upregulated vdUTPase activity to compensate for the low cellular dUTPase activity in the CNS of mice for efficient HSV-1 replication and virulence.
This is the first report, to our knowledge, directly showing that a virus-encoded dUTPase was able to compensate for low cellular dUTPase activity in host cells for efficient viral replication and pathogenicity in vivo. Since this conclusion is in agreement with the observations in cell cultures that we reported previously (22), one might argue that the present study simply confirmed the earlier observations in cell cultures. However, it is known that a viral replication phenotype exhibited in vitro is not always in agreement with the phenotype displayed in vivo. In fact, we previously reported that the S147A mutation in Us3 and the T887A mutation in HSV-1 envelope glycoprotein B (gB), which precludes autophosphorylation of Us3 at Ser-147 and Us3 phosphorylation of gB at Thr-887, respectively, had no effect on HSV-1 replication in cell cultures, although these mutations significantly reduced viral replication in mice (33–36). In contrast, we reported that the null mutation in HSV-1 alkaline nuclease UL12 reduced viral replication in cell cultures approximately 10,000-fold more than the enzyme-dead mutation in UL12 in cell cultures, but the null mutation in UL12 reduced viral neurovirulence in mice only approximately 10-fold more than the enzyme-dead mutation in UL12 (37). Therefore, confirmation that phosphorylation of vdUTPase Ser-187 compensated for low cellular dUTPase activity for efficient viral replication and pathogenicity in vivo is significant and provides insight into the roles of other virus-encoded dUTPases in viral replication and pathogenicity in vivo and into the mechanism of HSV-1 CNS-specific virulence. There has been an analogous report by Chen et al. (38) that HSV-1 thymidine kinase (vTK), which is another viral homolog of a host cell enzyme involved in nucleotide metabolism and is required for viral replication in ganglia of mice and for reactivation from the latency following ganglionic explant (39–42), seemed to compensate for endogenous cellular TK in ganglia in vivo, based on the observation that replacement of vTK with human TK was able to take over the vTK functions in the ganglia of infected mice (38). These results and those in this paper suggested that enzymes encoded by various viruses, which are homologs of host cell enzymes involved in nucleotide metabolism, may in general compensate for low activity of the homologous cellular enzymes for efficient viral replication in vivo.
Another striking feature in this study was that the vdUTPase S187A mutation increased the mutation frequency in progeny HSV-1 genomes in cellular dUTPase knockdown HEp-2 cells, whereas the mutation did not increase the mutation frequency in control HEp-2 cells. We also showed that the vdUTPase S187A mutation increased the mutation frequency in progeny HSV-1 genomes in the CNS of mice and that overexpression of cellular dUTPase attenuated the increase in the mutation frequency due to the vdUTPase S187A mutation. These results indicated that wild-type vdUTPase activity regulated by Us3 phosphorylation of vdUTPase Ser-187 compensated for insufficient cellular dUTPase activity for accurate HSV-1 genome replication in vitro and in vivo. Thus, HSV-1 dUTPase functioned as an antimutator. In agreement with this, Pyles and Thompson previously reported that loss of HSV-1 dUTPase increased the viral mutation frequency in mouse fibroblast NIH 3T3 cells (43). Furthermore, Lerner et al. reported that feline immunodeficiency virus dUTPase was required for reduction of the viral mutation frequency in macrophages in infected cats in which endogenous cellular dUTPase activity was low, but not in lymphocytes with higher endogenous cellular dUTPase activity (19).
We should note, however, that Pyles and Thompson previously reported that the loss of vdUTPase in the HSV-1 17syn+ strain increased the mutation frequency approximately 5-fold in NIH 3T3 cells as described above but had no effect on viral replication in these cells (43). That report contradicts our observation that the vdUTPase S187A mutation in wild-type HSV-1(F) increased the viral mutation frequency approximately 5-fold and significantly reduced viral replication in cellular dUTPase knockdown HEp-2 cells. This discrepancy may be due to differences in cell types, viral strains, and/or methods of detecting mutations in the two studies. A similar contradiction was also reported for vTK: Pyles and Thompson reported that vTK of the HSV-1 17syn+ strain had mutator activity (43), but Hwang et al. were not able to detect mutator activity of vTK in the HSV-1 KOS strain using a different method of detecting mutations (44). Specifically, Pyles and Thompson used the lacZ mutagenesis assay to examine the mutation frequency of the lacZ gene, when it was inserted into the viral genome, by observing the relative ratio of the number of white/blue plaques in plaque assays after X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) staining (43). Unlike the method used to detect mutations in the present study, the lacZ mutagenesis assay does not directly detect mutations in progeny viral genomes. It has been suggested elsewhere (45) that identifying plaques with mutated lacZ genes may not be as simple as expected, since there may be experimental bias using cells infected with a mixture of viruses with wild-type and mutant lacZ genes in which insertion of the lacZ gene at different locations in the viral genome of each recombinant virus, including those used to determine the baseline relative mutation frequency, may affect the replication fidelity.
