

ABSTRACT
Susceptibility to alphavirus infection is age dependent, and host maturation is associated with decreased virus replication and less severe encephalitis. To identify factors associated with maturation-dependent restriction of virus replication, we studied AP-7 rat olfactory bulb neuronal cells, which can differentiate in vitro. Differentiation was associated with a 150- to 1,000-fold decrease in replication of the alphaviruses Sindbis virus and Venezuelan equine encephalitis virus, as well as La Crosse bunyavirus. Differentiation delayed synthesis of SINV RNA and protein but did not alter the susceptibility of neurons to infection or virion maturation. Additionally, differentiation slowed virus-induced translation arrest and death of infected cells. Differentiation of uninfected AP-7 neurons was associated with changes in expression of antiviral genes. Expression of key transcription factors was increased, including interferon regulatory factor 3 and 7 (IRF-3 and IRF-7) and STAT-1, suggesting that neuronal maturation may enhance the capacity for antiviral signaling upon infection. IRF-7 produced by undifferentiated AP-7 neurons was exclusively the short dominant negative γ-isoform, while that produced by differentiated neurons was the full-length α-isoform. A similar switch in IRF-7 isoforms also occurred in the brains of maturing C57BL/6J mice. Silencing of IRF expression did not improve virus multiplication in differentiated neurons. Therefore, neuronal differentiation is associated with upregulation of transcription factors that activate antiviral signaling, but this alone does not account for maturation-dependent restriction of virus replication.
IMPORTANCE Viral encephalomyelitis is an important cause of age-dependent morbidity and mortality. Because mature neurons are not readily regenerated, recovery from encephalitis suggests that mature neurons utilize unique antiviral mechanisms to block infection and/or clear virus. To identify maturational changes in neurons that may improve outcome, we compared immature and mature cultured neurons for susceptibility to three encephalitic arboviruses and found that replication of Old World and New World alphaviruses and a bunyavirus was reduced in mature compared to immature neurons. Neuronal maturation was associated with increased baseline expression of interferon regulatory factor 3 and 7 mRNAs and production of distinct isoforms of interferon regulatory factor 7 protein. Overall, our studies identified maturational changes in neurons that likely contribute to assembly of immunoregulatory factors prior to infection, a more rapid antiviral response, increased resistance to virus infection, and improved survival.
INTRODUCTION
Development of age-dependent resistance to fatal disease is a characteristic of many virus infections of the central nervous system (CNS) (1–9). We have used Sindbis virus (SINV), the prototype alphavirus in the family Togaviridae, as a model system to understand maturation-mediated restriction of virus multiplication in neurons. SINV preferentially infects neurons and causes age-dependent encephalomyelitis in mice. Young animals are highly susceptible to SINV infection and succumb within 3 to 4 days. In contrast, adult animals infected with the same strain of SINV restrict virus replication in the CNS, are able to clear virus, and recover from infection (4). Age-dependent restriction of virus replication is not due to the maturation of the immune system, but rather to a decreased susceptibility of maturing neurons to infection (10, 11). The molecular basis for this maturation-dependent restriction of virus replication is unclear, but it has previously been attributed to decreased expression of viral receptors, proapoptotic molecules, and inflammatory response genes and to increased expression of fractalkine and interferon (IFN)-inducible genes (12, 13). Although changes in neuronal receptor expression may contribute to age-dependent susceptibility to SINV (14), the reduced production of virus by mature neurons compared to immature neurons suggests a postentry restriction of replication (15). Because neurons are terminally differentiated essential cells, for host recovery neural function must be preserved during viral clearance. Therefore, neurons are uniquely important cells for the study of antiviral responses (16, 17).
Type I IFN signaling constitutes the first line of host defense against many virus infections (18, 19). IFN treatment of cells prior to SINV infection blocks replication (20), and mice deficient in IFN-α/β signaling are highly susceptible to infection (21, 22). IFN signaling is vital for early control of SINV infection in vivo (21, 23, 24), and susceptibility of Venezuelan equine encephalitis virus (VEEV) to IFN is a major determinant of attenuation and age-dependent susceptibility (25, 26). Similarly, deletion of the IFN antagonist NSs (a nonstructural protein) from La Crosse virus (LACV) decreases virulence in mammals and allows neuronal production of type I IFN during infection (27–29). These studies suggest a potential role for IFN signaling in maturation-dependent neuronal restriction of neuronotropic virus replication. Neurons can induce IFN signaling upon infection (28, 30–33), and IFN expression is increased during maturation of cultured neurons (34). However, autocrine IFN signaling has not been implicated in maturation-dependent control of virus replication (33).
Canonically, the innate response to infection with RNA viruses is dependent on detection of viral RNA through surface interaction with Toll-like receptors or cytoplasmic interaction with RNA helicases, such as RIG-I or MDA-5 (35, 36). These signaling pathways activate constitutively expressed IFN regulatory factors (IRFs), which leads to expression of type I IFN and IFN-stimulated genes (ISGs). Thus, small amounts of IFN can stimulate increased ISG expression and facilitate an enhanced response to subsequent virus infection, a phenomenon known as IFN priming (37). Likewise, increased expression of proteins in the IFN induction pathway, such as RIG-I, increases resistance to virus infection (36, 38, 39).
Pathogen detection pathways converge on activation of IRF-3 and IRF-7, which are critical transcription factors for type I IFN gene expression. IRF-3 is ubiquitously expressed and is the primary regulator of IFN-β expression (40, 41). IRF-7 expression is regulated in a cell-type-dependent manner and is required for maximal type I IFN-α gene expression (42, 43). Loss of IRF-3 and/or IRF-7 greatly impairs the immune response to infections in a virus- and cell-type-specific manner (44–47). IRFs can stimulate ISG expression in response to virus infection independently of IFN, suggesting that increased IRF expression could also have a direct protective effect (48). For example, IRF-3 mediates innate immune signaling in neurons during Western equine encephalitis virus and St. Louis encephalitis virus infection independently of IFN signaling (49).
To investigate the molecular mechanisms underlying maturation-dependent restriction of neuronotropic virus replication, we used cultured AP-7 rat olfactory sensory neurons immortalized by a temperature-sensitive simian virus 40 (SV40) T antigen (50). Similar to the CSM14.1 nigral neuron system (15), these neurons can differentiate in vitro and thus allow comparison of virus replication and host antiviral responses in undifferentiated cycling (cAP-7) and differentiated nondividing (dAP-7) neurons. Rats are susceptible to SINV-induced encephalomyelitis (51), and cultured immature and mature primary rat dorsal root ganglion neurons show maturation-dependent restriction of SINV replication (52).
dAP-7 neurons have an intrinsic ability to restrict multiplication of three different neuronotropic viruses compared to cAP-7 neurons. To determine the molecular mechanisms that contribute to the intrinsic resistance of maturing neurons to neuronotropic viruses, we assessed the expression of cellular factors that induce and amplify the cellular antiviral response. We found that neural maturation was associated with increased expression of immunoregulatory transcription factors Irf-3 and Irf-7 and further induction upon infection.
MATERIALS AND METHODS
Cell culture.
Rat AP-7 Odora neurons, an olfactory-derived cell line immortalized with a temperature-sensitive SV40 T antigen (a gift from Dale Hunter, Tufts University, Boston, MA) (50), were grown at 33°C in 7% CO2 in Dulbecco's modified Eagle's medium (DMEM)–10% fetal bovine serum (FBS) supplemented with 100 U penicillin/ml, 100 μg streptomycin/ml, and 2 mM glutamine. At about 25% confluence, cells were differentiated for 5 to 7 days by shifting to 39°C and 5% CO2 in DMEM–10% FBS supplemented with 1 μg/ml insulin, 20 μM dopamine, 100 μM ascorbic acid, penicillin, streptomycin, and glutamine. CSM14.1 neurons were grown in DMEM–10% FBS with penicillin, streptomycin, and glutamine at 31°C in 5% CO2. For differentiation, CSM14.1 cells were shifted to the nonpermissive culture conditions of DMEM–1% FBS with penicillin, streptomycin, and glutamine at 39°C in 5% CO2 for at least 3 weeks as described previously (15). BHK-21 cells were grown at 37°C in 5% CO2 in DMEM–10% FBS with penicillin, streptomycin, and glutamine. Cells were monitored for mycoplasma with the MycoAlert mycoplasma detection kit (Lonza Rockland Inc., Rockland, ME) and determined to be free of contamination.
Viruses and infection of cells.
SINV strain TE (53), VEEV strain TC-83 (a gift from Ilya Frolov, University of Alabama, Birmingham, AL), and La Crosse virus strain La Crosse/original (54) (a gift from Andrew Pekosz, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD) were used. Virus stocks were grown in and titers were determined by plaque formation on BHK-21 cells. AP-7 or CSM14.1 monolayers were infected with viruses at the indicated multiplicity of infection (MOI) in DMEM–1% FBS for 1 h and washed with phosphate-buffered saline (PBS; pH 6.2), and medium was replaced.
To quantify one-step virus production, neuronal monolayers were inoculated as described for each experiment. At each time point, 100 μl of supernatant fluid was collected from each of 3 wells. Infectious virus was quantified by plaque assay on BHK-21 cells. Percentages of infected cells were determined by immunofluorescence microscopy by using monoclonal antibodies (MAb) to E2 of either SINV (MAb 209 [55]) or VEEV (1A3B7 [56]) with Alexa Fluor 594-conjugated anti-mouse IgG (Life Technologies). The number of infected cells was compared to the total number of cells as visualized by 4′,6-diamidino-2-phenylindole (DAPI)-stained nuclei (ProLong Gold; Life Technologies).
Transmission electron microscopy.
