

ABSTRACT
Viral RNA-dependent RNA polymerases are considered to be low-fidelity enzymes, providing high mutation rates that allow for the rapid adaptation of RNA viruses to different host cell environments. Fidelity is tuned to provide the proper balance of virus replication rates, pathogenesis, and tissue tropism needed for virus growth. Using our structures of picornaviral polymerase-RNA elongation complexes, we have previously engineered more than a dozen coxsackievirus B3 polymerase mutations that significantly altered virus replication rates and in vivo fidelity and also provided a set of secondary adaptation mutations after tissue culture passage. Here we report a biochemical analysis of these mutations based on rapid stopped-flow kinetics to determine elongation rates and nucleotide discrimination factors. The data show a spatial separation of fidelity and replication rate effects within the polymerase structure. Mutations in the palm domain have the greatest effects on in vitro nucleotide discrimination, and these effects are strongly correlated with elongation rates and in vivo mutation frequencies, with faster polymerases having lower fidelity. Mutations located at the top of the finger domain, on the other hand, primarily affect elongation rates and have relatively minor effects on fidelity. Similar modulation effects are seen in poliovirus polymerase, an inherently lower-fidelity enzyme where analogous mutations increase nucleotide discrimination. These findings further our understanding of viral RNA-dependent RNA polymerase structure-function relationships and suggest that positive-strand RNA viruses retain a unique palm domain-based active-site closure mechanism to fine-tune replication fidelity.
IMPORTANCE Positive-strand RNA viruses represent a major class of human and animal pathogens with significant health and economic impacts. These viruses replicate by using a virally encoded RNA-dependent RNA polymerase enzyme that has low fidelity, generating many mutations that allow the rapid adaptation of these viruses to different tissue types and host cells. In this work, we use a structure-based approach to engineer mutations in viral polymerases and study their effects on in vitro nucleotide discrimination as well as virus growth and genome replication fidelity. These results show that mutation rates can be drastically increased or decreased as a result of single mutations at several key residues in the polymerase palm domain, and this can significantly attenuate virus growth in vivo. These findings provide a pathway for developing live attenuated virus vaccines based on engineering the polymerase to reduce virus fitness.
INTRODUCTION
Positive-strand RNA viruses replicate by using virally encoded RNA-dependent RNA polymerases (RdRPs) that provide a direct RNA-to-RNA replication function not found in host cells. The structures of many RdRPs, primarily from picornaviruses, caliciviruses, and flaviviruses, have been solved to reveal a core structure composed of the classic right-hand arrangement of finger, palm, and thumb domains (Fig. 1A) (1–3). This core represents the complete polymerases of smaller RNA viruses, such as the picornaviruses, while the polymerases from larger viruses provide additional functions, such as de novo initiation via thumb domain extensions that bind a priming nucleotide triphosphate (NTP) in the product exit channel (4–7) or RNA processing activity via N-terminal methyltransferase domains, as seen in the structure of full-length NS5 from Japanese encephalitis virus (8). One notable aspect of the core RdRP structure is that the tip of the finger domain reaches over the active site to contact the top of the thumb in all structures solved thus far. This precludes the use of major swinging motions within the finger domain as a molecular mechanism for repositioning an NTP from an initial binding site into the “closed” active-site configuration needed for catalysis. Instead, positive-strand RNA virus RdRPs utilize a subtle rearrangement in their palm domains to fully structure the active site for catalysis only upon NTP binding (Fig. 1B) (9, 10). This method appears to be conserved among all positive-stranded RNA viruses based on a consistent “open” active-site conformation seen in polymerase and polymerase-RNA complex structures in the absence of bound NTP, while the “closed” conformation has been seen in poliovirus and norovirus polymerase complexes with RNA and a cognate nucleotide (9, 11).
FIG 1.
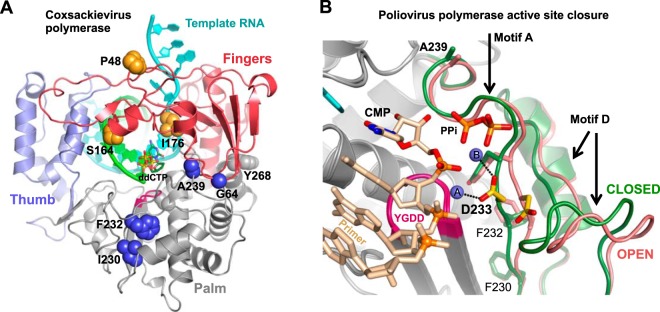
Structure-based modulation of viral RdRP fidelity. (A) Structure of the coxsackievirus B3 3Dpol elongation complex (10). The targeted fidelity-modulating residues are shown as spheres. (B) Detailed view of the molecular movement involved in positive-strand RNA virus RdRP active-site closure whereby poliovirus 3Dpol motifs A and D move toward the active site when transitioning from the open (red) to the closed (green) active-site conformation. Note how Asp233 (yellow) on motif A rotates to interact with both catalytic Mg2+ ions (spheres A and B) in the active site. The view is from atop the active site looking down onto the palm domain, and two strong fidelity-determinant residues, F230 and F232, located directly underneath Asp233 on the motif A β-strand, are shown as sticks.
The viral RdRPs are low-fidelity replicases, having mutation rates of 10−4 to 10−6 that result in a genetically heterogeneous “quasispecies” population wherein individual viruses differ from the consensus genome sequence at a few random sites (12–14). Thorough deep-sequencing analysis reveals differences in mutation frequencies among viruses, with coxsackieviruses and polioviruses having 3.8 and 6.1 mutations per 10 kb, respectively, in total newly synthesized RNA (15). Maintaining the proper genome heterogeneity is essential for virus fitness, as it allows rapid adaptation to different host environments, and viral pathogenesis can be attenuated by increasing or decreasing polymerase fidelity (16). This has been shown for poliovirus (PV), coxsackievirus B3 (CV), and enterovirus 71 (EV71) in mouse studies (17–21) and for chikungunya virus, where fidelity variants lose fitness in both vertebrate and invertebrate hosts (22, 23).
The structure-function relationships responsible for modulating RdRP fidelity are not well understood. Nuclear magnetic resonance (NMR) and molecular dynamics studies suggest that NTP sensing and catalysis involve long-range interaction networks (24, 25), which is consistent with fidelity variants mapping across many regions of the structures. Crystal structures of the PV polymerase elongation complex showed specific conformational changes associated with active-site closure for catalysis (9), and based on these changes, we previously engineered mutations in the homologous CV polymerase. This led to the identification of several palm domain substitutions that increased in vivo mutation frequencies as much as 2.6-fold and provided initial data to show that in vitro 2′-deoxy-NTP discrimination is correlated with observed virus mutation frequencies based on sequencing (19). That study also identified several adaptive mutations in the progeny virus populations, yielding a large set of fidelity variants that can be used to map structure-function relationships in picornaviral RdRPs. Here we present such a mapping study based on in vitro biochemical assays that provide a consistent experimental framework for directly comparing nucleotide discrimination by multiple RdRPs. The results showed that single point mutations can drastically alter the polymerase rate and nucleotide selectivity, suggesting that the positive-strand RNA virus polymerases have retained the unique palm domain-based active-site closure as a mechanism for evolutionarily fine-tuning replication fidelity and subsequent quasispecies distributions.
MATERIALS AND METHODS
Elongation rates and NTP discrimination.
CV and PV polymerases were expressed in and purified from Escherichia coli (10), using a ubiquitin fusion construct to generate the native N-terminal glycine residue (26) that is required for activity (27, 28). Polymerase elongation kinetics were measured by using a stopped-flow-based assay to measure how long it takes to elongate an extended heteropolymeric template by monitoring the fluorescence signal from an environmentally sensitive fluorescein label at the 5′ end of the template (29, 30). Preinitiated elongation complexes were assembled at a high concentration to drive RNA binding (15 μM 3Dpol, 5 μM RNA, 60 μM each ATP and GTP, 2 mM MgCl2) and then diluted to a 10 nM final RNA concentration for the stopped-flow kinetics experiments, which were done at 37°C in a solution containing 75 mM NaCl, 4 mM MgCl2, 50 mM HEPES (pH 6.5), and 2 mM Tris(2-carboxyethyl)phosphine (TCEP) as a reducing agent. Maximal polymerase elongation rates were determined by titrating an equimolar mixture of all four NTPs and by using Michaelis-Menten analysis of elongation rates versus NTP concentrations to determine maximal elongation rates (in nucleotides per second) and nucleotide Km values (19, 29). The temporal stability of stalled elongation complexes was determined by incubating preinitiated complexes at room temperature under high-salt conditions and periodically testing their ability to fully elongate the prebound template without reinitiation, as previously described (30).