We have also presented data here showing that a high mutation frequency in progeny HSV-1 genomes in cellular dUTPase knockdown HEp-2 cells and in the CNS of mice was linked to lower viral replication and/or virulence of viruses with the vdUTPase S187A mutation. Therefore, compensation for low cellular dUTPase activity by Us3 phosphorylation of vdUTPase Ser-187 for accurate viral genome replication during HSV-1 replication may be important for maintenance of viral replication and pathogenicity.
ACKNOWLEDGMENTS
We thank Tomoko Ando and Shihoko Koyama for excellent technical assistance.
This study was supported by the Funding Program for Next Generation World-Leading Researchers and Grants for Scientific Research from the Japan Society for the Promotion of Science (JSPS); a contract research fund for the Program of Japan Initiative for Global Research Network on Infectious Diseases (J-GRID) and a grant for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan; and grants from the Takeda Science Foundation and the Ichiro Kanehara Foundation.
REFERENCES
- 1.Kouzminova EA, Kuzminov A. 2004. Chromosomal fragmentation in dUTPase-deficient mutants of Escherichia coli and its recombinational repair. Mol Microbiol 51:1279–1295. doi: 10.1111/j.1365-2958.2003.03924.x. [DOI] [PubMed] [Google Scholar]
- 2.Galperin MY, Moroz OV, Wilson KS, Murzin AG. 2006. House cleaning, a part of good housekeeping. Mol Microbiol 59:5–19. doi: 10.1111/j.1365-2958.2005.04950.x. [DOI] [PubMed] [Google Scholar]
- 3.Mathews CK. 2006. DNA precursor metabolism and genomic stability. FASEB J 20:1300–1314. doi: 10.1096/fj.06-5730rev. [DOI] [PubMed] [Google Scholar]
- 4.Vertessy BG, Toth J. 2009. Keeping uracil out of DNA: physiological role, structure and catalytic mechanism of dUTPases. Acc Chem Res 42:97–106. doi: 10.1021/ar800114w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shlomai J, Kornberg A. 1978. Deoxyuridine triphosphatase of Escherichia coli. Purification, properties, and use as a reagent to reduce uracil incorporation into DNA. J Biol Chem 253:3305–3312. [PubMed] [Google Scholar]
- 6.Kunz BA, Kohalmi SE. 1991. Modulation of mutagenesis by deoxyribonucleotide levels. Annu Rev Genet 25:339–359. doi: 10.1146/annurev.ge.25.120191.002011. [DOI] [PubMed] [Google Scholar]
- 7.Sedwick WD, Brown OE, Glickman BW. 1986. Deoxyuridine misincorporation causes site-specific mutational lesions in the lacI gene of Escherichia coli. Mutat Res 162:7–20. doi: 10.1016/0027-5107(86)90066-7. [DOI] [PubMed] [Google Scholar]
- 8.Bessman MJ, Lehman IR, Adler J, Zimmerman SB, Simms ES, Kornberg A. 1958. Enzymatic synthesis of deoxyribonucleic acid. III. The incorporation of pyrimidine and purine analogues into deoxyribonucleic acids. Proc Natl Acad Sci U S A 44:633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elder JH, Lerner DL, Hasselkus-Light CS, Fontenot DJ, Hunter E, Luciw PA, Montelaro RC, Phillips TR. 1992. Distinct subsets of retroviruses encode dUTPase. J Virol 66:1791–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baldo AM, McClure MA. 1999. Evolution and horizontal transfer of dUTPase-encoding genes in viruses and their hosts. J Virol 73:7710–7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McClure MA. 2001. Evolution of the DUT gene: horizontal transfer between host and pathogen in all three domains of life. Curr Protein Pept Sci 2:313–324. doi: 10.2174/1389203013381062. [DOI] [PubMed] [Google Scholar]
- 12.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823-1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed. Lippincott-Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 13.McGeehan JE, Depledge NW, McGeoch DJ. 2001. Evolution of the dUTPase gene of mammalian and avian herpesviruses. Curr Protein Pept Sci 2:325–333. doi: 10.2174/1389203013380964. [DOI] [PubMed] [Google Scholar]