For transmission electron microscopy (TEM), samples were fixed in 2.5% glutaraldehyde, 3 mM CaCl2, 1% sucrose in 0.1 M sodium cacodylate buffer (pH 7.2) for 1 h at room temperature. After buffer rinse, samples were postfixed in 1% osmium tetroxide in buffer (1 h) on ice in the dark. Following a distilled water rinse, plates were stained with 2% aqueous uranyl acetate (0.22-μm filtered; 1 h, in the dark), dehydrated in a graded series of ethanol, and embedded in Eponate 12 (Ted Pella) resin. Samples were polymerized at 60°C overnight.
Thin sections, of 60 to 90 nm, were cut with a diamond knife on the Reichert-Jung Ultracut E ultramicrotome and picked up with naked 200-mesh copper grids. Grids were stained with 2% uranyl acetate in 50% methanol and observed with a Hitachi 7600 TEM at 80 kV. Images were captured with an AMT charge-coupled-device (1,000 by 1,000 pixels) camera.
Antisera and immunoblot analysis.
For immunoblot analysis, neuronal monolayers were washed with cold 1× PBS (pH 6.2), lysed with cold RIPA buffer (50 mM Tris, 150 mM NaCl, 1% SDS, 1% NP-40, 0.5% Na-deoxycholate, 1 mM EDTA), incubated on ice for 30 min, and cleared by centrifugation. Brains from 3-day-old or 6-week-old C57BL/6J mice were homogenized in 1× PBS and cleared by centrifugation. Ten micrograms of total protein was separated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to a nitrocellulose membrane (Bio-Rad). Immunodetection was conducted with the following antisera diluted 1:1,000 unless otherwise indicated: monoclonal anti-E2 (MAb 209; 1:2,000) (55), polyclonal anti-nsP3 (57), polyclonal anti-IRF-7 (Sigma), polyclonal anti-IRF-3 (Santa Cruz Biotechnology), polyclonal anti-phospho-STAT-1(Y701) (Cell Signaling), polyclonal anti-STAT-1 (Cell Signaling), and monoclonal anti-β-actin (1:5,000; Millipore). The membranes were incubated with horseradish peroxidase-conjugated donkey anti-rabbit or sheep anti-mouse immunoglobulin G (GE Healthcare) and developed using ECL Prime Western blotting reagents (GE Healthcare).
Measurement of RNA levels.
Total cellular RNA was isolated using Qiagen RNeasy or RNeasy Plus minikits per the manufacturer's directions. SINV positive-strand RNA was quantified as described previously (57). Briefly, cDNA from total cellular RNA was transcribed using the ABI high-efficiency reverse transcription kit with a SINV-specific primer, SINV9899R (5′-AGCATTGGCCGACCTAACGCAGCAC-3′). Quantitative PCR (qPCR) was performed on an ABI 7500 thermocycler with ABI qPCR mastermix and primers SINVE2F (5′-TGGGACGAAGCGGACGATAA-3′) and SINVE2R (5′-CTGCTCCGCTTTGGTCGTAT-3′) and TaqMan probe (5′–6-carboxyfluorescein–CGCATACAGACTTCCGCCCAGT–6-carboxytetramethylrhodamine–3′). Viral RNA was quantified by comparison to a standard of SINV genomic RNA.
Equal microgram amounts of RNA isolated from AP-7 neurons from 3 independent experiments were pooled and analyzed using the rat antiviral response qPCR array (PARN-122; SABiosciences) according to the manufacturer's directions.
cDNA was synthesized from cellular RNA using random primers and the ABI high-efficiency reverse transcription kit. qPCR to determine the threshold cycle (CT) and ΔΔCT was conducted in an ABI 7500 thermocycler with ABI qPCR mastermix. The following PrimeTime standard qPCR assays from IDT were used: Irf-3, Rn.PT.47.14307929; Irf-7, Rn.PT.47.7921156.
Cell survival.
Neuronal monolayers were mock infected or infected with SINV (MOI, 5) or VEEV (MOI, 50). Cell viability was determined microscopically by using trypan blue exclusion at the indicated times after infection and is reported as the percent viability.
Protein radiolabeling.
At the indicated times after infection, the neuronal monolayer growth medium was replaced with 1× PBS (pH 6.2) containing 50 μCi/ml of Trans35S-label (1,175 Ci/mmol; 70% methionine, ≤15% cystein; MP Biomedical, LLC). After 1 h, the cells were dislodged, collected by centrifugation, and lysed with cold RIPA buffer. The lysates were subjected to SDS-PAGE and autoradiography.
Transfection of double-stranded RNA (dsRNA).
Four days into the differentiation process, RNA duplexes (40 pmol total) specific to three nonoverlapping regions of the cDNA of interest (IDT trifecta RNA) were transfected into AP-7 neurons by using RNAiMAX (Invitrogen). The neurons were infected 96 h after transfection.
Nucleotide sequence accession numbers.
The sequences for Irf-3 and Irf-7 were deposited with GenBank and assigned accession numbers NM_001006969 and NM_001033691, respectively.
RESULTS
Alphavirus multiplication is restricted in differentiated neurons.
To define AP-7 neurons as a faithful model of in vivo infection, we first compared alphavirus multiplication in immature cAP-7 and mature dAP-7 neurons. After differentiation of AP-7 cells for 7 days, cell division ceases, processes extend from the cell body, and neuronal markers are expressed by the mature dAP-7 neurons (50). cAP-7 neurons produced new infectious SINV by 6 h after infection with an increase to 108.6 PFU/ml by 18 h (Fig. 1A). In contrast, dAP-7 neurons did not produce new SINV until 9 h, and virus titers increased more slowly, to a 50-fold-lower peak of 106.9 PFU/ml at 18 h (Fig. 1A). Production of infectious virus then continued at the same levels through 48 h for both cAP-7 and dAP-7 neurons.
FIG 1.

Alphavirus multiplication is restricted in differentiated AP-7 neurons. cAP-7 (dashed lines) or dAP-7 (solid lines) neurons were infected with the TE strain of SINV (MOI, 5) (A) or TC-83 strain of VEEV (MOI, 50) (B). Viral titers in supernatant fluids collected at the indicated times after infection were measured by plaque formation in BHK cells and are expressed as the mean log10 PFU/ml ± standard deviation of triplicate samples from a representative of three experiments. (C) Percentages of cAP-7 and dAP-7 neurons expressing detectable E2 glycoprotein at the indicated times after infection were determined by immunofluorescence. E2-positive cells compared to the total number of DAPI-positive cells were determined in three nonoverlapping fields.
Infectious virus production by the New World alphavirus VEEV (TC-83 strain) was similarly affected by neuronal differentiation. In cAP-7 neurons, VEEV accumulated through 24 h, with peak titers of 107.8 PFU/ml (Fig. 1B). In contrast, VEEV production by dAP-7 neurons was not detectable until 12 h and increased only modestly through 24 h, with a peak titer of 105.2 PFU/ml.
Additionally, mature neurons were less susceptible to infection with VEEV than with SINV. By 8 h after infection with SINV at an MOI of 5, more cAP-7 cells (58%) were positive for E2 glycoprotein than dAP-7 cells (28%), as determined by immunofluorescence microscopy (Fig. 1C). However, similar numbers of cAP-7 and dAP-7 neurons were positive by 24 h after infection. Upon infection at an MOI of 50, fewer dAP-7 (52%) than cAP-7 (84%) neurons were positive by 8 h after infection, but levels of infection (>90%) were similar 24 to 48 h after infection. This indicates that all cells in culture were susceptible to SINV infection regardless of maturation state. Immature neurons were susceptible to VEEV at either MOI tested, with greater than 90% of cells infected by 24 h after infection. In contrast, only 40% of dAP-7 neurons were positive for E2 glycoprotein through 48 h after VEEV infection at an MOI of 5. At an MOI of 50, 70% of dAP-7 neurons were infected by 8 h after infection and greater than 90% were infected by 24 h. This suggests that maturation of dAP-7 neurons confers protection to VEEV infection. Therefore, studies using VEEV were conducted at an MOI of 50 to ensure infection of all cells and to focus on factors restricting intracellular replication. During both SINV and VEEV infection, cAP-7 neurons displayed a brighter fluorescent signal at 8 h after infection than dAP-7 neurons, suggesting greater synthesis of glycoprotein on a per cell basis.
A defect in any step of virus replication or in assembly of virus particles could reduce infectious virus production. To determine if progeny virus from dAP-7 neurons was morphologically distinct from that produced by cAP-7 neurons, we imaged budding virus 24 h after infection by using TEM. The majority of virions produced by both cAP-7 and dAP-7 neurons were single particles, with only occasionally observed multiple particles packaged into one envelope (Fig. 2A). Virus particles from both cAP-7 and dAP-7 neurons were comparable in size and electron density, indicating that virion assembly is not likely to be responsible for reduced infectious virus production.
FIG 2.

SINV intracellular replication is delayed in differentiated AP-7 neurons. AP-7 neurons were infected with the TE strain of SINV (MOI, 5). (A) cAP-7 and dAP-7 neurons were fixed at 24 h after infection, and ultrastructure analysis was performed by transmission electron microscopy of thin sections. Bar, 100 nM. (B) Total cellular RNA was collected at various times after infection. cDNA was produced using SINV-specific primers and quantified by qPCR compared to standard SINV genomic DNA. SINV RNA levels in cAP-7 (dashed lines) or dAP-7 (solid lines) neurons are expressed as the mean SINV copy number (log10) ± standard deviation of triplicate samples from a representative of two experiments. (C) Immunoblot analysis of total cell lysates prepared at the indicated times from mock-infected (M) or SINV-infected cAP-7 (left) or dAP-7 (right) neurons with anti-nsP3 (top), anti-E2 (middle), or anti-β-actin (bottom) antibodies. A representative of 2 experiments is shown. Significant differences between cAP-7 and dAP-7 at each time point were determined by two-way ANOVA with Bonferroni's posttest. ***, P < 0.001.
