

ABSTRACT
The recent global resurgence of arthritogenic alphaviruses, in particular chikungunya virus (CHIKV), highlights an urgent need for the development of therapeutic intervention strategies. While there has been significant progress in defining the pathophysiology of alphaviral disease, relatively little is known about the mechanisms involved in CHIKV-induced arthritis or potential therapeutic options to treat the severe arthritic symptoms associated with infection. Here, we used microcomputed tomographic (μCT) and histomorphometric analyses to provide previously undescribed evidence of reduced bone volume in the proximal tibial epiphysis of CHIKV-infected mice compared to the results for mock controls. This was associated with a significant increase in the receptor activator of nuclear factor-κB ligand/osteoprotegerin (RANKL/OPG) ratio in infected murine joints and in the serum of CHIKV patients. The expression levels of the monocyte chemoattractant proteins (MCPs), including MCP-1/CCL2, MCP-2/CCL8, and MCP-3/CCL7, were also highly elevated in joints of CHIKV-infected mice, accompanied by increased cellularity within the bone marrow in tibial epiphysis and ankle joints. Both this effect and CHIKV-induced bone loss were significantly reduced by treatment with the MCP inhibitor bindarit. Collectively, these findings demonstrate a unique role for MCPs in promoting CHIKV-induced osteoclastogenesis and bone loss during disease and suggest that inhibition of MCPs with bindarit may be an effective therapy for patients affected with alphavirus-induced bone loss.
IMPORTANCE Arthritogenic alphaviruses, including chikungunya virus (CHIKV) and Ross River virus (RRV), cause worldwide outbreaks of polyarthritis, which can persist in patients for months following infection. Previous studies have shown that host proinflammatory soluble factors are associated with CHIKV disease severity. Furthermore, it is established that chemokine (C-C motif) ligand 2 (CCL2/MCP-1) is important in cellular recruitment and inducing bone-resorbing osteoclast (OC) formation. Here, we show that CHIKV replicates in bone and triggers bone loss by increasing the RANKL/OPG ratio. CHIKV infection results in MCP-induced cellular infiltration in the inflamed joints, and bone loss can be ameliorated by treatment with an MCP-inhibiting drug, bindarit. Taken together, our data reveal a previously undescribed role for MCPs in CHIKV-induced bone loss: one of recruiting monocytes/OC precursors to joint sites and thereby favoring a pro-osteoclastic microenvironment. This suggests that bindarit may be an effective treatment for alphavirus-induced bone loss and arthritis in humans.
INTRODUCTION
Arthritogenic Old World alphaviruses, such as Sindbis virus (SINV), o'nyong-nyong virus (ONNV), Ross River virus (RRV), and chikungunya virus (CHIKV), are mosquito-borne viruses that cause myalgia and debilitating polyarthritis (reviewed in reference 1). During the last decade, severe CHIKV outbreaks involving millions of cases have occurred in countries neighboring the Indian Ocean, including India, La Reunion, Thailand, Indonesia, and Singapore (2). Recently, CHIKV established itself among several Caribbean islands, where an estimated 170,000 suspected or confirmed autochthonous cases have been reported (Pan America Health Organization, 2014).
CHIKV is a positive-sense RNA virus that causes chikungunya fever (CHIKF), a self-limiting and febrile illness with clinical manifestations that include fever, headache, fatigue, rash, conjunctivitis, and incapacitating polyarthralgias that can persist for months to years (3, 4). Following the 2005-2006 epidemic in Indian Ocean islands, radiographic evidence emerged of bone pathologies similar to those in rheumatoid arthritis (RA) in CHIKV-infected patients (5, 6). Recently, bone pathologies in joints of CHIKV patients have been reported, providing clinical evidence that alphaviruses may also affect skeletal health (7). In RA joints, inflammation in the bone microenvironment triggers the activation of osteoclast (OC)-mediated bone resorption and inhibits bone formation by osteoblasts (OBs) (reviewed in reference 8). Differentiation of mature OCs is triggered by the binding of receptor activator of nuclear factor-κB (NF-κB) ligand (RANKL) to its signaling receptor RANK, which is expressed on monocytic precursors (9, 10). Cells from the osteoblastic lineage and nonosteoblastic cells, such as T cells, NK cells, and synoviocytes, express RANKL in inflammatory arthritis (11–17). The activity of RANKL can be blocked by its endogenous soluble decoy receptor, osteoprotegerin (OPG) (reviewed in reference 18). Hence, the RANKL/OPG ratio within the local bone microenvironment regulates OC differentiation and function, with an increase in the ratio leading to bone pathology (19–22).
During alphavirus infection, the expression of RANKL/OPG by OBs is also modified (23, 24). Recently, we reported that RRV infection increases OB lineage support of OC formation and contributes to pathological bone loss in an established acute RRV disease (RRVD) mouse model. In that study, the RANKL/OPG ratio was increased in RRV-infected mice (24).
Increased infiltration of macrophages in joints has been reported in mouse models of RRV (25) and CHIKV infection (26, 27). Depletion of macrophages in an acute RRVD mouse model resulted in a prominent reduction in cellular infiltrates and disease signs (25). In a primate model of CHIKV infection, persistently infected macrophages were detected in the joints, muscles, lymphoid organs, and liver, suggesting macrophages as a primary cellular reservoir of virus during the chronic phase of infection (28). These findings concur with a recent study of CHIKV patients at 18 months postinfection, where CHIKV persisted in perivascular synovial macrophages (29). Collectively, it can be postulated that, following an arthritogenic alphaviral infection, the recruitment of monocytes which may serve as OC precursors and their differentiation are promoted by increased RANKL expression by cells in the local bone microenvironment, thereby contributing to bone loss.
Several chemokines have been associated with chemotaxis of OC precursors (30) and osteoclastogenesis in pathological bone conditions, including multiple myeloma (31, 32), RA (33), and juvenile idiopathic arthritis (34). These include CXC chemokine ligand 1 (CXCL1) (KC), CC chemokine ligand 3 (CCL3) (macrophage inflammatory protein-1α [MIP-1α]), CCL4 (MIP-1β), CCL11 (eotaxin), CCL5 (RANTES), and CCL2 (monocyte chemotactic protein-1 [MCP-1]). CCL8 (MCP-2) and CCL7 (MCP-3) are potent chemotactic cytokines involved in the egress of monocytic cells and play crucial roles in maintaining the leukocyte homeostasis (35, 36). High levels of CCL8 and CCL7 have been detected in synovial tissues of individuals suffering from RA, osteoarthritis, and reactive arthritis (37). Studies in CCL2-deficient mice and human peripheral blood mononuclear cells (PBMCs) have shown CCL2 to be an osteoclastogenic factor (38, 39). PBMCs from RA patients showed a higher osteoclastogenic potential than those of healthy controls, and this correlated positively to serum levels of CCL2 (40). In RRV and CHIKV infections, MCPs have an indispensable pathogenic role in mediating alphavirus-induced arthritis and myositis (41, 42). MCPs may also contribute to the exacerbated bone loss observed in these infections.
Bindarit is a small-molecule indazolic derivative with anti-inflammatory properties. Bindarit has been reported to reduce symptoms in several animal models of human disease, such as adjuvant-induced arthritis (43), breast cancer (44), autoimmune encephalitis (45), severe acute pancreatitis (46), and lupus nephritis (47), through potent suppression of CCL2 transcription. Although CCL2 is the best characterized target of bindarit, it also inhibits other inflammatory mediators, such as CCL8, CCL7, and interleukin-12 (IL-12) (48). A recent study has also identified a modulatory effect of bindarit on the classical NF-κB pathway, where bindarit specifically inhibits p65 and p65/p50-mediated CCL2 promoter activation (48).
In this study, the role of MCPs in modulating alphavirus-induced bone loss was investigated. We demonstrate that CHIKV infection stimulates osteoclastogenesis, leading to significant bone loss. Recruitment of monocytic OC precursor cells to the ankle joint and tibial epiphysis was mediated in part by MCPs. Bindarit treatment both attenuated the joint swelling and prevented bone loss. These results strongly suggest bindarit as a potential therapy for bone loss caused by alphaviruses and other infectious agents and for other diseases featuring localized increasing bone resorption, such as RA.
MATERIALS AND METHODS
Ethics statement.
Animal experiments were approved by the Animal Ethics Committee of Griffith University (BDD/04/11/AEC and GLY/06/13/AEC). All procedures involving animals conformed to the National Health and Medical Research Council Australian code for the care and use of animals for scientific purposes (49). All CHIKV human serum samples had been submitted to the Centre for Infectious Diseases and Microbiology Laboratory Services (CIDMLS), Westmead Hospital, for diagnostic testing and laboratory investigation of Chikungunya virus with written and oral informed patient consent. Serum from healthy individuals was provided by the Australian Red Cross with written and oral informed patient consent, approved by Griffith University Human Research Ethics Committee (BDD/01/12/HREC). No new human samples were collected as part of this study. Serum samples were deidentified before being used in the research project.
Virus.
The CHIKV variant expressing the red fluorescent protein mCherry (CHIKV-mCherry) was constructed using a full-length infectious cDNA clone of the La Reunion CHIKV isolate LR2006-OPY1 as described previously (50).