Nucleotide triphosphate substrate preference was measured by using a template strand with a unique guanosine nucleotide such that the incorporation of a single cytosine becomes the rate-limiting step for the polymerase to reach the 5′ end of the template and trigger a fluorescein fluorescence change (Fig. 2). We used 2′-dCTP to mimic a noncognate nucleotide and calculated an NTP discrimination factor as the ratio of the catalytic efficiencies of CTP and 2′-dCTP incorporation, i.e., (kpol/Km)CTP/(kpol/Km)dCTP, based on Michaelis-Menten analysis of rate-versus-NTP concentration data. The remaining NTPs were held at 20, 1, and 1 μM concentrations for ATP, GTP, and UTP, respectively. Note that in the study presented here, we moved the unique guanosine to be 8 nucleotides (nt) from the 5′ end of the template, and thus, the entire fluorescence change was CTP dependent, whereas in the previous study, it was located only 4 nucleotides from the end and accounted for the final ≈60% of the fluorescence change (19). The new nonterminal positioning of the guanosine more accurately reflects rapid processive elongation by avoiding a slowing of the polymerase associated with nucleotide addition near the end of the template (29). The change in rates numerically changes the observed NTP discrimination factors, but the relative effects among the different polymerases scale linearly when different RNA substrates are compared.
FIG 2.

Stopped-flow fluorescence-based elongation and fidelity assays. (A) Structure of the hairpin primer-template RNA used in fidelity experiments. The change in fluorescence of the 5′-terminal fluorescein label requires that the polymerase incorporate CTP or 2′-dCTP to extend past the unique guanosine in the template. The stopped-flow data traces illustrate the CTP-dependent fluorescence changes associated with elongation on templates that were preinitiated with GTP and ATP to stall complexes after the CUCU template sequence. The ≈7-s lag phase in the data reflects processive elongation up to the unique guanosine, at which point further elongation becomes dependent on the CTP concentration. (B) Michaelis-Menten analysis of CV 3Dpol A239G fluorescence change (ΔFl) rates as a function of CTP and 2′-dCTP concentrations. The resulting Km and kpol values were used to calculate the nucleotide discrimination factor, which provides an in vitro assessment of polymerase fidelity. (C) Temporal stabilities of stalled CV 3Dpol-RNA elongation complexes (EC) determined by monitoring the fraction of preinitiated RNA that can be elongated to the full-length product as a function of incubation time in the presence of 300 mM NaCl, which prevents the rebinding of any RNA that dissociates from the polymerase. Values represent the time constants and standard errors from a single exponential fit of the data.
Stopped-flow elongation experiments were carried out with an Applied Photophysics SX-20 instrument, where equal volumes of preformed polymerase-RNA elongation complex and NTP solutions were mixed to initiate the reaction. Fluorescence excitation was at 492 nm from a monochromator source with bandwidth set to 9.3 nm, and fluorescein emission was detected by using a 515-nm high-pass Schott glass filter. Experiments at 30°C were done with a Bio-Logic SFM-4000 titrating stopped-flow instrument with an MOS-500 spectrometer and a 496/40 nm long-pass filter (Semrock, Inc.) on the detection channel.
The gel-based assay to determine how efficiently the CV G64S mutant incorporated 2′-dCTP compared to CTP was performed by using an RNA template with a unique guanosine 5 nucleotides from the 5′ end and an IRdye 800RS (Li-Cor Biosciences) imaging label in the hairpin loop. Preinitiated complexes were formed by incubating polymerase, RNA, and GTP for 30 min at room temperature; the samples were moved to 37°C; and 20 μM ATP, 1 μM each GTP and UTP, and various concentrations of either CTP or 2′-dCTP were added. Elongation was stopped 1 min later by the addition of EDTA, and RNA products were resolved on 20% acrylamide–7 M urea denaturing gels, which were imaged with a Li-Cor Odyssey 9120 scanner and quantified by using Li-Cor ImageStudio software.
G-U mismatch incorporation.
To measure the ability of 3Dpol enzymes to incorporate a natural nucleotide mismatch, we carried out initiation and elongation experiments with a CUCUGGUAAG template sequence in the presence of high concentrations of GTP as the only nucleotide (see Fig. 6A). A total of 15 μM 3Dpol was incubated at 37°C with 5 μM RNA in 75 mM NaCl (pH 6.5) and either 40 μM or 1 mM GTP with 4 mM excess MgCl2, samples were quenched at various time points up to 1 h, and the RNA products were visualized on 15% acrylamide denaturing urea gels (Fig. 6B). To directly compare misincorporation by different polymerases during processive elongation, we first formed ∼50% preinitiated complexes in the presence of 40 μM GTP at 75 mM NaCl at room temperature, as described above for the stopped-flow experiments, and then diluted these reaction mixtures 10-fold into a 1 mM GTP solution with 300 mM NaCl to prevent further initiation or rebinding of any preinitiated RNAs that dissociate from 3Dpol. Reaction mixtures were incubated at 37°C; samples were removed and quenched with EDTA after 5, 10, 20, 30, and 60 min; and the product RNAs were visualized on higher-resolution 20% acrylamide denaturing gels (Fig. 6C). The total amount of RNA in all bands larger than the +1 preinitiated product was found to increase linearly as a function of time in the presence of 1 mM GTP (data not shown).
FIG 6.
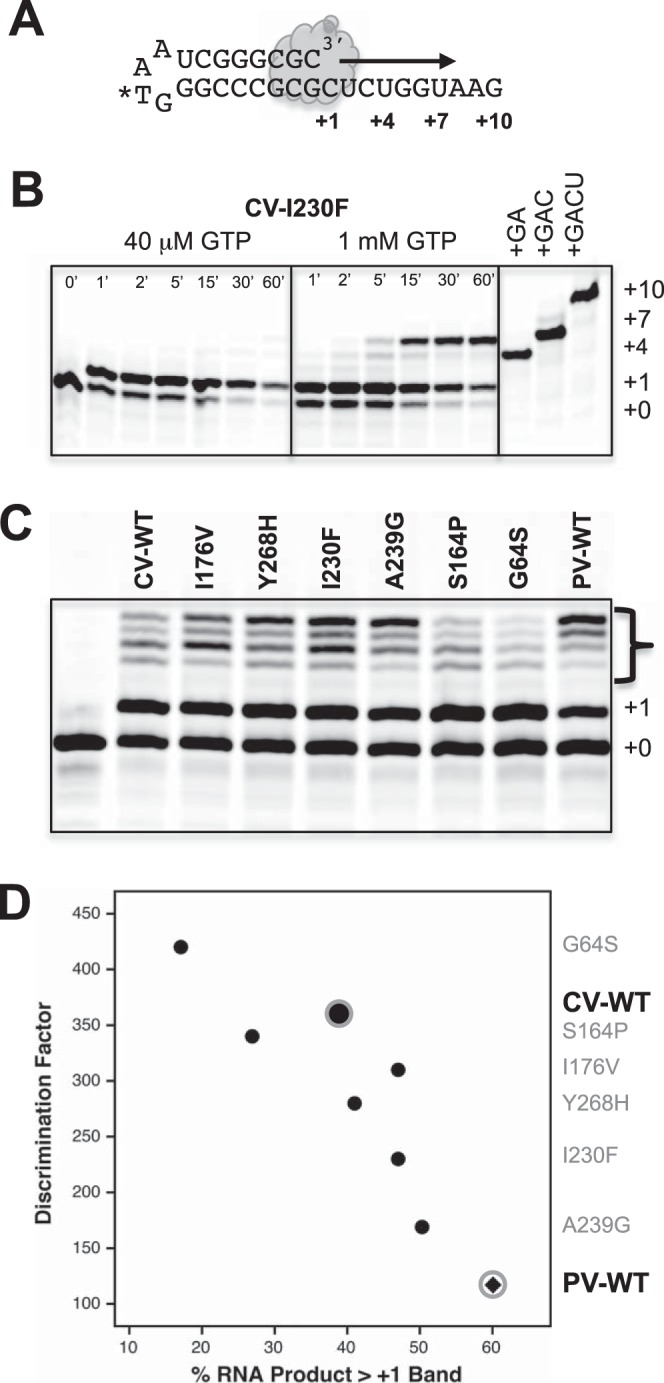
Guanosine-uracil misincorporation data. (A) Schematic of the self-priming hairpin RNA used for G-U misincorporation assays. (B) Initiation time course with CV I230F 3Dpol at 37°C in the presence of either 40 μM GTP or 1 mM GTP and a final salt concentration of 75 mM NaCl. Elongation stops at the expected +1 product at the low GTP concentration but readily extends to longer products via G-U mismatch incorporation at 1 mM GTP. (C) Direct comparison of mismatch incorporation by a set of CV polymerases as well as wild-type PV 3Dpol. Reactions were first initiated for 10 min at room temperature with 40 μM GTP to form a ≈50% +1 product, and the mixtures were then diluted 10-fold into a 1 mM GTP solution containing 300 mM NaCl to prevent RNA rebinding and further initiation. Reactions were quenched after 30 min at 37°C, and products were separated on a 20% acrylamide urea gel. (D) Plot comparing the amounts of misincorporation products (bracketed bands in panel C) with the 2′-dCTP discrimination factors from the stopped-flow-based elongation assays. The data show that guanosine misincorporation is correlated with 2′-dCTP discrimination for the wild type and six coxsackievirus 3Dpol mutants and that wild-type poliovirus 3Dpol has the highest guanosine misincorporation level of all the polymerases tested.