To compare viral RNA synthesis between cycling and differentiated AP-7 neurons, viral RNA was measured using qRT-PCR (Fig. 2B). The synthesis and accumulation of viral RNA was slower in dAP-7 neurons. However, levels of viral RNA at 24 h were similar in cAP-7 and dAP-7 neurons, potentially due to decreased infectious virus production. Delayed SINV RNA synthesis in dAP-7 neurons may reduce the amount of genomic RNA available to be packaged into new virions or the RNA available for translation of viral proteins needed for virion assembly.
SINV nonstructural proteins (nsP1 to -4) are translated from both incoming viral genomic RNA and newly synthesized full-length, positive-strand RNA. Structural proteins (C, E1, pE2, and 6K) are translated from subgenomic mRNA produced later in infection. Thus, delayed RNA replication could also affect the timing and levels of viral protein production. Comparison of SINV protein production in cAP-7 and dAP-7 neurons showed a delay in production of both nonstructural and structural proteins by dAP-7 cells (Fig. 2C). nsP3 was detected in cAP-7 neurons at 4 h after infection and increased through 8 h, but it was not detectable until 12 h in dAP-7 neurons (Fig. 2C, top).
The envelope glycoprotein E2 is produced as a precursor pE2 (54 kDa) that is cleaved to E2 (47 kDa) and E3 (7 kDa) prior to virion maturation. In cAP-7 neurons, pE2 was detected by 5 h after infection with cleavage to E2 detected at 7 h with both pE2 and E2 accumulating in similar amounts through 24 h (Fig. 2C middle). In dAP-7 neurons, neither pE2 nor E2 were detected until 24 h after infection. Therefore, multiple steps of SINV multiplication are delayed in differentiated neurons, all of which probably contribute to a reduced production of infectious virus.
Cell survival after alphavirus infection is extended in differentiated neurons.
In addition to reduced virus multiplication in vivo, host maturation is associated with decreased morbidity and mortality upon SINV TE and VEEV TC-83 infection in mice (25, 26, 53). SINV induces apoptosis of immature neurons both in vivo and in cultured cells (58–61). Cytopathic effects (CPE), including cell rounding, detachment, and ultimately production of cytoplasmic blebs, were first apparent between 12 and 18 h in both SINV- and VEEV-infected cAP-7 neurons. Two days after infection, less than 10% of cAP-7 neurons were viable, as determined by trypan blue exclusion (Fig. 3). In contrast, 62% of SINV-infected and 73% of VEEV-infected dAP-7 neurons were viable at this time, and reduction in survival to 10% did not occur until 4 days after infection. Therefore, AP-7 differentiation extended survival after alphavirus infection, potentially the result of reduced virus replication, increased cellular survival signaling, or a combination of both.
FIG 3.
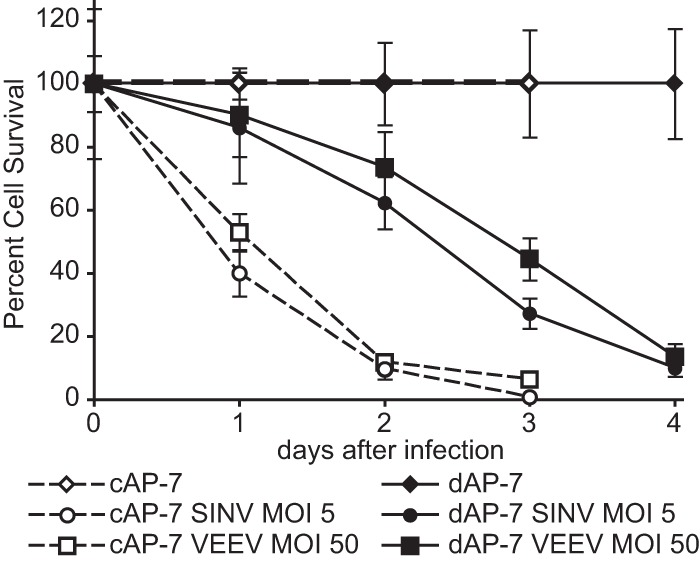
Virus-induced cell death is delayed in differentiated AP-7 neurons. cAP-7 (dashed lines) and dAP-7 (solid lines) neurons were mock infected or infected with either SINV (MOI, 5) or VEEV (MOI, 50). Live cells, as determined by trypan blue exclusion, were counted at the indicated days after infection. Survival of cells is expressed as a percentage of the survival of mock-infected cells at the corresponding time point ± the standard deviation of triplicate samples. Representative data from three independent experiments are presented.
Shutoff host protein synthesis is delayed upon infection of differentiated neurons.
Alphavirus infection causes significant changes in cellular processes, including inhibition of host protein synthesis. To determine if effects on host translation differ during SINV or VEEV infection of cAP-7 and dAP-7 neurons, we monitored incorporation of radiolabeled Cys/Met throughout infection (Fig. 4). In SINV-infected cAP-7 neurons, reduced host protein synthesis was evident by 6 h, and most translation of cellular proteins was shut off by 12 h. In contrast, in SINV-infected dAP-7 neurons, shutoff host protein synthesis occurred between 12 and 24 h and correlated with delayed nsP3 synthesis (Fig. 2D). Compared to SINV infection, translational arrest induced by VEEV infection of cAP-7 neurons was slower, occurring between 24 and 48 h after infection, although induction of cell death was similar (Fig. 3). During VEEV infection of dAP-7 neurons, host protein synthesis was minimally affected through 48 h. In agreement with immunoblotting studies (Fig. 2D), structural proteins were visible by 6 h in SINV- and VEEV-infected cAP-7 neurons but were not apparent until 24 h after infection in dAP-7 neurons. Therefore, changes in AP-7 neurons associated with differentiation delayed alphavirus-mediated translational arrest, potentially providing a longer window for synthesis of host antiviral proteins after infection.
FIG 4.

Alphavirus-mediated shutoff of host protein synthesis is delayed in differentiated AP-7 neurons. Mock-, SINV (MOI, 5)-, or VEEV (MOI, 50)-infected cAP-7 (upper) and dAP-7 (lower) neurons were radiolabeled for 1 h with 35S-labeled cysteine/methionine at the indicated times after infection. Protein synthesis (equal cpm per lane) was analyzed by SDS-PAGE autoradiography. Viral proteins are denoted to the right. Results of a representative experiment of three experiments are shown.
Bunyavirus replication is reduced in differentiated AP-7 neurons.
To determine if dAP-7 cell-mediated restriction of virus multiplication was unique to alphaviruses or a more general phenomenon for neuronotropic viruses, we assessed progeny virus production of the unrelated encephalitic La Crosse virus (original strain; family Bunyaviridae). At an MOI of 5, LACV was produced rapidly in cAP-7 neurons, with peak titers of 105.5 PFU/ml at 6 h after infection (Fig. 5). In dAP-7 neurons, LACV titers increased less than 10-fold in 24 h. We were unable to test the effect of higher MOIs for LACV infection due to low stock titers, and we cannot rule out that LACV would replicate somewhat better with infection of dAP-7 neurons at a higher MOI. Overall, these studies indicated that neuronal differentiation confers inherent characteristics that restrict replication of three different neuronotropic viruses.
FIG 5.
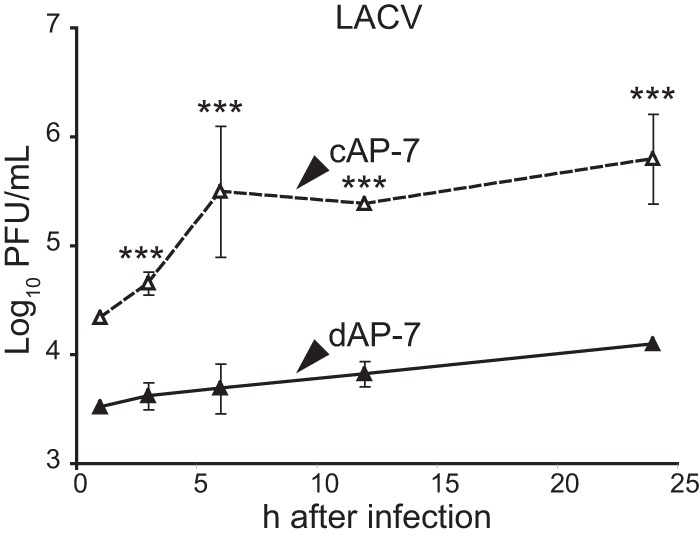
Differentiated AP-7 neurons also restrict multiplication of LACV. cAP-7 (dashed lines) or dAP-7 (solid lines) neurons were infected with the original strain of LACV (MOI, 5). Viral titers in supernatant fluids collected at the indicated times after infection were measured by plaque formation on BHK cells and are expressed as the mean log10 PFU/ml ± standard deviation of triplicate samples from a representative of two experiments. Significant differences between cAP-7 and dAP-7 at each time point were determined by two-way ANOVA with Bonferroni's posttest. ***, P < 0.001.
AP-7 differentiation is associated with the upregulation of antiviral gene expression.