Mice.
C57BL/6 wild-type (WT) mice were obtained from the Animal Resource Centre (Perth, Australia), and CSF1R-EGFP (MacGreen) mice, expressing enhanced green fluorescent protein (EGFP)-labeled colony-stimulating factor 1 receptor (CSF1R), were purchased from the University of Queensland. All mice were maintained in the animal facilities of Griffith University (Gold Coast, Australia). Twenty-five-day-old male and female mice, of equal distribution, were inoculated subcutaneously (s.c.) in the ventral side of the foot with 105 PFU of CHIKV-mCherry diluted in phosphate-buffered saline (PBS) to a volume of 20 μl. Mock-infected mice were inoculated with PBS alone. Treatment with bindarit (Nanjing Chemlin Chemical Industry Co., Nanjing, China), also known as 2-methyl-2-{[1-(phenylmethyl)-1H-indazol-3yl] methoxy} propanoic acid, was given to mice intraperitoneally twice daily at 100 mg/kg of body weight in 0.5% methyl cellulose (vehicle). Mice were monitored daily for diet and well-being. Mice were weighed, and CHIKV-induced footpad swelling was assessed by measuring the height (H) and width (W) of the perimetatarsal area of the hind foot using Kinchrome digital vernier calipers every 24 h as described previously (26). Disease score was expressed as the increase of the ankle joint measurement relative to the measurement on day 0 postinfection (0dpi), calculated as [H(xdpi) × W(xdpi)] − {[H(0dpi) × W(0dpi)]/[H(0dpi) × W(0dpi)]}.
Human serum samples.
Serum samples from 14 serologically positive CHIKV patients (age range, 30 to 68 years; 8 females, 6 males) were collected between 3 and 22 weeks after onset of symptoms and were provided by CIMMLS (Westmead Hospital, Sydney, Australia). Serum samples from 7 healthy individuals (age range, 18 to 65 years; 4 females, 3 males) were provided by the Australian Red Cross.
ELISA.
The concentrations of human RANKL (PeproTech, Rocky Hill, NJ, USA) and OPG (R&D Systems, Minneapolis, MN, USA) were determined using enzyme-linked immunosorbent assay (ELISA) development kits. The concentrations of mouse RANKL and OPG were determined using mouse ELISA development kits (R&D Systems, Minneapolis, MN, USA). To determine the concentrations of CCL2, CXCL1, CCL3, CCL4, CCL11, and CCL5, joint tissues of mice were homogenized in PBS and centrifuged, and supernatant was collected and used for multiplex bead array kits (Bio-Plex pro mouse cytokine 23-plex kits; Bio-Rad, Hercules, CA). The concentrations of cytokines relative to total protein concentration for each tissue lysate are reported and are expressed as pg/mg protein. All ELISAs were performed according to the manufacturer's instructions.
Histology.
Mice were sacrificed, and the hind limbs were collected, fixed in 4% paraformaldehyde (PFA), decalcified in 14% EDTA, and embedded in paraffin. Five-micrometer sections were dewaxed, rehydrated, and stained with hematoxylin and eosin (H&E), tartrate-resistant acid phosphatase (TRAP), or Masson's trichrome. Images were taken using a Nikon Eclipse TS100 inverted microscope. OCs were identified as TRAP-positive (TRAP+) multinucleated cells (more than 3 nuclei) and were counted in 8 different tibial sections per mouse at the distal epiphysis, distal primary spongiosa, and proximal epiphysis (tibial-talar joint) using ImageJ software. The nomenclature and units used in the analysis are as recommended by the Nomenclature Committee of the American Society for Bone and Mineral Research (51).
μCT.
Microcomputed tomography (μCT) analyses were performed on murine hind limbs with the Skyscan 1076 μCT system (Skyscan, Aartselaar, Belgium). Bone volume fraction (bone volume per total volume [BV/TV]), trabecular thickness (Tb.Th), trabecular separation (Tb.Sp), and trabecular number (Tb.N) were determined using CTAN (Skyscan). The settings for data acquisition were as follows: 9-μm voxel resolution, 0.5-mm aluminum filter, 48-kV voltage, 100-μA current, 2,600-ms exposure, rotation of 0.5° across 180°, and frame averaging of 1. Reconstruction was performed using NRecon (version 1.6.3.1) with the following parameters: compressed sensing to image conversion (dynamic range), 0.0 to 0.09; ring artifact, 6; pixel defect mask, 5%; and beam hardening correction, 35%. Analysis was performed using the CT Analyser (CTAn, 1.12.0.0) on coronal data sets for epiphyseal bone generated using Dataviewer (version 1.4.4). Regions of interest (ROI) were identified as follows: epiphyseal bone, including cortical and trabecular bone above the growth plate, was outlined across 1-mm ROI (anterior to posterior region of the joint). Greyscale thresholds for quantitation of structural parameters were determined for the experimental groups using the automatic otsu algorithm within CTAn. The algorithms used for structural analysis were as follows: epiphyseal trabecular bone adaptive (mean and maximum) with prethreshold of 44 to 255, radius of 7, and constant of 1. Both 2-dimensional (2-D) and 3-dimensional (3-D) data were generated for all analyses. 3-D models were generated from raw (unthresholded) data using CTVox (version 2.4.0) and pseudocolored according to greyscale intensity, which reflects tissue mineralization.
Plaque assay.
Vero cells were seeded into 24-well plates at a density of 1 × 105 cells per well and cultured to confluence. Serially diluted samples (10−1 to 10−6) were added to cell monolayers and incubated for 1 h at 37°C in a 5% CO2 incubator. The cells were then overlaid with Opti-MEM (Life Technologies, Victoria, Australia) containing 3% heat-inactivated fetal calf serum (HI-FCS) and 1% agarose (Sigma-Aldrich, Sydney, Australia) and incubated at 37°C for 48 h in a 5% CO2 incubator. The cells were fixed with 1% formalin, and plaques were visualized by staining with 0.1% crystal violet. Viral titers were expressed as PFU per milliliter per gram of tissue.
Total RNA extraction and quantitative real-time PCR (qRT-PCR).
RNA was prepared from cell pellets using TRIzol (Life Technologies, Victoria, Australia) according to the manufacturer's instructions. Eluted RNA was stored at −80°C. Quantification of total RNA was performed with a NanoDrop 1000 spectrophotometer (Thermo Scientific, Victoria, Australia). Extracted total RNA (20 ng/μl) was reverse transcribed using an oligo(dT) primer and reverse transcriptase (Sigma-Aldrich, Sydney, Australia) according to the manufacturer's instructions. SYBR green real-time PCR was performed using 10 ng of template cDNA on a CFX96 Touch real-time PCR system in 96-well plates, using QuantiTect primer assay kits (Qiagen, Hilden, Germany) with the following conditions: (i) PCR initial activation step of 95°C for 15 min, 1 cycle, and (ii) 3-step cycling of 94°C for 15 s, followed by 55°C for 30 s and 72°C for 30 s, 40 cycles. Amplification specificity was evaluated by a melting curve analysis of PCR products. The fold change in mRNA expression relative to the expression in mock-infected samples for each gene was calculated with the cycle threshold (ΔΔCT) method, with normalization to the level of the housekeeping gene HPRT, encoding hypoxanthine phosphoribosyltransferase. Briefly, ΔΔCT = ΔCT (CHIKV-infected) – ΔCT (mock-infected), where ΔCT = CT(gene of interest) – CT(HPRT). The fold change for each gene was calculated as 2−ΔΔCT.
Immunofluorescence.
Mice were sacrificed and perfused with PBS. The femur, tibia, patella, and foot from the hind limb were collected, fixed in 4% PFA, decalcified in 14% EDTA, embedded in optimal cutting temperature (OCT) compound, and cryosectioned. Five-micrometer sections were prepared, followed by 4′,6-diamidino-2-phenylindole (DAPI) staining, and were visualized on an FV1000 confocal microscope (Olympus, Victoria, Australia). For quantification of CSF1R-positive (CSF1R+) cells, the total numbers of CSF1R+ cells were quantified and expressed over the total nucleus count (n = 100 to 200 cells) as percentages.
Statistical analysis.
ELISA data for patients' serum samples, ELISA/multiplex protein analysis of CHIKV-infected mouse knee joint homogenate, bone morphometric indices, viral titer analyses of mouse specimens, and TRAP+ cell and CSF1R+ cell numberss were statistically analyzed by Mann-Whitney U test. For comparison between vehicle- and bindarit-treated groups, the disease score was analyzed by two-way ANOVA with Bonferroni posttest. TRAP+ cell numbers, bone morphometric indices, and the results of qRT-PCR of mouse specimens were analyzed by one-way ANOVA with Tukey's posttest. Longitudinal qRT-PCR analyses of mouse specimens were performed using one-way ANOVA with Dunnett's posttest. All data were assessed for Gaussian distribution using the D'Agostino-Pearson normality test before analysis with these parametric tests. Statistical analyses were performed with GraphPad Prism 5.02.
RESULTS
Disruption of serum RANKL/OPG ratio in CHIKV patients.