Virus growth and mutation rate assessments.
Coxsackievirus B3 infectious virus studies, high-throughput sequencing of progeny virus genomes, and mutation frequency calculations were carried out as previously described (19).
Structural biology.
CV 3Dpol enzymes without bound RNA were crystallized at 16°C by sitting-drop or hanging-drop vapor diffusion in the previously established ammonium sulfate crystal form (27) and in a new NaCl-based crystal form. Crystallization drops consisted of one part 5 mg/ml protein and one part precipitant solution, the contents of which are footnoted in Table 1, and crystals grew in 1 to 2 weeks. Despite the different conditions, the CV 3Dpol crystals had the same space group and unit cell dimensions. Magnesium soaking experiments were performed with 10-μl drops of reservoir solutions with added 10 mM MgCl2 at 16°C for 5 h. Crystals were harvested and frozen in liquid nitrogen, and diffraction data were collected at Advanced Light Source beamline 4.2.2 (Berkeley, CA), using shutterless data collection (1,800 frames covering 180° in 180 s) on a Taurus detector (RDI, Inc.). Diffraction data were processed with XDS, and all structures were determined by molecular replacement using PHASER and the wild-type (WT) CV structure (PDB accession number 3DDK) as the search model. Model building, refinement, and validation were performed with Coot, Phenix, and MolProbity, respectively, as distributed via the SBgrid consortium (31).
TABLE 1.
Crystallographic data collection and refinement statistics
| Parameter | Value for polymerasec |
||
|---|---|---|---|
| CV 3Dpol F232L NaCl forma (PDB accession no. 4WFX) | CV 3Dpol F232L AmSO4 formb (PDB accession no. 4WFY) | CV 3Dpol WT NaCl forma (PDB accession no. 4WFZ) | |
| Data collection statistics | |||
| Space group | P43212 | P43212 | P43212 |
| Cell dimensions | |||
| a, b, c (Å) | 74.36, 74.36, 288.05 | 74.36, 74.36, 288.05 | 74.42, 74.42, 288.76 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 52.6–1.81 (1.87–1.81) | 52.6–2.06 (2.13–2.06) | 58.9–1.80 (1.86–1.80) |
| Rmerge | 0.103 (1.06) | 0.1283 (0.7495) | 0.1815 (44.34) |
| CC½ | 0.999 (0.596) | 0.999 (0.796) | 0.998 (0.129) |
| CC* | 1 (0.864) | 1 (0.942) | 0.999 (0.478) |
| Mean I/σI | 18.97 (1.53) | 18.65 (3.1) | 12.81 (0.27) |
| Completeness (%) | 99.4 (94.3) | 99.3 (93.3) | 98.2 (84.5) |
| Multiplicity | 12.5 (9.3) | 13.9 (12.7) | 13.4 (8.3) |
| Refinement statistics | |||
| Resolution (Å) | 52–1.8 (1.87–1.81) | 52.6–2.06 (2.13–2.06) | 58.9–1.80 (1.86–1.80) |
| Total no. of reflections | 933,365 (64,255) | 710,311 (59,923) | 1,009,535 (52,999) |
| No. of unique reflections | 74,782 (6,932) | 51,243 (4,732) | 75,393 (6,381) |
| Rwork/Rfree (%) | 19.8/23.3 (27.3/28.9) | 18.5/22.0 (24.2/29.7) | 20.0/23.1 (41.2/47.0) |
| No. of atoms | |||
| Protein | 3,687 | 3,687 | 3,690 |
| Ligand/ion | 2 | 33 | 1 |
| Water | 795 | 536 | 497 |
| B-factor | |||
| Protein | 21.3 | 25.5 | 31.6 |
| Ligand/ion | 24.1 | 41.2 | 24.8 |
| Water | 31.7 | 33.9 | 39.1 |
| RMS deviationd | |||
| Bond lengths (Å) | 0.007 | 0.007 | 0.008 |
| Bond angles (°) | 1.06 | 1.06 | 1.14 |
| Ramachandran plot (%) | 99/100 | ||
| Favored regions | 99 | 98 | 98 |
| Outliers | 0 | 0 | 0.4 |
The NaCl form was grown in a solution containing 0.085 M HEPES (pH 7.5), 3.5 to 3.7 M NaCl, and 15% glycerol.
The AmSO4 form was grown in a solution containing 0.085 M Tris (pH 7.5), 1.1 to 1.3 M (NH4)2SO4, and 25% glycerol.
Values in parentheses are for the highest-resolution shell.
RMS, root mean square.
RESULTS
We analyzed 20 mutations of CV 3Dpol to map structure-function relationships responsible for controlling replication fidelity and processive elongation rates and then tested a subset of these mutations in poliovirus 3Dpol to examine if the effects would cross virus species. Initial characterization experiments showed that all the mutant polymerases initiated on primer-template hairpin RNA substrates with high efficiency to form stable stalled elongation complexes whose RNA replication could be studied by using a stopped-flow rapid-kinetics assay (Fig. 2). The CV Y268H and F232Y mutations were hyperstable (>2,000 min), and the CV G64S-containing mutants were drastically destabilized at 37°C but were only slightly less stable than the wild type at room temperature.
Structural rearrangement within motif D.
Crystal structures of the CV WT, I176V, I230F, and Y268H polymerases retained the same overall conformation as the original WT structure solved in the ammonium sulfate crystal form (27), while the F232L structure in a new high-[NaCl] crystal form revealed a significant rearrangement in motif D (Fig. 3 and Table 1). The rearrangement is centered on a movement of Phe364 that leads to a major rearrangement of the 10-residue Thr356-Asn365 loop at the C-terminal end of motif D (Fig. 3A). In the wild-type structure, Phe364 is tucked into a hydrophobic pocket with the motif D helix below the phenylalanine ring, Pro357 above the phenylalanine ring, and Phe232 at the base of the pocket. In the F232L mutant structure, the Phe364 side chain flips out of its pocket and moves ≈11.5 Å to become tucked up against the thumb domain, and Pro357 drops down into the position previously occupied by the Phe364 ring. Lys360 is no longer pointed toward the NTP entry channel, and its new conformation is stabilized by an interaction between the side-chain CH2 groups and the repositioned Pro357. The mutated F232L residue moves slightly toward the former Phe364 binding pocket but retains the open active-site conformation of motif A. The conformational rearrangement in the F232L structure is accompanied by two bound Na+ ions, one interacting with the motif D loop and the second binding between Asp233 and Asp330 in the same site as the prebound “metal A” Mg2+ ion seen in the coxsackievirus polymerase elongation complex structure (10). Crystal soaking with 10 mM magnesium chloride showed that this metal A site sodium ion was readily displaced by Mg2+ in the F232L mutant based on water coordination distances that were reduced from 2.4 to 2.5 Å down to 2.2 to 2.3 Å.
FIG 3.
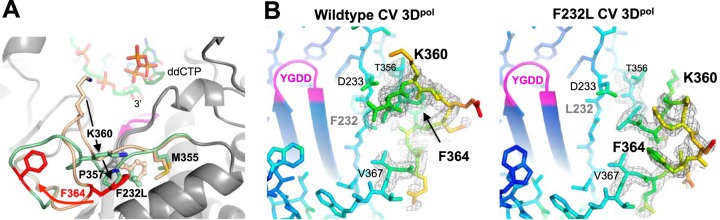
Structural rearrangement within motif D of the CV F232L mutant. (A) Comparison of the wild-type (tan) and F232L (green) loop conformations in the high-NaCl crystal form. Phe364 (red) undergoes a major movement, Pro357 drops down to fill the Phe364 position, and Lys360 moves away from the NTP entry channel. RNA primer and ddCTP are from a superimposed poliovirus elongation complex structure (PDB accession number 3OLB) and are shown to indicate the location of the catalytic site. (B) 5000K simulated annealing omit maps contoured at 2.0 σ to show electron density in the motif D loop region of the NaCl crystal form structures. Atoms are colored according to B-factors, with this loop containing the highest values for the entire structure.
Structure-function mapping of 3Dpol fidelity variants.