We hypothesized that neurons may upregulate expression of cellular defense genes during differentiation, allowing an expedited innate immune response upon pathogen detection. We compared the gene expression levels of 84 antiviral genes in uninfected dAP-7 neurons to those in uninfected cAP-7 neurons by using an antiviral response qPCR array. Expression of 22 genes was upregulated 2-fold or more during differentiation (Table 1). These included genes at all stages of the innate immune signaling pathway, including the dsRNA receptors tlr3 (11.78-fold increase) and ddx58/rig-I (3.65-fold), transcription factors stat1 (5.35-fold) and Irf-7 (3.59-fold), and effector genes ifnb1 (4.67-fold) and isg15 (5.91-fold). Expression of two genes, cxcl10 and spp1, was reduced more than 2-fold during differentiation.
TABLE 1.
Gene products with a ≥2-fold change in expression in dAP-7 neurons compared to cAP-7 neurons
| Gene product | Reference (accession no.) | Fold change |
|---|---|---|
| Cd80 | NM_012926 | 152.58 |
| Il6 | NM_012589 | 95.22 |
| Casp1 | NM_012762 | 33.48 |
| Nlrp3 | XM_220513 | 14.63 |
| ctsb (cathepsin B) | NM_022597 | 14.36 |
| Tlr3 | NM_198791 | 11.78 |
| Dhx58 (LGP2) | NM_001106645 | 10.11 |
| Azi2 | NM_001025705 | 9.45 |
| Tlr9 | NM_198131 | 8.62 |
| Isg15 | XM_216605 | 5.91 |
| Stat1 | NM_032612 | 5.35 |
| Il18 | NM_019165 | 4.90 |
| Atg12 | NM_001038495 | 4.69 |
| Ifnb1 | NM_019127 | 4.67 |
| Irak4 | NM_001106791 | 4.26 |
| ctss (cathepsin S) | NM_017320 | 3.90 |
| ctsl1 (cathepsin L1) | NM_013156 | 3.77 |
| Ddx58 (RIG-I) | NM_001106645 | 3.65 |
| Irf7 | NM_001033691 | 3.59 |
| Il15 | NM_013129 | 3.52 |
| Tbkbp1 | NM_172021 | 3.37 |
| Dak | NM_001039031 | 3.10 |
| Cxcl10 | NM_139089 | −4.64 |
| Spp1 | NM_012881 | −4.35 |
As STAT-1 is an important determinant for outcome after SINV infection (21), we determined if protein levels were increased in AP-7 neurons during differentiation. STAT-1 levels were higher in uninfected dAP-7 neurons compared to cAP-7 neurons and remained unchanged in both cell types during infection (Fig. 6A). Classically, activation of downstream signaling through the JAK/STAT pathway requires phosphorylation of STAT1. Thus, to compare signaling in undifferentiated and differentiated AP-7 neurons, we investigated the phosphorylation state of STAT1. Phosphorylated STAT1 (Y701) was not detected before or after SINV infection (Fig. 6A), although the cells responded to recombinant IFN treatment with STAT1 phosphorylation (Fig. 6B). This confirmed results of previous studies showing a lack of STAT1 phosphorylation during SINV infection of AP-7 or CSM14.1 neurons (62).
FIG 6.
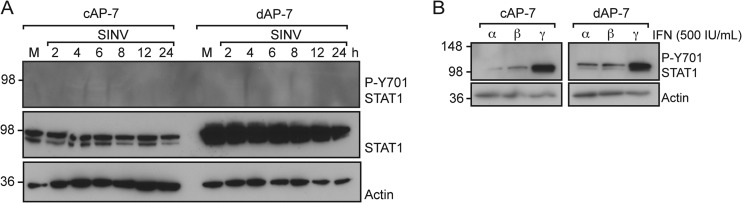
Stat1 is not phosphorylated during SINV infection of AP-7 neurons. (A) cAP-7 or dAP-7 neurons were mock infected (M) or infected with SINV (MOI, 5). Total cell lysates prepared at the indicated times after infection were analyzed with anti-pSTAT1 (P-Y701) (top row), anti-STAT1 (middle row), or anti-β-actin (bottom row) antibodies. Results of a representative experiment of 2 experiments are shown. (B) cAP-7 or dAP-7 neurons were treated with 500 IU/ml of recombinant rIFN-α, -β, or -γ. Total cell lysates prepared 4 h after treatment were analyzed by immunoblotting using anti-STAT1 (P-Y701) (top row) or anti-β-actin (bottom row) antibodies. Results of a representative experiment of 2 experiments are shown.
Differentiated neurons upregulate IRF-7 expression.
IRFs comprise a family of transcription factors that upon activation regulate expression of IFN and ISG mRNAs. IRF-3 and IRF-7 are central regulators of the cellular innate immune response and the convergence point for integration of pathogen detection pathways. IRF-3 is constitutively expressed and found inactive in the cytoplasm, whereas IRF-7 oftentimes is produced in response to IFN signaling. Upon activation, IRF-3 and IRF-7 are phosphorylated, translocate to the nucleus, and stimulate ISG expression. In a positive feedback loop, IFN upregulates expression of Irf-3 and Irf-7 mRNAs. We compared expression of Irf-3 and Irf-7 during AP-7 differentiation by using quantitative reverse transcription-PCR (qRT-PCR) (Fig. 7A). Day zero corresponds to cAP-7 neurons, as these samples were not supplemented with differentiation medium, nor were they incubated at the nonpermissive temperature. Gene expression throughout the time course is represented as the fold change compared to day zero levels. Irf-3 mRNA levels increased steadily through the differentiation process and were 4-fold higher on day 7. Irf-7 mRNA levels increased 13-fold on day 3 and were sustained at an elevated level with an average 11-fold increase on day 7.
FIG 7.

Irf-3 and Irf-7 mRNAs and IRF-7 protein increase during differentiation. (A) Total cellular RNA was collected at various times after plating of AP-7 neurons. Neurons were incubated at the permissive temperature in DMEM–10% FBS, 1 day prior to day 0 collection, corresponding to cAP-7 neurons. After day 0, neurons were maintained in differentiation medium at the nonpermissive temperature as described for the differentiation procedures. cDNA was produced using random primers, and Irf-3 and Irf-7 mRNAs were measured by qPCR and normalized to glyceraldehyde 3-phosphate dehydrogenase levels. mRNA levels are expressed as the mean value compared to levels in uninfected day 0 cAP-7 neurons ± standard deviations of six samples. (B) Total RNA was collected from day 0 (9 replicates) and day 7 (12 replicates) AP-7 neuron cultures or from triplicate cultures of day 0 or day 28 CSM14.1 neurons. mRNA levels, measured as described for panel A, are expressed as the mean value of differentiated cultures compared to levels in day 0 AP-7 or CSM14.1 neurons ± standard deviations. (C) Total cell lysates prepared from AP-7 neurons during differentiation were analyzed by immunoblot analysis with anti-IRF-7 (top row), anti-IRF-3 (middle row), or anti-β-actin (bottom row) antibodies. Multiple isoforms of IRF-7 are detected by the IRF-7 antibody. (D) Total cell lysates prepared from CSM neurons at the indicated days during differentiation were analyzed by immunoblot analysis with anti-IRF-7 (top row) or anti-β-actin (bottom row) antibodies. (E) Brain homogenates (10%) from 3-day-old or 6-week-old C57BL/6J mice were analyzed by immunoblot analysis with anti-IRF-7 (top row), anti-IRF-3 (middle row), or anti-β-actin (bottom row) antibodies. Results of a representative experiment of at least 2 experiments are shown. Significant differences between cAP-7 and dAP-7 at each time point were determined by one-way ANOVA with Dunnett's posttest. ***, P < 0.001.
We confirmed the effect of differentiation on Irf expression by using CSM14.1 neurons, an immortalized rat nigral neuron cell line that restricts SINV infection similarly to dAP-7 neurons when differentiated (15) (Fig. 7B). Similar increases in Irf-3 and Irf-7 transcripts were detected in dCSM14.1 neurons as in cCSM14.1 cells. These studies showed that expression of Irf-3 and Irf-7 mRNAs is upregulated during differentiation of two distinct neuronal cell lines.
We investigated the levels of IRF-3 and IRF-7 proteins in cycling and differentiated AP-7 neurons (Fig. 7C). No changes in IRF-3 protein levels were detected during differentiation. At day zero, a single IRF-7 band was visible by immunoblot analysis. Upon differentiation, a slower-mobility band accumulated, and a third band of intermediate mobility was visible upon longer exposures. These bands corresponded in sizes to mouse IRF-7 isoforms α, β, and γ (63). All three isoforms contain exons 8 and 9, and so the corresponding mRNAs would be detected by qRT-PCR (Fig. 7A and B). In dCSM14.1 neurons, we detected a single IRF-7 band corresponding in size to the α-isoform, while no IRF-7 was detected in cCSM14.1 neurons (Fig. 7D). Importantly, IRF-7 α, the full-length isoform expressed by differentiated neurons, is a potent transactivator, whereas IRF-7 γ, the primary isoform expressed by undifferentiated neurons, inhibits IRF-7 α/β transactivation (63). Thus, in two distinct neuronal cell lines, total IRF-7 protein levels are increased upon differentiation, in agreement with increased mRNA levels, and differentiation is associated with increased levels of the active full-length α-isoform of IRF-7.
To investigate whether IRF-3 or IRF-7 levels were changed during development in vivo, we compared IRF-3 and IRF-7 in brain homogenates from 3-day-old and 6-week-old C57BL/6J mice. In 3-day-old mice, IRF-3 protein migrated as would be expected for a protein with a predicted molecular mass of 62 kDa. However, IRF-3 was detected in a high-molecular-mass complex in the brains of 6-week-old mice, suggesting that it is in a tight complex that was not dissociated by SDS–β-mercaptoethanol and high heat. In brain homogenates from 3-day-old mice, IRF-7 was predominately in the γ-isoform. In contrast, the α-isoform was dominant in brain homogenates from 6-week-old mice. Thus, the IRF-7 isoforms switched from the dominant negative γ-isoform to the active α-isoform upon maturation of cultured neurons and in the mouse brain, possibly priming neurons for a rapid response upon infection.