The serum RANKL levels were higher in CHIKV patients than in healthy control subjects (Fig. 1A). There was no significant difference in the serum OPG levels of patients and controls. The resulting serum RANKL/OPG ratio was significantly higher in CHIKV patients than in healthy control subjects, suggesting that pro-osteoclastic conditions exist in response to CHIKV infection (Fig. 1B).
FIG 1.
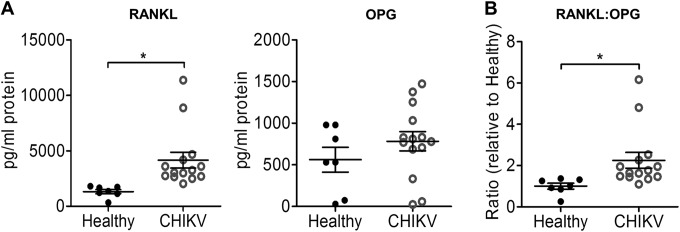
CHIKV infection perturbs bone homeostasis through an upregulated RANKL/OPG ratio in humans. Serum samples from CHIKV-infected patients (n = 14) or healthy controls (n = 7) were analyzed by ELISA for RANKL and OPG protein levels (A), and the RANKL/OPG ratios are shown (B). Data are presented as means ± standard errors of the means (SEM). *, P < 0.05; Mann-Whitney U test.
Bone homeostasis is disrupted at peak disease in an acute CHIKV mouse model.
To determine whether CHIKV infection can affect bone homeostasis, we next examined the RANKL/OPG ratio in a mouse model of CHIKV described previously (52). Twenty-five-day-old C57BL/6 mice were infected s.c. in the ventral side of the foot with 105 PFU of CHIKV-mCherry. At day 3 postinfection (p.i.), peak swelling was observed in the ankle joint of CHIKV-infected mice, and the swelling was fully resolved by day 10 p.i. (Fig. 2A). The RANKL transcript levels in the joint were significantly higher at days 1, 3, and 15 p.i. but not on day 7 p.i. compared to the RANKL transcript levels in PBS-injected control mice (Fig. 2B). At days 3 and 15 p.i. but not on days 1 and 7 p.i., the OPG transcript levels were significantly elevated compared to the OPG transcript levels in PBS-injected control mice (Fig. 2C). The RANKL/OPG ratio was significantly elevated at day 1 p.i. (Fig. 2D), suggesting that CHIKV infection leads to a pro-osteoclastic microenvironment within the infected tissues at early acute infection.
FIG 2.

CHIKV infection rapidly and transiently increases the RANKL/OPG ratio. Twenty-five-day-old C57BL/6 WT mice (n = 4 per group) were subcutaneously injected with 105 PFU of CHIKV or PBS in the ventral side of the foot. (A) Disease score quantified by measurement of joint swelling in mock- and CHIKV-infected mice up to day 10 p.i. (B to D) Transcriptional profiles of RANKL (B), OPG (C), and RANKL/OPG ratio (D) in ankle joints of C57BL/6 WT mock- and CHIKV-infected mice (n = 4 per group) at days 1, 3, 7, and 15 p.i., determined using qRT-PCR. Data are normalized to the results for HPRT and shown as fold expression relative to the expression levels in the mock-infected group. Data are presented as means ± SEM. *, P < 0.05; one-way ANOVA, Dunnett's posttest, compared to PBS-injected controls.
CHIKV replicates in murine bone and induces bone loss.
To investigate whether CHIKV can replicate in bone in vivo, CHIKV-infected WT mice were sacrificed and the femur, tibia, patella, and foot collected for virus titer measurement using plaque assays. At peak swelling (day 3 p.i.), high viral titers were detected in each of these sites (Fig. 3A). Similar to the serum results in CHIKV patients, the RANKL/OPG ratios were elevated in knee joints of CHIKV-infected mice at day 3 p.i. (Fig. 3B). Consistent with the increased RANKL/OPG ratio, there were increased numbers of TRAP+ cells in CHIKV-infected mice compared to the numbers in mock-infected mice in tibial trabecular bone (Fig. 4A), and this was associated with low trabecular bone mass in the secondary spongiosa of tibias in CHIKV-infected mice as evidenced by histology, suggesting that CHIKV-induced bone loss may be mediated through enhanced osteoclastogenesis (Fig. 4A). In addition, CHIKV-infected mice showed reduced growth plate width, indicating growth impairment due to infection (Fig. 4A). μCT analysis of the trabecular and cortical bone within the tibial epiphysis revealed significant bone loss in CHIKV-infected mice compared to the results for PBS-injected control mice (Fig. 4B). In CHIKV-infected mice, the bone volume fraction (BV/TV) was reduced by approximately 25%, associated with reductions in trabecular thickness (Tb. Th) and trabecular bone numbers (Tb. N) and increased trabecular separation (Tb. Sp) (Fig. 4C). The elevated RANKL/OPG ratio, increased number of TRAP+ cells, and reduced bone volume were consistent with the observed bone loss following CHIKV infection.
FIG 3.
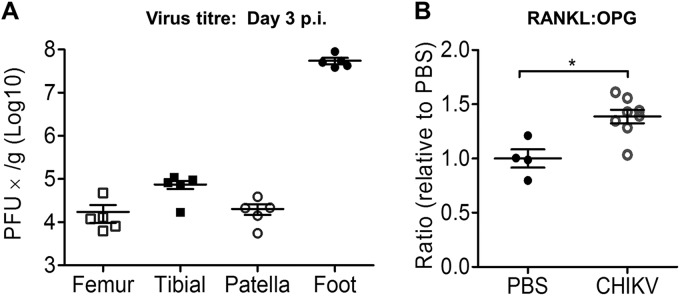
CHIKV replicates in bones of murine model. Twenty-five-day-old C57BL/6 WT mice (n = 5 per group) were subcutaneously injected with 105 PFU of CHIKV-mCherry or PBS in the ventral side of the foot. (A) At day 3 p.i., the femur, tibia, patella, and foot were collected and homogenized, and the presence of infectious virus determined by plaque assays. (B) RANKL and OPG protein levels in the knee joints were analyzed by ELISA, and the RANKL/OPG ratios are shown. Data are presented as means ± SEM. *, P < 0.05; Mann-Whitney U test.
FIG 4.

CHIKV infection induces bone pathology in the mouse model. (A) Paraffin sections (5 μm) from proximal tibia of mock- and CHIKV-infected WT mice were stained with Masson's trichrome and TRAP stain at day 3 after infection (magnification, ×20). Representative images for 4 to 5 mice per group are shown. The numbers of TRAP+ multinucleated (>3 nuclei) osteoclasts per bone perimeter (N.Oc/B.Pm) were quantified. Data are presented as means ± SEM. *, P < 0.05; Mann-Whitney U test. (B) Volume-rendered 3-D images of raw (unthresholded) data pseudocolored according to gray scale intensity, which is indicative of tissue mineralization of the hind limbs of C57BL/6 WT PBS-injected and CHIKV-infected mice harvested at day 3 p.i. Color-coded 3-D model is indicative of tissue mineralization; low mineralization is represented in red, which changes to yellow and blue when mineralization increases. Representative images for 4 to 5 mice per group are shown. (C) Results of quantitation of bone volume fraction (BV/TV), trabecular number (Tb. N), trabecular spacing (Tb. Sp), and trabecular thickness (Tb. Th) in the proximal tibial epiphysis of PBS-injected and CHIKV-infected mice (n = 4 or 5 per group) at day 3 p.i. are shown. Data are presented as means ± SEM. *, P < 0.05; Mann-Whitney U test.
Specific upregulation of MCPs in response to CHIKV infection.
Recruitment of inflammatory cellular infiltrates to the joint has been associated with bone loss in inflammatory arthritis, including RA (8). To investigate the involvement of infiltrates of the monocyte/macrophage lineage during CHIKV-induced bone loss, C57BL/6 CSF1R-EGFP (MacGreen) mice were inoculated with CHIKV-mCherry. Increased numbers of CSF1R+ cells were observed within the bone marrow in the ankle joints of CHIKV-infected mice at day 3 p.i. (Fig. 5A and B). To further identify the immune mediators responsible for CHIKV-elicited cellular infiltration, the protein expression profiles of several chemokines (CCL2, CXCL1, CCL3, CCL4, CCL11, and CCL5) were investigated at day 3 p.i. by multiplex cytokine assay. CCL2 was significantly elevated in CHIKV-infected knee joints, while no significant change was observed for the other chemokines (Fig. 5C). In a separate experiment, the mRNA levels of CCL2 (Fig. 6A), CCL8 (Fig. 6B), and CCL7 (Fig. 6C) were found to be significantly increased at peak disease in ankle joints of CHIKV-infected mice compared to their levels in PBS-injected control mice and to have resolved by day 15 p.i. along with the resolution of joint swelling. Hence, our results identify the MCPs (CCL2, CCL7, and CCL8) as potential candidates that may mediate the recruitment of CSF1R+ cells and contribute to the increased osteoclastogenesis during the early phase of CHIKV infection.
FIG 5.

