

ABSTRACT
Murine cytomegalovirus (MCMV) is a betaherpesvirus of the house mouse, Mus musculus domesticus. It is a common infectious agent of wild mice and a highly studied pathogen of the laboratory mouse. Betaherpesviruses are specific to their hosts, and it is not known if other Mus taxa carry MCMV or if it is restricted to M. m. domesticus. We sampled mice over a 145-km transect of Bavaria-Bohemia crossing a hybrid zone between M. m. domesticus and Mus musculus musculus in order to investigate the occurrence of MCMV in two Mus subspecies and to test the limits of the specificity of the virus for its host. We hypothesized that if the two subspecies carry MCMV and if the virus is highly specific to its host, divergent MCMV lineages would have codiverged with their hosts and would have a geographical distribution constrained by the host genetic background. A total of 520 mice were tested by enzyme-linked immunosorbent assay (ELISA) and/or nested PCR targeting the M94 gene. Seropositive and PCR-positive individuals were found in both Mus subspecies. Seroprevalence was high, at 79.4%, but viral DNA was detected in only 41.7% of mice. Sequencing revealed 20 haplotypes clustering in 3 clades that match the host genetic structure in the hybrid zone, showing 1 and 2 MCMV lineages in M. m. domesticus and M. m. musculus, respectively. The estimated time to the most recent common ancestor (1.1 million years ago [Mya]) of the MCMVs matches that of their hosts. In conclusion, MCMV has coevolved with these hosts, suggesting that its diversity in nature may be underappreciated, since other members of the subgenus Mus likely carry different MCMVs.
IMPORTANCE Murine cytomegalovirus (MCMV) is a betaherpesvirus of the house mouse, Mus musculus domesticus, an important lab model for human cytomegalovirus (HCMV) infection. The majority of lab studies are based on only two strains of MCMVs isolated from M. m. domesticus, Smith and K181, the latter derived from repeated passage of Smith in mouse submaxillary glands. The presence of MCMV in other members of the Mus subgenus had not even been investigated. By screening mouse samples collected in the European house mouse hybrid zone between M. m. domesticus and M. m. musculus, we show that MCMV is not restricted to the M. m. domesticus subspecies and that MCMVs likely codiverged with their Mus hosts. Thus, the diversity of MCMV in nature may be seriously underappreciated, since other members of the subgenus Mus likely carry their own MCMV lineages.
INTRODUCTION
Cytomegaloviruses (CMVs) are enveloped double-stranded DNA viruses (family Herpesviridae, subfamily Betaherpesvirinae) that have coevolved with their vertebrate hosts. Murine cytomegalovirus 1 (MCMV), also called Murid herpesvirus 1 (MuHV-1), is a betaherpesvirus of the house mouse and one of its most studied pathogens, because it serves as a laboratory model for human cytomegalovirus (HCMV) infection, which can be highly pathogenic for immunocompromised individuals (1). Almost all laboratory research on MCMV uses either the Smith or the K181 strain of the virus (2). The Smith strain was originally isolated from salivary gland tissue of laboratory mice in the United States in 1954 (3), and the K181 strain was selected by repeated passage of the prototype Smith strain in mouse submaxillary glands (2). These strains have been serially passaged in vivo and in vitro—in the case of the Smith virus, for more than 50 years. Apart from the two standard MCMV laboratory strains, several strains have been isolated from wild house mice in Australia, Beacon Island, and Macquarie Island, and their whole genomes have been sequenced (2, 4, 5). These strains are genetically different from strains Smith and K181 (4) and show in vivo replication kinetics different from those of the standard strains (5), suggesting that the patterns of host resistance to MCMV described for inbred mice are not applicable to all MCMV strains (2). All of these strains have been isolated either from a single subspecies of mouse, Mus musculus domesticus, or from lab mice, which are derived mostly from M. m. domesticus stocks (6).
Several studies have investigated the occurrence of antibodies against MCMV in natural M. m. domesticus populations in North America, Australia, and Australasian islands (for a review, see reference 1) and in the United Kingdom (7). In these studies, a high proportion of mice (60 to 90%) were seropositive for MCMV, indicating that the circulation of this virus is very common in wild mice. To our knowledge, only one study has investigated the occurrence of MCMV directly by PCR, on Boullanger and Macquarie Islands (Australia), reporting 42 to 86% prevalence of this virus in wild mice (8). MCMV infection in wild M. m. domesticus mice is dependent on mouse population density (9, 10) and is positively correlated with the age of the individuals, suggesting cumulative exposure over time (11). Multistrain MCMV infections are common in free-living mice (5, 12). The strains are acquired by immunocompetent mice through simultaneous or successive infections (12). A study on enclosure populations of M. m. domesticus has shown that MCMV had minimal or no impact on mouse survival or breeding when individuals were infected with a single strain but that young males infected with two viral strains had a 20% reduction in survival (13).
