

ABSTRACT
Adenoviruses are frequent causes of pediatric myocarditis. Little is known about the pathogenesis of adenovirus myocarditis, and the species specificity of human adenoviruses has limited the development of animal models, which is a significant barrier to strategies for prevention or treatment. We have developed a mouse model of myocarditis following mouse adenovirus 1 (MAV-1) infection to study the pathogenic mechanisms of this important cause of pediatric myocarditis. Following intranasal infection of neonatal C57BL/6 mice, we detected viral replication and induction of interferon gamma (IFN-γ) in the hearts of infected mice. MAV-1 caused myocyte necrosis and induced substantial cellular inflammation that was composed predominantly of CD3+ T lymphocytes. Depletion of IFN-γ during acute infection reduced cardiac inflammation in MAV-1-infected mice without affecting viral replication. We observed decreased contractility during acute infection of neonatal mice, and persistent viral infection in the heart was associated with cardiac remodeling and hypertrophy in adulthood. IFN-γ is a proinflammatory mediator during adenovirus-induced myocarditis, and persistent adenovirus infection may contribute to ongoing cardiac dysfunction.
IMPORTANCE Studying the pathogenesis of myocarditis caused by different viruses is essential in order to characterize both virus-specific and generalized factors that contribute to disease. Very little is known about the pathogenesis of adenovirus myocarditis, which is a significant impediment to the development of treatment or prevention strategies. We used MAV-1 to establish a mouse model of human adenovirus myocarditis, providing the means to study host and pathogen factors contributing to adenovirus-induced cardiac disease during acute and persistent infection. The MAV-1 model will enable fundamental studies of viral myocarditis, including IFN-γ modulation as a therapeutic strategy.
INTRODUCTION
Acute myocarditis is a significant cause of morbidity and mortality in childhood. Myocarditis has been identified in 16 to 21% of sudden deaths in children (1). The course of viral myocarditis is often more severe in neonates and infants than in older patients, with up to 67% mortality in newborns and 55% in infants less than 1 year of age, compared to 20 to 25% in older children and 38% in adults (2, 3). Associations between myocarditis and adenovirus infection are well established (3–5). Persistent human adenovirus (HAdV) infections of the myocardium have been implicated in the development of dilated cardiomyopathy and cardiac dysfunction (6–8). Detection of HAdV genomes in myocardial biopsy specimens of pediatric heart transplant recipients is predictive of coronary artery vasculopathy and transplant loss (9). The histopathology of hearts in patients with HAdV myocarditis can be milder than that in patients with myocarditis caused by other viruses (3), suggesting that mechanisms underlying HAdV-mediated disease may differ from those involved in myocarditis caused by other viruses.
Much of what is known about viral myocarditis has come from the study of myocarditis resulting from coxsackievirus B3 (CVB3) and reovirus. The pathogenesis of viral myocarditis involves many interrelated processes. Direct damage to cardiac myocytes can occur by viral lysis, inhibition of host protein synthesis, or cleavage of host proteins such as dystrophin by viral proteases (10). Immune responses to acute infection contribute to the clearance of virus but can also cause further damage, either directly or due to cross-reaction between viral and cardiac antigens that leads to autoimmune myocardial injury (11). Interferon alpha (IFN-α) and IFN-β, the type I IFNs, are essential for limiting viral replication in cardiac myocytes and are protective in a neonatal mouse model of reovirus myocarditis (12). IFN-γ, the type II IFN, is upregulated and is often protective in models of viral myocarditis (12–14), although other reports suggest that it can promote myocarditis associated with CVB3 (15, 16), and prolonged expression of IFN-γ may lead to a chronic inflammatory cardiomyopathy (17). Myocyte apoptosis induced by viral infection and virus-induced inflammation can also contribute to progressive cardiac damage (18). Ongoing injury can lead to myocardial remodeling and fibrosis accompanied by ventricular dilation and cardiac dysfunction.
We have previously established intranasal (i.n.) mouse adenovirus 1 (MAV-1) infection of C57BL/6 and BALB/c mice as a model to study the pathogenesis of adenovirus respiratory infection (19). One previous report demonstrated that intraperitoneal (i.p.) MAV-1 infection of neonatal outbred Swiss Webster mice led to cardiac inflammation, myocardial fibrosis, and calcification (20), and a related virus (MAV-3) was detected in the heart following intravenous infection of adult C57BL/6N mice (21). However, MAV-1 and MAV-3 have yet to be used in detailed studies of myocarditis pathogenesis. Here, we demonstrate that intranasal MAV-1 infection of neonatal mice led to viral replication and induction of IFN-γ expression in the heart that correlated with cellular inflammation and acute myocyte necrosis. Depletion of IFN-γ during acute infection reduced cardiac inflammation in MAV-1-infected mice without affecting viral replication. Long-term persistence of viral DNA was associated with increased heart mass and cellular hypertrophy.
MATERIALS AND METHODS
Cardiac myocyte primary culture and infection.
Cardiac myocytes were isolated as previously described (22), plated on laminin-coated coverslips, cultured in the presence of a 25 μM concentration of the contraction inhibitor S-(−)-blebbistatin (Toronto Research Chemicals), and infected at a multiplicity of infection (MOI) of 5. At 24 or 48 h, supernatant was collected and the cells were incubated in 0.5 ml of TRIzol (Invitrogen) for 5 min. RNA and DNA were isolated according to the manufacturer's protocol and resuspended in high-pressure liquid chromatography (HPLC)-grade water. Infectious virus in supernatant was quantified by plaque assay as previously described (23).
Mice.
All work was approved by the University of Michigan Committee on Use and Care of Animals. C57BL/6 mothers with litters of neonatal mice and male C57BL/6 mice (4 to 6 weeks of age) were obtained from The Jackson Laboratory. All mice were maintained under specific-pathogen-free conditions.
Virus and infections.
MAV-1 was grown and its titers determined on NIH 3T6 fibroblasts as previously described (23). Neonates (7 days old) were manually restrained and infected intranasally (i.n.) with 105 PFU in 10 μl of sterile phosphate-buffered saline (PBS). Adult mice were anesthetized with ketamine and xylazine and infected i.n. with 105 PFU of MAV-1 in 40 μl of sterile PBS. Control mice were mock infected i.n. with conditioned medium at an equivalent dilution in sterile PBS. Mice were euthanized by pentobarbital overdose at the indicated time points. Blood was collected from the posterior vena cava of euthanized animals and incubated on ice for 15 to 30 min. Samples were centrifuged in a tabletop microcentrifuge at 17,000 × g for 10 min at 4°C. Serum was transferred to a new microcentrifuge tube and stored at −80°C until assayed. After blood collection, hearts were harvested, snap-frozen in dry ice, and stored at −80°C until processed further. One-third to one-half of each heart (∼20 mg) was homogenized in 1 ml of TRIzol (Invitrogen) using sterile glass beads in a mini-Beadbeater (Biospec Products) for 30 s. RNA and DNA were isolated from the homogenates according to the manufacturer's protocol.
Analysis of viral loads by PCR.
MAV-1 viral loads were measured in organs and in cardiac myocytes infected ex vivo using quantitative real-time PCR (qPCR) as previously described (24). The primers and probe used to detect a 59-bp region of the MAV-1 E1A gene are listed in Table 1. Five microliters of extracted DNA was added to reaction mixtures containing TaqMan II universal PCR mix with uracil N-glycosylase (UNG) (Applied Biosystems), forward and reverse primers (each at a 200 nM final concentration), and probe (20 nM final concentration) in a 25-μl reaction volume. Analysis on an ABI Prism 7300 machine (Applied Biosystems) consisted of 40 cycles of 15 s at 90°C and 60 s at 60°C. Standard curves generated using known amounts of plasmid containing the MAV-1 E1A gene were used to convert threshold cycle values for experimental samples to copy numbers of E1A DNA. Results were standardized to the nanogram amount of input DNA. Each sample was assayed in triplicate.