CCL2 is involved in cellular infiltration during CHIKV infection. Twenty-five-day-old C57BL/6 CSF1R-EGFP (MacGreen) mice (n = 5 per group) were subcutaneously injected with 105 PFU of CHIKV-mCherry or PBS (mock) in the ventral side of the foot. (A) At day 3 p.i., hind limbs of mock- and CHIKV-infected mice were harvested, fixed, decalcified, and either paraffin sectioned (5 μm) or cryosectioned (5 μm) for H&E and immunofluorescence staining, respectively. CSF1R (green), replicating CHIKV-mCherry (red), and nuclei (blue) were visualized by confocal microscopy. Magnification, ×20 (H&E) and ×60 (immunofluorescence). Scale bar, 30 μm. Images are representative of 2 independent experiments. (B) The percentages of CSF1R+ cells in 100 to 200 cells per group were quantified. Data are presented as means ± SEM. *, P < 0.05; Mann-Whitney U test. (C) Protein concentrations of CCL2 (MCP-1), CXCL1 (KC), CCL3 (MIP-1α), CCL4 (MIP-1β), CCL11 (eotaxin-1), and CCL5 (RANTES) were determined by using a multiplex cytokine assay. Data are presented as means ± SEM. *, P < 0.05; Mann-Whitney U test.
FIG 6.

CCL2, CCL7, and CCL8 mRNA transcripts are highly expressed at CHIKV peak disease. Longitudinal transcriptional profiles of CCL2 (MCP-1) (A), CCL8 (MCP-2) (B), and (C) CCL7 (MCP-3) (C) in ankle joints of C57BL/6 WT PBS-injected and CHIKV-infected mice (n = 4 per group) at days 1, 3, 7, and 15 p.i. were determined by qRT-PCR. Data are normalized to the results for HPRT and shown as fold expression relative to the expression in the PBS-injected group. Data are means ± SEM. *, P < 0.05; one-way ANOVA, Dunnett's posttest, compared to mock-infected group.
Inhibition of MCP production with bindarit ameliorates disease and reduces virus replication in ankle joints.
To investigate the role of MCPs in CHIKV-induced bone loss, treatment with bindarit, a potent inhibitor of MCPs, was tested. At peak disease, inhibition of CCL2 transcript expression was confirmed in ankle joints of bindarit-treated CHIKV-infected mice by using qRT-PCR (Fig. 7A). Transcriptional inhibition was also observed in other members of the MCP family, including CCL8 (Fig. 7B) and CCL7 (Fig. 7C). Bindarit treatment of CHIKV-infected mice resulted in a significant amelioration of acute joint swelling at days 2 and 3 p.i. compared to the results for vehicle-treated CHIKV-infected mice (Fig. 7D), with a corresponding reduction of infectious virus recovered from the ankle joint (Fig. 7E). However, no difference in virus titers was observed in distant sites, such as the tibia and patella, suggesting that bindarit has no direct antiviral effect (Fig. 7E). Similarly, reduced numbers of CHIKV-infected cells and reduced CSF1R+ cell recruitment were observed in the ankle joint and tibial epiphysis of bindarit-treated CHIKV-infected mice compared to the results for vehicle-treated controls (Fig. 8A and B). In addition, bindarit treatment of CHIKV-infected mice suppressed the expression of the proinflammatory cytokine IL-6 (Fig. 9A). However, no significant reduction of IL-1β, tumor necrosis factor (TNF), or IL-12β/p40 expression was observed in bindarit-treated CHIKV-infected mice compared to their expression in vehicle-treated CHIKV-infected mice (Fig. 9B, C, and D).
FIG 7.

Inhibition of MCPs with bindarit suppresses CHIKV-induced chemotactic responses and reduces disease. Twenty-five-day-old C57BL/6 CSF1R-EGFP (MacGreen) mice (n = 4 or 5 per group) were subcutaneously injected with 105 PFU of CHIKV-mCherry or PBS (mock infection) in the ventral side of the foot. Treatment with methyl cellulose (vehicle) or 100 mg/kg bindarit was given 3 h prior to infection and then twice daily through intraperitoneal injection. Mice were sacrificed at day 3 p.i., and ankle joints and hind limbs were harvested. Transcriptional profiles of CCL2 (MCP-1) (A), CCL8 (MCP-2) (B), and CCL7 (MCP-3) (C) in ankle joints of vehicle- or bindarit-treated C57BL/6 WT PBS-injected and CHIKV-infected mice (n = 4 or 5 per group) at day 3 p.i. were determined by qRT-PCR. Data are normalized to the results for HPRT and shown as fold expression relative to the expression in the PBS-injected vehicle-treated group. Data are means ± SEM. *, P < 0.05; one-way ANOVA, Tukey's posttest. (D) Quantification of disease score was performed through measurement of joint swelling in vehicle- or bindarit-treated, PBS-injected and CHIKV-infected mice up to peak disease (day 3 p.i.). (E) Tibia, patella, and foot of mice were collected and homogenized, and the presence of infectious virus determined by plaque assays. Data are presented as means ± SEM. *, P < 0.05; Mann-Whitney U test.
FIG 8.

Bindarit treatment reduced cellular infiltration during CHIKV infection. Twenty-five-day-old C57BL/6 CSF1R-EGFP (MacGreen) mice (n = 4 or 5 per group) were subcutaneously injected with 105 PFU of CHIKV-mCherry or PBS (mock) in the ventral side of the foot. Treatment with methyl cellulose (vehicle) or 100 mg/kg bindarit was given twice daily through intraperitoneal injection. Mice were sacrificed at day 3 p.i., and ankle joints and hind limbs were harvested. (A) Hind limbs of vehicle- or bindarit-treated, CHIKV-infected mice were fixed, decalcified, and either paraffin sectioned (5 μm) or cryosectioned (5 μm) for H&E and immunofluorescence staining, respectively. CSF1R (green), replicating CHIKV-mCherry (red), and nuclei (blue) were visualized by confocal microscopy. Magnification, ×20 (H&E) and ×60 (immunofluorescence). Scale bar, 30 μm. Images are representative of 2 independent experiments. (B) The percentages of CSF1R+ cells in 100 to 200 cells per group were quantified. Data are presented as means ± SEM. *, P < 0.05; Mann-Whitney U test.
FIG 9.

Bindarit treatment differentially modulates CHIKV-induced proinflammatory responses. Transcriptional profiles of IL-6 (A), IL-1β (B), TNF (C), and IL-12β/p40 (D) in ankle joints of vehicle- or bindarit-treated C57BL/6 WT PBS-injected and CHIKV-infected mice (n = 4 or 5 per group) at day 3 p.i. were determined by qRT-PCR. Data are normalized to the results for HPRT and shown as fold expression relative to the expression in the PBS-injected, vehicle-treated group. Data are presented as means ± SEM. *, P < 0.05; one-way ANOVA, Tukey's posttest.
Bindarit prevents CHIKV-induced bone loss by protecting the RANKL/OPG ratio.
Having shown that bindarit treatment reduces both CHIKV disease and virus titers, we further investigated the impact of bindarit on CHIKV-induced bone loss. An increased RANKL/OPG ratio was evident in vehicle-treated CHIKV-infected knee joints, whereas bindarit treatment of CHIKV-infected mice prevented this increase (Fig. 10A), suggesting that bindarit-treated CHIKV-infected mice have reduced capacity to support osteoclastogenesis. A decrease in TRAP+ multinucleated OCs in the tibial epiphysis, trabecular bone, and tibial-talar joint was observed in CHIKV-infected mice treated with bindarit, confirming reduced osteoclastogenesis (Fig. 10B and C).
FIG 10.

Bindarit treatment suppresses CHIKV-induced disruption of RANKL/OPG ratio and osteoclastogenesis. (A) Transcriptional profiles of RANKL/OPG ratio in knee joint of vehicle- or bindarit-treated C57BL/6 WT PBS-injected and CHIKV-infected mice were determined using qRT-PCR. Data are normalized to the results for HPRT and shown as fold expression relative to the expression in the PBS-injected vehicle-treated group. Data are presented as means ± SEM. *, P < 0.05; one-way ANOVA, Tukey's posttest. (B) Paraffin sections (5 μm) from proximal tibia of bindarit-treated, PBS-injected or CHIKV-infected WT mice were stained with TRAP stain at day 3 after infection. Representative images for 4 to 5 mice per group are shown. Magnification, ×20. Scale bar, 30 μm. (C) The numbers of TRAP+ multinucleated (>3 nuclei) osteoclasts per bone perimeter (N.Oc/B.Pm) were quantified. Data are presented as means ± SEM. *, P < 0.05; one-way ANOVA, Tukey's posttest.
To characterize the bone architecture of bindarit-treated CHIKV-infected mice, μCT analysis was performed. Bone loss in the tibial epiphysis was evident in the vehicle-treated CHIKV-infected mice, as indicated by lower BV/TV, Tb. N, and Tb. Th and greater Tb. Sp values compared to the results for noninfected mice. Treatment with bindarit prevented this CHIKV-induced bone loss in the tibial epiphysis, preserving BV/TV, Tb. N, Tb. Sp, and Tb. Th (Fig. 11A and B). This suggests that MCPs may be a major mediator of virus-induced bone loss during CHIKV infection.