The Mus musculus species group comprises 5 species (Mus musculus, Mus spretus, Mus spicilegus, Mus macedonicus, and Mus cypriacus) (14). Within the species Mus musculus, the “house mouse,” 5 subspecies are recognized: Mus musculus musculus, Mus musculus domesticus, Mus musculus castaneus, Mus musculus gentilulus, and Mus musculus molossinus. So far, MCMV has been investigated and isolated in only one of these subspecies, Mus musculus domesticus (see above), leaving the presence of MCMV in other members of this taxon an open question (1). The subspecies M. m. domesticus is one of the most successful invasive mammals and is now present worldwide owing to its synanthropy. It seems that MCMV has been its fellow traveler, since the virus is found in wild M. m. domesticus populations on all continents and even in remote areas such as the sub-Antarctic Kerguelen Islands (8). In Europe, M. m. domesticus meets another subspecies of the house mouse, Mus musculus musculus, along a 2,500-km-long front stretching from Scandinavia to the Black Sea (15–17). At this front, the subspecies form a narrow hybrid zone (Fig. 1) maintained by selection against hybrids (see, e.g., references18 and 19). The initial contact of this hybrid zone likely occurred a few thousand years ago (20). The common ancestor of these two mouse subspecies originated in South Central Asia. DNA-DNA hybridization and sequencing of a number of loci suggest that M. m. domesticus and M. m. musculus likely diverged between 350,000 years ago and 1 million years ago (Mya) (21–25). M. m. domesticus and M. m. musculus colonized Europe by different routes around the Mediterranean and Black Seas in association with human shifts from hunting and gathering to sedentary farming (15, 26). During their divergence, the subspecies appear to have undergone a long isolation period followed by gene flow around 200,000 generations ago, presumably in the Fertile Crescent, long before the advent of the current hybrid zone (20). Such multiple secondary contacts and interaction with Mus musculus castaneus during interglacial periods may have favored pathogen transmission or gene flow. This complex history of M. m. domesticus is now reflected in the genetic makeup of lab mice, which originates in large part from M. m. domesticus but contains M. m. musculus and M. m. castaneus introgressions (6). Given this context, it seems pertinent to investigate the diversity of MCMV in wild house mouse populations other than M. m. domesticus, because this diversity could be directly relevant to understanding the pathogenesis and immunobiology of the MCMV strains currently used in lab mice.
FIG 1.

(A) The European house mouse hybrid zone. The black line depicts the course of the zone; the rectangle delineates the study area. (B) Detail of the study area with sample localities. Distances are given in kilometers. The gray line represents the Czech-German border, and the dashed line marks the consensus hybrid zone center as estimated by Macholán et al. in 2008 (51).
The goal of our study was to investigate the prevalence of MCMV infection in house mice across the European house mouse hybrid zone (HMHZ) in order to (i) find out if another member of the Mus subgenus could carry MCMV and (ii) test the limits of the specificity of this virus for its host. Congruence in branching patterns and consistency in divergence between hosts and viruses suggest that betaherpesviruses have coevolved with their vertebrate hosts for millions of years (27). We hypothesize that if each subspecies harbors MCMV from their common ancestor, after some hundreds of thousands of years of separation in their travels to Europe (see above), M. m. domesticus and M. m. musculus are very likely to carry divergent strains of MCMV. If specificity is strong, we thus expect that the genetic structure of these MCMV strains should reflect the genetic (and geographic) structure of the house mice in the European HMHZ.
MATERIALS AND METHODS
Sampling.
Between 2006 and 2011, at the same period in each year (10 days centered around the end of September), house mice were trapped with metal and wooden live traps in farms across a 145-km-long and 50-km-wide belt stretching from northeastern Bavaria (Germany) to western Bohemia (Czech Republic) (Fig. 1). This is where the ranges of M. m. domesticus and M. m. musculus meet, forming a secondary-contact hybrid zone. All trapped mice were housed individually overnight and were euthanized the day after capture with a halothane overdose. After the carotid arteries were cut, blood was collected in an Eppendorf tube and was allowed to clot for a couple of hours before being centrifuged for 10 min at 3,000 × g to collect the serum, which was subsequently stored at −20°C. We collected tissue samples (liver and spleen) in ethanol for molecular genotyping and lung tissues in liquid nitrogen, with storage at −80°C, for molecular screening for pathogens, including the yeast-like fungus Pneumocystis murina and MCMV. All work followed the experimental protocol (no. 27/2007) approved by the Institutional Committee and Czech Academy of Sciences Committee for animal welfare according to Czech law.
Serological and molecular screening.
We screened a total of 520 mice from 128 localities for MCMV. We screened 291 samples collected from 100 localities in 2008 to 2011 for anti-MCMV antibodies by enzyme-linked immunosorbent assay (ELISA) (kit IM-811C-EB; BioCat, Heidelberg, Germany) and 319 lung tissue samples collected from 97 localities in 2006 to 2011 for MCMV DNA by nested PCR. A subset of 90 mice was screened both serologically and molecularly. For the ELISA, serum was first diluted 1:50, and 100 μl of the diluted serum was used for the reaction according to the manufacturer's instructions. Reaction results were read using Magellan software and the Sunrise absorbance reader (Tecan). DNA from a small piece of lung tissue was isolated using a QIAamp DNA minikit (Qiagen, Venlo, Netherlands) and was eluted in the 100 μl of kit buffer AE. Although the volume of lung tissue used for DNA extraction was standardized by eye for each individual, we did not further standardize the DNA concentration before the amplification step. We performed a nested PCR targeting 737 bp (71%) of the M94 gene, which is essential to viral morphogenesis (28). We chose M94 instead of genes more commonly used for phylogeny, such as those encoding glycoprotein B or DNA polymerase, because M94 is more conserved among the sequences of the 11 MCMV strains from M. m. domesticus available in GenBank. The primers used for the first round were M94-F1 (5′-GTTTGAGAAGCATCAGTAGG-3′) and M94-R1 (5′-AAGAGCTGCCAGGGCTCGGT-3′), and those used for the second round were M94-F2 (5′-GCTACCGAGAGTTCCGCGCC-3′) and M94-R2 (5′-CTTTCCTGCCGTCGTAGCGC-3′); all primers were designed on the basis of MCMV sequences available in GenBank. The PCR for the first round was carried out in a 10-μl volume containing 5 μl of the Multiplex PCR kit master mix (Qiagen, Venlo, Netherlands), 1 μM each primer, and 1 μl of DNA. The second-round mixture was the same as that for the first round but with 0.5 μl of the first-round mixture used as the template. The PCR started with an initial activation step at 95°C (15 min), followed by 38 cycles of 92°C (60 s), 61°C (90 s), and 72°C (60 s) and a final extension at 72°C (10 min). We took special care during the nested-PCR step: after the first PCR round, amplification products were handled in a different room than the PCR preparation room, and PCRs were monitored using negative and positive controls included in each run. PCR products from the second PCR rounds were visualized on 1.4% agarose gels. The PCR products were diluted by half, purified, and sequenced by the VIB Genetic Service Facility (Antwerp, Belgium) using the nested primers.