Data from stopped-flow elongation assays were used to determine CTP-versus-2′-dCTP nucleotide discrimination as an in vitro measure of polymerase fidelity (Fig. 2). The assay is based on our structural biology finding that the 2′-hydroxyl group plays a critical role in stabilizing the closed conformation of the active site for catalysis (9). A direct comparison of these nucleotide discrimination values with mutation frequencies obtained by high-throughput sequencing of progeny genomes shows a high correlation, with an R value of 0.88 and a P value of <0.01 (Fig. 4A). These data clearly demonstrate that the ability of a given polymerase to incorporate and translocate past a nonnative 2′-deoxy-NTP in vitro is highly predictive of that enzyme's tendency to make a variety of natural misincorporation mutations during in vivo virus replication. Importantly, this biochemical nucleotide discrimination assay allows us to rapidly assess RdRP fidelity, and we can use it to study highly mutagenic polymerases that do not support virus growth. A linear extrapolation of the correlation data indicates that single point mutations in the polymerase can increase mutation frequencies >3-fold, from 4.3 to ≈15 mutations per 10 kb (Fig. 4A, open circles), which is well above the ≈11-mutation maximum observed for viable progeny viruses. This leads us to propose that a value of ≈11 mutations per 10 kb defines an in vivo extinction threshold for coxsackievirus B3.
FIG 4.
Fidelity modulation of coxsackievirus B3 3Dpol. (A) Correlation plot comparing in vitro coxsackievirus 3Dpol CTP-versus-2′-dCTP discrimination factors with in vivo mutation rates obtained by sequencing of progeny genomes. A linear interpolation of data from viable viruses (black circles) was used to predict in vivo mutation rates of several nonviable variants (open circles) that appeared fully functional in the biochemical assays, leading to the proposed CV extinction threshold of ≈11 mutations per 104 nt (see Table 2 for a complete list of residue identities). (B) Correlation plot comparing the in vitro NTP discrimination factors of CV 3Dpol mutants with their maximal in vitro elongation rates to show that the low-fidelity polymerases have higher elongation rates. Mutants in the palm domain are shown as solid circles, while those in the fingers are shown as open circles. (C) Discrimination factor-versus-rate plot showing that the G64S mutant is a slightly high-fidelity variant of CV 3Dpol based on data obtained at 30°C and pH 7. Several other mutants were also tested at this temperature for direct comparison, as indicated. Note that the x axis is the lag-phase elongation rate observed with nonsaturating 60 μM ATP and 10 μM each GTP and UTP and not the maximal rate from an all-NTP titration as for panel B.
Further mechanistic insights are provided by a comparison of discrimination factors with overall processive elongation rates. These are also highly correlated among the CV polymerases studied, with higher elongation rates being characteristic of lower-fidelity enzymes (Fig. 4B). The magnitudes of the speed and fidelity effects map to distinct regions of the RdRP structure: mutations in the palm domain have the largest effects on both fidelity and the elongation rate, while mutations in the finger domain primarily affect the elongation rate and have smaller effects on fidelity. The finding that palm and finger domain mutations have differential effects on rate and fidelity led us to generate a series of CV 3Dpol double mutants to test if these regions of the structure function independently of each other. The data indicate that this can be the case (Fig. 5 and Table 2); for example, combining the palm domain I230F mutation with the finger domain I176V mutation resulted in a polymerase with an even higher elongation rate that retained the lower I230F nucleotide discrimination, and similarly, the addition of the I176V mutation increased the elongation rate of the slow G64S polymerase. Combining the I230F and F232Y mutations, two motif A palm mutations which both have significant fidelity effects on their own, results in an even lower-fidelity polymerase. However, combining the I230F motif A mutation with another palm domain mutation that is not involved in the active-site closure step, S299T, located near the top of the motif B helix, resulted in intermediate fidelity and elongation rates.
FIG 5.

Effects of double mutations on CV 3Dpol function. Shown is a bar graph representation of the CTP-versus-2′-dCTP discrimination factor (top) and elongation rate (bottom) data for a subset of the CV 3Dpol single mutants and their combination as double mutants. The dashed line represents the elongation rate of wild-type 3Dpol.
TABLE 2.
In vivo mutation rates and in vitro elongation data
| 3Dpol | Location | No. of mutations/10 kba | Max elongation rate (nt/s) ± SEb | CTP Km (nM) ± SEc | CTP-vs-dCTP discrimination factor ± SEc |
|---|---|---|---|---|---|
| Coxsackievirus B3 | |||||
| Wild type | 4.3 | 22.8 ± 1.5 | 115 ± 7 | 360 ± 40 | |
| I230F | Palm/motif A | 11.2 | 30.4 ± 1.0 | 116 ± 7 | 230 ± 30 |
| I230W | Palm/motif A | NV | 26 ± 4d | 93 ± 7 | 330 ± 40 |
| I230L | Palm/motif A | ND | 34 ± 5d | 88 ± 7 | 240 ± 30 |
| F232Y | Palm/motif A | 11.2 | 28 ± 5 | ND | 270 ± 50e |
| F232L | Palm/motif A | Rev | 34.6 ± 1.5 | 128 ± 5 | 174 ± 8 |
| F232V | Palm/motif A | Rev | 29.9 ± 1.4 | ND | 240 ± 40e |
| S299T | Palm/motif B | 7.0 | 19.7 ± 0.7 | 92 ± 6 | 290 ± 30 |
| K360R | Palm/motif D | NV | 0.6 ± 0.1 | ND | ND |
| G64S | Palm/N terminus | IVT | 12.6 ± 0.5 (37°C) | ND | 420 ± 70 (30°C) |
| A239G | Palm/N terminus | 8.2 | 37 ± 3 | 116 ± 7 | 169 ± 12 |
| Y268W | Fingers-palm | 8.9 | 25 ± 3 | 101 ± 5 | 310 ± 30 |
| Y268H | Fingers-palm | 9.2 | 28 ± 2 | 103 ± 10 | 280 ± 40 |
| P48K | Fingers | 6.8 | 28.1 ± 1.0 | 79 ± 3 | 320 ± 60 |
| S164P | Fingers | 6.7 | 21 ± 3 | 91 ± 3 | 340 ± 18 |
| I176V | Fingers-RNA | 5.6 | 30 ± 2 | 89 ± 8 | 310 ± 30 |
| CV double mutants | |||||
| I230F + S299T | Motif A + palm | 7.2 | 25.7 ± 1.6 | 106 ± 7 | 255 ± 20 |
| I230F + F232Y | Motif A + motif A | ND | 37 ± 5d | 114 ± 10 | 190 ± 20 |
| I230F + I176V | Motif A + fingers | ND | 38 ± 3 | 77 ± 3 | 234 ± 17 |
| G64S + I176V | N terminus + fingers | Rev | 17.2 ± 1.0 | ND | ND |
| G64S + Y268H | N terminus + fingers-palm | NV | 14.6 ± 1.9 (37°C) | ND | 350 ± 50 (30°C) |
| Poliovirus | |||||
| Wild type | 88 ± 5 | 85 ± 7 | 117 ± 11 | ||
| G64S | Palm/N terminus | ND | 46 ± 2 | 66 ± 5 | 310 ± 30 |
| F230I | Palm/motif A | ND | 75 ± 8 | 73 ± 3 | 152 ± 9 |
| F232I | Palm/motif A | ND | 56 ± 16 | 92 ± 2 | 200 ± 13 |
| H273R | Fingers | ND | 86 ± 14 | 123 ± 7 | 57 ± 19 |
| K359R | Palm/motif D | ND | 3.0 ± 0.1 | 34 ± 4 | 1,300 ± 500 |
From sequencing of progeny virus genomes as described previously by Gnadig et al. (19). NV, virus was not viable; IVT, insufficient virus titer for mutational analysis; ND, not determined; Rev, virus was viable but reverted to the wild-type residue.
Maximal rate from Michaelis-Menten analysis of the lag phase in 37°C all-NTP stopped-flow assays.
From Michaelis-Menten analysis of CTP and 2′-dCTP titration data with propagation of fractional errors in the curve-fitted parameters (see Fig. 4C and Table 3 for further details regarding 30°C data).
Obtained from the length of the lag phase in the discrimination assay.
Scaled from data obtained with the original RNA template used by Gnadig et al. (19).
G-U mismatch incorporation.
As a more direct test of a native nucleotide misincorporation event, we tested the ability of several 3Dpol enzymes to utilize GTP as a substrate on a self-priming hairpin RNA with a 3′-CUCUGGUAAG-5′ template sequence (Fig. 6A). In the presence of GTP at low concentrations (40 μM), we observed elongation by only a single nucleotide, as expected based on the single cytosine at the beginning of the template, and no further extension even after 1 h of incubation (Fig. 6B). However, at a 1 mM GTP concentration, we observed elongation past the +1 site in as little as 5 min, indicative of G-U mismatch incorporation to yield a +4 product as well as possible G-G mismatches to yield an apparent +7 product based on gel migration. Note that incomplete denaturation of the elongated hairpin primer-templates can affect gel migration, and the effects of G-U and possible G-G mismatches on duplex stability were not explicitly studied. It is thus possible that the final band in our gels is not exactly a +7 product, but it is certainly longer than the cognate +1 endpoint seen with 40 μM GTP.