TABLE 1.
Primers and probes used for real-time PCR analysis
| Target | Oligonucleotide | Sequence (5′ to 3′) |
|---|---|---|
| MAV-1 E1A genomic | Forward primer | GCACTCCATGGCAGGATTCT |
| Reverse primer | GGTCGAAGCAGACGGTTCTTC | |
| Probe | TACTGCCACTTCTGC | |
| GAPDH | Forward primer | TGCACCACCAACTGCTTAG |
| Reverse primer | GGATGCAGGGATGATGTTC | |
| IFN-γ | Forward primer | AAAGAGATAATCTGGCTCTGC |
| Reverse primer | GCTCTGAGACAATGAACGCT | |
| TNF-α | Forward primer | CCACCACGCTCTTCTGTCTAC |
| Reverse primer | AGGGTCTGGGCCATAGAACT | |
| MAV-1 E1A | Forward primer | AATGGGTTTTGCAGTCTGTGTTAC |
| Reverse primer | CGCCTGAGGCAGCAGATC | |
| MAV-1 hexon | Forward primer | GGCCAACACTACCGACACTT |
| Reverse primer | TTTTGTCCTGTGGCATTTGA |
Nested PCR detection of viral DNA.
Primers used in nested PCR to detect a final 246-bp region of MAV-1 E1A are listed in Table 2. Five hundred nanograms of extracted DNA was added to reaction mixtures containing 10× standard Taq buffer (New England BioLabs), 4 mM MgCl2, 0.8 mM deoxynucleoside triphosphate (Promega), Taq polymerase (New England BioLabs), and 100 nM forward and reverse primers in a 100-μl reaction volume. The first PCR amplification was carried out with 1 cycle at 94°C for 10 min, 35 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s, and 1 cycle at 72°C for 7 min on an Eppendorf thermocycler. After the initial round of PCR, 20 μl of primary PCR product was added to a fresh PCR mixture and amplified in a second round of PCR. The second PCR round was conducted under the same conditions with the exception of 200 nM primer concentrations instead of 100 nM and 30 cycles instead of 35.
TABLE 2.
Primers used for nested PCR analysis
| Target and round | Oligonucleotide | Sequence (5′ to 3′) |
|---|---|---|
| MAV-1 E1A | ||
| 1 | Forward primer | ATGTCGCGGCTCCTACG |
| Reverse primer | CAACGAACCATAAAAAGACATCAT | |
| 2 | Forward primer | ATGGGATGGTTCGCCTACTT |
| Reverse primer | CACCGCAGATCCATGTCCTCAA | |
| Actin | Forward primer | CCTAAGGCCAACCGTGAAAAGATG |
| Reverse primer | ACCGCTCGTTGCCAATAGTGATG |
Real-time PCR analysis of gene expression.
Cytokine gene expression was quantified using reverse transcriptase qPCR (RT-qPCR) as previously described (25, 26). First, 2.5 μg of RNA was reverse transcribed using Moloney murine leukemia virus (MMLV) reverse transcriptase (Invitrogen) in 20-μl reaction mixtures according to the manufacturer's instructions. Water was added to the cDNA product to bring the total volume to 50 μl. cDNA was amplified using duplexed gene expression assays for mouse CCL5 and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Applied Biosystems). Five microliters of cDNA was added to reaction mixtures containing TaqMan universal PCR mix and 1.25 μl each of 20× gene expression assay mixtures for the target cytokine and GAPDH. Primers used to detect IFN-γ, tumor necrosis factor alpha (TNF-α), GAPDH, early region 1A (E1A), and hexon are listed in Table 1. For these measurements, 5 μl of cDNA was added to reaction mixtures containing Power SYBR green PCR mix (Applied Biosystems) and forward and reverse primers (each at a 200 nM final concentration) in a 25-μl reaction volume. In all cases, RT-qPCR analysis consisted of 40 cycles of 15 s at 90°C and 60 s at 60°C. Quantification of target gene mRNA was normalized to GAPDH and expressed in arbitrary units as 2−ΔCT, where CT is the threshold cycle and ΔCT = CT(target) − CT(GAPDH).
IFN-γ neutralization.
Rabbit anti-mouse IFN-γ (α-IFN-γ) polyclonal antibody (27) and nonimmune rabbit serum were generously provided by Steven Kunkel (University of Michigan) and purified using protein A column purification (Thermo Scientific). Beginning on the first day postinfection (p.i.), mice were treated with 50 μg of α-IFN-γ antibody or nonimmune rabbit IgG given i.p. every other day beginning at 1 day p.i. Neutralizing activity of α-IFN-γ was confirmed by its capacity to block IFN-γ-mediated repression of MAV-1 replication in vitro (data not shown).
Measurement of IFN-γ Protein.
In some experiments, heart tissue (approximately half of a neonatal heart) was homogenized in PBS containing protease inhibitor (complete, Mini, EDTA-free tablets; Roche Applied Science) and 1% Triton X-100 (Fisher Scientific) at a concentration of 50 mg lung tissue per 1 ml homogenization buffer. Tissue was homogenized using sterile glass beads in a mini-Beadbeater (Biospec Products) for 2 30-s cycles, with resting on ice between cycles. After homogenization, tissue was spun twice at 17,000 × g for 15 min at 4°C, and supernatant was stored at −80°C until assayed. IFN-γ protein concentrations in heart homogenates were determined by enzyme-linked immunosorbent assay (ELISA) (Duoset kits; R&D Systems) according to the manufacturer's protocol.
Histology.
Hearts were fixed in 10% formalin and embedded in paraffin. Five-micrometer sections were stained with hematoxylin and eosin to evaluate cellular infiltrates. Separate sections were stained with anti-CD3 antibody (Thermo Scientific). CD3+ cells were quantified at a magnification of ×40, using the average from at least three independent fields per sample. Results are expressed as the number of CD3+ cells per high-power field (HPF). In some experiments, hearts were removed and immediately frozen in Tissue-Tek OCT compound (Sakura Finetek), and 5 μm sections were stained with antibodies to CD4 and CD8 (Serotec and BD Pharmingen, respectively). Sectioning and immunohistochemical staining were performed by the University of Michigan Unit for Laboratory Animal Medicine Pathology Cores for Animal Research. Heart sections were viewed through a Laborlux 12 microscope (Leitz). Digital images were obtained with an EC3 digital imaging system (Leica Microsystems) using Leica Acquisition Suite software (Leica Microsystems). Images were assembled using Adobe Illustrator (Adobe Systems). To quantify cellular inflammation in the hearts, slides were examined in a blinded fashion to determine a pathology index score for the size/intensity of cellular infiltrate and the extent of involvement in the heart (Table 3).
TABLE 3.
Quantification of cellular inflammation in histological specimens
| Score | Description |
|---|---|
| Severitya | |
| 0 | No inflammatory infiltrates |
| 1 | Inflammatory cells present without discrete foci |
| 2 | Larger foci of 10 to 100 inflammatory cells |
| 3 | Larger foci of >100 inflammatory cells |
| Extent | |
| 1 | Mild (<25% of section involved) |
| 2 | Moderate (∼25–75% of section involved) |
| 3 | Severe (>75% of section involved) |
A score from 0 to 3 was given for the size/intensity of cellular infiltrates. The score was then multiplied by a number reflecting the extent of involvement in the specimen to reach a single final severity score, resulting in a total score that could range from 0 to 9.
To visualize cardiomyocyte cell membranes, paraffin-free left ventricular (LV) heart sections were incubated with 100 μg/ml fluorescein isothiocyanate (FITC)-conjugated wheat germ agglutinin (Sigma) in PBS for 2 h and then washed 3 times with PBS. Slides were mounted using Prolong Gold Antifade reagent with DAPI (4,6-diamidine-2-phenylindole) (Invitrogen). Fluorescent images were viewed through an Olympus BX41 microscope, and digital images were processed using Olympus DP Manager software. Cross-sectional areas were calculated from fluorescent images using NIH ImageJ software (28).