Analyses of MCMV infection across the HMHZ.
We calculated the prevalence of anti-MCMV antibody or infection (proportion of MCMV-positive individuals) with 95% confidence intervals (95% CI) in Quantitative Parasitology, version 3.0, using the Sterne exact method (29, 30). We used the chi-square test to determine whether the prevalence of anti-MCMV antibody or infection differed between the two sides of the HMHZ. To determine if mice were M. m. domesticus or M. m. musculus, we used the hybrid index (HI) (see reference 31), i.e., the proportion of M. m. musculus alleles based on 1,401 (32) or 0.62 million (33) single-nucleotide-polymorphism (SNP) markers. For the purposes of the current study, individuals were considered M. m. domesticus-like if the HI was ≥0 and <0.5 and M. m. musculus-like if the HI was >0.5 and ≤1. HI estimates were available for 441/520 of the mice studied.
Genetic analyses.
The viral sequences were corrected and aligned in Geneious, version 7.1.4. During the checking process, clear double peaks (distinct from the baseline noise) were visible on the sequencing chromatograms for several of the samples. These were likely the result of mixed infections. The uncertainty thus introduced could be encoded by replacing double-peak positions by undetermined nucleotides (e.g., R for A or G, Y for T or C). However, because several mixed sequences seemed to result from coinfections with very different MCMV strains, this encoding could produce substantial noise during phylogenetic reconstruction. We thus assumed coinfection when more than 5 double peaks occurred on the chromatograms (15.8% of hosts) and removed these sequences from the data for subsequent analyses. These double-peak chromatograms could have been resolved by cloning. However, since they were consistent with heterozygotes between “clean” (single-peak) sequences already observed, cloning would have added little or no information about MCMV diversity. Committing resources to cloning was therefore not justified. The number of haplotypes and haplotype diversity (h) were calculated in DnaSP, version 5 (34). The different MCMV variants were aligned at the amino acid level against other MCMV sequences and the rat CMV Maastricht strain (RCMV-M) sequence, available in GenBank, by using MUSCLE (35). The English strain of rat CMV (RCMV-E) was used as an outgroup, since it represents a distinct murine CMV lineage (36, 37). Regions containing indels were removed from the final alignment, resulting in 716 nucleotides that were used in subsequent analyses. We used RDP4 to test for the presence of recombination (38). We estimated evolutionary divergence using the p-distance method in MEGA, version 5.2.2 (39). jModelTest, version 0.1.1 (40, 41), was used to evaluate the fit of 40 nested models of nucleotide substitution to the sequences by using the Bayesian information criterion (BIC). The BIC indicated that the substitution model best fitting the data was the HKY85 model assuming a gamma-distributed rate variation among sites. Phylogenetic analyses were performed using model-based inference procedures: maximum likelihood (ML) methods implemented in MEGA, version 5.2.2, and Bayesian methods implemented in MrBayes, version 3.2.2 (42). For the ML tree, branch support was evaluated by 1,000 replicate bootstraps. We were interested in estimating the time of most recent common ancestor (tMRCA) for M. m. domesticus and M. m. musculus MCMVs by using the Bayesian inference approach and assuming that these viruses have coevolved with their murine hosts, based on node calibrations. We chose a strict clock model with uniform branch lengths. For the clock rate prior, we used a normal distribution with 0.35 as the mean and 0.17 as the standard deviation based on the estimation of a substitution rate in herpesviruses of 3.5 × 10−8 substitution/site/year (43). The tree age prior was set as gamma (100, 100), and we calibrated the node corresponding to the common ancestor for RCMV-M and MCMV by setting the prior as uniform (10–12), since the time of Mus-Rattus divergence has been estimated as 10 to 12 Mya (44). For the other parameters, we used the default priors. Two independent runs were conducted with 4 × 106 generations per run, and trees and parameters were sampled every 500 generations. Runs were initiated from random trees, and three hot chains plus one cold chain were used in all analyses. Convergence was assessed by examining the average standard deviation of split frequencies and the potential scale reduction factor. For each run, the first 25% of trees sampled were discarded as burn-in. Bayesian posterior probabilities were used to assess branch support. Trees were annotated in FigTree, version 1.4.1 (http://tree.bio.ed.ac.uk/software/figtree/).
Nucleotide sequence accession numbers.
The sequences determined in this study were deposited in GenBank under accession numbers KM360065 to KM360084.