Because dAP-7 neurons had decreased SINV multiplication (Fig. 1A) and increased levels of Irf-3 and Irf-7 expression (Fig. 7B), we assessed whether medium from uninfected dAP-7 neurons could confer these characteristics on cAP-7 neurons. cAP-7 neurons were incubated for 24 h before infection with normal medium (DMEM–10% FBS), differentiation medium (normal medium plus dopamine, insulin, and ascorbic acid), or conditioned differentiation medium from 7- to 9-day dAP-7 neurons (Fig. 8A). No differences (determined by one-way analysis of variance [ANOVA]) in SINV multiplication were detected. Additionally, pretreatment did not alter expression of either Irf-3 or Irf-7. Thus, the antiviral properties observed in dAP-7 neurons could not be conferred to cAP-7 neurons by the differentiation medium alone or by soluble factors produced by dAP-7 neurons.
FIG 8.
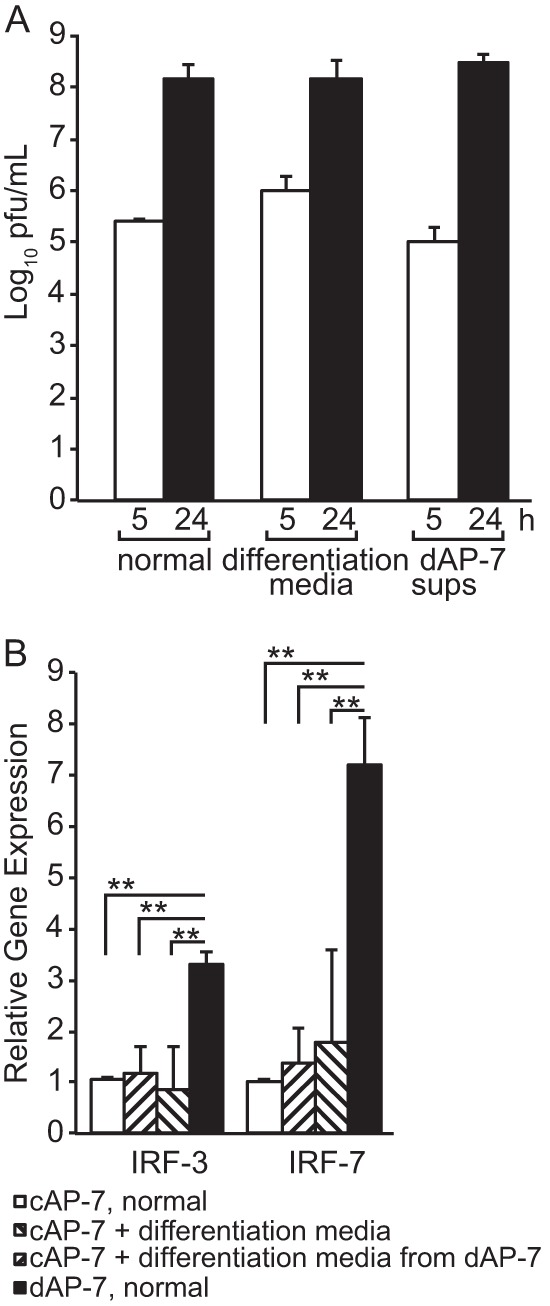
Medium exchange does not confer antiviral properties. cAP-7 neurons were incubated in normal medium (DMEM–10% FBS), differentiation medium, or supernatant fluids from dAP-7 neurons for 24 h. (A) Extracellular virus collected at 5 and 24 h after SINV infection (MOI, 5) infection was measured by plaque assay. Titers are expressed as the mean log10 PFU/ml ± standard deviation of triplicate samples. (B) Total cellular RNA was collected, and cDNA was produced using random primers. Gene expression was measured by qPCR and normalized to glyceraldehyde-3-phosphate dehydrogenase levels. RNA levels are expressed as the mean value compared to levels in cAP-7 neurons in normal medium ± standard deviation of six samples. Significant differences by Student's t test or one-way ANOVA with Dunnett's posttest are shown. **, P < 0.01.
IRF-3 and IRF-7 expression is increased after infection of dAP-7 neurons.
In many cell types, activation of innate immune signaling results in increased Irf-7 gene expression. Thus, we compared Irf-3 and Irf-7 gene expression levels in cAP-7 and dAP-7 cells following infection with SINV, VEEV, or LACV. Irf-3 and Irf-7 gene expression increased less than 3-fold after infection of cAP-7 neurons with SINV, VEEV, or LACV (Fig. 9A and B). In contrast, Irf-3 and Irf-7 gene expression was rapidly and substantially increased in dAP-7 neurons after infection (Fig. 9A and B). By 1 h after infection with all three viruses, Irf-3 was increased 10-fold and Irf-7 was increased approximately 25-fold. Irf-3 continued to increase through 24 h after SINV infection, while VEEV- or LACV-infected dAP7 cells did not show further increases (Fig. 9A). Irf-7 expression was further increased at 24 h after infection with all three viruses (Fig. 9B).
FIG 9.

Expression of Irf-3 and Irf-7 is upregulated during infection of differentiated AP-7 neurons. cAP-7 or dAP-7 neurons were infected with SINV (MOI, 5), VEEV (MOI, 50), or LACV (MOI, 5). Total cellular RNA was collected at 1, 12, or 24 h after infection, and cDNA was produced using random primers. Irf-3 (A) or Irf-7 (B) gene expression was measured by qPCR and normalized to glyceraldehyde-3-phosphate dehydrogenase levels. RNA levels are expressed as the mean value compared to levels in mock-infected cAP-7 neurons ± standard deviation of triplicate samples. Results of a representative experiment of 2 experiments are shown. Significant differences by two-way ANOVA with Bonferroni's posttest are shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) cAP-7 or dAP-7 neurons were mock infected or infected with SINV (MOI, 5). Total cell lysates were prepared at the indicated times after infection and analyzed by immunoblotting using anti-IRF-7 (top) or anti-β-actin (bottom) antibodies. Results of a representative experiment of 2 experiments are shown.
To determine if increased mRNA levels resulted in increased protein levels, we investigated protein accumulation in SINV-infected cells by immunoblot analysis. In cAP-7 neurons, total levels of IRF-7 protein increased during infection (Fig. 9C, left). However, the isoform that increased was the dominant negative γ-form, indicating that even after infection, cAP-7 neurons do not produce the functional α or β IRF-7 isoforms. No change in total IRF-7 protein levels was detected in dAP-7 neurons during SINV infection, and isoform α remained the most abundant form throughout the time course (Fig. 9C, right). IRF-7 is reported to have a short-half-life of approximately 30 min in most cells, but the half-life in neurons is unknown and virus infection has a variable effect on IRF-7 stability (64). Therefore, increased gene expression may be necessary to maintain IRF-7 protein levels during infection.
To determine if IRF-3 and IRF-7 are critical for restricting virus multiplication during alphavirus infection of dAP-7 cells, we silenced Irf-3 and Irf-7 alone or together during SINV and VEEV infection of dAP-7 neurons. dsRNA was transfected into AP-7 neurons on day 4 of differentiation. Replacement of differentiation medium at this stage did not affect the neurons morphologically or the restriction of virus multiplication (Fig. 10D and E, none versus dAP-7). In uninfected neurons, Irf-3 expression was decreased 8- to 71-fold when silenced alone and 7- to 53-fold when silenced along with Irf-7 (Fig. 10A). Irf-7 expression was decreased 25- to 42-fold when silenced alone and 15- to 36-fold when silenced with Irf-3 (Fig. 10B).
FIG 10.

Virus replication is unchanged upon silencing of IRF-3 or IRF-7 during infection of differentiated AP-7 neurons. On day 4 of differentiation, dAP-7 neurons were transfected with dsRNA specific to Irf-3 (3), Irf-7 (7), both (3/7), or enhanced green fluorescent protein (E), or not transfected, with medium change (N). For comparison, dAP-7 neurons were differentiated normally, without medium changes (d). At 96 h after transfection, dAP-7 neurons were infected with SINV (MOI, 5) or VEEV (MOI, 50). Total cellular RNA was collected at 0 or 24 h after infection, and cDNA was produced using random primers. Irf-3 (A) and Irf-7 (B) gene expression was measured by qPCR and normalized to glyceraldehyde-3-phosphate dehydrogenase levels. RNA levels are expressed as the mean value compared to levels in mock-infected, 0-h dAP-7 neurons ± standard deviation of duplicate samples. Significant differences by one-way ANOVA with Dunnet's posttest are shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) Total cell lysates were prepared at the indicated times after infection and analyzed by immunoblotting using anti-IRF-7 (top), anti-IRF-3 (middle), or anti-β-actin (bottom) antibodies. Viral titers in supernatant fluids collected at the indicated times after SINV (D) or VEEV (E) infection were measured by plaque formation in BHK cells and are expressed as the mean log10 PFU/ml ± standard deviation of triplicate samples. No significant differences by one-way ANOVA with Dunnet's posttest. Results of a representative experiment of 2 experiments are shown.