Determination of serum cardiac troponin levels.
Cardiac troponin I (cTnI) concentrations were measured using the Ultra Sensitive Mouse Cardiac Troponin-I ELISA kit (Life Diagnostics) according to the manufacturer's instructions. Samples were assayed in duplicate.
Echocardiography.
In vivo echocardiography was performed as previously described (29), consistent with guidelines of the American Society of Echocardiography. Mice were anesthetized by inhaled isoflurane, chest hair was removed with Nair (Church & Dwight), and imaging was performed using a Vevo770 ultrasound system (Visual Sonics Inc.). Transducers used were an RMV707B (15 to 45 MHz) for adult mice or an RMV706 (20 to 60 MHz) for neonatal mice. Imaging and analysis were performed by a single blinded sonographer. Left ventricular (LV) mass was calculated by measuring the LV internal diameter (LVID), the LV posterior wall thickness (LVPW), and interventricular septal thickness (IVS) in diastole (d), using the following formula: LV mass = 1.053 × [(LVIDd + LVPWd + IVSd)3 − LVIDd3. LV end systolic (LVs) and LV end diastolic (LVd) dimensions, as well as systolic and diastolic wall thickness, were measured from M-mode tracings to calculate fractional shortening and ejection fraction, assuming a spherical LV geometry [(LVd3 − LVs3)/LVd3 × 100)].
Statistics.
Analysis for statistical significance was conducted using Prism 6 for Macintosh (GraphPad Software, Inc.). Differences between more than two groups at a single time point were analyzed using one-way analysis of variance (ANOVA). Differences between groups at multiple time points were analyzed using two-way ANOVA followed by Bonferroni's multiple-comparison tests. For viral load data, differences in log-transformed viral loads between groups at a given time point were analyzed using the Mann-Whitney test. Survival analysis was performed using the log rank (Mantel-Cox) test. P values of less than 0.05 were considered statistically significant.
RESULTS
MAV-1 infects primary cardiac myocytes ex vivo and hearts in vivo.
To determine whether MAV-1 can productively infect cardiac myocytes, we isolated cardiac myocytes from adult mice and inoculated them with MAV-1. We used RT-qPCR to quantify the expression of the MAV-1 E1A and hexon genes and qPCR to quantify viral DNA in infected myocytes. Expression of E1A, a nonstructural protein expressed early in infection, and hexon, a virion structural protein expressed late, increased between 24 and 48 h p.i. (Fig. 1A). Likewise, viral DNA increased between 24 and 48 h p.i. (Fig. 1B), further suggesting productive viral replication in these cells. To confirm that infectious progeny were produced from cardiac myocytes, we performed plaque assays with supernatants of infected cardiac myocyte cultures. Viral titers in the supernatants of infected cells increased slightly over time (Fig. 1C), further indicating that cardiac myocytes were productively infected by MAV-1.
FIG 1.

MAV-1 infects cardiac myocytes ex vivo and neonatal hearts in vivo. (A) Primary cardiac myocytes from adult C57BL/6 mice were infected with MAV-1 (MOI = 5), and expression of the viral E1A and hexon genes was measured by RT-qPCR, shown standardized to GAPDH in arbitrary units (A.U.). (B) DNA was extracted from primary cardiac myocytes, and qPCR was used to quantify copies of MAV-1 genome. (C) Supernatants were harvested from infected primary cardiac myocytes and viral titers measured by plaque assay. Data are shown as means ± standard errors of the means (SEM) for three samples per time point. (D) Neonatal mice were infected with MAV-1. qPCR was used to quantify viral loads in hearts. DNA viral loads are expressed as copies of MAV-1 genome per 100 ng of input DNA. Individual circles represent values for individual mice, and horizontal bars represent means for each group. (E and F) Expression of the viral E1A (E) and hexon (F) genes in the heart was measured by RT-qPCR, shown in arbitrary units and standardized to GAPDH. Combined data from 4 to 9 mice per group are presented as means ± SEM.
To verify that MAV-1 replicates in hearts in vivo, we infected 7-day-old C57BL/6 mice i.n. with MAV-1. We harvested hearts at multiple time points and used qPCR to quantify DNA viral loads. MAV-1 DNA was readily detectable at 4 days p.i. (Fig. 1D). Viral loads in the heart increased to peak levels between 7 and 10 days p.i. and were reduced at 21 days p.i. We also detected expression of MAV-1 E1A and hexon genes in the hearts of infected mice using RT-qPCR (Fig. 1E and F). Expression of both E1A and hexon peaked at 7 and 10 days p.i. and then decreased over time, although we detected persistent expression at 21 days p.i. The presence of viral gene expression and the logarithmic increases and subsequent decreases in heart viral loads over the time course suggest that MAV-1 replicates in the hearts of mice following i.n. infection.
MAV-1 induces cellular inflammation in the heart.
Cardiac inflammation was reported following i.p. MAV-1 infection of neonatal outbred Swiss Webster mice (20), but this inflammatory response was not characterized in detail. We evaluated the histological appearance of hearts obtained from inbred mice at various times postinfection. We observed no differences between hearts of mock-infected and infected mice at 4 days p.i. (data not shown) or at 7 days p.i. (Fig. 2A). By 10 days p.i., numerous focal accumulations of inflammatory cells were present, scattered throughout the myocardium (Fig. 2A). In many areas, these foci were associated with necrotic cardiac myocytes. Cellular inflammation was less severe at 14 and 21 days p.i., although hearts of infected mice contained scattered focal accumulations of mononuclear inflammatory cells and necrotic cardiac myocytes (Fig. 2A and data not shown).
FIG 2.

Cellular inflammation in hearts of infected neonatal mice. Mice were infected with MAV-1 or mock infected with conditioned medium. (A) Hematoxylin- and eosin-stained or CD3-stained sections were prepared from paraffin-embedded sections. Scale bars, 100 μm. (B) CD3 staining was quantified by counting the number of CD3+ cells per high-power field, averaging three fields per individual mouse. Combined data from 3 to 5 mice per group are presented as means ± SEM. (C) CD4- and CD8-stained sections were prepared from hearts of infected neonatal mice at 10 days p.i. Scale bars, 100 μm. (D) Serum cTnI levels were measured by ELISA. Combined data from 4 to 6 mice per group are presented as means ± SEM. ***, P < 0.001, comparing mock to MAV-1 infection.
We used immunohistochemistry to evaluate and quantify recruitment of inflammatory cells to hearts following MAV-1 infection. Increased numbers of CD3+ T lymphocytes were first detected in hearts of infected mice at 7 days p.i. (Fig. 2B). By 10 days p.i., we observed further increases in the number of CD3+ cells. At this time, recruited CD3+ cells were clustered around blood vessels and distributed throughout the myocardium (Fig. 2A and B). Fewer CD3+ cells were detected at later times, but they were still present in greater numbers in the hearts of infected mice than in those of mock-infected mice at 14 and 21 days p.i. (Fig. 2B). Both CD4+ and CD8+ cells were detected in the hearts of infected mice at 10 days p.i., although CD8+ cells were more abundant than CD4+ cells (Fig. 2C). We also observed recruitment of F4/80+ macrophages to the heart after MAV-1 infection (data not shown).
Histological evaluation suggested that MAV-1 infection and/or MAV-1-induced cardiac inflammation induces myocyte necrosis. To determine whether the histological findings described above correlated with other markers of cardiac injury during acute infection, we measured cTnI in the sera of mice after infection. We detected increased concentrations of cTnI in the sera of infected mice at 10 days p.i. (Fig. 2D) but not at other times or in mock-infected mice.
IFN-γ and other proinflammatory cytokines are upregulated in hearts of infected neonatal mice.