RESULTS
ELISA was performed on samples from 291 mice from 100 localities across the HMHZ. The HI was known for 251 of these, identifying 126 mice as M. m. domesticus-like and 125 as M. m. musculus-like. The localities with seropositive individuals are presented in Fig. 2A. The prevalences of MCMV-seropositive individuals were 79.4% (95% CI, 74.3 to 83.7%) overall, 84.1% (95% CI, 76.7 to 89.8%) for M. m. domesticus, and 76.0% (95% CI, 67.7 to 82.9%) for M. m. musculus, but no significant difference in seropositivity was found between M. m. domesticus and M. m. musculus (χ2 = 2.598; P = 0.107). Screening for the detection of MCMV DNA in the lungs was performed for 319 mice from 97 localities. The HI was known for 272 of these, identifying 127 mice as M. m. domesticus-like and 145 as M. m. musculus-like. The localities with individuals positive for MCMV DNA are presented in Fig. 2B. The total number of PCR positive-individuals was 133, representing prevalences of 41.7% (95% CI, 36.4 to 47.2%) overall, 47.2% (95% CI, 38.6 to 55.9%) for M. m. domesticus, and 35.9% (95% CI, 28.2 to 44.1%) for M. m. musculus, with a potential difference in the prevalence of PCR-positive individuals between M. m. domesticus and M. m. musculus (χ2 = 3.621; P = 0.057). Thus, in the HMHZ, both antibodies against MCMV and the presence of MCMV DNA in the lungs were detected in both subspecies of the house mouse, with a tendency toward higher prevalence for M. m. domesticus than for M. m. musculus. For the 90 samples screened both by ELISA and by nested PCR, the ELISA outcome did not predict the outcome of the nested PCR (χ2 = 0.058; P = 0.81): of 22 ELISA-negative individuals, half were PCR positive and half were PCR negative; of 68 ELISA-positive individuals, 36 were PCR positive and 32 were PCR negative.
FIG 2.

Individuals were tested for the presence of anti-MCMV antibodies by ELISA (A) and for the presence of MCMV DNA by nested PCR (B). Filled circles, positive individuals; open circles, negative individuals. Distances are given in kilometers. The gray line represents the Czech-German border, and the dashed line marks the consensus hybrid zone center as estimated by Macholán et al. in 2008 (51).
After editing, we obtained 114 sequences of 737 bp containing 58 polymorphic sites, which clustered into 20 different haplotypes with a total haplotype diversity (h) of 0.676. No recombination event was detected in the M94 gene. Phylogeny estimation using ML and Bayesian methods produced trees that differed in only one nontrivial node. The 20 haplotypes of MCMV obtained in this study clustered in three well-supported clades labeled 1, 2 and 3 (ML bootstrap values, 96, 91, and 93%, respectively; the Bayesian posterior probability was 1 for all clades) (Fig. 3). In the ML tree, clades 1 and 2 shared a common ancestor but with weak support (bootstrap value, 30%), while in the Bayesian tree, clades 2 and 3 shared a common ancestor (posterior probability, 0.88). A few additional nodes differed between the Bayesian and ML trees at some tips of the tree, but they were not supported (bootstrap values, <50%). All existing MCMV sequences (those isolated from M. m. domesticus populations in Australia and lab strains Smith and K181) were within clade 1. When we plotted members of the three clades on a map of the HMHZ field area, a clear phylogeographic structure appeared: clade 1 was found on the western side of the HMHZ (i.e., in M. m. domesticus territory), and clades 2 and 3 were found only on the eastern side of the HMHZ (i.e., in M. m. musculus territory) (Fig. 4). Only 4/60 (6.7%) clade 1 MCMV strains were found in the “wrong” territory, i.e., in M. m. musculus-like mice (see Fig. 4). All four were found in localities close to the consensus hybrid zone center. No clade 2 or 3 MCMV strains were found in M. m. domesticus-like mice.
FIG 3.

Phylogenetic tree estimated from Bayesian analysis of the M94 gene of murid CMV, including MCMV strains sampled in the European house mouse hybrid zone in this study, the two MCMV lab strains (Smith and K181), MCMV strains isolated from wild M. m. domesticus mice in Australia, and the Maastricht isolate of rat CMV (RCMV-M). The English isolate of rat CMV (RCMV-E) was used to root the tree. The main clades, indicated by brackets, are identified on the right. Numbers above branches represent Bayesian posterior probability/ML bootstrap support. The small scale bar represents the number of nucleotide substitutions per site. The larger scale bar represents the time in Mya.
FIG 4.
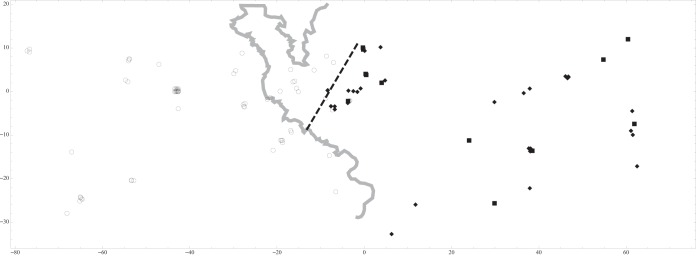
Map of the three MCMV M94 genetic lineages in the European house mouse hybrid zone. Open circles, lineage 1; filled squares, lineage 2: filled diamonds, lineage 3. Distances are given in kilometers. The gray line represents the Czech-German border, and the dashed line marks the consensus hybrid zone center as estimated by Macholán et al. in 2008 (51).