FIG 11.

Bindarit treatment ameliorates CHIKV-induced bone pathology. (A) Volume-rendered 3-D images of raw (unthresholded) data pseudocolored according to gray scale intensity (indicative of tissue mineralization) of the hind limbs of vehicle- or bindarit-treated C57BL/6 WT PBS-injected and CHIKV-infected mice harvested at day 3 p.i. Color-coded 3-D model indicates levels of tissue mineralization; low mineralization is represented in red, which changes to yellow and blue when mineralization increases, as indicated by the key at the bottom of the panel. Representative images from a total of 4 to 5 mice per group are shown. (B) Trabecular bone volume fraction (BV/TV), trabecular number (Tb. N), trabecular spacing (Tb. Sp), and trabecular thickness (Tb. Th) in the proximal tibial epiphysis of mock- and CHIKV-infected mice (n = 4 or 5 per group) at day 3 p.i. are shown. Data are presented as means ± SEM. *, P < 0.05; one-way ANOVA, Tukey's posttest.
DISCUSSION
Arthritogenic alphaviruses, such as CHIKV, cause arthritis in humans, but the underlying mechanism responsible for joint swelling and subsequent arthralgia is still largely unknown. High levels of urinary hydroxyproline, which has been used as a marker for bone resorption in metabolic bone diseases (53), have been reported in CHIKV patients (54), suggesting elevated bone resorption. Several cases with bone pathology, including bone erosions, joint space narrowing, and periarticular osteopenia, have been reported for CHIKV patients negative for RA-associated factors (anti-cyclic citrullinated peptide antibody and rheumatoid factor) (6, 7, 55). Whether there is a link between RA and CHIKV arthritis is an area that requires further study, since there are other examples in the literature of CHIKV cases positive for rheumatoid factor that qualify under American College of Rheumatology (ACR) criteria for a diagnosis of RA. In addition, radiolucent bone lesions have been reported in the shoulder joint (head of humerus) and tibial intercondylar region in CHIKV patients, indicating that infection may lead to osteolysis at joint sites (7).
Several cytokines and local mediators, together with many cell types, including OBs, OCs, and inflammatory cells, are involved in the bone remodelling process (56). We have previously reported that CHIKV-infected primary human OBs express a higher RANKL/OPG ratio in response to infection (23), in addition to the potential for nonosteoblastic cells, such as NK cells (12), T cells (15), and synovial fibroblasts (14), to also stimulate osteoclastogenesis by RANKL production. CHIKV-infected primary human fibroblastlike synoviocytes were also reported to contribute to osteoclastogenesis by secreting osteotropic factors, such as IL-6 and CCL2 (57). Although our longitudinal gene expression study showed elevated RANKL and OPG transcript levels, the RANKL/OPG ratio was only significantly elevated at day 1 p.i., prior to peak disease. Hence, the dysregulation in the RANKL/OPG ratio in CHIKV-infected joints favors an osteoclastogenic microenvironment, resulting in differentiation of TRAP+ multinucleated OCs. These results are consistent with human serum data from CHIKV patients, where we observed an increase in the serum RANKL/OPG ratio, indicating a systemic pro-osteoclastogenic condition in response to CHIKV infection. We observed an approximate 2-fold increase in the RANKL/OPG ratio using serum specimens from CHIKV patients. This is similar in magnitude to the increases in RANKL/OPG ratios reported for patients with postmenopausal osteoporosis (1.8-fold) (58), osteoarthritis patients (∼1.5-fold) (59), and children with osteoporosis due to glucocorticoid therapy for idiopathic nephrotic syndrome (∼1.5-fold) (60).
In this study, we provide evidence of CHIKV-induced bone loss in the proximal tibial epiphysis due in part to increased OC-mediated bone resorption associated with an increased RANKL/OPG ratio and increased MCP expression. This is consistent with the periarticular bone loss occurring in several chronic inflammatory diseases, such as RA and systemic lupus erythematosus (SLE) (61, 62). We have recently reported periarticular and systemic bone loss in an established RRV murine model and demonstrated that RRV-induced bone loss is in part a result of an IL-6-dependent increase in the RANKL/OPG ratio and the subsequent increase in OC differentiation and bone resorption (24). In addition to IL-6, several proinflammatory factors, such as CCL2, are upregulated in response to alphaviral infection (26, 63, 64). During inflammation, CCL2 is thought to be an important chemoattractant for monocytic precursors (65).
We demonstrated recruitment of CSF1R+ monocytic cells to the inflamed ankle and knee joints during CHIKV disease in mice. Chemokines such as CCL2, CCL3, CCL4, CCL5, CCL11, and CXCL1 are involved in modulating chemotaxis of OBs and OCs during many arthritic conditions (30–34, 66) and are elevated in plasma specimens from CHIKV-infected patients (63). Here, we showed that CCL2, CCL8, and CCL7 are highly elevated within the inflamed joints of CHIKV-infected mice, suggesting that MCPs may contribute to alphavirus-induced bone loss. Hence, our data are in agreement with prior reports that MCPs are highly expressed during alphaviral infection (25, 41).
In the present study, inhibition of the expression of CCL2 and other members of the MCP family, including CCL8 and CCL7, by bindarit dampened the influx of CSF1R+ cells to the inflamed joints, reducing joint inflammation and virus replication in ankle joints. Several in vitro studies have identified active infection of human monocytes and macrophages during CHIKV infection, which may assist in virus dissemination (28, 67, 68). In addition, an in vivo study conducted in nonhuman primates demonstrated persistence of CHIKV within macrophages in lymphoid, liver, muscle, and joint tissues (28). Collectively, these studies suggest that macrophages may be a primary cellular target during in vivo CHIKV infection. Since cells of the monocyte/macrophage lineage are a cellular target of CHIKV, inhibition of their infiltration to inflamed ankle joints (also the direct site of infection) may in part account for the reduction in virus recovered after bindarit treatment. The lack of reduced viral replication in distant sites of infection, such as the patella and tibia, of CHIKV-infected mice suggests that bindarit does not play any direct antiviral role during alphaviral infection, consistent with a previous study conducted in a mouse model of RRV (41).
We have previously demonstrated that bindarit treatment ameliorates alphavirus-induced muscle damage and cellular infiltration in ankle joints, identifying MCPs as pivotal pathogenic factors in alphaviral inflammatory disease (41, 42). Here, we further characterize MCPs as key players in the underlying pathology of alphavirus-induced bone loss. Bindarit treatment during CHIKV infection suppressed the alphavirus-induced disruption of the RANKL/OPG ratio, inhibiting the osteoclastogenesis elicited by CHIKV. More importantly, treatment with bindarit also alleviated bone damage in the tibial epiphysis of CHIKV-infected mice. This finding is, at first glance, inconsistent with a recent report of disease exacerbation, including cartilage damage, during the chronic stage of disease in CCR2-deficient mice infected with CHIKV (69). However, it is worth noting that CCR2 is a promiscuous receptor that interacts not only with CCL2 but also with seven other ligands, including CCL7 and CCL12 (70), which may suggest that other ligands for CCR2 play an inhibitory role in CHIKV, particularly in terms of cartilage damage. Notably, while low-dose, monoclonal antibody treatment targeting CCR2 (MC-21) efficiently reduced murine collagen-induced arthritis symptoms, high doses exacerbated disease (71).
The efficacy of bindarit in inhibiting CCL2 expression has been demonstrated in vitro and in vivo (43, 47, 72, 73). In adjuvant-induced arthritis in rats, bindarit treatment led to a drastic reduction of radiological scores assigned based on joint effusion and bone erosion, reflective of a protective effect (43, 74, 75). The safety of bindarit has been demonstrated in an SLE clinical trial: bindarit treatment was safe and well-tolerated in patients, and there was no evidence of immunosuppressive effects on acquired immunity (76). Although bindarit has also been associated with inhibition of other immune mediators, including TNF (43) and IL-12 (48), CCL2 remains the best-characterized target of bindarit. A previous in vivo study conducted with bindarit demonstrated an inhibitory effect on TNF but no effect on IL-6 or IL-1 (43). In the present study, CHIKV-infected mice showed reduced expression of IL-6 but not of IL-1β, TNF, or IL-12β/p40 after treatment with bindarit. These observations likely represent a secondary effect of reduced cellular recruitment rather than a direct inhibitory effect of bindarit on these cytokines. The downstream suppressive effect on IL-6 may also contribute to protection from inflammatory bone loss.
In summary, CHIKV infection stimulates a rapid, transient increase in the RANKL/OPG ratio, leading to a chronic enhancement in osteoclastogenesis and pathological bone loss. A similar increase in the RANKL/OPG ratio in human patients after peak infection suggests a risk of systemic bone loss in patients with CHIKV infection, a topic requiring further investigation. We have also identified CCL2 as a major chemokine contributing to this increased osteoclastogenesis that acts by promoting recruitment of CSF1R+ monocyte-macrophage cells, which may serve as a reservoir of osteoclast precursors. The capacity of bindarit to prevent alphavirus-induced bone loss not only highlights the potential of this drug as a candidate for therapy of alphavirus-induced bone loss but also suggests it may be applicable to other infectious arthritides and destructive inflammatory arthritic conditions, such as RA.