Because we observed recruitment of T cells to the heart following MAV-1 infection, we used RT-qPCR to measure expression of proinflammatory chemokines and cytokines in the heart. We detected increased expression of IFN-γ in the hearts of infected mice beginning at 7 days p.i. (Fig. 3A). IFN-γ upregulation peaked at 10 days p.i. and then decreased in magnitude but persisted in infected mice at 21 days p.i. We observed no significant changes in expression of IFN-β, interleukin-4 (IL-4), or IL-17 (data not shown). We detected increased expression of the chemokine CCL5 starting at 10 days p.i., which persisted in infected mice until 21 days p.i. (Fig. 3B). Expression of TNF-α (Fig. 3C) and IL-1β (Fig. 3D) was also increased in infected mice, whereas we detected no significant changes in expression of IL-6 (data not shown).
FIG 3.

Induction of cytokines in hearts. Mice were infected with MAV-1 or mock infected with conditioned medium. RT-qPCR was used to quantify IFN-γ (A), CCL5 (B), TNF-α (C), and IL-1β (D) expression, shown standardized to GAPDH in arbitrary units (A.U.). Combined data from 4 to 13 mice per group are presented as means ± SEM. ***, P < 0.001; **, P < 0.01 (comparing mock to MAV-1 infection at a given time point).
MAV-1-induced cardiac disease is less pronounced in adult mice.
We have previously described increased susceptibility to MAV-1 respiratory infection and blunted lung IFN-γ responses in neonatal mice compared to adults (26). To determine whether this is also the case in the heart, we compared viral loads and inflammatory responses in the hearts of neonates and adult mice. We detected peak viral loads in the hearts of adults at 10 days p.i. (Fig. 4A). There were no statistically significant differences between heart viral loads in neonates and adults at any time point. There was substantially less cellular inflammation and evidence of myocyte necrosis (data not shown) and fewer CD3+ cells (Fig. 4B) in adult hearts. cTnI was not detected in the sera of infected adult mice at any time point (data not shown). IFN-γ (Fig. 4C), CCL5 (Fig. 4D), and TNF-α (Fig. 4E) were upregulated in the hearts of infected adult mice, but the magnitudes of these responses were substantially lower than and the kinetics were delayed compared to those in neonates, in contrast to our previous findings in the lungs (26). We detected marked increases in the expression of both IL-1β (Fig. 4F) and IL-6 (Fig. 4G) in adult mice at 14 days p.i., but expression of both had decreased to baseline levels by 21 days p.i. Peak levels of IL-1β and IL-6 were greater in adults than in neonates at 14 days p.i. However, there was a minimal difference between neonates and adults when comparing the fold change in mRNA levels relative to those in mock-infected animals at 14 days p.i. for IL-1β (neonates, 1.83 ± 0.29; adults, 4.64 ± 2.26; P = 0.44) or IL-6 (neonates, 1.56 ± 0.67; adults, 4.92 ± 2.67; P = 0.34).
FIG 4.

Age-based differences in MAV-1 myocarditis. Adult and neonatal mice were infected with MAV-1. (A) qPCR was used to quantify MAV-1 genome copies in heart DNA. DNA viral loads are expressed as copies of MAV-1 genome per 100 ng of input DNA. Individual circles represent values for individual mice, and horizontal bars represent means for each group. (B) CD3 staining was quantified by counting the number of CD3+ cells per high-power field, averaging three fields per individual mouse. Combined data from 2 to 5 mice per group are presented as means ± SEM. ***, P < 0.001; **, P < 0.01, comparing neonate to adult mice at a given time point. (C to G) RT-qPCR was used to quantify expression of IFN-γ (C), CCL5 (D), TNF-α (E), IL-1β (F), and IL-6 (G), all shown standardized to GAPDH in arbitrary units (A.U.). Combined data from 4 to 13 mice per group are presented as means ± SEM. Neonatal data from Fig. 1D, 2B, and 3A to D are included in panels A, B, and C to F, respectively, for reference.
MAV-1 infection is associated with cardiac dysfunction in neonatal mice.
Acute viral myocarditis often results in decreased cardiac function (30, 31). To determine whether our histological observations correlated with measurements of cardiac function, we assessed heart function by echocardiography. In mice infected as neonates, there were no differences in the left ventricular ejection fraction or cardiac output between mock- and MAV-1-infected mice at 5 days p.i. (Fig. 5A and B). However, the left ventricular ejection fraction and cardiac output were significantly lower in infected neonates than in mock-infected controls at 10 days p.i. MAV-1 infection of neonatal mice did not cause left ventricle dilation (Fig. 5C), and the heart rate did not differ between groups (Fig. 5D). MAV-1 infection of adult mice had no effect on the left ventricular ejection fraction, cardiac output, left ventricle dilation, or heart rate at 10 days p.i. (Fig. 5E to H).
FIG 5.

Cardiac dysfunction following MAV-1 infection. Neonatal and adult mice were infected with MAV-1, and echocardiography was performed to measure ejection fraction (A and E), cardiac output (B and F), left ventricle internal diameter (C and G), and heart rate (D and H). Combined data from 4 to 11 mice per group are presented as means ± SEM. *, P < 0.05, comparing mock to MAV-1 infection at a given time point.
IFN-γ is proinflammatory during MAV-1 myocarditis.
Type I IFN (IFN-α and IFN-β) and type II IFN (IFN-γ) are upregulated and are often protective in other models of viral myocarditis (12–14). Although we did not observe virus-induced changes in IFN-β expression (data not shown), we did detect significant upregulation of IFN-γ in hearts after MAV-1 infection (Fig. 3). To further define the role of IFN-γ during MAV-1 myocarditis, we depleted IFN-γ beginning on the day after infection. Due to accelerated mortality in the infected IFN-γ-depleted mice beginning at 9 days after infection (Fig. 6A), mice were euthanized at 9 days p.i. Heart viral loads did not differ between IFN-γ-depleted and control mice at 9 days p.i. (Fig. 6B). The severity of myocarditis was quantified by blinded scoring of histological sections of hearts. As before, substantial cellular inflammation was present in the hearts of control mice infected with MAV-1. In contrast, virus-induced cardiac inflammation was significantly lower in IFN-γ-depleted mice, equivalent to that in mock-infected animals (Fig. 6C). Likewise, IFN-γ depletion reduced virus-induced CCL5, TNF-α, and IL-1β upregulation (Fig. 6D to F). We confirmed IFN-γ depletion using ELISA to measure IFN-γ protein in heart homogenate (Fig. 6G).
FIG 6.

Role of IFN-γ in MAV-1 myocarditis. Mice were infected with MAV-1 or mock infected with conditioned medium and treated every other day with control IgG or anti-IFN-γ antibody beginning at 1 day p.i. (A) Survival of infected animals was compared using the log rank (Mantel-Cox) test. (B) qPCR was used to quantify MAV-1 genome copies in heart DNA. Viral loads are expressed as copies of MAV-1 genome per 100 ng of input DNA. Individual circles represent values for individual mice, and horizontal bars represent means for each group. (C) Pathology index scores were generated to quantify cellular inflammation. (D to F) RT-qPCR was used to quantify CCL5 (D), TNF-α (E), and IL-1β (F) expression, shown standardized to GAPDH in arbitrary units (A.U.). Combined data from 4 to 6 mice per group are presented as means ± SEM. (G) ELISA was used to measure IFN-γ protein in heart homogenate. IFN-γ data from 3 to 5 mice per group are standardized to mg of heart tissue and are presented as means ± SEM. ***, P < 0.001; **, P < 0.01; *, P < 0.05 (comparing mock to MAV-1 infection within a given condition. †††, P < 0.001; ††, P < 0.001; †, P < 0.05 (comparing MAV-1-infected control IgG- to anti-IFN-γ-treated mice).