To directly compare G-U mismatch incorporation by different mutant polymerases, we set up a second experiment where 40 μM GTP was first used to generate a ∼50% preinitiated cognate +1 product. The elongation complexes were then diluted into a 1 mM GTP solution containing 300 mM NaCl to prevent further RNA binding or reinitiation of dissociated RNA, and reaction products were quenched at various time points by the addition of EDTA. Gel analysis showed the time-dependent appearance of longer products from only the preinitiated +1 band, and a comparison gel of the 30-min-time-point samples showed that the CV polymerases differed greatly in their abilities to misincorporate GTP on this template (Fig. 6C). For example, the I230F and A239G mutants made more elongation products than the WT, while the CV 3Dpol G64S mutant clearly made fewer. As shown in Fig. 6D, the amount of GTP misincorporation was correlated with the stopped-flow-based CTP-versus-2′-dCTP discrimination factors, providing another direct demonstration that 2′-dNTP discrimination is a valid indirect measure of polymerase fidelity.
The G64S mutant is a high-fidelity coxsackievirus variant.
The G64S mutant is a known high-fidelity mutant of poliovirus polymerase, where it was selected for in the presence of the nucleoside analog ribavirin (20, 32). However, the mutation was not viable in a coxsackievirus background, precluding an assessment of its fidelity by sequencing of progeny genomes (19), and the CV G64S elongation complex is too unstable to measure nucleotide discrimination by the stopped-flow assay. However, the GTP mismatch incorporation data (Fig. 6C and D) suggest that the G64S mutant is a high-fidelity variant of CV 3Dpol, and we found that by reducing the assay temperature to 30°C, we could biochemically assess nucleotide selectivity by both the stopped-flow assay (Table 3) and gel-based analysis of products from elongation reactions (Fig. 7). The data obtained by both methods further indicate that the G64S mutant is in fact a high-fidelity variant polymerase of CV 3Dpol. In the stopped-flow assay, the G64S mutant had a discrimination factor of 420, which is higher than the discrimination factor of 370 observed for the WT at 30°C (Table 3). In the gel-based analysis, there was rapid formation of a +7 product upon nucleotide addition, analogous to the lag phase in the stopped-flow experiments, and this was followed by the CTP or dCTP concentration-dependent appearance of the full-length product (Fig. 7A). The gel assay-based discrimination number calculated by taking the ratios of the CTP and 2′-dCTP concentrations needed for half-maximal incorporation ([NTP]1/2) also indicates that the G64S polymerase has a higher fidelity than WT 3Dpol (Fig. 7B). Finally, in vitro biochemical data for G64S I176V and G64S Y268H double mutants suggested that second-site mutations could potentially remedy the elongation rate (Table 2) and elongation complex stability (Fig. 2C) defects in the CV G64S 3Dpol mutant. This could perhaps restore virus growth, but for unknown reasons, neither of these double mutant nor a G64S I176V Y268H triple mutant virus was viable.
TABLE 3.
CV 3Dpol G64S NTP discrimination
| CV 3Dpol | Mean discrimination factor ± SDa |
|
|---|---|---|
| 30°C | 37°C | |
| WT | 370 ± 80 | 360 ± 40 |
| G64S | 420 ± 70 | ND |
| G64S + Y268H | 350 ± 50 | ND |
| Y268H | 250 ± 60 | 280 ± 40 |
| I230F | 200 ± 30 | 230 ± 30 |
ND, not determined.
FIG 7.
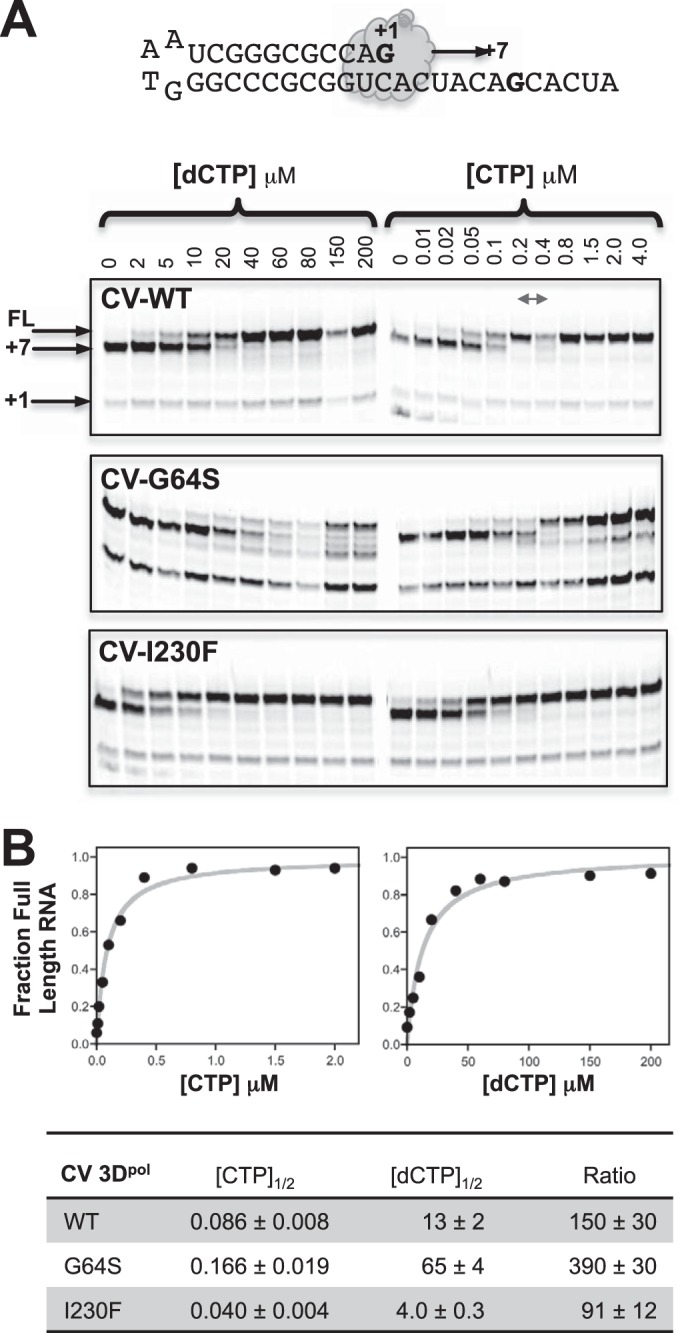
Gel-based analysis of CV 3Dpol G64S nucleotide discrimination. (A) Primer-template RNA used and gel analysis of elongation products formed by WT, G64S, and I230F CV polymerases at 37°C as a function of the CTP or 2′-dCTP concentration. All three polymerases readily carry out cognate elongation to the +7 product but then differ in how they proceed past the unique guanosine in the template; G64S incorporates 2′-dCTP less efficiently than the WT, while the low-fidelity I230F mutant does so more efficiently than the WT. Note that the G64S mutant also yields abortive products, suggesting that there may be destabilization of the elongation complex during or after the 2′-dCTP addition followed by RNA degradation. The double-headed arrow refers to two lanes that were swapped in the CV WT CTP gel only. FL, full-length product. (B) Plots of NTP concentration dependence for elongation to a full-length product with curve fits to the equation [NTP]/([NTP]1/2 + [NTP]), which were used to determine [NTP]1/2 values for CTP and 2′-dCTP incorporation. The ratios of these concentrations also show that the G64S variant has higher nucleotide discrimination than the wild-type polymerase, while the I230F variant has lower discrimination.
Extension to poliovirus 3Dpol.
We extended this study to the highly homologous poliovirus 3Dpol by making a subset of analogous point mutations and studying their effects by in vitro biochemistry. The gel data in Fig. 6C show GTP misincorporation opposite uracil by the wild-type PV and CV polymerases, which suggests that wild-type PV 3Dpol has significantly lower fidelity than wild-type CV 3Dpol, and in fact, it appears to have a lower fidelity than even the lowest-fidelity CV A239G mutation (Fig. 6D). This was further borne out by stopped-flow assay data, where the two wild-type enzymes had drastically different nucleotide discrimination factors and maximal elongation rates (Fig. 8). Wild-type PV 3Dpol has a discrimination factor of only 117, versus 360 for CV 3Dpol, and it has a much higher maximal elongation rate of 88 nt/s versus 23 nt/s for CV 3Dpol on this template RNA (Fig. 8A and Table 2). Most interestingly, when introduced into PV 3Dpol, the palm domain mutations exhibited an overall range of fidelity variation similar to that seen in CV 3Dpol but with the notable difference that they tended to slow the inherently faster PV enzyme and increase its replication fidelity, the exact opposite of the effects seen with CV 3Dpol. As a result, the discrimination factors of the highest-fidelity PV mutants are comparable to those of wild-type CV, while those of the lowest-fidelity CV mutants are comparable to those of wild-type PV (Fig. 8B). We also examined the recently identified PV 3Dpol H273R variant in the nucleotide discrimination assay and obtained a discrimination factor of 57 (Fig. 8B and Table 2), which is much lower than the discrimination factor of 117 seen for wild-type PV 3Dpol and consistent with its mutator phenotype virus and low fidelity replication based on sequencing and guanidine resistance (33). Combined with data for the established PV G64S high-fidelity variant, this demonstrates that 2′-dCTP discrimination is correlated with both high- and low-fidelity variants in PV as well as CV polymerases.