The level of silencing was then determined during infection. Compared to neurons that had not been transfected with silencing RNA, Irf-3 expression alone was decreased 28-fold during SINV infection and 72-fold during VEEV infection. When silenced along with Irf-7, Irf-3 levels were further decreased 26- to 111-fold during alphavirus infection (Fig. 10A). IRF-3 protein levels were decreased to the level of detection when silenced alone or with Irf-7 (Fig. 10C, middle). When silenced alone, Irf-7 mRNA levels decreased 11-fold after SINV infection and 19-fold when cosilenced with Irf-3 (Fig. 10B). After VEEV infection, there was a 29- to 33-fold decrease in Irf-7 mRNA levels compared to untransfected dAP-7 neurons when silenced alone or with Irf-3. However, IRF-7 protein was still detectable regardless of virus infection or silencing conditions (Fig. 10C, top), and we were unable to identify an RNA silencing strategy that consistently reduced IRF-7 protein levels at the time of or throughout infection. It is possible that IRF-7 in a stable transcriptionally active complex is less susceptible to rapid turnover. Levels of Irf-3 and Irf-7 mRNAs changed less than 4-fold in neurons transfected with irrelevant dsRNAs, indicating that neither transfection nor medium changes affected expression.
To determine the impact of Irf-3 and Irf-7 dsRNA on virus multiplication in dAP-7 neurons, we compared the levels of infectious SINV and VEEV produced by dAP-7 neurons with and without Irf silencing prior to infection (Fig. 10D and E). There was no effect on replication of either virus when Irf-7 transcript levels were decreased alone or simultaneously with Irf-3. Furthermore, reduction in IRF-3 protein alone did not increase production of either SINV or VEEV compared to untreated dAP-7 neurons (determined by one-way ANOVA). Thus, loss of IRF-3 is not sufficient to reverse restriction of alphavirus replication in differentiated AP-7 neurons.
DISCUSSION
Host maturity is an important factor determining the outcome of viral encephalitis (1, 3–9). We have previously demonstrated maturation-dependent restriction of SINV replication in CSM14.1 neuronal cells (15). In this study, we have shown maturation-dependent restricted replication of the encephalitic arboviruses SINV, VEEV, and LACV in olfactory bulb-derived AP-7 neurons. During SINV infection, maturation did not affect neuronal susceptibility to infection or virion maturation. However, maturation did reduce susceptibility to VEEV infection. When equal percentages of immature and mature neurons were infected with either alphavirus, restriction was associated with delayed synthesis of viral RNA and protein, decreased production of infectious virus, delayed shutoff of host protein synthesis, and improved viability of infected neurons. During maturation from immature cycling cells to differentiated neurons, expression of Stat-1 and the immunoregulatory transcription factors Irf-3 and Irf-7 was increased. The IRF-7 protein isoforms changed from production of the inhibitory γ short form to the transcriptionally competent α long form. Silencing of Irf-3 and Irf-7 did not improve viral replication in mature neurons, indicating that there are additional maturation-dependent mechanisms for restriction of virus replication. However, we were unable to consistently reduce IRF-7 protein levels by RNA silencing, so IRF-7 may yet prove to be important for restricting virus replication.
To counter host antiviral signaling, viruses encode IFN antagonists that circumvent the host antiviral response by distinct mechanisms, often by inhibiting host transcription and translation (65). SINV nsP2 shuts off both host transcription and translation, whereas VEEV capsid is responsible for transcriptional arrest (66–68). LACV NSs inhibits RNA polymerase II-dependent transcription to block IFN signaling (69). In our studies, host protein synthesis was sustained longer during infection of dAP-7 neurons than in cAP-7 neurons during alphavirus infection. Additionally, alphavirus protein synthesis was delayed in dAP-7 neurons. Thus, active translation of antiviral proteins in dAP-7 neurons could likely support survival and reduce virus multiplication prior to accumulation of viral proteins required for cellular translation arrest.
The protective role of IFNs against encephalitic virus infections has been demonstrated in other in vitro systems and in vivo. Pretreatments of a variety of cultured cells, including neurons, with IFN-α, -β, or -γ prevents more than 99% of infectious SINV production, significantly reduces synthesis of viral proteins, and preserves cellular viability and protein synthesis (20, 62, 70). The G3A mutation in VEEV TC-83 renders this strain responsive to IFN-α/β priming and is correlated with decreased mortality in weanling mice (25, 26, 70). Mice that lack the α-chain of the IFN-α/β receptor, IFN-β, or the IFN-activated STAT1 transcription factor are more susceptible to SINV, VEEV TC-83, and LACV (21, 24, 26, 27, 70, 71). Subprotective levels of IFN-β are sufficient to stimulate antiviral signaling during SINV infection and allow for signaling during the first hours of infection, which determine the outcome of infection (23). Consistent with these previous studies, STAT1 was not phosphorylated in either dAP-7 or cAP-7 neurons, and transfer of dAP-7-dervied medium to cAP-7 neurons failed to confer protection, suggesting that if IFN were present, it was not signaling detectably through the classical JAK/STAT pathway.
Canonically, IRFs activate ISG expression in response to IFN as well as stimulate IFN expression. However, the control of innate immune signaling varies according to tissue and virus (72–75). Increased susceptibility of IRF-7-deficient mice to viruses such as West Nile virus, Chikungunya virus, and SINV demonstrates the central importance of IRF-7 in orchestrating IFN signaling (44, 47, 76–78). Noncanonically, IRF-1, -3, and -7 can upregulate ISG expression in the absence of measurable IFN (48, 79–82). Utilization of noncanonical, STAT1-independent antiviral signaling by neurons has been demonstrated in neurons during measles virus, WEEV, and St. Louis encephalitis virus infection (49, 83). These studies support the importance of higher levels of Irf-7 and Irf-3 to enable differentiated neurons to orchestrate a prompt and effective antiviral response. Additionally, Irf-7 is constitutively expressed in the brains of naive, weanling mice and is upregulated in neurons during LACV infection (28). By analyzing protein as well as mRNA, we found that distinct IRF-7 isoforms are produced in brains of newborn and weanling mice, with the functional α-isoform prominent in weanling mice.
Our studies are consistent with a model in which preassembled IRFs mediate rapid intracellular signaling upon pathogen detection to induce the antiviral response. Production of a distinct isoform of IRF-7 may contribute to the improved IFN response in differentiated rodent neurons. Conversely, the presence of the dominant negative isoform in immature rodent neurons may contribute to the failure of these cells to mount an antiviral response. In the absence of Irf-3 and Irf-7, and presumably consequent ISG expression, viral titers did not increase to those measured in cAP-7 neurons. We cannot rule out that residual IRF-7 was sufficient to support antiviral signaling during infection. However, IRF-mediated signaling may play a greater role in vivo to control virus spread, orchestrate virus clearance, or modulate the adaptive immune response in the CNS.
Recently, advances have been made in understanding the active role neurons play in the response to neuronotropic virus infection. Along with our studies, these findings collectively suggest that the CNS has evolved specialized control of the cellular response to pathogens, possibly due to the limited regenerative properties and specific function of neurons. Collectively, maturation of neurons is associated with increased levels of IFN signaling pathway components (33, 34). Mature neurons are able to produce an innate immune response upon virus infection (28, 30–33). Increased basal levels of Irf-3 and Irf-7 expression could contribute to an enhanced host response to infection. However, innate immune signaling does not fully explain the improved outcome for mature neurons following virus infection (33, 45). Further comparison of antiviral gene expression in mature and immature neurons may identify other factors that make greater contributions to age-dependent susceptibility to encephalitic viruses.
ACKNOWLEDGMENTS
These studies were funded in part by research grant R01 NS038932 (D.E.G.) and training grants T32 AI007247 (K.L.W.S.) and T32 GM07309 (P.S.V.) from the National Institutes of Health.
We thank Andrew Pekosz, Wei Li, and Kirsten Kulcsar for many helpful discussions. We acknowledge help from the Johns Hopkins Microscopy Core and production of anti-E2 monoclonal antibody 209 by the Johns Hopkins University School of Medicine Department of Neuroscience Monoclonal Antibody Core (NS050274).