Persistent MAV-1 infection is associated with cardiac hypertrophy.
Both HAdV and MAV-1 establish persistent infections (32, 33). To determine whether MAV-1 persists in the heart, we infected mice at 7 days of age and harvested hearts from surviving mice at approximately 9 weeks postinfection, when the mice had grown into adulthood. We detected viral genome in all infected mice by both nested PCR (Fig. 7A) and qPCR (data not shown). To determine whether persistent MAV-1 infection affects cardiac function, we performed echocardiography at 9 weeks postinfection. Although there was a trend toward a lower left ventricle ejection fraction in mice that were infected as neonates than in mock-infected mice (data not shown), the difference was not statistically significant, suggesting that the acute functional deficits of MAV-1 infection were at least partially recovered. We observed a significant increase in the ratio of left ventricle mass (as measured by echocardiography) to tibia length in MAV-1-infected mice compared to mock-infected mice (Fig. 7B). There were no statistically significant differences between groups in tibia length (data not shown). To further assess long-term effects of infection, we quantified the cardiomyocyte cross-sectional area in cardiac sections stained with wheat germ agglutinin. Mice that were infected as neonates displayed a significant increase in cardiomyocyte size, consistent with cellular hypertrophy (Fig. 7C).
FIG 7.
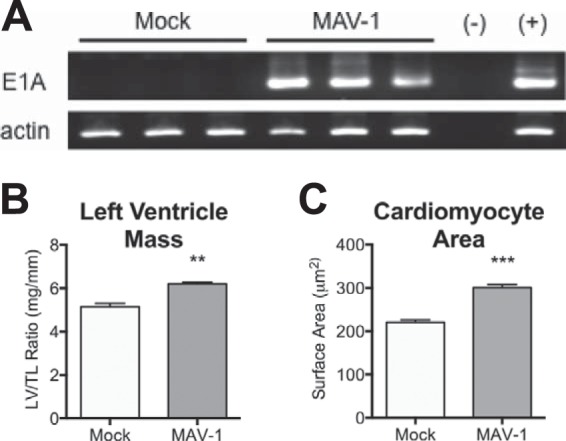
MAV-1 persistence and cardiac hypertrophy. Mice were infected with MAV-1 and hearts harvested at 9 weeks postinfection. (A) Nested PCR was used to evaluate the presence of MAV-1 DNA (top) and β-actin (bottom). (B) Left ventricular mass (LV) was measured by echocardiography and normalized to tibia length (TL). (C) Heart sections were stained with FITC-conjugated wheat germ agglutinin to outline cell borders. The cardiomyocyte cross-sectional area was measured from digital images using NIH ImageJ software. Combined data from >350 cells for each condition are presented as means ± SEM. ***, P < 0.001; **, P < 0.01 (comparing mock to MAV-1 infection).
DISCUSSION
HAdVs are common causes of viral myocarditis (3, 4). They are implicated in the development of dilated cardiomyopathy (7) and cardiac dysfunction (6, 34). The present study is the first to examine the pathogenesis of adenovirus myocarditis in detail, using MAV-1 to study an adenovirus in its natural host. We demonstrated that i.n. MAV-1 infection of neonatal C57BL/6 mice caused myocardial inflammation and tissue damage. Maximal viral loads, induction of IFN-γ, and recruitment of CD3+ T cells correlated with markers of cardiomyocyte damage and cardiac dysfunction. Neutralization of IFN-γ during acute MAV-1 infection reduced cardiac inflammation without affecting viral replication. Long-term persistence of MAV-1 in hearts was associated with increased LV mass and cellular hypertrophy.
Although it is possible that virus present in residual blood in hearts harvested from mice contributed to the measured viral loads in our study, increases in viral gene expression and logarithmic increases in viral loads between 4 and 7 days p.i. strongly suggest that MAV-1 replicates in hearts of mice after infection. Further, our in vitro data showing MAV-1 replication in primary cardiac myocytes suggest that cardiac myocytes are a likely cellular target of the virus in vivo, consistent with a previous report demonstrating the presence of MAV-1 in cardiac myocytes using electron microscopy (20). In that study, virions appeared to be present in other cell types as well, including cardiac fibroblasts and endothelial cells. Cardiac fibroblasts may serve as another target of MAV-1 replication in vivo, and we are currently investigating this possibility. HAdV DNA is often detected at high levels in lymphocytes of human tonsillar and adenoid tissue (35), and MAV-1 has been reported to replicate in macrophages (36), raising the possibility that MAV-1 could also infect resident or recruited immune cells in the heart.
CD3+ T cells and F4/80+ macrophages were recruited to the hearts of neonatal mice after MAV-1 infection. This is consistent with observations that hearts of patients with HAdV myocarditis are infiltrated with T cells and macrophages (34, 37). The T cell infiltrates following MAV-1 infection were mainly CD8+ T cells, although CD4+ T cells were also present. Following i.p. MAV-1 infection of adult mice, either CD4+ or CD8+ T cells are required for viral clearance from brain, and perforin (Pfn) contributes to signs of acute encephalomyelitis (38). A previous study demonstrated a role for both CD4+ and CD8+ T cells in the development of CVB3 myocarditis (39), and Pfn is a major contributor to severe tissue damage during CVB3 myocarditis (40). T cells likely contribute to both tissue damage and control of viral replication in the heart during acute MAV-1 infection. Specific mechanisms regulating the effects of T cells in the heart during MAV-1 myocarditis have not yet been defined.
CD8+ T cells may play both inflammatory and cytolytic roles during infection, either by secretion of cytokines, such as IFN-γ or TNF-α, or through release of cytolytic granules such as Pfn (41). Both type I and type II IFN can play protective roles in other models of viral myocarditis (12–14). IFN-γ and Pfn play antagonistic roles during chronic myocarditis caused by Trypanosoma cruzi, with Pfn+ CD8+ T cells implicated in causing tissue damage and IFN-γ+ CD8+ T cells preventing tissue damage (41). In the current study, we identified robust induction of IFN-γ in the heart after MAV-1 infection, consistent with previous studies showing induction of IFN-γ in MAV-1-infected lungs (25, 26). During acute MAV-1 respiratory infection of adult mice, CD4+ and CD8+ T cells are the primary producers of IFN-γ in the lung (24). IFN-γ induced during MAV-1 myocarditis is likely produced by infiltrating CD4+ and/or CD8+ T cells, because the robust IFN-γ induction that we observe correlates closely with recruitment of these cells to the myocardium. Cells not evaluated in this study, such as natural killer cells, may also contribute to early IFN-γ production in the heart during MAV-1 myocarditis.
Although IFN-γ is induced in the lung during MAV-1 respiratory infection, it is not essential for control of MAV-1 replication in the lungs or for survival of adult mice following i.n. inoculation (26). Similarly, IFN-γ does not appear to be critical for control of MAV-1 replication in the hearts of neonatal mice, since IFN-γ depletion did not lead to increased viral replication in the heart. IFN-γ-depleted mice developed significantly less cardiac inflammation after MAV-1 infection, suggesting that IFN-γ plays a proinflammatory role in the heart, as it does in some studies of CVB3 myocarditis (15, 16). At the same time, IFN-γ-depleted mice became moribund sooner than controls following infection. In addition, we detected equivalent viral loads in neonates and adults despite substantially greater IFN-γ induction in neonates. MAV-1 can be detected in many different organs following both i.n. and i.p. inoculation (42). We speculate that IFN-γ has organ-specific effects on MAV-1 pathogenesis. IFN-γ depletion might lead to increased viral loads and/or increased immunopathology in other organs, in particular the lungs, due to the inoculation route used in this study, and brain, as central nervous system disease is a cause of mortality in susceptible mouse strains following i.p. inoculation (43, 44). Our findings indicate that IFN-γ exerts an important proinflammatory effect in the heart during MAV-1 myocarditis in neonatal mice and that inflammation induced by IFN-γ signaling rather than direct antiviral effects of IFN-γ may be important for survival. Similarly, IFN-γ overexpression (13) or administration (45) ameliorates myocarditis in CVB3 and encephalomyocarditis virus models, respectively. However, this effect is likely due to the direct suppression of viral replication by IFN-γ or IFN-γ-mediated activation of natural killer cells (46).