The average percentage of nucleotide differences per site between M. m. domesticus MCMV (clade 1) and M. m. musculus MCMV clade 2 or 3 was 5.4% or 5.3%, respectively (Table 1). The average percentage of amino acid differences per site between M. m. domesticus MCMV (clade 1) and M. m. musculus MCMV clade 2 or 3 was 5.8% or 6.5%, respectively. The average percentages of nucleotide and amino acid differences within M. m. domesticus MCMV clade 1 (including not only M. m. domesticus MCMVs from the HMHZ but also those from Australia as well as the two lab strains Smith and K181) were 1.13 and 1.25%, respectively. In comparison, the average percentages of nucleotide and amino acid differences among all M. m. musculus MCMV haplotypes were twice those within clade 1 (2.40 and 2.86%, respectively), although these differences were relatively limited within each clade of M. m. musculus MCMV (0.25 and 0.17% in clade 2 and 0.35 and 0.39% in clade 3, respectively) (Table 1). In summary, the diversity of M. m. musculus MCMVs is higher than that of M. m. domesticus MCMVs for the M94 gene. These differences were small, however, compared to the nucleotide and amino acid differences between MCMV and RMCV-M (36 and 45.4%, respectively).
TABLE 1.
Percentages of nucleotide and amino acid differences (p distances) in murid CMV M94 sequences
| Virusa | Data for:b |
||||||
|---|---|---|---|---|---|---|---|
| RCMV-E | RCMV-M | MCMVs (all) | MCMV clade 1 (all M. m. domesticus MCMVs) |
M. m. musculus MCMVs |
|||
| All | Clade 2 | Clade 3 | |||||
| RCMV-E | 50.8 | 51.7 | 51.8 | 51.5 | 51.7 | 51.3 | |
| RCMV-M | 47.4 | 45.4 | 45.4 | 45.5 | 45.0 | 46.01 | |
| MCMVs (all) | 45.7 | 36.0 | 3.26/3.76 | ||||
| MCMV clade 1 (all M. m. domesticus MCMVs) | 45.6 | 36.2 | 1.13/1.25 | 6.1 | 5.8 | 6.4 | |
| M. m. musculus MCMVs | |||||||
| All | 45.8 | 35.5 | 5.32 | 2.40/2.86 | |||
| Clade 2 | 45.7 | 35.7 | 5.4 | 0.25/0.17 | 5.0 | ||
| Clade 3 | 45.8 | 35.3 | 5.3 | 4.2 | 0.35/0.39 | ||
Clades 1, 2 and 3 are shown in Fig. 3.
Diagonal, number of nucleotide/amino acid differences per site, averaging over all sequence pairs within each group. Below diagonal, nucleotides. Above diagonal, amino acids.
We calibrated the Bayesian tree based on the time of Mus-Rattus divergence (10 to 12 Mya), assuming that murine CMVs have coevolved with their hosts, to estimate the tMRCA of all MCMV diversity found across the two mouse subspecies. The Bayesian Markov chain Monte Carlo (MCMC) inference resulted in a tMRCA estimation of 1.12 Mya (95% highest posterior density [HPD] interval, 0.80 to 1.52 Mya) (Fig. 3). The root is placed at 21.33 Mya (95% HPD interval, 16.33 to 27.19 Mya) (this is the tMRCA of the two lineages of CMV proposed to have evolved separately in Murinae [36, 37]).
DISCUSSION
To date, MCMV has been described only in a single mouse subspecies, Mus musculus domesticus (1). We used the European HMHZ to investigate whether another subspecies of the house mouse, Mus musculus musculus, could also carry MCMV and how specific MCMV was for its host. We found that MCMV infection was very frequent among mice on both sides of the HMHZ. Our genetic analysis revealed that three different lineages were present in the sampling area, one in M. m. domesticus and two in M. m. musculus, and that these lineages were structured in relation to the host genetics. Finally, the estimated tMRCA for M. m. domesticus and M. m. musculus MCMVs was consistent with the time of divergence of their hosts. Our results have several implications for the biology and evolutionary history of MCMV in wild mouse populations.
MCMV infection has been shown to be highly prevalent in natural populations of M. m. domesticus on several continents (Australia, United States) and islands (1), and our study confirms that this is also the case for European M. m. domesticus populations. We report for the first time that MCMV infection can also be found in another subspecies of the house mouse, M. m. musculus, and also at a high prevalence (76% antibody prevalence and 35.9% viral DNA prevalence), reflecting transmission at high frequency. The high levels of infection detected in M. m. domesticus and M. m. musculus, the fact that each subspecies of mice carries its own lineage(s) of viruses, and the match between the hosts' divergence time and the tMRCA of M. m. domesticus and M. m. musculus MCMVs (∼1 Mya) are in favor of a long-term association or coevolution of MCMV with its Mus musculus subspecies. The high prevalence of MCMV in a subspecies other than M. m. domesticus warrants future investigations into the occurrence of MCMV in other species and subspecies of Mus. It is likely that several members of this subgenus carry their own lineages of MCMV, so the true diversity of MCMV in the wild is very much underappreciated.