REFERENCES
- 1.Ben-Hur T, Hadar J, Shtram Y, Gilden DH, Becker Y. 1983. Neurovirulence of herpes simplex virus type 1 depends on age in mice and thymidine kinase expression. Arch Virol 78:303–308. doi: 10.1007/BF01311326. [DOI] [PubMed] [Google Scholar]
- 2.Bennett RS, Cress CM, Ward JM, Firestone C-Y, Murphy BR, Whitehead SS. 2008. La Crosse virus infectivity, pathogenesis, and immunogenicity in mice and monkeys. Virol J 5:25. doi: 10.1186/1743-422X-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ikegami H, Takeda M, Doi K. 1997. An age-related change in susceptibility of rat brain to encephalomyocarditis virus infection. Int J Exp Pathol 78:101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson RT, McFarland HF, Levy SE. 1972. Age-dependent resistance to viral encephalitis: studies of infections due to Sindbis virus in mice. J Infect Dis 125:257–262. doi: 10.1093/infdis/125.3.257. [DOI] [PubMed] [Google Scholar]
- 5.McJunkin JE, de los Reyes EC, Irazuzta JE, Caceres MJ, Khan RR, Minnich LL, Fu KD, Lovett GD, Tsai T, Thompson A. 2001. La Crosse encephalitis in children. N Engl J Med 344:801–807. doi: 10.1056/NEJM200103153441103. [DOI] [PubMed] [Google Scholar]
- 6.McKendall RR, Woo W. 1987. Possible neural basis for age-dependent resistance to neurologic disease from herpes simplex virus. J Neurol Sci 81:227–237. doi: 10.1016/0022-510X(87)90098-0. [DOI] [PubMed] [Google Scholar]
- 7.Sigel MM. 1952. Influence of age on susceptibility to virus infections with particular reference to laboratory animals. Annu Rev Microbiol 6:247–280. doi: 10.1146/annurev.mi.06.100152.001335. [DOI] [PubMed] [Google Scholar]
- 8.Taguchi F, Aiuchi M, Fujiwara K. 1977. Age-dependent response of mice to a mouse hepatitis virus, MHV-S. Jpn J Exp Med 47:109–115. [PubMed] [Google Scholar]
- 9.Tardieu M, Powers ML, Weiner HL. 1983. Age dependent susceptibility to reovirus type 3 encephalitis: role of viral and host factors. Ann Neurol 13:602–607. doi: 10.1002/ana.410130604. [DOI] [PubMed] [Google Scholar]
- 10.Griffin DE. 1976. Role of the immune response in age-dependent resistance of mice to encephalitis due to Sindbis virus. J Infect Dis 133:456–464. doi: 10.1093/infdis/133.4.456. [DOI] [PubMed] [Google Scholar]
- 11.Oliver KR, Scallan MF, Dyson H, Fazakerley JK. 1997. Susceptibility to a neurotropic virus and its changing distribution in the developing brain is a function of CNS maturity. J Neurovirol 3:38–48. doi: 10.3109/13550289709015791. [DOI] [PubMed] [Google Scholar]
- 12.Kunin CM. 1962. Virus-tissue union and the pathogenesis of enterovirus infections. J Immunol 88:556–569. [PubMed] [Google Scholar]
- 13.Labrada L, Liang XH, Zheng W, Johnston C, Levine B. 2002. Age-dependent resistance to lethal alphavirus encephalitis in mice: analysis of gene expression in the central nervous system and identification of a novel interferon-inducible protective gene, mouse ISG12. J Virol 76:11688–11703. doi: 10.1128/JVI.76.22.11688-11703.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ubol S, Griffin DE. 1991. Identification of a putative alphavirus receptor on mouse neural cells. J Virol 65:6913–6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vernon PS, Griffin DE. 2005. Characterization of an in vitro model of alphavirus infection of immature and mature neurons. J Virol 79:3438–3447. doi: 10.1128/JVI.79.6.3438-3447.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Binder GK, Griffin DE. 2003. Immune-mediated clearance of virus from the central nervous system. Microbes Infect 5:439–448. doi: 10.1016/S1286-4579(03)00047-9. [DOI] [PubMed] [Google Scholar]
- 17.Chakraborty S, Kaushik DK, Gupta M, Basu A. 2010. Inflammasome signaling at the heart of central nervous system pathology. J Neurosci Res 88:1615–1631. doi: 10.1002/jnr.22343. [DOI] [PubMed] [Google Scholar]
- 18.Maher SG, Romero-Weaver AL, Scarzello AJ, Gamero AM. 2007. Interferon: cellular executioner or white knight? Curr Med Chem 14:1279–1289. doi: 10.2174/092986707780597907. [DOI] [PubMed] [Google Scholar]
- 19.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol 89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- 20.Després P, Griffin JW, Griffin DE. 1995. Antiviral activity of alpha interferon in Sindbis virus-infected cells is restored by anti-E2 monoclonal antibody treatment. J Virol 69:7345–7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Byrnes AP, Durbin JE, Griffin DE. 2000. Control of Sindbis virus infection by antibody in interferon-deficient mice. J Virol 74:3905–3908. doi: 10.1128/JVI.74.8.3905-3908.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 23.Frolov I, Akhrymuk M, Akhrymuk I, Atasheva S, Frolova EI. 2012. Early events in alphavirus replication determine the outcome of infection. J Virol 86:5055–5066. doi: 10.1128/JVI.07223-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryman KD, Klimstra WB, Nguyen KB, Biron CA, Johnston RE. 2000. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J Virol 74:3366–3378. doi: 10.1128/JVI.74.7.3366-3378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kinney RM, Chang GJ, Tsuchiya KR, Sneider JM, Roehrig JT, Woodward TM, Trent DW. 1993. Attenuation of Venezuelan equine encephalitis virus strain TC-83 is encoded by the 5′-noncoding region and the E2 envelope glycoprotein. J Virol 67:1269–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.White LJ, Wang JG, Davis NL, Johnston RE. 2001. Role of alpha/beta interferon in Venezuelan equine encephalitis virus pathogenesis: effect of an attenuating mutation in the 5′ untranslated region. J Virol 75:3706–3718. doi: 10.1128/JVI.75.8.3706-3718.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blakqori G, Delhaye S, Habjan M, Blair CD, Sánchez-Vargas I, Olson KE, Attarzadeh-Yazdi G, Fragkoudis R, Kohl A, Kalinke U, Weiss S, Michiels T, Staeheli P, Weber F. 2007. La Crosse bunyavirus nonstructural protein NSs serves to suppress the type I interferon system of mammalian hosts. J Virol 81:4991–4999. doi: 10.1128/JVI.01933-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delhaye S, Paul S, Blakqori G, Minet M, Weber F, Staeheli P, Michiels T. 2006. Neurons produce type I interferon during viral encephalitis. Proc Natl Acad Sci U S A 103:7835–7840. doi: 10.1073/pnas.0602460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lienenklaus S, Cornitescu M, Zietara N, Łyszkiewicz M, Gekara N, Jabłónska J, Edenhofer F, Rajewsky K, Bruder D, Hafner M, Staeheli P, Weiss S. 2009. Novel reporter mouse reveals constitutive and inflammatory expression of IFN-beta in vivo. J Immunol 183:3229–3236. doi: 10.4049/jimmunol.0804277. [DOI] [PubMed] [Google Scholar]
- 30.Dionne KR, Galvin JM, Schittone SA, Clarke P, Tyler KL. 2011. Type I interferon signaling limits reoviral tropism within the brain and prevents lethal systemic infection. J Neurovirol 17:314–326. doi: 10.1007/s13365-011-0038-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chopy D, Detje CN, Lafage M, Kalinke U, Lafon M. 2011. The type I interferon response bridles rabies virus infection and reduces pathogenicity. J Neurovirol 17:353–367. doi: 10.1007/s13365-011-0041-6. [DOI] [PubMed] [Google Scholar]
- 32.Préhaud C, Mégret F, Lafage M, Lafon M. 2005. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J Virol 79:12893–12904. doi: 10.1128/JVI.79.20.12893-12904.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castorena KM, Peltier DC, Peng W, Miller DJ. 2008. Maturation-dependent responses of human neuronal cells to western equine encephalitis virus infection and type I interferons. Virology 372:208–220. doi: 10.1016/j.virol.2007.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farmer JR, Altschaefl KM, O'Shea KS, Miller DJ. 2013. Activation of the type I interferon pathway is enhanced in response to human neuronal differentiation. PLoS One 8:e58813. doi: 10.1371/journal.pone.0058813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kato H, Takahasi K, Fujita T. 2011. RIG-I-like receptors: cytoplasmic sensors for non-self RNA. Immunol Rev 243:91–98. doi: 10.1111/j.1600-065X.2011.01052.x. [DOI] [PubMed] [Google Scholar]
- 36.Levy DE, Marié IJ. 2004. RIGging an antiviral defense: it's in the CARDs. Nat Immunol 5:699–701. doi: 10.1038/ni0704-699. [DOI] [PubMed] [Google Scholar]
- 37.Taniguchi T, Takaoka A. 2001. A weak signal for strong responses: interferon-alpha/beta revisited. Nat Rev Mol Cell Biol 2:378–386. doi: 10.1038/35073080. [DOI] [PubMed] [Google Scholar]
- 38.Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE. 2004. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci U S A 101:17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 40.Barnes BJ. 2003. Virus-induced heterodimer formation between IRF-5 and IRF-7 modulates assembly of the IFNA enhanceosome in vivo and transcriptional activity of IFNA genes. J Biol Chem 278:16630–16641. doi: 10.1074/jbc.M212609200. [DOI] [PubMed] [Google Scholar]
- 41.Juang YT, Lowther W, Kellum M, Au WC, Lin R, Hiscott J, Pitha PM. 1998. Primary activation of interferon A and interferon B gene transcription by interferon regulatory factor 3. Proc Natl Acad Sci U S A 95:9837–9842. doi: 10.1073/pnas.95.17.9837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Au WC, Moore PA, LaFleur DW, Tombal B, Pitha PM. 1998. Characterization of the interferon regulatory factor-7 and its potential role in the transcription activation of interferon A genes. J Biol Chem 273:29210–29217. doi: 10.1074/jbc.273.44.29210. [DOI] [PubMed] [Google Scholar]
- 43.Marié I, Durbin JE, Levy DE. 1998. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J 17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schilte C, Buckwalter MR, Laird ME, Diamond MS, Schwartz O, Albert ML. 2012. Cutting edge: independent roles for IRF-3 and IRF-7 in hematopoietic and nonhematopoietic cells during host response to Chikungunya infection. J Immunol 188:2967–2971. doi: 10.4049/jimmunol.1103185. [DOI] [PubMed] [Google Scholar]