ACKNOWLEDGMENTS
This work was supported by Australian National Health and Medical Research Council (NHMRC) grants (628011 and 1033068) to S.M. A.T. is the recipient of a NHMRC Peter Doherty Early Career Fellowship, L.J. is the recipient of an Australian Research Council Discovery Early Career Researcher Award, and S.M. is the recipient of an NHMRC Senior Research Fellowship.
We gratefully acknowledge Brett A. Tonkin (SVIMR, Australia) for his assistance with μCT scanning and Wendy Kelly and Brad Paterson for histotechnical assistance.
REFERENCES
- 1.Foo SS, Chen W, Herrero L, Bettadapura J, Narayan J, Dar L, Broor S, Mahalingam S. 2011. The genetics of alphaviruses. Future Virol 6:1407–1422. doi: 10.2217/fvl.11.123. [DOI] [Google Scholar]
- 2.Josseran L, Paquet C, Zehgnoun A, Caillere N, Le Tertre A, Solet JL, Ledrans M. 2006. Chikungunya disease outbreak, Reunion Island. Emerg Infect Dis 12:1994–1995. doi: 10.3201/eid1212.060710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Larrieu S, Pouderoux N, Pistone T, Filleul L, Receveur MC, Sissoko D, Ezzedine K, Malvy D. 2010. Factors associated with persistence of arthralgia among Chikungunya virus-infected travellers: report of 42 French cases. J Clin Virol 47:85–88. doi: 10.1016/j.jcv.2009.11.014. [DOI] [PubMed] [Google Scholar]
- 4.Borgherini G, Poubeau P, Staikowsky F, Lory M, Moullec NL, Becquart JP, Wengling C, Michault A, Paganin F. 2007. Outbreak of Chikungunya on Reunion Island: early clinical and laboratory features in 157 adult patients. Clin Infect Dis 44:1401–1407. doi: 10.1086/517537. [DOI] [PubMed] [Google Scholar]
- 5.Bouquillard E, Combe B. 2009. Rheumatoid arthritis after Chikungunya fever: a prospective follow-up study of 21 cases. Ann Rheum Dis 68:1505–1506. doi: 10.1136/ard.2008.097626. [DOI] [PubMed] [Google Scholar]
- 6.Malvy D, Ezzedine K, Mamani-Matsuda M, Autran B, Tolou H, Receveur MC, Pistone T, Rambert J, Moynet D, Mossalayi D. 2009. Destructive arthritis in a patient with chikungunya virus infection with persistent specific IgM antibodies. BMC Infect Dis 9:200. doi: 10.1186/1471-2334-9-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manimunda SP, Vijayachari P, Uppoor R, Sugunan AP, Singh SS, Rai SK, Sudeep AB, Muruganandam N, Chaitanya IK, Guruprasad DR. 2010. Clinical progression of chikungunya fever during acute and chronic arthritic stages and the changes in joint morphology as revealed by imaging. Trans R Soc Trop Med Hyg 104:392–399. doi: 10.1016/j.trstmh.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 8.Walsh NC, Gravallese EM. 2010. Bone remodeling in rheumatic disease: a question of balance. Immunol Rev 233:301–312. doi: 10.1111/j.0105-2896.2009.00857.x. [DOI] [PubMed] [Google Scholar]
- 9.Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, Armstrong A, Shen V, Bain S, Cosman D, Anderson D, Morrissey PJ, Peschon JJ, Schuh J. 1999. RANK is essential for osteoclast and lymph node development. Genes Dev 13:2412–2424. doi: 10.1101/gad.13.18.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li J, Sarosi I, Yan X-Q, Morony S, Capparelli C, Tan H-L, McCabe S, Elliott R, Scully S, Van G, Kaufman S, Juan S-C, Sun Y, Tarpley J, Martin L, Christensen K, McCabe J, Kostenuik P, Hsu H, Fletcher F, Dunstan CR, Lacey DL, Boyle WJ. 2000. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci U S A 97:1566–1571. doi: 10.1073/pnas.97.4.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walsh NC, Alexander KA, Manning CA, Karmakar S, Wang JF, Weyand CM, Pettit AR, Gravallese EM. 2013. Activated human T cells express alternative mRNA transcripts encoding a secreted form of RANKL. Genes Immun 14:336–345. doi: 10.1038/gene.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soderstrom K, Stein E, Colmenero P, Purath U, Muller-Ladner U, de Matos CT, Tarner IH, Robinson WH, Engleman EG. 2010. Natural killer cells trigger osteoclastogenesis and bone destruction in arthritis. Proc Natl Acad Sci U S A 107:13028–13033. doi: 10.1073/pnas.1000546107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyce BF, Xing L. 2008. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys 473:139–146. doi: 10.1016/j.abb.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim H-R, Kim K-W, Kim B-M, Jung H-G, Cho M-L, Lee S-H. 2014. Reciprocal activation of CD4+ T cells and synovial fibroblasts by stromal cell-derived factor 1 promotes RANKL expression and osteoclastogenesis in rheumatoid arthritis. Arthritis Rheum 66:538–548. doi: 10.1002/art.38286. [DOI] [PubMed] [Google Scholar]
- 15.Horwood NJ, Kartsogiannis V, Quinn JM, Romas E, Martin TJ, Gillespie MT. 1999. Activated T lymphocytes support osteoclast formation in vitro. Biochem Biophys Res Commun 265:144–150. doi: 10.1006/bbrc.1999.1623. [DOI] [PubMed] [Google Scholar]
- 16.Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S, Wong T, Campagnuolo G, Moran E, Bogoch ER, Van G, Nguyen LT, Ohashi PS, Lacey DL, Fish E, Boyle WJ, Penninger JM. 1999. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 402:304–309. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- 17.Fumoto T, Takeshita S, Ito M, Ikeda K. 2014. Physiological functions of osteoblast lineage and T cell-derived RANKL in bone homeostasis. J Bone Miner Res 29:830–842. doi: 10.1002/jbmr.2096. [DOI] [PubMed] [Google Scholar]
- 18.Theoleyre S, Wittrant Y, Tat SK, Fortun Y, Redini F, Heymann D. 2004. The molecular triad OPG/RANK/RANKL: involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev 15:457–475. doi: 10.1016/j.cytogfr.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 19.Ainola M, Mandelin J, Liljestrom M, Konttinen YT, Salo J. 2008. Imbalanced expression of RANKL and osteoprotegerin mRNA in pannus tissue of rheumatoid arthritis. Clin Exp Rheumatol 26:240–246. [PubMed] [Google Scholar]
- 20.van Tuyl LH, Voskuyl AE, Boers M, Geusens P, Landewe RB, Dijkmans BA, Lems WF. 2010. Baseline RANKL:OPG ratio and markers of bone and cartilage degradation predict annual radiological progression over 11 years in rheumatoid arthritis. Ann Rheum Dis 69:1623–1628. doi: 10.1136/ard.2009.121764. [DOI] [PubMed] [Google Scholar]
- 21.Crotti TN, Smith MD, Weedon H, Ahern MJ, Findlay DM, Kraan M, Tak PP, Haynes DR. 2002. Receptor activator NF-kappaB ligand (RANKL) expression in synovial tissue from patients with rheumatoid arthritis, spondyloarthropathy, osteoarthritis, and from normal patients: semiquantitative and quantitative analysis. Ann Rheum Dis 61:1047–1054. doi: 10.1136/ard.61.12.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pettit AR, Walsh NC, Manning C, Goldring SR, Gravallese EM. 2006. RANKL protein is expressed at the pannus-bone interface at sites of articular bone erosion in rheumatoid arthritis. Rheumatology (Oxford) 45:1068–1076. doi: 10.1093/rheumatology/kel045. [DOI] [PubMed] [Google Scholar]
- 23.Noret M, Herrero L, Rulli N, Rolph M, Smith PN, Li RW, Roques P, Gras G, Mahalingam S. 2012. Interleukin 6, RANKL, and osteoprotegerin expression by chikungunya virus-infected human osteoblasts. J Infect Dis 206:455–459. doi: 10.1093/infdis/jis368. [DOI] [PubMed] [Google Scholar]
- 24.Chen W, Foo SS, Rulli NE, Taylor A, Sheng K, Herrero LJ, Herring BL, Lidbury BA, Li RW, Walsh NC, Sims NA, Smith PN, Mahalingam S. 2014. Arthritogenic alphaviral infection perturbs osteoblast function and triggers pathologic bone loss. Proc Natl Acad Sci U S A 111:6040–6045. doi: 10.1073/pnas.1318859111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lidbury BA, Rulli NE, Suhrbier A, Smith PN, McColl SR, Cunningham AL, Tarkowski A, van Rooijen N, Fraser RJ, Mahalingam S. 2008. Macrophage-derived proinflammatory factors contribute to the development of arthritis and myositis after infection with an arthrogenic alphavirus. J Infect Dis 197:1585–1593. doi: 10.1086/587841. [DOI] [PubMed] [Google Scholar]