Acute MAV-1 infection caused cardiac dysfunction in neonatal mice, similar to decreased cardiac function seen after infection with influenza A virus or CVB3 (30, 31). The significantly decreased cardiac function in MAV-1-infected mice that we observed at 10 days p.i. correlated with the greatest degree of virus-induced IFN-γ expression and also with peak levels of viral replication and cellular inflammation. It may be that cardiac dysfunction observed during MAV-1 myocarditis is due to direct cardiomyocyte damage caused by MAV-1 infection itself or by cytotoxic effects of virus-induced immune responses. However, because viral replication in hearts of adult mice was not associated with substantial inflammation, evidence of cardiac myocyte damage, or echocardiographic changes, it seems likely that host responses to viral infection are the most important contributors to cardiac dysfunction following MAV-1 infection. For instance, IFN-γ itself contributes to contractile dysfunction in immune-mediated myocarditis (47). Given its pronounced upregulation during MAV-1 myocarditis, it seems possible that IFN-γ contributes to the physiological abnormalities that we detected in infected mice. Other cytokines induced in the heart by MAV-1 infection, including TNF-α, IL-1β, and IL-6, have also been reported to impair cardiac contractility (48–52) and may therefore also contribute to cardiac dysfunction during MAV-1 infection.
Our results suggest that MAV-1 replicates equally well in hearts of neonatal and adult mice, but only neonatal mice are susceptible to MAV-1-induced myocarditis. Immune responses to many different pathogens differ between neonates and adults. Neonates are generally thought to mount inefficient Th1 responses (53, 54), although there is evidence that neonatal T cells can mount protective CD8+ T cell responses equivalent to those of adults (55). We have recently observed differences between neonatal and adult BALB/c mice in susceptibility to MAV-1 respiratory infection that correlated with less exuberant IFN-γ responses in neonatal lungs (26). Our results in the present study using neonate and adult C57BL/6 mice, however, demonstrate that neonatal mice have an exaggerated IFN-γ response in heart tissue compared to adult mice after MAV-1 infection that correlates with infiltration of CD3+ T cells and cardiac myocyte damage. This suggests that the response to acute MAV-1 infection is both age and organ specific. Supporting this possibility, a previous study demonstrated that susceptibility of mice to group B coxsackieviruses differs by age of mice, organ, and virus type (56).
The exuberant immune response observed in the hearts of neonatal mice, but not adult mice, after MAV-1 infection could be due to organ- and age-specific differences in cytokine receptor expression levels or function of antigen-presenting cells. Age-based differences in the activity of intracellular signaling pathways could also lead to the age-specific outcomes we observe during MAV-1 myocarditis. For instance, a previous study demonstrated high expression of components of the IL-1 receptor (IL-1R)/Toll-like receptor (TLR) signaling pathway in the neonatal period that is rapidly downregulated by 12 months of age (57). Another study demonstrated hypersensitivity of neonatal mice to TLR stimulation compared to adult mice, which may increase susceptibility of neonates to infection (58). Interestingly, we detected a different pattern in overall levels of IL-1β and IL-6 mRNA late during infection, both of which were higher in adults than in neonates at 14 days p.i. (Fig. 4). Contributions of these cytokines to the control of MAV-1 replication and to MAV-1-induced disease in the heart or in other organs have not yet been described. These cytokines may serve to limit viral replication in adult mice, minimizing deleterious virus-induced inflammation and subsequent cardiac dysfunction. Our data emphasize both the complicated interplay of various immune responses that is likely to occur during MAV-1 myocarditis and the changes in immune function that occur with age.
Persistent HAdV infections have been implicated in the development of dilated cardiomyopathy (DCM) and cardiac dysfunction (6–8, 34). A wide range of viral genomes (enterovirus, HAdV, parvovirus B19, or human herpesvirus 6) were detected in endomyocardial biopsy specimens from patients with clinically suspected myocarditis or dilated cardiomyopathy (6). We demonstrated that MAV-1 DNA persisted to at least 9 weeks postinfection in hearts of mice infected with MAV-1 as neonates, and MAV-1 persistence was associated with cardiac hypertrophy. It is unclear whether long-term persistent MAV-1 replication occurs in the heart and in which cells MAV-1 persists. Using RT-qPCR, we were unable to detect viral gene expression in hearts at 9 weeks postinfection (data not shown), although it is possible that this technique is not sensitive enough to detect isolated replication in a very small number of cells. Long-term effects of MAV-1 infection on cardiac remodeling could be due to chronic inflammation induced by persistence of virus in the absence of active replication. Long-term effects of infection on cardiac function can be caused by an autoimmune process, as occurs with autoreactive T cells that arise following CV3B infection (59). To our knowledge, no reports document this type of response during HAdV or MAV-1 infection, but we are in the process of characterizing virus-specific and autoreactive T cells in the context of MAV-1 myocarditis.
In summary, our findings demonstrate that IFN-γ is a proinflammatory mediator during adenovirus-induced myocarditis, and they suggest that persistent adenovirus infection may contribute to ongoing cardiac dysfunction and cardiac remodeling. The MAV-1 model will enable fundamental studies of adenovirus myocarditis and facilitate investigation of therapeutic strategies such as modulation of IFN-γ and other host responses.
ACKNOWLEDGMENTS
We thank Katherine Spindler and Michael Imperiale for helpful reviews of the manuscript. We appreciate the assistance of Paula Arrowsmith with histology, which was performed in the Pathology Cores for Animal Research in the University of Michigan Unit for Laboratory Management. We thank Kimber Converso-Baran for assistance with echocardiography, which was performed in the Physiology Phenotyping Core in the University of Michigan Department of Molecular and Integrative Physiology.
This work was supported by NIH grants R21 AI103452 (J.B.W.) and R01 HL093338 (S.M.D.), American Heart Association grant 13GRNT17110004 (J.B.W.), and a University of Michigan Department of Pediatrics Amendt-Heller Award for Newborn Research (J.B.W.).