FIG 8.
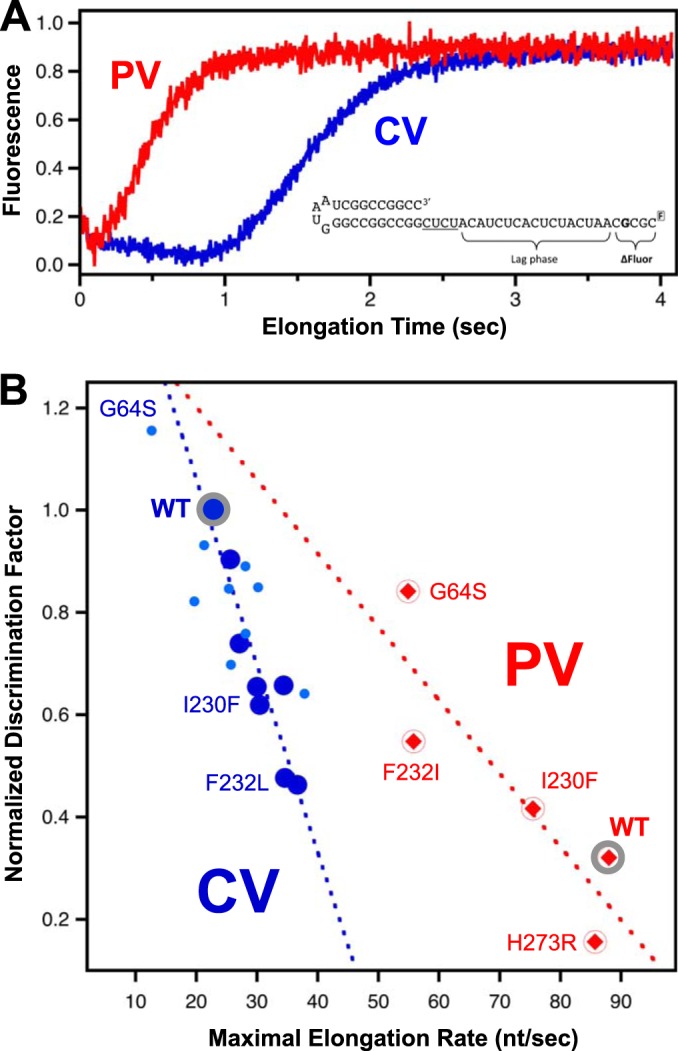
Comparison of coxsackievirus and poliovirus polymerases. (A) Stopped-flow kinetics data show that PV 3Dpol has higher elongation rates than CV 3Dpol. Traces reflect maximal elongation rates observed with equimolar mixtures of 300 (PV) and 400 (CV) μM each NTP. The 5′-fluorescein RNA used to determine maximal elongation rates by titrating an equimolar mixture of all four NTPs is also shown. (B) Correlation plot of in vitro discrimination factors versus elongation rates for wild-type and variant CV (blue circles) (palm mutants are larger) and PV (red diamonds) polymerases. Notably, the wild-type CV polymerase has significantly higher fidelity than the wild-type PV polymerase, yet single point mutations drastically change the fidelity of either polymerase over comparable ranges. Mutations tend to decrease the fidelity of the higher-fidelity CV 3Dpol while increasing that of the lower-fidelity PV 3Dpol. The dashed lines represent a linear curve fit to the data from the palm domain mutants of each polymerase.
DISCUSSION
The crystal structures of poliovirus polymerase elongation complexes capable of in-crystal catalysis showed that the positive-strand RNA virus polymerases utilize a unique palm domain-based movement to close their active sites for catalysis (9). Based on this movement, we previously engineered 50 mutations in coxsackievirus 3Dpol and showed that 7 of these mutations resulted in viable viruses that had lower fidelity than did wild-type polymerases, as determined by deep sequencing of progeny genomes, and were attenuated for growth in vivo (19). In the study presented here, we build on these results with an expanded library of mutations and rapid in vitro fidelity assays to further address structure-function relationships that control the replication fidelity of picornaviral RdRPs. The use of polymerase structure-function relationships to directly mutate key interactions involved in fidelity modulation has allowed us to identify both high- and low-fidelity variants, which is a distinct advantage over mutagen-based approaches, which select for only high-fidelity variants. Using in vitro assays, we can also biochemically characterize polymerases that prove to be lethal in the context of an infectious virus, allowing us to further map protein structure-function relationships important for RdRP replication fidelity and quasispecies diversity.
The structural observation that the NTP 2′-hydroxyl group is a key determinant of active-site closure (9) led to the development of an in vitro biochemical fidelity assay based on the discrimination between CTP and 2′-dCTP substrates (19, 29). In this assay, polymerase fidelity is measured as a discrimination factor that reflects the ratio of the two nucleotide incorporation efficiencies: (kpol/Km)CTP/(kpol/Km)dCTP. The data are obtained via a single cytosine incorporation event during processive replication of an extended single-stranded RNA (ssRNA) template (Fig. 2). While the 2′-dNTP discrimination assay is not a formal measure of fidelity in terms of nucleotide mismatching, our data clearly show that 2′-dCTP incorporation is well correlated with overall in vivo mutation frequencies, as measured by both deep sequencing of progeny virus genomes (Fig. 4A) and the misincorporation of guanosine opposite a templating uracil (Fig. 6D). The finding that utilization of this one nonnative modified nucleotide can reflect all the possible nucleotide mismatches encompassed by the sequencing results is also consistent with data showing that PV 3Dpol misincorporation rates do not differ greatly among different noncomplementary nucleotides (34). This in vitro discrimination assay has the advantages of measuring the inherent tendencies of purified polymerases to incorporate a nonnative NTP in a processive elongation context on extended RNA templates. It also does so without selective pressures imposed by the in vivo requirement for functional RNA genomes and viral proteins, effects which can reduce the observed mutation rates in a virus-dependent manner depending on quasispecies distributions, genome robustness, and host factor interactions (14, 15, 35).
Overall, our nucleotide discrimination and fidelity data illustrate the biochemical plasticity of the viral RdRP replication mechanism by showing that even single amino acid changes in the polymerase can drastically increase or decrease the replication fidelity and elongation rate. The wild-type CV and PV polymerases have CTP/dCTP discrimination factors that differ by 3.5-fold, with the CV enzyme being of higher fidelity than the PV enzyme, but single point mutations can almost convert the fidelity of either polymerase to that of the other (Fig. 8B). For example, F232L or A239G mutants of CV 3Dpol lowered the discrimination factor by 2.1-fold, while the G64S mutant of PV 3Dpol increased its discrimination factor 2.6-fold. Similarly, converting the native isoleucine 230 in CV 3Dpol to a phenylalanine (I230F) lowered fidelity, while the converse F230I mutation in PV 3Dpol increased fidelity. The finding that CV and PV polymerases have drastically different fidelities likely explains the ease of selection for higher-fidelity PV variants with mutagenic nucleoside analogs such as ribavirin, but this approach has proven to be difficult with CV because its polymerase already has fairly high fidelity (19).
The crystal structures of several fidelity-variant CV polymerases showed a native 3Dpol conformation in the absence of RNA and bound NTP, suggesting that the mutations act by subtly altering the kinetics and energetics of the motif A/D movement that takes place during active-site closure (Fig. 1B). The exception to this is the CV F232L variant, whose structure showed a significant rearrangement of the loop located at the C-terminal end of motif D into a new conformation that has not yet been observed in any RdRP. Solution NMR studies of PV 3Dpol have shown that the 13CH3 chemical shift from Met354, which is analogous to CV Met355, is significantly perturbed upon correct NTP binding to the 3Dpol-RNA complex (36). This methyl group is located ∼7 Å from the Phe364 aromatic ring, and the repacking of its immediate environment when Phe364 and Pro357 move may explain the chemical shift change (Fig. 3A). The Lys360 side chain, thought to be involved in NTP transport into the active site (37), where it may serve as a proton donor during catalysis (38), is also repositioned in the F232L structure and no longer points toward the active site. This suggests that this newly observed motif D loop conformation is likely adopted after NTP delivery and catalysis when Lys360 is no longer needed near the active site.
Detailed biochemical studies of the RdRP catalytic cycle have shown that the overall elongation reaction is rate limited by a conformational change step that precedes catalysis and by the phosphoryl transfer step itself (39). Our results showing that mutations in motif A greatly alter elongation rates are consistent with the rate-limiting precatalysis step being the NTP-induced movement of motif A/D in the palm domain that closes the active site for catalysis. We further found that mutations in the finger domain primarily affect the elongation rate while having smaller effects on fidelity, and they likely function by affecting the postcatalysis translocation step via altering interactions between the template RNA and its entry channel on the polymerase surface (9, 10, 40). Consistent with this, data from double mutations in both the palm and finger domains show that rate increases can be additive, while fidelity is dominated by the effects of the palm domain mutation (Fig. 5 and Table 2). For example, the CV I230F palm domain mutant is a high-fidelity and rate-variant mutant, while the I176V mutant in the finger domain primarily affects the rate, and the combination of these two mutants retains the fidelity of the I230F mutant but increases the elongation rate from 30 to 38 nt/s. Similarly, the CV G64S mutant is a slightly high-fidelity variant that decreases the elongation rate from 23 to 13 nt/s, and the double mutant with the I176V mutation partially restores the rate to 17 nt/s.