We found almost double the number of positive individuals by ELISA as by nested PCR. CMV establishes latent infection in some tissues of the host after recovery from acute infection, and lungs have been shown to be one of the main organ sites for latency (45). Since we examined lung tissue, we expected that the number of positive nested-PCR samples would be higher. Possible explanations for the low PCR-positive count include the following: (i) a number of mice had acute infections with viruses present in organs such as salivary glands but not in the lungs; (ii) the latent virus remained undetectable in the lungs due to a lack of sensitivity of our molecular assay (46); (iii) the ELISA was not specific to MCMV and detected antibodies from other herpesviruses. Recently, a new betaherpesvirus that clusters with the English isolate of RCMV was found in the house mouse, suggesting that paired distinct CMV lineages exist in both Mus musculus and Rattus norvegicus (37). The other surprising aspect of these results is that 11 ELISA-negative individuals were found positive for MCMV DNA in the lungs. Possible explanations are that these individuals could have been sampled just after infection but before mounting an immune response or that there was cross-contamination of MCMV in the lab (an elevated risk with nested PCR). Two considerations make us think that cross-contamination is unlikely: (i) we took extra care during all steps of the molecular screening and in particular during the nested-PCR step, and (ii) the three lineages show a clear genetic structure consistent with the geographic origins of their sampling. Cross-contamination would erase such signals. Finally, we note that this type of “discrepancy” between negative serology and positive CMV DNA detection has already been reported for other betaherpesviruses (e.g., HCMV [47]).
The complex multiple-secondary-contact (reticulate) history of M. m. domesticus and M. m. musculus, and likely also Mus musculus castaneus (20), may have influenced the evolutionary history of MCMV strains. Our analyses indicate that M. m. musculus carries two lineages of MCMV for the M94 gene, but our phylogenetic estimates from the two model-based methods contradict each other with regard to tree topology. We suggest that more data are necessary to resolve the source of this diversity: we should be careful not to overinterpret results based only on a single well-conserved gene, M94 (2). Longer genomic fragments of these new MCMV strains encompassing conserved and more diverse genes could help to resolve the evolutionary history of MCMV in M. m. musculus.
The new strains from M. m. musculus show at least 5.3% nucleotide divergence from strain K181 for the M94 gene, while K181 and other strains isolated from M. m. domesticus show almost no divergence for this gene (1.13%). It would be interesting to test if the specificity of MCMV strains observed in wild M. m. domesticus and M. m. musculus populations also applies to inbred lab mice. Inbred lab mice are of mixed origin, with elements of M. m. domesticus, M. m. musculus, and M. m. castaneus genomes (6). If the new M. m. musculus MCMV strains described here are capable of infecting inbred lab mice, they may show very different kinetics and a very different biology of infection from those of the standard strains used in research, and thus, they may be useful complementary models.
MCMV is the second example suggesting that the mouse hybrid zone may act as a barrier to pathogen gene flow. A recent study on the apicomplexan protozoan Cryptosporidium tyzzeri showed that different subspecies of mice harbor different lineages of this parasite, which have likely coevolved with their hosts (48). Our study is based on the M94 gene, which is essential to viral morphogenesis (28) and is relatively conserved between MCMV strains (2). The cryptosporidium study was also based on conserved genes (48). It would be interesting to investigate how genes that are more variable and are involved in the evasion of the host immune system, such as the MCMV m02 and m145 gene families, involved in immune subversion via NK cell-mediated immune escape (49), behave at the contact zone between the two mouse subspecies. If we expect host immune genes, such as the major histocompatibility complex (MHC) genes, to introgress into the genetic background of the parental taxa in hybrid zones, as proposed recently (31, 50), we might expect that pathogen genes involved in the subversion of the host immune system will do the same.
ACKNOWLEDGMENTS
Collecting such a large body of data required contributions from many colleagues and local farmers, and we warmly acknowledge their help.
This work was supported by Czech Science Foundation grants 14-35009S and 206/08/0640.
REFERENCES
- 1.Shellam GR, Redwood AJ, Smith LM, Gorman S. 2007. Murine cytomegalovirus and other herpesviruses,p 1–48. In Fox JG, Barthold SW, Davisson MT, Newcomer CE, Quimby FW, Smith AL (ed), The mouse in biomedical research, 2nd ed, vol II Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 2.Smith LM, McWhorter AR, Masters LL, Shellam GR, Redwood AJ. 2008. Laboratory strains of murine cytomegalovirus are genetically similar to but phenotypically distinct from wild strains of virus. J Virol 82:6689–6696. doi: 10.1128/JVI.00160-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith MG. 1954. Propagation of salivary gland virus of the mouse in tissue cultures. Proc Soc Exp Biol Med 86:435–440. [DOI] [PubMed] [Google Scholar]