- 14.Preston VG, Fisher FB. 1984. Identification of the herpes simplex virus type 1 gene encoding the dUTPase. Virology 138:58–68. doi: 10.1016/0042-6822(84)90147-8. [DOI] [PubMed] [Google Scholar]
- 15.Kato A, Tsuda S, Liu Z, Kozuka-Hata H, Oyama M, Kawaguchi Y. 2014. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral dUTPase and regulates its catalytic activity in infected cells. J Virol 88:655–666. doi: 10.1128/JVI.02710-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kato A, Shindo K, Maruzuru Y, Kawaguchi Y. 2014. Phosphorylation of a herpes simplex virus 1 dUTPase by a viral protein kinase, Us3, dictates viral pathogenicity in the central nervous system but not at the periphery. J Virol 88:2775–2785. doi: 10.1128/JVI.03300-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Payne SL, Elder JH. 2001. The role of retroviral dUTPases in replication and virulence. Curr Protein Pept Sci 2:381–388. doi: 10.2174/1389203013381008. [DOI] [PubMed] [Google Scholar]
- 18.Threadgill DS, Steagall WK, Flaherty MT, Fuller FJ, Perry ST, Rushlow KE, Le Grice SF, Payne SL. 1993. Characterization of equine infectious anemia virus dUTPase: growth properties of a dUTPase-deficient mutant. J Virol 67:2592–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lerner DL, Wagaman PC, Phillips TR, Prospero-Garcia O, Henriksen SJ, Fox HS, Bloom FE, Elder JH. 1995. Increased mutation frequency of feline immunodeficiency virus lacking functional deoxyuridine-triphosphatase. Proc Natl Acad Sci U S A 92:7480–7484. doi: 10.1073/pnas.92.16.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ladner RD, McNulty DE, Carr SA, Roberts GD, Caradonna SJ. 1996. Characterization of distinct nuclear and mitochondrial forms of human deoxyuridine triphosphate nucleotidohydrolase. J Biol Chem 271:7745–7751. doi: 10.1074/jbc.271.13.7745. [DOI] [PubMed] [Google Scholar]
- 21.Turelli P, Petursson G, Guiguen F, Mornex JF, Vigne R, Querat G. 1996. Replication properties of dUTPase-deficient mutants of caprine and ovine lentiviruses. J Virol 70:1213–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kato A, Hirohata Y, Arii J, Kawaguchi Y. 2014. Phosphorylation of herpes simplex virus 1 dUTPase upregulated viral dUTPase activity to compensate for low cellular dUTPase activity for efficient viral replication. J Virol 88:7776–7785. doi: 10.1128/JVI.00603-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin-Belmonte F, Perez-Moreno M. 2012. Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer 12:23–38. doi: 10.1038/nrc3169. [DOI] [PubMed] [Google Scholar]
- 24.Jakobsson J, Lundberg C. 2006. Lentiviral vectors for use in the central nervous system. Mol Ther 13:484–493. doi: 10.1016/j.ymthe.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J Virol 77:1382–1391. doi: 10.1128/JVI.77.2.1382-1391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J Gen Virol 2:357–364. doi: 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- 27.Tanaka M, Kato A, Satoh Y, Ide T, Sagou K, Kimura K, Hasegawa H, Kawaguchi Y. 2012. Herpes simplex virus 1 VP22 regulates translocation of multiple viral and cellular proteins and promotes neurovirulence. J Virol 86:5264–5277. doi: 10.1128/JVI.06913-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Imai T, Arii J, Minowa A, Kakimoto A, Koyanagi N, Kato A, Kawaguchi Y. 2011. Role of the herpes simplex virus 1 Us3 kinase phosphorylation site and endocytosis motifs in the intracellular transport and neurovirulence of envelope glycoprotein B. J Virol 85:5003–5015. doi: 10.1128/JVI.02314-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams MV, Cheng Y. 1979. Human deoxyuridine triphosphate nucleotidohydrolase. Purification and characterization of the deoxyuridine triphosphate nucleotidohydrolase from acute lymphocytic leukemia. J Biol Chem 254:2897–2901. [PubMed] [Google Scholar]