We hypothesize that the finger domain dynamics associated with RNA translocation may be inherently faster in PV 3Dpol than in CV 3Dpol as a molecular explanation for the observation that PV polymerases always have higher elongation rates than CV polymerases with comparable discrimination factors (Fig. 8). The high-fidelity PV 3Dpol K359R variant (41) was not viable as the homologous K360R variant in the CV background (19), and the in vitro elongation data suggest that this may be due to a very low processive elongation rate; the mutation has similar 35-fold effects on the elongation rates of both polymerases, but this lowers the rate of the inherently slower CV enzyme to only 0.6 nt/s, which likely explains the nonviability of the mutant virus. The adaptive P48K and S164P mutations are located in the ring finger and were found in CV isolates wherein Gly64 was mutated to a number of different amino acids in an effort to mimic the high-fidelity phenotype of PV G64S 3Dpol (19). The desired Gly64 mutations proved to be genetically unstable and reverted to the wild type within three passages, but in the process, the virus acquired stable P48K or S164P mutations that by themselves did not provide any strong growth advantage or disadvantage for the wild-type virus. However, we suspect that the P48K and S164P finger domain mutations provided an elongation rate boost and a slight growth advantage in early passages that allowed the desired Gly64 mutant to replicate albeit slowly. However, once the Gly64 reversion did occur, the faster-growing revertant virus quickly became dominant in the population, as was also observed for PV G64S 3Dpol (17).
The observation that CV 3Dpol has a lower elongation rate and a significantly higher fidelity than PV 3Dpol is interesting in the context of the pathogenesis of the two viruses and the ability to select high-fidelity variants by growth in the presence of mutagens. PV exists on the edge of error catastrophe such that even minor increases in the mutation frequency significantly decrease infectivity (42). This means that few low-fidelity PV variants are viable, and only the H273R variant, which increases the number of mutations per genome from 1.9 to 3.0, has been identified thus far (33), but higher-fidelity variants such as the 3Dpol G64S mutant can readily be recovered by growth on mutagens such as ribavirin (32). This is the exact opposite of what is seen with coxsackievirus, where high-fidelity variants have not been obtained by growth on mutagens but engineered low-fidelity variants are viable and grow at essentially wild-type virus titers in tissue culture despite almost 3-fold-higher mutation frequencies (19). The G64S mutation increased the in vitro nucleotide selectivity of CV 3Dpol (Fig. 4C), but the effect on the already high-fidelity polymerase was much smaller than what was seen with PV 3Dpol, and it lowered the polymerase elongation rate almost 2-fold, which likely explains the 100-fold reduction in the replication of the CV G64S virus (19).
The study presented here involved only two highly homologous picornaviral polymerases, but similar fidelity modulation effects have been observed for many RNA viruses. Ribavirin has been used to select high-fidelity variants of EV71 (21, 43), foot-and-mouth disease virus (FMDV) (44, 45), and chikungunya virus (22), suggesting that these viruses occupy the lower end of the fidelity spectrum. One might expect that the need to retain cross-species infectivity in zoonotic arboviruses necessitates higher-fidelity polymerases, as implied by yellow fever virus fidelity data (46), although data from dengue virus polymerase suggest that it has a fidelity comparable to those of PV and FMDV RdRPs based on single nucleotide incorporation assays (47). Mutations in motif A of West Nile virus polymerase that were designed to stabilize the active-site structure diminish virus growth (48), suggesting that this virus has some tolerance for slowing the polymerase, but biochemical data to assess the effects on the polymerase rate and fidelity are not available. Together, these studies certainly indicate that the fidelity of many RdRPs can be readily tuned and adapted to facilitate virus growth. However, it is not yet clear if our data showing vastly different nucleotide discrimination factors for CV and PV polymerases represent the extremes of the viral RdRP fidelity spectrum. The full extent of fidelity effects could be addressed experimentally with further in vitro studies carried out under consistent experimental conditions to facilitate direct comparisons of different polymerases.
In conclusion, the data from this structure-function study show that viral RdRP fidelity and elongation rate are closely linked but are also somewhat independently controlled by distinct regions of the polymerase structure. Using both high- and low-fidelity CV and PV enzymes, we show that point mutations in the palm domain significantly alter nucleotide discrimination in vitro and that this is correlated with deep-sequencing-based mutation frequencies obtained in vivo. These effects can be varied over comparable ranges in both coxsackievirus and poliovirus polymerases despite very different wild-type characteristics. Processive elongation rates are further controlled by finger domain interactions that likely involve RNA binding and translocation. The data support the idea that viral RdRPs evolved and retained the unique palm domain-based movement for active-site closure as a mechanism for fine-tuning replication fidelity to ensure virus adaptation and fitness in vivo.
ACKNOWLEDGMENTS
This work was supported by NIH grant R01 AI059130 to O.B.P. and European Research Council starting grant no. 242719 to M.V.
REFERENCES
- 1.Ferrer-Orta C, Arias A, Escarmis C, Verdaguer N. 2006. A comparison of viral RNA-dependent RNA polymerases. Curr Opin Struct Biol 16:27–34. doi: 10.1016/j.sbi.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Ng KK, Arnold JJ, Cameron CE. 2008. Structure-function relationships among RNA-dependent RNA polymerases. Curr Top Microbiol Immunol 320:137–156. doi: 10.1007/978-3-540-75157-1_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lescar J, Canard B. 2009. RNA-dependent RNA polymerases from flaviviruses and Picornaviridae. Curr Opin Struct Biol 19:759–767. doi: 10.1016/j.sbi.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 4.Mosley RT, Edwards TE, Murakami E, Lam AM, Grice RL, Du J, Sofia MJ, Furman PA, Otto MJ. 2012. Structure of HCV polymerase in complex with primer-template RNA. J Virol 86:6503–6511. doi: 10.1128/JVI.00386-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. 1999. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat Struct Biol 6:937–943. doi: 10.1038/13305. [DOI] [PubMed] [Google Scholar]
- 6.Malet H, Egloff MP, Selisko B, Butcher RE, Wright PJ, Roberts M, Gruez A, Sulzenbacher G, Vonrhein C, Bricogne G, Mackenzie JM, Khromykh AA, Davidson AD, Canard B. 2007. Crystal structure of the RNA polymerase domain of the West Nile virus non-structural protein 5. J Biol Chem. 282:10678–10689. doi: 10.1074/jbc.M607273200. [DOI] [PubMed] [Google Scholar]
- 7.Yap TL, Xu T, Chen YL, Malet H, Egloff MP, Canard B, Vasudevan SG, Lescar J. 2007. Crystal structure of the dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J Virol 81:4753–4765. doi: 10.1128/JVI.02283-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu G, Gong P. 2013. Crystal structure of the full-length Japanese encephalitis virus NS5 reveals a conserved methyltransferase-polymerase interface. PLoS Pathog 9:e1003549. doi: 10.1371/journal.ppat.1003549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gong P, Peersen OB. 2010. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc Natl Acad Sci U S A 107:22505–22510. doi: 10.1073/pnas.1007626107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gong P, Kortus MG, Nix JC, Davis RE, Peersen OB. 2013. Structures of coxsackievirus, rhinovirus, and poliovirus polymerase elongation complexes solved by engineering RNA mediated crystal contacts. PLoS One 8:e60272. doi: 10.1371/journal.pone.0060272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zamyatkin DF, Parra F, Alonso JM, Harki DA, Peterson BR, Grochulski P, Ng KK. 2008. Structural insights into mechanisms of catalysis and inhibition in Norwalk virus polymerase. J Biol Chem 283:7705–7712. doi: 10.1074/jbc.M709563200. [DOI] [PubMed] [Google Scholar]
- 12.Salas-Rojas M, Galvez-Romero G, Anton-Palma B, Acevedo R, Blanco-Favela F, Aguilar-Setien A. 2014. The coelomic fluid of the sea urchin Tripneustes depressus shows antiviral activity against Suid herpesvirus type 1 (SHV-1) and rabies virus (RV). Fish Shellfish Immunol 36:158–163. doi: 10.1016/j.fsi.2013.10.025. [DOI] [PubMed] [Google Scholar]