- 45.Daffis S, Samuel MA, Keller BC, Gale M, Diamond MS. 2007. Cell-specific IRF-3 responses protect against West Nile virus infection by interferon-dependent and -independent mechanisms. PLoS Pathog 3:e106. doi: 10.1371/journal.ppat.0030106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13:539–548. doi: 10.1016/S1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 47.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 48.Bego MG, Mercier J, Cohen EA. 2012. Virus-activated interferon regulatory factor 7 upregulates expression of the interferon-regulated BST2 gene independently of interferon signaling. J Virol 86:3513–3527. doi: 10.1128/JVI.06971-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peltier DC, Lazear HM, Farmer JR, Diamond MS, Miller DJ. 2013. Neurotropic arboviruses induce interferon regulatory factor 3-mediated neuronal responses that are cytoprotective, interferon independent, and inhibited by Western equine encephalitis virus capsid. J Virol 87:1821–1833. doi: 10.1128/JVI.02858-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murrell JR, Hunter DD. 1999. An olfactory sensory neuron line, odora, properly targets olfactory proteins and responds to odorants. J Neurosci 19:8260–8270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kerr DA, Lladó J, Shamblott MJ, Maragakis NJ, Irani DN, Crawford TO, Krishnan C, Dike S, Gearhart JD, Rothstein JD. 2003. Human embryonic germ cell derivatives facilitate motor recovery of rats with diffuse motor neuron injury. J Neurosci 23:5131–5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Levine B, Huang Q, Isaacs JT, Reed JC, Griffin DE. 1993. Conversion of lytic to persistent alphavirus infection by the bcl-2 cellular oncogene. Nature 361:739–742. doi: 10.1038/361739a0. [DOI] [PubMed] [Google Scholar]
- 53.Lustig S, Jackson AC, Hahn CS, Griffin DE, Strauss EG, Strauss JH. 1988. Molecular basis of Sindbis virus neurovirulence in mice. J Virol 62:2329–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Janssen R, Gonzalez-Scarano F. 1984. Mechanisms of bunyavirus virulence. Comparative pathogenesis of a virulent strain of La Crosse and an avirulent strain of Tahyna virus. Lab Invest 50:447–455. [PubMed] [Google Scholar]
- 55.Stanley J, Cooper SJ, Griffin DE. 1985. Alphavirus neurovirulence: monoclonal antibodies discriminating wild-type from neuroadapted Sindbis virus. J Virol 56:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roehrig JT, Mathews JH. 1985. The neutralization site on the E2 glycoprotein of Venezuelan equine encephalomyelitis (TC-83) virus is composed of multiple conformationally stable epitopes. Virology 142:347–356. doi: 10.1016/0042-6822(85)90343-5. [DOI] [PubMed] [Google Scholar]
- 57.Park E, Griffin DE. 2009. Interaction of Sindbis virus non-structural protein 3 with poly(ADP-ribose) polymerase 1 in neuronal cells. J Gen Virol 90:2073–2080. doi: 10.1099/vir.0.012682-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Levine B, Goldman JE, Jiang HH, Griffin DE, Hardwick JM. 1996. Bc1-2 protects mice against fatal alphavirus encephalitis. Proc Natl Acad Sci U S A 93:4810–4815. doi: 10.1073/pnas.93.10.4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lewis J, Wesselingh SL, Griffin DE, Hardwick JM. 1996. Alphavirus-induced apoptosis in mouse brains correlates with neurovirulence. J Virol 70:1828–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nava VE, Rosen A, Veliuona MA, Clem RJ, Levine B, Hardwick JM. 1998. Sindbis virus induces apoptosis through a caspase-dependent, CrmA-sensitive pathway. J Virol 72:452–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ubol S, Park S, Budihardjo I, Desnoyers S, Montrose MH, Poirier GG, Kaufmann SH, Griffin DE. 1996. Temporal changes in chromatin, intracellular calcium, and poly(ADP-ribose) polymerase during Sindbis virus-induced apoptosis of neuroblastoma cells. J Virol 70:2215–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Burdeinick-Kerr R, Govindarajan D, Griffin DE. 2009. Noncytolytic clearance of Sindbis virus infection from neurons by gamma interferon is dependent on Jak/Stat signaling. J Virol 83:3429–3435. doi: 10.1128/JVI.02381-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marié I, Smith E, Prakash A, Levy DE. 2000. Phosphorylation-induced dimerization of interferon regulatory factor 7 unmasks DNA binding and a bipartite transactivation domain. Mol Cell Biol 20:8803–8814. doi: 10.1128/MCB.20.23.8803-8814.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Prakash A, Levy DE. 2006. Regulation of IRF7 through cell type-specific protein stability. Biochem Biophys Res Commun 342:50–56. doi: 10.1016/j.bbrc.2006.01.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hollidge BS, Weiss SR, Soldan SS. 2011. The role of interferon antagonist, non-structural proteins in the pathogenesis and emergence of arboviruses. Viruses 3:629–658. doi: 10.3390/v3060629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gorchakov R, Frolova E, Frolov I. 2005. Inhibition of transcription and translation in Sindbis virus-infected cells. J Virol 79:9397–9409. doi: 10.1128/JVI.79.15.9397-9409.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Garmashova N, Atasheva S, Kang W, Weaver SC, Frolova E, Frolov I. 2007. Analysis of Venezuelan equine encephalitis virus capsid protein function in the inhibition of cellular transcription. J Virol 81:13552–13565. doi: 10.1128/JVI.01576-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garmashova N, Gorchakov R, Volkova E, Paessler S, Frolova E, Frolov I. 2007. The Old World and New World alphaviruses use different virus-specific proteins for induction of transcriptional shutoff. J Virol 81:2472–2484. doi: 10.1128/JVI.02073-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Verbruggen P, Ruf M, Blakqori G, Overby AK, Heidemann M, Eick D, Weber F. 2011. Interferon antagonist NSs of La Crosse virus triggers a DNA damage response-like degradation of transcribing RNA polymerase II. J Biol Chem 286:3681–3692. doi: 10.1074/jbc.M110.154799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yin J, Gardner CL, Burke CW, Ryman KD, Klimstra WB. 2009. Similarities and differences in antagonism of neuron alpha/beta interferon responses by Venezuelan equine encephalitis and Sindbis alphaviruses. J Virol 83:10036–10047. doi: 10.1128/JVI.01209-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hefti HP, Frese M, Landis H, Di Paolo C, Aguzzi A, Haller O, Pavlovic J. 1999. Human MxA protein protects mice lacking a functional alpha/beta interferon system against La crosse virus and other lethal viral infections. J Virol 73:6984–6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Christensen JE, Fenger C, Issazadeh-Navikas S, Krug A, Liljestrøm P, Goriely S, Paludan SR, Finsen B, Christensen JP, Thomsen AR. 2012. Differential impact of interferon regulatory factor 7 in initiation of the type I interferon response in the lymphocytic choriomeningitis virus-infected central nervous system versus the periphery. J Virol 86:7384–7392. doi: 10.1128/JVI.07090-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Suthar MS, Brassil MM, Blahnik G, McMillan A, Ramos HJ, Proll SC, Belisle SE, Katze MG, Gale M. 2013. A systems biology approach reveals that tissue tropism to West Nile virus is regulated by antiviral genes and innate immune cellular processes. PLoS Pathog 9:e1003168. doi: 10.1371/journal.ppat.1003168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Clarke P, Leser JS, Bowen RA, Tyler KL. 2014. Virus-induced transcriptional changes in the brain include the differential expression of genes associated with interferon, apoptosis, interleukin 17 receptor A, and glutamate signaling as well as flavivirus-specific upregulation of tRNA synthetases. mBio 5(2):e00902-14. doi: 10.1128/mBio.00902-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fensterl V, Wetzel JL, Ramachandran S, Ogino T, Stohlman SA, Bergmann CC, Diamond MS, Virgin HW, Sen GC. 2012. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog 8:e1002712. doi: 10.1371/journal.ppat.1002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Colina R, Costa-Mattioli M, Dowling RJO, Jaramillo M, Tai L-H, Breitbach CJ, Martineau Y, Larsson O, Rong L, Svitkin YV, Makrigiannis AP, Bell JC, Sonenberg N. 2008. Translational control of the innate immune response through IRF-7. Nature 452:323–328. doi: 10.1038/nature06730. [DOI] [PubMed] [Google Scholar]
- 77.Esen N, Blakely PK, Rainey-Barger EK, Irani DN. 2012. Complexity of the microglial activation pathways that drive innate host responses during lethal alphavirus encephalitis in mice. ASN Neuro 4:207–221. doi: 10.1042/AN20120016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Daffis S, Samuel MA, Suthar MS, Keller BC, Gale M, Diamond MS. 2008. Interferon regulatory factor IRF-7 induces the antiviral alpha interferon response and protects against lethal West Nile virus infection. J Virol 82:8465–8475. doi: 10.1128/JVI.00918-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bandyopadhyay SK, Leonard GT, Bandyopadhyay T, Stark GR, Sen GC. 1995. Transcriptional induction by double-stranded RNA is mediated by interferon-stimulated response elements without activation of interferon-stimulated gene factor 3. J Biol Chem 270:19624–19629. doi: 10.1074/jbc.270.33.19624. [DOI] [PubMed] [Google Scholar]
- 80.Di Domizio J, Blum A, Gallagher-Gambarelli M, Molens J-P, Chaperot L, Plumas J. 2009. TLR7 stimulation in human plasmacytoid dendritic cells leads to the induction of early IFN-inducible genes in the absence of type I IFN. Blood 114:1794–1802. doi: 10.1182/blood-2009-04-216770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lin R, Noyce RS, Collins SE, Everett RD, Mossman KL. 2004. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J Virol 78:1675–1684. doi: 10.1128/JVI.78.4.1675-1684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ning S, Huye LE, Pagano JS. 2005. Regulation of the transcriptional activity of the IRF7 promoter by a pathway independent of interferon signaling. J Biol Chem 280:12262–12270. doi: 10.1074/jbc.M404260200. [DOI] [PubMed] [Google Scholar]
- 83.O'Donnell LA, Conway S, Rose RW, Nicolas E, Slifker M, Balachandran S, Rall GF. 2012. STAT1-independent control of a neurotropic measles virus challenge in primary neurons and infected mice. J Immunol 188:1915–1923. doi: 10.4049/jimmunol.1101356. [DOI] [PMC free article] [PubMed] [Google Scholar]