- 4.Smith LM, McWhorter AR, Shellam GR, Redwood AJ. 2013. The genome of murine cytomegalovirus is shaped by purifying selection and extensive recombination. Virology 435:258–268. doi: 10.1016/j.virol.2012.08.041. [DOI] [PubMed] [Google Scholar]
- 5.Booth TWM, Scalzo AA, Carrello C, Lyons PA, Farrell H, Singleton GR, Shellam GR. 1993. Molecular and biological characterization of new strains of murine cytomegalovirus isolated from wild mice. Arch Virol 132:209–220. doi: 10.1007/BF01309855. [DOI] [PubMed] [Google Scholar]
- 6.Yang H, Wang JR, Didion JP, Buus RJ, Bell TA, Welsh CE, Bonhomme F, Yu AH-T, Nachman MW, Pialek J, Tucker P, Boursot P, McMillan L, Churchill GA, de Villena FP-M. 2011. Subspecific origin and haplotype diversity in the laboratory mouse. Nat Genet 43:648–655. doi: 10.1038/ng.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Becker SD, Bennett M, Stewart JP, Hurst JL. 2007. Serological survey of virus infection among wild house mice (Mus domesticus) in the UK. Lab Anim 41:229–238. doi: 10.1258/002367707780378203. [DOI] [PubMed] [Google Scholar]
- 8.Moro D, Lawson MA, Hobbs RP, Thompson RC. 2003. Pathogens of house mice on arid Boullanger Island and subantarctic Macquarie Island, Australia. J Wildl Dis 39:762–771. doi: 10.7589/0090-3558-39.4.762. [DOI] [PubMed] [Google Scholar]
- 9.Singleton GR, Smith AL, Krebs CJ. 2000. The prevalence of viral antibodies during a large population fluctuation of house mice in Australia. Epidemiol Infect 125:719–727. doi: 10.1017/S0950268800004945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singleton GR, Smith AL, Shellam GR, Fitzgerald N, Muller WJ. 1993. Prevalence of viral antibodies and helminths in field populations of house mice (Mus domesticus) in southeastern Australia. Epidemiol Infect 110:399–417. doi: 10.1017/S0950268800068345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacob J, Sutherland DR. 2004. Murine cytomegalovirus (MCMV) infections in house mice: a matter of age or sex? Wildl Res 31:369–373. doi: 10.1071/WR03130. [DOI] [Google Scholar]
- 12.Gorman S, Harvey NL, Moro D, Lloyd ML, Voigt V, Smith LM, Lawson MA, Shellam GR. 2006. Mixed infection with multiple strains of murine cytomegalovirus occurs following simultaneous or sequential infection of immunocompetent mice. J Gen Virol 87:1123–1132. doi: 10.1099/vir.0.81583-0. [DOI] [PubMed] [Google Scholar]
- 13.Farroway LN, Singleton GR, Lawson MA, Jones DA. 2002. The impact of murine cytomegalovirus (MCMV) on enclosure populations of house mice (Mus domesticus). Wildl Res 29:11–17. doi: 10.1071/WR01066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Auffray J-C, Britton-Davidian J. 2012. The house mouse and its relatives: systematics and taxonomy, p 1–34. In Machoĺan M, Baird SJE, Munclinger P, Píalek J (ed), The evolution of the house mouse. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 15.Boursot P, Auffray J-C, Britton-Davidian J, Bonhomme F. 1993. The evolution of house mice. Annu Rev Ecol Syst 24:119–152. doi: 10.1146/annurev.es.24.110193.001003. [DOI] [Google Scholar]
- 16.Jones EP, Van Der Kooij J, Solheim R, Searle JB. 2010. Norwegian house mice (Mus musculus musculus/domesticus): distributions, routes of colonization and patterns of hybridization. Mol Ecol 19:5252–5264. doi: 10.1111/j.1365-294X.2010.04874.x. [DOI] [PubMed] [Google Scholar]
- 17.Ďureje L, Macholán M, Baird SJE, Piálek J. 2012. The mouse hybrid zone in Central Europe: from morphology to molecules. Fol Zool 61:308–318. [Google Scholar]
- 18.Albrechtová J, Albrecht T, Baird SJE, Macholán M, Rudolfsen G, Munclinger P, Tucker PK, Piálek J. 2012. Sperm-related phenotypes implicated in both maintenance and breakdown of a natural species barrier in the house mouse. Proc Biol Sci 279:4803–4810. doi: 10.1098/rspb.2012.1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turner LM, Schwahn DJ, Harr B. 2012. Reduced male fertility is common but highly variable in form and severity in a natural house mouse hybrid zone. Evolution 66:443–458. doi: 10.1111/j.1558-5646.2011.01445.x. [DOI] [PubMed] [Google Scholar]
- 20.Duvaux L, Belkhir K, Boulesteix M, Boursot P. 2011. Isolation and gene flow: inferring the speciation history of European house mice. Mol Ecol 20:5248–5264. doi: 10.1111/j.1365-294X.2011.05343.x. [DOI] [PubMed] [Google Scholar]
- 21.Geraldes A, Basset P, Gibson B, Smith KL, Harr B, Yu H-T, Bulatova N, Ziv Y, Nachman MW. 2008. Inferring the history of speciation in house mice from autosomal, X-linked, Y-linked and mitochondrial genes. Mol Ecol 17:5349–5363. doi: 10.1111/j.1365-294X.2008.04005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.She JX, Bonhomme F, Boursot P, Thaler L, Catzeflis F. 1990. Molecular phylogenies in the genus Mus: comparative analysis of electrophoretic, scnDNA hybridization, and mtDNA RFLP data. Biol J Linnean Soc 41:83–103. doi: 10.1111/j.1095-8312.1990.tb00823.x. [DOI] [Google Scholar]
- 23.Boursot P, Din W, Anand R, Darviche D, Dod B, Von Deimling F, Talwar GP, Bonhomme F. 1996. Origin and radiation of the house mouse: mitochondrial DNA phylogeny. J Evol Biol 9:391–415. doi: 10.1046/j.1420-9101.1996.9040391.x. [DOI] [Google Scholar]