- 30.Pyles RB, Sawtell NM, Thompson RL. 1992. Herpes simplex virus type 1 dUTPase mutants are attenuated for neurovirulence, neuroinvasiveness, and reactivation from latency. J Virol 66:6706–6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nie C, Sato K, Misawa N, Kitayama H, Fujino H, Hiramatsu H, Heike T, Nakahata T, Tanaka Y, Ito M, Koyanagi Y. 2009. Selective infection of CD4+ effector memory T lymphocytes leads to preferential depletion of memory T lymphocytes in R5 HIV-1-infected humanized NOD/SCID/IL-2Rgammanull mice. Virology 394:64–72. doi: 10.1016/j.virol.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 32.Morimoto T, Arii J, Akashi H, Kawaguchi Y. 2009. Identification of multiple sites suitable for insertion of foreign genes in herpes simplex virus genomes. Microbiol Immunol 53:155–161. doi: 10.1111/j.1348-0421.2008.00104.x. [DOI] [PubMed] [Google Scholar]
- 33.Imai T, Sagou K, Arii J, Kawaguchi Y. 2010. Effects of phosphorylation of herpes simplex virus 1 envelope glycoprotein B by Us3 kinase in vivo and in vitro. J Virol 84:153–162. doi: 10.1128/JVI.01447-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sagou K, Imai T, Sagara H, Uema M, Kawaguchi Y. 2009. Regulation of the catalytic activity of herpes simplex virus 1 protein kinase Us3 by autophosphorylation and its role in pathogenesis. J Virol 83:5773–5783. doi: 10.1128/JVI.00103-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kato A, Arii J, Shiratori I, Akashi H, Arase H, Kawaguchi Y. 2009. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J Virol 83:250–261. doi: 10.1128/JVI.01451-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J Virol 82:6172–6189. doi: 10.1128/JVI.00044-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujii H, Mugitani M, Koyanagi N, Liu Z, Tsuda S, Arii J, Kato A, Kawaguchi Y. 2014. Role of the nuclease activities encoded by herpes simplex virus 1 UL12 in viral replication and neurovirulence. J Virol 88:2359–2364. doi: 10.1128/JVI.03621-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen SH, Cook WJ, Grove KL, Coen DM. 1998. Human thymidine kinase can functionally replace herpes simplex virus type 1 thymidine kinase for viral replication in mouse sensory ganglia and reactivation from latency upon explant. J Virol 72:6710–6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coen DM, Kosz-Vnenchak M, Jacobson JG, Leib DA, Bogard CL, Schaffer PA, Tyler KL, Knipe DM. 1989. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc Natl Acad Sci U S A 86:4736–4740. doi: 10.1073/pnas.86.12.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tenser RB, Miller RL, Rapp F. 1979. Trigeminal ganglion infection by thymidine kinase-negative mutants of herpes simplex virus. Science 205:915–917. doi: 10.1126/science.224454. [DOI] [PubMed] [Google Scholar]
- 41.Jacobson JG, Ruffner KL, Kosz-Vnenchak M, Hwang CB, Wobbe KK, Knipe DM, Coen DM. 1993. Herpes simplex virus thymidine kinase and specific stages of latency in murine trigeminal ganglia. J Virol 67:6903–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gordon YJ, Gilden DM, Becker Y. 1983. HSV-1 thymidine kinase promotes virulence and latency in the mouse. Invest Ophthalmol Vis Sci 24:599–602. [PubMed] [Google Scholar]
- 43.Pyles RB, Thompson RL. 1994. Mutations in accessory DNA replicating functions alter the relative mutation frequency of herpes simplex virus type 1 strains in cultured murine cells. J Virol 68:4514–4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hwang YT, Wang YA, Lu Q, Hwang CB. 2003. Thymidine kinase of herpes simplex virus type 1 strain KOS lacks mutator activity. Virology 305:388–396. doi: 10.1006/viro.2002.1776. [DOI] [PubMed] [Google Scholar]
- 45.Hwang CB. 2011. DNA replication fidelity of herpes simplex virus. In Jelena KT. (ed), DNA replication and related cellular processes. InTech, Rijeka, Croatia. doi: 10.5772/23548. [DOI] [Google Scholar]