- 26.Gardner J, Anraku I, Le TT, Larcher T, Major L, Roques P, Schroder WA, Higgs S, Suhrbier A. 2010. Chikungunya virus arthritis in adult wild-type mice. J Virol 84:8021–8032. doi: 10.1128/JVI.02603-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morrison TE, Oko L, Montgomery SA, Whitmore AC, Lotstein AR, Gunn BM, Elmore SA, Heise MT. 2011. A mouse model of chikungunya virus-induced musculoskeletal inflammatory disease: evidence of arthritis, tenosynovitis, myositis, and persistence. Am J Pathol 178:32–40. doi: 10.1016/j.ajpath.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Labadie K, Larcher T, Joubert C, Mannioui A, Delache B, Brochard P, Guigand L, Dubreil L, Lebon P, Verrier B, de Lamballerie X, Suhrbier A, Cherel Y, Le Grand R, Roques P. 2010. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. J Clin Invest 120:894–906. doi: 10.1172/JCI40104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoarau JJ, Jaffar Bandjee MC, Krejbich Trotot P, Das T, Li-Pat-Yuen G, Dassa B, Denizot M, Guichard E, Ribera A, Henni T, Tallet F, Moiton MP, Gauzere BA, Bruniquet S, Jaffar Bandjee Z, Morbidelli P, Martigny G, Jolivet M, Gay F, Grandadam M, Tolou H, Vieillard V, Debre P, Autran B, Gasque P. 2010. Persistent chronic inflammation and infection by Chikungunya arthritogenic alphavirus in spite of a robust host immune response. J Immunol 184:5914–5927. doi: 10.4049/jimmunol.0900255. [DOI] [PubMed] [Google Scholar]
- 30.Onan D, Allan EH, Quinn JM, Gooi JH, Pompolo S, Sims NA, Gillespie MT, Martin TJ. 2009. The chemokine Cxcl1 is a novel target gene of parathyroid hormone (PTH)/PTH-related protein in committed osteoblasts. Endocrinology 150:2244–2253. doi: 10.1210/en.2008-1597. [DOI] [PubMed] [Google Scholar]
- 31.Abe M, Hiura K, Ozaki S, Kido S, Matsumoto T. 2009. Vicious cycle between myeloma cell binding to bone marrow stromal cells via VLA-4-VCAM-1 adhesion and macrophage inflammatory protein-1alpha and MIP-1beta production. J Bone Miner Metab 27:16–23. doi: 10.1007/s00774-008-0012-z. [DOI] [PubMed] [Google Scholar]
- 32.Choi SJ, Cruz JC, Craig F, Chung H, Devlin RD, Roodman GD, Alsina M. 2000. Macrophage inflammatory protein 1-alpha is a potential osteoclast stimulatory factor in multiple myeloma. Blood 96:671–675. [PubMed] [Google Scholar]
- 33.Hueber W, Tomooka BH, Zhao X, Kidd BA, Drijfhout JW, Fries JF, van Venrooij WJ, Metzger AL, Genovese MC, Robinson WH. 2007. Proteomic analysis of secreted proteins in early rheumatoid arthritis: anti-citrulline autoreactivity is associated with up regulation of proinflammatory cytokines. Ann Rheum Dis 66:712–719. doi: 10.1136/ard.2006.054924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Jager W, Hoppenreijs EP, Wulffraat NM, Wedderburn LR, Kuis W, Prakken BJ. 2007. Blood and synovial fluid cytokine signatures in patients with juvenile idiopathic arthritis: a cross-sectional study. Ann Rheum Dis 66:589–598. doi: 10.1136/ard.2006.061853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. 2007. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest 117:902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Damme J, Proost P, Lenaerts JP, Opdenakker G. 1992. Structural and functional identification of two human, tumor-derived monocyte chemotactic proteins (MCP-2 and MCP-3) belonging to the chemokine family. J Exp Med 176:59–65. doi: 10.1084/jem.176.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haringman JJ, Smeets TJ, Reinders-Blankert P, Tak PP. 2006. Chemokine and chemokine receptor expression in paired peripheral blood mononuclear cells and synovial tissue of patients with rheumatoid arthritis, osteoarthritis, and reactive arthritis. Ann Rheum Dis 65:294–300. doi: 10.1136/ard.2005.037176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyamoto K, Ninomiya K, Sonoda KH, Miyauchi Y, Hoshi H, Iwasaki R, Miyamoto H, Yoshida S, Sato Y, Morioka H, Chiba K, Egashira K, Suda T, Toyama Y, Miyamoto T. 2009. MCP-1 expressed by osteoclasts stimulates osteoclastogenesis in an autocrine/paracrine manner. Biochem Biophys Res Commun 383:373–377. doi: 10.1016/j.bbrc.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 39.Kim MS, Day CJ, Morrison NA. 2005. MCP-1 is induced by receptor activator of nuclear factor-{kappa}B ligand, promotes human osteoclast fusion, and rescues granulocyte macrophage colony-stimulating factor suppression of osteoclast formation. J Biol Chem 280:16163–16169. doi: 10.1074/jbc.M412713200. [DOI] [PubMed] [Google Scholar]
- 40.Ikic M, Jajic Z, Lazic E, Ivcevic S, Grubisic F, Marusic A, Kovacic N, Grcevic D. 2014. Association of systemic and intra-articular osteoclastogenic potential, pro-inflammatory mediators and disease activity with the form of inflammatory arthritis. Int Orthop 38:183–192. doi: 10.1007/s00264-013-2121-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rulli NE, Guglielmotti A, Mangano G, Rolph MS, Apicella C, Zaid A, Suhrbier A, Mahalingam S. 2009. Amelioration of alphavirus-induced arthritis and myositis in a mouse model by treatment with bindarit, an inhibitor of monocyte chemotactic proteins. Arthritis Rheum 60:2513–2523. doi: 10.1002/art.24682. [DOI] [PubMed] [Google Scholar]
- 42.Rulli NE, Rolph MS, Srikiatkhachorn A, Anantapreecha S, Guglielmotti A, Mahalingam S. 2011. Protection from arthritis and myositis in a mouse model of acute chikungunya virus disease by bindarit, an inhibitor of monocyte chemotactic protein-1 synthesis. J Infect Dis 204:1026–1030. doi: 10.1093/infdis/jir470. [DOI] [PubMed] [Google Scholar]
- 43.Guglielmotti A, D'Onofrio E, Coletta I, Aquilini L, Milanese C, Pinza M. 2002. Amelioration of rat adjuvant arthritis by therapeutic treatment with bindarit, an inhibitor of MCP-1 and TNF-alpha production. Inflamm Res 51:252–258. doi: 10.1007/PL00000301. [DOI] [PubMed] [Google Scholar]
- 44.Steiner JL, Davis JM, McClellan JL, Guglielmotti A, Murphy EA. 2014. Effects of the MCP-1 synthesis inhibitor bindarit on tumorigenesis and inflammatory markers in the C3(1)/SV40Tag mouse model of breast cancer. Cytokine 66:60–68. doi: 10.1016/j.cyto.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ge S, Shrestha B, Paul D, Keating C, Cone R, Guglielmotti A, Pachter JS. 2012. The CCL2 synthesis inhibitor bindarit targets cells of the neurovascular unit, and suppresses experimental autoimmune encephalomyelitis. J Neuroinflammation 9:171. doi: 10.1186/1742-2094-9-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou GX, Zhu XJ, Ding XL, Zhang H, Chen JP, Qiang H, Zhang HF, Wei Q. 2010. Protective effects of MCP-1 inhibitor on a rat model of severe acute pancreatitis. Hepatobiliary Pancreat Dis Int 9:201–207. [PubMed] [Google Scholar]
- 47.Zoja C, Corna D, Benedetti G, Morigi M, Donadelli R, Guglielmotti A, Pinza M, Bertani T, Remuzzi G. 1998. Bindarit retards renal disease and prolongs survival in murine lupus autoimmune disease. Kidney Int 53:726–734. doi: 10.1046/j.1523-1755.1998.00804.x. [DOI] [PubMed] [Google Scholar]
- 48.Mora E, Guglielmotti A, Biondi G, Sassone-Corsi P. 2012. Bindarit: an anti-inflammatory small molecule that modulates the NFkappaB pathway. Cell Cycle 11:159–169. doi: 10.4161/cc.11.1.18559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.National Health and Medical Research Council. 2013. Australian code for the care and use of animals for scientific purposes, 8th ed. National Health and Medical Research Council, Canberra, Australia: https://www.nhmrc.gov.au/_files_nhmrc/publications/attachments/ea28_code_care_use_animals_131209.pdf. [Google Scholar]
- 50.Tsetsarkin K, Higgs S, McGee CE, De Lamballerie X, Charrel RN, Vanlandingham DL. 2006. Infectious clones of Chikungunya virus (La Reunion isolate) for vector competence studies. Vector Borne Zoonotic Dis 6:325–337. doi: 10.1089/vbz.2006.6.325. [DOI] [PubMed] [Google Scholar]