- 13.Acevedo A, Brodsky L, Andino R. 2014. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature 505:686–690. doi: 10.1038/nature12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lauring AS, Frydman J, Andino R. 2013. The role of mutational robustness in RNA virus evolution. Nat Rev Microbiol 11:327–336. doi: 10.1038/nrmicro3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graci JD, Gnadig NF, Galarraga JE, Castro C, Vignuzzi M, Cameron CE. 2012. Mutational robustness of an RNA virus influences sensitivity to lethal mutagenesis. J Virol 86:2869–2873. doi: 10.1128/JVI.05712-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borderia AV, Stapleford KA, Vignuzzi M. 2011. RNA virus population diversity: implications for inter-species transmission. Curr Opin Virol 1:643–648. doi: 10.1016/j.coviro.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 17.Pfeiffer JK, Kirkegaard K. 2005. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog 1:e11. doi: 10.1371/journal.ppat.0010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pfeiffer JK, Kirkegaard K. 2006. Bottleneck-mediated quasispecies restriction during spread of an RNA virus from inoculation site to brain. Proc Natl Acad Sci U S A 103:5520–5525. doi: 10.1073/pnas.0600834103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gnadig NF, Beaucourt S, Campagnola G, Borderia AV, Sanz-Ramos M, Gong P, Blanc H, Peersen OB, Vignuzzi M. 2012. Coxsackievirus B3 mutator strains are attenuated in vivo. Proc Natl Acad Sci U S A 109:E2294–E2303. doi: 10.1073/pnas.1204022109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arnold JJ, Vignuzzi M, Stone JK, Andino R, Cameron CE. 2005. Remote site control of an active site fidelity checkpoint in a viral RNA-dependent RNA polymerase. J Biol Chem 280:25706–25716. doi: 10.1074/jbc.M503444200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng T, Kwang J. 2014. Attenuation of human enterovirus 71 high-replication-fidelity variants in AG129 mice. J Virol 88:5803–5815. doi: 10.1128/JVI.00289-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coffey LL, Beeharry Y, Borderia AV, Blanc H, Vignuzzi M. 2011. Arbovirus high fidelity variant loses fitness in mosquitoes and mice. Proc Natl Acad Sci U S A 108:16038–16043. doi: 10.1073/pnas.1111650108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rozen-Gagnon K, Stapleford KA, Mongelli V, Blanc H, Failloux AB, Saleh MC, Vignuzzi M. 2014. Alphavirus mutator variants present host-specific defects and attenuation in mammalian and insect models. PLoS Pathog 10:e1003877. doi: 10.1371/journal.ppat.1003877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang X, Welch JL, Arnold JJ, Boehr DD. 2010. Long-range interaction networks in the function and fidelity of poliovirus RNA-dependent RNA polymerase studied by nuclear magnetic resonance. Biochemistry 49:9361–9371. doi: 10.1021/bi100833r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moustafa IM, Shen H, Morton B, Colina CM, Cameron CE. 2011. Molecular dynamics simulations of viral RNA polymerases link conserved and correlated motions of functional elements to fidelity. J Mol Biol 410:159–181. doi: 10.1016/j.jmb.2011.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gohara DW, Ha CS, Kumar S, Ghosh B, Arnold JJ, Wisniewski TJ, Cameron CE. 1999. Production of “authentic” poliovirus RNA-dependent RNA polymerase (3D(pol)) by ubiquitin-protease-mediated cleavage in Escherichia coli. Protein Expr Purif 17:128–138. doi: 10.1006/prep.1999.1100. [DOI] [PubMed] [Google Scholar]
- 27.Campagnola G, Weygandt M, Scoggin K, Peersen O. 2008. Crystal structure of coxsackievirus B3 3Dpol highlights the functional importance of residue 5 in picornavirus polymerases. J Virol 82:9458–9464. doi: 10.1128/JVI.00647-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson AA, Peersen OB. 2004. Structural basis for proteolysis-dependent activation of the poliovirus RNA-dependent RNA polymerase. EMBO J 23:3462–3471. doi: 10.1038/sj.emboj.7600357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gong P, Campagnola G, Peersen OB. 2009. A quantitative stopped-flow fluorescence assay for measuring polymerase elongation rates. Anal Biochem 391:45–55. doi: 10.1016/j.ab.2009.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hobdey SE, Kempf BJ, Steil BP, Barton DJ, Peersen OB. 2010. Poliovirus polymerase residue 5 plays a critical role in elongation complex stability. J Virol 84:8072–8084. doi: 10.1128/JVI.02147-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, Sliz P. 2013. Collaboration gets the most out of software. eLife 2:e01456. doi: 10.7554/eLife.01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pfeiffer JK, Kirkegaard K. 2003. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc Natl Acad Sci U S A 100:7289–7294. doi: 10.1073/pnas.1232294100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korboukh VK, Lee CA, Acevedo A, Vignuzzi M, Xiao Y, Arnold JJ, Hemperly S, Graci JD, August A, Andino R, Cameron CE. 2014. RNA virus population diversity: an optimum for maximal fitness and virulence. J Biol Chem 289:29531–29544. doi: 10.1074/jbc.M114.592303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ward CD, Stokes MA, Flanegan JB. 1988. Direct measurement of the poliovirus RNA polymerase error frequency in vitro. J Virol 62:558–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lauring AS, Acevedo A, Cooper SB, Andino R. 2012. Codon usage determines the mutational robustness, evolutionary capacity, and virulence of an RNA virus. Cell Host Microbe 12:623–632. doi: 10.1016/j.chom.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang X, Smidansky ED, Maksimchuk KR, Lum D, Welch JL, Arnold JJ, Cameron CE, Boehr DD. 2012. Motif D of viral RNA-dependent RNA polymerases determines efficiency and fidelity of nucleotide addition. Structure 20:1519–1527. doi: 10.1016/j.str.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen H, Sun H, Li G. 2012. What is the role of motif D in the nucleotide incorporation catalyzed by the RNA-dependent RNA polymerase from poliovirus? PLoS Comput Biol 8:e1002851. doi: 10.1371/journal.pcbi.1002851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castro C, Smidansky ED, Arnold JJ, Maksimchuk KR, Moustafa I, Uchida A, Gotte M, Konigsberg W, Cameron CE. 2009. Nucleic acid polymerases use a general acid for nucleotidyl transfer. Nat Struct Mol Biol 16:212–218. doi: 10.1038/nsmb.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arnold JJ, Cameron CE. 2004. Poliovirus RNA-dependent RNA polymerase (3Dpol): pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mg2+. Biochemistry 43:5126–5137. doi: 10.1021/bi035212y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kortus MG, Kempf BJ, Haworth KG, Barton DJ, Peersen OB. 2012. A template RNA entry channel in the fingers domain of the poliovirus polymerase. J Mol Biol 417:263–278. doi: 10.1016/j.jmb.2012.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weeks SA, Lee CA, Zhao Y, Smidansky ED, August A, Arnold JJ, Cameron CE. 2012. A polymerase mechanism-based strategy for viral attenuation and vaccine development. J Biol Chem 287:31618–31622. doi: 10.1074/jbc.C112.401471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crotty S, Cameron CE, Andino R. 2001. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc Natl Acad Sci U S A 98:6895–6900. doi: 10.1073/pnas.111085598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sadeghipour S, Bek EJ, McMinn PC. 2013. Ribavirin-resistant mutants of human enterovirus 71 express a high replication fidelity phenotype during growth in cell culture. J Virol 87:1759–1769. doi: 10.1128/JVI.02139-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arias A, Arnold JJ, Sierra M, Smidansky ED, Domingo E, Cameron CE. 2008. Determinants of RNA-dependent RNA polymerase (in)fidelity revealed by kinetic analysis of the polymerase encoded by a foot-and-mouth disease virus mutant with reduced sensitivity to ribavirin. J Virol 82:12346–12355. doi: 10.1128/JVI.01297-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sierra M, Airaksinen A, Gonzalez-Lopez C, Agudo R, Arias A, Domingo E. 2007. Foot-and-mouth disease virus mutant with decreased sensitivity to ribavirin: implications for error catastrophe. J Virol 81:2012–2024. doi: 10.1128/JVI.01606-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pugachev KV, Guirakhoo F, Ocran SW, Mitchell F, Parsons M, Penal C, Girakhoo S, Pougatcheva SO, Arroyo J, Trent DW, Monath TP. 2004. High fidelity of yellow fever virus RNA polymerase. J Virol 78:1032–1038. doi: 10.1128/JVI.78.2.1032-1038.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin Z, Deval J, Johnson KA, Swinney DC. 2011. Characterization of the elongation complex of dengue virus RNA polymerase: assembly, kinetics of nucleotide incorporation, and fidelity. J Biol Chem 286:2067–2077. doi: 10.1074/jbc.M110.162685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Slyke GA, Ciota AT, Willsey GG, Jaeger J, Shi PY, Kramer LD. 2012. Point mutations in the West Nile virus (Flaviviridae; Flavivirus) RNA-dependent RNA polymerase alter viral fitness in a host-dependent manner in vitro and in vivo. Virology 427:18–24. doi: 10.1016/j.virol.2012.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]