REFERENCES
- 1.Grist NR, Reid D. 1988. General pathogenicity and epidemiology, p 241–252. In Bendinelli M, Friedman H (ed), Coxsackieviruses: a general update. Plenum, New York, NY. [Google Scholar]
- 2.Amabile N, Fraisse A, Bouvenot J, Chetaille P, Ovaert C. 2006. Outcome of acute fulminant myocarditis in children. Heart 92:1269–1273. doi: 10.1136/hrt.2005.078402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bowles NE, Ni J, Kearney DL, Pauschinger M, Schultheiss H-P, McCarthy R, Hare J, Bricker JT, Bowles KR, Towbin JA. 2003. Detection of viruses in myocardial tissues by polymerase chain reaction. evidence of adenovirus as a common cause of myocarditis in children and adults. J Am Coll Cardiol 42:466–472. doi: 10.1016/S0735-1097(03)00648-X. [DOI] [PubMed] [Google Scholar]
- 4.Martin AB, Webber S, Fricker FJ, Jaffe R, Demmler G, Kearney D, Zhang YH, Bodurtha J, Gelb B, Ni J. 1994. Acute myocarditis. Rapid diagnosis by PCR in children. Circulation 90:330–339. [DOI] [PubMed] [Google Scholar]
- 5.Feldman AM, McNamara D. 2000. Myocarditis. N Engl J Med 343:1388–1398. doi: 10.1056/NEJM200011093431908. [DOI] [PubMed] [Google Scholar]
- 6.Kühl U, Pauschinger M, Seeberg B, Lassner D, Noutsias M, Poller W, Schultheiss H-P. 2005. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation 112:1965–1970. doi: 10.1161/CIRCULATIONAHA.105.548156. [DOI] [PubMed] [Google Scholar]
- 7.Tátrai E, Hartyánszky I, Lászik A, Acsády G, Sótonyi P, Hubay M. 2011. The role of viral infections in the development of dilated cardiomyopathy. Pathol Oncol Res 17:229–235. doi: 10.1007/s12253-010-9302-6. [DOI] [PubMed] [Google Scholar]
- 8.Pauschinger M, Bowles NE, Fuentes-Garcia FJ, Pham V, Kühl U, Schwimmbeck PL, Schultheiss HP, Towbin JA. 1999. Detection of adenoviral genome in the myocardium of adult patients with idiopathic left ventricular dysfunction. Circulation 99:1348–1354. doi: 10.1161/01.CIR.99.10.1348. [DOI] [PubMed] [Google Scholar]
- 9.Shirali GS, Ni J, Chinnock RE, Johnston JK, Rosenthal GL, Bowles NE, Towbin JA. 2001. Association of viral genome with graft loss in children after cardiac transplantation. N Engl J Med 344:1498–1503. doi: 10.1056/NEJM200105173442002. [DOI] [PubMed] [Google Scholar]
- 10.Dennert R, Crijns HJ, Heymans S. 2008. Acute viral myocarditis. Eur Heart J 29:2073–2082. doi: 10.1093/eurheartj/ehn296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rose NR, Hill SL. 1996. The pathogenesis of postinfectious myocarditis. Clin Immunol Immunopathol 80:S92–S99. doi: 10.1006/clin.1996.0146. [DOI] [PubMed] [Google Scholar]
- 12.Sherry B, Torres J, Blum MA. 1998. Reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures correlate with induction of myocarditis and are determined by viral core proteins. J Virol 72:1314–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henke A, Jarasch N, Martin U, Zell R, Wutzler P. 2008. Characterization of the protective capability of a recombinant coxsackievirus B3 variant expressing interferon-gamma. Viral Immunol 21:38–48. doi: 10.1089/vim.2007.0077. [DOI] [PubMed] [Google Scholar]
- 14.Irvin SC, Zurney J, Ooms LS, Chappell JD, Dermody TS, Sherry B. 2012. A single-amino-acid polymorphism in reovirus protein μ2 determines repression of interferon signaling and modulates myocarditis. J Virol 86:2302–2311. doi: 10.1128/JVI.06236-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frisancho-Kiss S, Nyland JF, Davis SE, Frisancho JA, Barrett MA, Rose NR, Fairweather D. 2006. Sex differences in coxsackievirus B3-induced myocarditis: IL-12Rbeta1 signaling and IFN-gamma increase inflammation in males independent from STAT4. Brain Res 1126:139–147. doi: 10.1016/j.brainres.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 16.Huber SA, Graveline D, Born WK, O'Brien RL. 2001. Cytokine production by Vgamma(+)-T-cell subsets is an important factor determining CD4(+)-Th-cell phenotype and susceptibility of BALB/c mice to coxsackievirus B3-induced myocarditis. J Virol 75:5860–5869. doi: 10.1128/JVI.75.13.5860-5869.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torzewski M, Wenzel P, Kleinert H, Becker C, El-Masri J, Wiese E, Brandt M, Pautz A, Twardowski L, Schmitt E, Münzel T, Reifenberg K. 2012. Chronic inflammatory cardiomyopathy of interferon γ-overexpressing transgenic mice is mediated by tumor necrosis factor-α. Am J Pathol 180:73–81. doi: 10.1016/j.ajpath.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 18.DeBiasi RL, Robinson BA, Sherry B, Bouchard R, Brown RD, Rizeq M, Long C, Tyler KL. 2004. Caspase inhibition protects against reovirus-induced myocardial injury in vitro and in vivo. J Virol 78:11040–11050. doi: 10.1128/JVI.78.20.11040-11050.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weinberg JB, Stempfle GS, Wilkinson JE, Younger JG, Spindler KR. 2005. Acute respiratory infection with mouse adenovirus type 1. Virology 340:245–254. doi: 10.1016/j.virol.2005.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blailock ZR, Rabin ER, Melnick JL. 1968. Adenovirus myocarditis in mice. An electron microscopic study. Exp Mol Pathol 9:84–96. [DOI] [PubMed] [Google Scholar]
- 21.Klempa B, Kruger DH, Auste B, Stanko M, Krawczyk A, Nickel KF, Uberla K, Stang A. 2009. A novel cardiotropic murine adenovirus representing a distinct species of mastadenoviruses. J Virol 83:5749–5759. doi: 10.1128/JVI.02281-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kabaeva Z, Zhao M, Michele DE. 2008. Blebbistatin extends culture life of adult mouse cardiac myocytes and allows efficient and stable transgene expression. Am J Physiol Heart Circ Physiol 294:H1667–H1674. doi: 10.1152/ajpheart.01144.2007. [DOI] [PubMed] [Google Scholar]
- 23.Cauthen A, Welton A, Spindler KR. 2007. Construction of mouse adenovirus type 1 mutants. Methods Mol Med 130:41–59. doi: 10.1385/1-59745-166-5:41. [DOI] [PubMed] [Google Scholar]
- 24.McCarthy MK, Zhu L, Procario MC, Weinberg JB. 2014. IL-17 contributes to neutrophil recruitment but not to control of viral replication during acute mouse adenovirus type 1 respiratory infection. Virology 456–457:259–267. doi: 10.1016/j.virol.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCarthy MK, Levine RE, Procario MC, McDonnell PJ, Zhu L, Mancuso P, Crofford LJ, Aronoff DM, Weinberg JB. 2013. Prostaglandin E2 induction during mouse adenovirus type 1 respiratory infection regulates inflammatory mediator generation but does not affect viral pathogenesis. PLoS One 8:e77628. doi: 10.1371/journal.pone.0077628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Procario MC, Levine RE, McCarthy MK, Kim E, Zhu L, Chang C-H, Hershenson MB, Weinberg JB. 2012. Susceptibility to acute mouse adenovirus type 1 respiratory infection and establishment of protective immunity in neonatal mice. J Virol 86:4194–4203. doi: 10.1128/JVI.06967-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chensue SW, Ruth JH, Warmington K, Lincoln P, Kunkel SL. 1995. In vivo regulation of macrophage IL-12 production during type 1 and type 2 cytokine-mediated granuloma formation. J Immunol 155:3546–3551. [PubMed] [Google Scholar]