- 24.Suzuki H, Shimada T, Terashima M, Tsuchiya K, Aplin K. 2004. Temporal, spatial, and ecological modes of evolution of Eurasian Mus based on mitochondrial and nuclear gene sequences. Mol Phylogenet Evol 33:626–646. doi: 10.1016/j.ympev.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 25.Geraldes A, Basset P, Smith KL, Nachman MW. 2011. Higher differentiation among subspecies of the house mouse (Mus musculus) in genomic regions with low recombination. Mol Ecol 20:4722–4736. doi: 10.1111/j.1365-294X.2011.05285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guénet J-L, Bonhomme F. 2003. Wild mice: an ever-increasing contribution to a popular mammalian model. Trends Genet 19:24–31. doi: 10.1016/S0168-9525(02)00007-0. [DOI] [PubMed] [Google Scholar]
- 27.McGeoch DJ, Dolan A, Ralph AC. 2000. Toward a comprehensive phylogeny for mammalian and avian herpesviruses. J Virol 74:10401–10406. doi: 10.1128/JVI.74.22.10401-10406.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maninger S, Bosse JB, Lemnitzer F, Pogoda M, Mohr CA, von Einem J, Walther P, Koszinowski UH, Ruzsics Z. 2011. M94 is essential for the secondary envelopment of murine cytomegalovirus. J Virol 85:9254–9267. doi: 10.1128/JVI.00443-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reiczigel J. 2003. Confidence intervals for the binomial parameter: some new considerations. Stat Med 22:611–621. doi: 10.1002/sim.1320. [DOI] [PubMed] [Google Scholar]
- 30.Rózsa L, Reiczigel J, Majoros G. 2000. Quantifying parasites in samples of hosts. J Parasitol 86:228–232. doi: 10.2307/3284760. [DOI] [PubMed] [Google Scholar]
- 31.Baird SJE, Ribas A, Macholán M, Albrecht T, Piálek J, Goüy de Bellocq J. 2012. Where are the wormy mice? a reexamination of hybrid parasitism in the European house mouse hybrid zone. Evolution 66:2757–2772. doi: 10.1111/j.1558-5646.2012.01633.x. [DOI] [PubMed] [Google Scholar]
- 32.Wang L, Luzynski K, Pool J, Janoušek V, Dufková P, Vyskocilová M, Teeter K, Nachman M, Munclinger P, Macholán M, Piálek J, Tucker PK. 2011. Measures of linkage disequilibrium among neighbouring SNPs indicate asymmetries across the house mouse hybrid zone. Mol Ecol 20:2985–3000. doi: 10.1111/j.1365-294X.2011.05148.x. [DOI] [PubMed] [Google Scholar]
- 33.Yang H, Ding Y, Hutchins LN, Szatkiewicz J, Bell TA, Paigen BJ, Graber JH, de Villena FP-M, Churchill GA. 2009. A customized and versatile high-density genotyping array for the mouse. Nat. Methods 6:663–666. doi: 10.1038/nmeth.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 35.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ettinger J, Geyer H, Nitsche A, Zimmermann A, Brune W, Sandford GR, Hayward GS, Voigt S. 2012. Complete genome sequence of the English isolate of rat cytomegalovirus (Murid herpesvirus 8). J Virol 86:13838. doi: 10.1128/JVI.02614-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teterina A, Richter D, Matuschka F-R, Ehlers B, Voigt S. 2009. Identification of a novel betaherpesvirus in Mus musculus. Virol J 6:225. doi: 10.1186/1743-422X-6-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. 2010. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 41.Posada D. 2008. jModelTest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- 42.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakaoka H, Kurita K, Iida Y, Takada S, Umene K, Kim YT, Ren CS, Nahmias AJ. 1994. Quantitative analysis of genomic polymorphism of herpes simplex virus type 1 strains from six countries: studies of molecular evolution and molecular epidemiology of the virus. J Gen Virol 75:513–527. doi: 10.1099/0022-1317-75-3-513. [DOI] [PubMed] [Google Scholar]
- 44.Lecompte E, Aplin K, Denys C, Catzeflis F, Chades M, Chevret P. 2008. Phylogeny and biogeography of African Murinae based on mitochondrial and nuclear gene sequences, with a new tribal classification of the subfamily. BMC Evol Biol 8:199. doi: 10.1186/1471-2148-8-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Balthesen M, Messerle M, Reddehase MJ. 1993. Lungs are a major organ site of cytomegalovirus latency and recurrence. J Virol 67:5360–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurz S, Steffens HP, Mayer A, Harris JR, Reddehase MJ. 1997. Latency versus persistence or intermittent recurrences: evidence for a latent state of murine cytomegalovirus in the lungs. J Virol 71:2980–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Tommaso A, Andrade P, Costa S, Escanhoela C, Hessel G. 2005. High frequency of human cytomegalovirus DNA in the liver of infants with extrahepatic neonatal cholestasis. BMC Infect Dis 5:108. doi: 10.1186/1471-2334-5-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kváč M, McEvoy J, Loudová M, Stenger B, Sak B, Květoňová D, Ditrich O, Rašková V, Moriarty E, Rost M, Macholán M, Piálek J. 2013. Coevolution of Cryptosporidium tyzzeri and the house mouse (Mus musculus). Int J Parasitol 43:805–817. doi: 10.1016/j.ijpara.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oliveira SA, Park S-H, Lee P, Bendelac A, Shenk TE. 2002. Murine cytomegalovirus m02 gene family protects against natural killer cell-mediated immune surveillance. J Virol 76:885–894. doi: 10.1128/JVI.76.2.885-894.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goüy de Bellocq J, Ribas A, Baird SJE. 2012. New insights into parasitism in the house mouse hybrid zone, p 455–481. In Machoĺan M, Baird SJE, Munclinger P, Píalek J (ed), The evolution of the house mouse. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 51.Macholán M, Baird SJE, Munclinger P, Dufková P, Bimová B, Piálek J. 2008. Genetic conflict outweighs heterogametic incompatibility in the mouse hybrid zone? BMC Evol Biol 8:271. doi: 10.1186/1471-2148-8-271. [DOI] [PMC free article] [PubMed] [Google Scholar]