- 51.Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, Parfitt AM. 2013. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 28:2–17. doi: 10.1002/jbmr.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herrero LJ, Sheng K-C, Jian P, Taylor A, Her Z, Herring BL, Chow A, Leo Y-S, Hickey MJ, Morand EF, Ng LFP, Bucala R, Mahalingam S. 2013. Macrophage migration inhibitory factor receptor CD74 mediates alphavirus-induced arthritis and myositis in murine models of alphavirus infection. Arthritis Rheum 65:2724–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kraenzlin M, Taylor A, Baylink D. 1989. Biochemical markers for bone formation and bone resorption, p 289 In Lindh E, Thorell JI (ed), Clinical impact of bone and connective tissue markers. Pharmacia Diagnostics Clinical Symposia I. Academic Press, Ltd., London, United Kingdom. [Google Scholar]
- 54.Lokireddy S, Vemula S, Vadde R. 2008. Connective tissue metabolism in chikungunya patients. Virol J 5:31. doi: 10.1186/1743-422X-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bouquillard E, Combe B. 2009. A report of 21 cases of rheumatoid arthritis following chikungunya fever. A mean follow-up of two years. Joint Bone Spine 76:654–657. doi: 10.1016/j.jbspin.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 56.Redlich K, Smolen JS. 2012. Inflammatory bone loss: pathogenesis and therapeutic intervention. Nat Rev Drug Discov 11:234–250. doi: 10.1038/nrd3669. [DOI] [PubMed] [Google Scholar]
- 57.Phuklia W, Kasisith J, Modhiran N, Rodpai E, Thannagith M, Thongsakulprasert T, Smith DR, Ubol S. 2013. Osteoclastogenesis induced by CHIKV-infected fibroblast-like synoviocytes: a possible interplay between synoviocytes and monocytes/macrophages in CHIKV-induced arthralgia/arthritis. Virus Res 177:179–188. doi: 10.1016/j.virusres.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 58.Xu X-J, Shen L, Yang Y-P, Zhu R, Shuai B, Li C-G, Wu M-X. 2013. Serum β-catenin levels associated with the ratio of RANKL/OPG in patients with postmenopausal osteoporosis. Int J Endocrinol 2013:534352. doi: 10.1155/2013/534352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pilichou A, Papassotiriou I, Michalakakou K, Fessatou S, Fandridis E, Papachristou G, Terpos E. 2008. High levels of synovial fluid osteoprotegerin (OPG) and increased serum ratio of receptor activator of nuclear factor-κB ligand (RANKL) to OPG correlate with disease severity in patients with primary knee osteoarthritis. Clin Biochem 41:746–749. doi: 10.1016/j.clinbiochem.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 60.Wasilewska A, Rybi-Szuminska A, Zoch-Zwierz W. 2010. Serum RANKL, osteoprotegerin (OPG), and RANKL/OPG ratio in nephrotic children. Pediatr Nephrol 25:2067–2075. doi: 10.1007/s00467-010-1583-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saidenberg-Kermanac'h N, Corrado A, Lemeiter D, deVernejoul MC, Boissier MC, Cohen-Solal ME. 2004. TNF-α antibodies and osteoprotegerin decrease systemic bone loss associated with inflammation through distinct mechanisms in collagen-induced arthritis. Bone 35:1200–1207. doi: 10.1016/j.bone.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 62.Tanaka K, Hashizume M, Mihara M, Yoshida H, Suzuki M, Matsumoto Y. 2014. Anti-interleukin-6 receptor antibody prevents systemic bone mass loss via reducing the number of osteoclast precursors in bone marrow in a collagen-induced arthritis model. Clin Exp Immunol 175:172–180. doi: 10.1111/cei.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chow A, Her Z, Ong EKS, Chen J-M, Dimatatac F, Kwek DJC, Barkham T, Yang H, Rénia L, Leo Y-S, Ng LFP. 2011. Persistent arthralgia induced by chikungunya virus infection is associated with interleukin-6 and granulocyte macrophage colony-stimulating factor. J Infect Dis 203:149–157. doi: 10.1093/infdis/jiq042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kelvin AA, Banner D, Silvi G, Moro ML, Spataro N, Gaibani P, Cavrini F, Pierro A, Rossini G, Cameron MJ, Bermejo-Martin JF, Paquette SG, Xu L, Danesh A, Farooqui A, Borghetto I, Kelvin DJ, Sambri V, Rubino S. 2011. Inflammatory cytokine expression is associated with chikungunya virus resolution and symptom severity. PLoS Negl Trop Dis 5:e1279. doi: 10.1371/journal.pntd.0001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pavkova Goldbergova M, Lipkova J, Pavek N, Gatterova J, Vasku A, Soucek M, Nemec P. 2012. RANTES, MCP-1 chemokines and factors describing rheumatoid arthritis. Mol Immunol 52:273–278. doi: 10.1016/j.molimm.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 66.Wintges K, Beil FT, Albers J, Jeschke A, Schweizer M, Claass B, Tiegs G, Amling M, Schinke T. 2013. Impaired bone formation and increased osteoclastogenesis in mice lacking chemokine (C-C motif) ligand 5 (Ccl5). J Bone Miner Res 28:2070–2080. doi: 10.1002/jbmr.1937. [DOI] [PubMed] [Google Scholar]
- 67.Her Z, Malleret B, Chan M, Ong EK, Wong SC, Kwek DJ, Tolou H, Lin RT, Tambyah PA, Renia L, Ng LF. 2010. Active infection of human blood monocytes by Chikungunya virus triggers an innate immune response. J Immunol 184:5903–5913. doi: 10.4049/jimmunol.0904181. [DOI] [PubMed] [Google Scholar]
- 68.Sourisseau M, Schilte C, Casartelli N, Trouillet C, Guivel-Benhassine F, Rudnicka D, Sol-Foulon N, Le Roux K, Prevost MC, Fsihi H, Frenkiel MP, Blanchet F, Afonso PV, Ceccaldi PE, Ozden S, Gessain A, Schuffenecker I, Verhasselt B, Zamborlini A, Saib A, Rey FA, Arenzana-Seisdedos F, Despres P, Michault A, Albert ML, Schwartz O. 2007. Characterization of reemerging chikungunya virus. PLoS Pathog 3:e89. doi: 10.1371/journal.ppat.0030089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Poo YS, Nakaya H, Gardner J, Larcher T, Schroder WA, Le TT, Major LD, Suhrbier A. 2014. CCR2 deficiency promotes exacerbated chronic erosive neutrophil-dominated chikungunya virus arthritis. J Virol 88:6862–6872. doi: 10.1128/JVI.03364-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Berchiche YA, Gravel S, Pelletier M-E, St-Onge G, Heveker N. 2011. Different effects of the different natural CC chemokine receptor 2b ligands on β-arrestin recruitment, Gαi signaling, and receptor internalization. Mol Pharmacol 79:488–498. doi: 10.1124/mol.110.068486. [DOI] [PubMed] [Google Scholar]
- 71.Brühl H, Cihak J, Plachý J, Kunz-Schughart L, Niedermeier M, Denzel A, Rodriguez Gomez M, Talke Y, Luckow B, Stangassinger M, Mack M. 2007. Targeting of Gr-1+,CCR2+ monocytes in collagen-induced arthritis. Arthritis Rheum 56:2975–2985. doi: 10.1002/art.22854. [DOI] [PubMed] [Google Scholar]
- 72.Bhatia M, Landolfi C, Basta F, Bovi G, Ramnath RD, de Joannon AC, Guglielmotti A. 2008. Treatment with bindarit, an inhibitor of MCP-1 synthesis, protects mice against trinitrobenzene sulfonic acid-induced colitis. Inflamm Res 57:464–471. doi: 10.1007/s00011-008-7210-y. [DOI] [PubMed] [Google Scholar]
- 73.Bhatia M, Ramnath RD, Chevali L, Guglielmotti A. 2005. Treatment with bindarit, a blocker of MCP-1 synthesis, protects mice against acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 288:G1259–G1265. doi: 10.1152/ajpgi.00435.2004. [DOI] [PubMed] [Google Scholar]
- 74.Saso L, Silvestrini B, Zwain I, Guglielmotti A, Luparini MR, Cioli V, Cheng CY. 1992. Abnormal glycosylation of hemopexin in arthritic rats can be blocked by bindarit. J Rheumatol 19:1859–1867. [PubMed] [Google Scholar]
- 75.Cioli V, Ciarniello MG, Guglielmotti A, Luparini MR, Durando L, Martinelli B, Catanese B, Fava L, Silvestrini B. 1992. A new protein antidenaturant agent, bindarit, reduces secondary phase of adjuvant arthritis in rats. J Rheumatol 19:1735–1742. [PubMed] [Google Scholar]
- 76.Ble A, Mosca M, Di Loreto G, Guglielmotti A, Biondi G, Bombardieri S, Remuzzi G, Ruggenenti P. 2011. Antiproteinuric effect of chemokine C-C motif ligand 2 inhibition in subjects with acute proliferative lupus nephritis. Am J Nephrol 34:367–372. doi: 10.1159/000330685. [DOI] [PubMed] [Google Scholar]