- 28.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zolov SN, Bridges D, Zhang Y, Lee W-W, Riehle E, Verma R, Lenk GM, Converso-Baran K, Weide T, Albin RL, Saltiel AR, Meisler MH, Russell MW, Weisman LS. 2012. In vivo, Pikfyve generates PI(3,5)P2, which serves as both a signaling lipid and the major precursor for PI5P. Proc Natl Acad Sci U S A 109:17472–17477. doi: 10.1073/pnas.1203106109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pan H-Y, Yamada H, Chida J, Wang S, Yano M, Yao M, Zhu J, Kido H. 2011. Up-regulation of ectopic trypsins in the myocardium by influenza A virus infection triggers acute myocarditis. Cardiovasc Res 89:595–603. doi: 10.1093/cvr/cvq358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheung C, Marchant D, Walker EK-Y, Luo Z, Zhang J, Yanagawa B, Rahmani M, Cox J, Overall C, Senior RM, Luo H, McManus BM. 2008. Ablation of matrix metalloproteinase-9 increases severity of viral myocarditis in mice. Circulation 117:1574–1582. doi: 10.1161/CIRCULATIONAHA.107.733238. [DOI] [PubMed] [Google Scholar]
- 32.Horwitz MS. 2001. Adenovirus immunoregulatory genes and their cellular targets. Virology 279:1–8. doi: 10.1006/viro.2000.0738. [DOI] [PubMed] [Google Scholar]
- 33.Smith K, Brown CC, Spindler KR. 1998. The role of mouse adenovirus type 1 early region 1A in acute and persistent infections in mice. J Virol 72:5699–5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kühl U, Pauschinger M, Noutsias M, Seeberg B, Bock T, Lassner D, Poller W, Kandolf R, Schultheiss H-P. 2005. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation 111:887–893. doi: 10.1161/01.CIR.0000155616.07901.35. [DOI] [PubMed] [Google Scholar]
- 35.Garnett CT, Erdman D, Xu W, Gooding LR. 2002. Prevalence and quantitation of species C adenovirus DNA in human mucosal lymphocytes. J Virol 76:10608–10616. doi: 10.1128/JVI.76.21.10608-10616.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ashley SL, Welton AR, Harwood KM, Rooijen NV, Spindler KR. 2009. Mouse adenovirus type 1 infection of macrophages. Virology 390:307–314. doi: 10.1016/j.virol.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Treacy A, Carr MJ, Dunford L, Palacios G, Cannon GA, O'Grady A, Moran J, Hassan J, Loy A, Connell J, Devaney D, Kelehan P, Hall WW. 2010. First report of sudden death due to myocarditis caused by adenovirus serotype 3. J Clin Microbiol 48:642–645. doi: 10.1128/JCM.00815-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moore ML, Brown CC, Spindler KR. 2003. T cells cause acute immunopathology and are required for long-term survival in mouse adenovirus type 1-induced encephalomyelitis. J Virol 77:10060–10070. doi: 10.1128/JVI.77.18.10060-10070.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henke A, Huber S, Stelzner A, Whitton JL. 1995. The role of CD8+ T lymphocytes in coxsackievirus B3-induced myocarditis. J Virol 69:6720–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gebhard JR, Perry CM, Harkins S, Lane T, Mena I, Asensio VC, Campbell IL, Whitton JL. 1998. Coxsackievirus B3-induced myocarditis: perforin exacerbates disease, but plays no detectable role in virus clearance. Am J Pathol 153:417–428. doi: 10.1016/S0002-9440(10)65585-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silverio JC, Pereira IR, Cipitelli MdC, Vinagre NF, Rodrigues MM, Gazzinelli RT, Lannes-Vieira J. 2012. CD8(+) T-cells expressing interferon gamma or perforin play antagonistic roles in heart injury in experimental Trypanosoma cruzi-elicited cardiomyopathy. PLoS Pathog 8:e1002645. doi: 10.1371/journal.ppat.1002645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kajon AE, Brown CC, Spindler KR. 1998. Distribution of mouse adenovirus type 1 in intraperitoneally and intranasally infected adult outbred mice. J Virol 72:1219–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guida JD, Fejer G, Pirofski LA, Brosnan CF, Horwitz MS. 1995. Mouse adenovirus type 1 causes a fatal hemorrhagic encephalomyelitis in adult C57BL/6 but not BALB/c mice. J Virol 69:7674–7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spindler KR, Fang L, Moore ML, Hirsch GN, Brown CC, Kajon A. 2001. SJL/J mice are highly susceptible to infection by mouse adenovirus type 1. J Virol 75:12039–12046. doi: 10.1128/JVI.75.24.12039-12046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamamoto N, Shibamori M, Ogura M, Seko Y, Kikuchi M. 1998. Effects of intranasal administration of recombinant murine interferon-gamma on murine acute myocarditis caused by encephalomyocarditis virus. Circulation 97:1017–1023. doi: 10.1161/01.CIR.97.10.1017. [DOI] [PubMed] [Google Scholar]
- 46.Godeny EK, Gauntt CJ. 1987. Murine natural killer cells limit coxsackievirus B3 replication. J Immunol 139:913–918. [PubMed] [Google Scholar]
- 47.Pérez Leirós C, Goren N, Sterin-Borda L, Borda ES. 1997. Myocardial dysfunction in an experimental model of autoimmune myocarditis: role of IFN-gamma. Neuroimmunomodulation 4:91–97. [DOI] [PubMed] [Google Scholar]
- 48.Janssen SP, Gayan-Ramirez G, Van den Bergh A, Herijgers P, Maes K, Verbeken E, Decramer M. 2005. Interleukin-6 causes myocardial failure and skeletal muscle atrophy in rats. Circulation 111:996–1005. doi: 10.1161/01.CIR.0000156469.96135.0D. [DOI] [PubMed] [Google Scholar]
- 49.Kapadia SR, Oral H, Lee J, Nakano M, Taffet GE, Mann DL. 1997. Hemodynamic regulation of tumor necrosis factor-alpha gene and protein expression in adult feline myocardium. Circ Res 81:187–195. doi: 10.1161/01.RES.81.2.187. [DOI] [PubMed] [Google Scholar]
- 50.Okusawa S, Gelfand JA, Ikejima T, Connolly RJ, Dinarello CA. 1988. Interleukin 1 induces a shock-like state in rabbits. Synergism with tumor necrosis factor and the effect of cyclooxygenase inhibition. J Clin Invest 81:1162–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Tassell BW, Seropian IM, Toldo S, Mezzaroma E, Abbate A. 2013. Interleukin-1beta induces a reversible cardiomyopathy in the mouse. Inflamm Res 62:637–640. doi: 10.1007/s00011-013-0625-0. [DOI] [PubMed] [Google Scholar]
- 52.Yokoyama T, Vaca L, Rossen RD, Durante W, Hazarika P, Mann DL. 1993. Cellular basis for the negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian heart. J Clin Invest 92:2303–2312. doi: 10.1172/JCI116834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adkins B, Bu Y, Guevara P. 2002. Murine neonatal CD4+ lymph node cells are highly deficient in the development of antigen-specific Th1 function in adoptive adult hosts. J Immunol 169:4998–5004. doi: 10.4049/jimmunol.169.9.4998. [DOI] [PubMed] [Google Scholar]
- 54.Adkins B, Leclerc C, Marshall-Clarke S. 2004. Neonatal adaptive immunity comes of age. Nat Rev Immunol 4:553–564. doi: 10.1038/nri1394. [DOI] [PubMed] [Google Scholar]
- 55.Zhang J, Silvestri N, Whitton JL, Hassett DE. 2002. Neonates mount robust and protective adult-like CD8(+)-T-cell responses to DNA vaccines. J Virol 76:11911–11919. doi: 10.1128/JVI.76.23.11911-11919.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khatib R, Chason JL, Silberberg BK, Lerner AM. 1980. Age-dependent pathogenicity of group B coxsackieviruses in Swiss-Webster mice: infectivity for myocardium and pancreas. J Infect Dis 141:394–403. doi: 10.1093/infdis/141.3.394. [DOI] [PubMed] [Google Scholar]
- 57.Martino D, Holt P, Prescott S. 2012. A novel role for interleukin-1 receptor signaling in the developmental regulation of immune responses to endotoxin. Pediatr Allergy Immunol 23:567–572. doi: 10.1111/j.1399-3038.2012.01287.x. [DOI] [PubMed] [Google Scholar]
- 58.Zhao J, Kim KD, Yang X, Auh S, Fu Y-X, Tang H. 2008. Hyper innate responses in neonates lead to increased morbidity and mortality after infection. Proc Natl Acad Sci U S A 105:7528–7533. doi: 10.1073/pnas.0800152105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gangaplara A, Massilamany C, Brown DM, Delhon G, Pattnaik AK, Chapman N, Rose N, Steffen D, Reddy J. 2012. Coxsackievirus B3 infection leads to the generation of cardiac myosin heavy chain-α-reactive CD4 T cells in A/J mice. Clin Immunol 144:237–249. doi: 10.1016/j.clim.2012.07.003. [DOI] [PubMed] [Google Scholar]