

ABSTRACT
It has been shown in various infection models that CD4+ T cell help (TH) is necessary for the conditioning, maintenance, and/or recall responses of memory CD8+ T cells (CD8M). Yet, in the case of vaccinia virus (VACV), which constitutes the vaccine used to eradicate smallpox and is a candidate vector for other infectious diseases, the issue is controversial because different groups have shown either TH dependence or independence of CD8M conditioning, maintenance, and/or recall response. In agreement with some of these groups, we show that TH plays a role in, but is not essential for, the maintenance, proliferation, and effector differentiation of polyclonal memory CD8+ T cells after infection with wild-type VACV strain Western Reserve. More important, we show that unhelped and helped anti-VACV memory CD8+ T cells are similarly efficient at protecting susceptible mice from lethal mousepox, the mouse equivalent of human smallpox. Thus, TH is not essential for the conditioning and maintenance of memory CD8+ T cells capable of mounting a recall response strong enough to protect from a lethal natural pathogen. Our results may partly explain why the VACV vaccine is so effective.
IMPORTANCE We used vaccinia virus (VACV)—a gold standard vaccine—as the immunogen and ectromelia virus (ECTV) as the pathogen to demonstrate that the conditioning and maintenance of anti-VACV memory CD8+ T cells and their ability to protect against an orthopoxvirus (OPV) infection in its natural host can develop in the absence of CD4+ T cell help. Our results provide important insight to our basic knowledge of the immune system. Further, because VACV is used as a vaccine in humans, our results may help us understand how this vaccine induces protective immunity in this species. In addition, this work may partly explain why VACV is so effective as a vaccine.
INTRODUCTION
Following primary viral infection or vaccination, naive antiviral CD8+ T cells (CD8N) contribute to virus control by expanding and becoming effectors (CD8E) that kill infected cells and produce antiviral cytokines such as gamma interferon (IFN-γ) (1). If the virus is eliminated, most CD8E die but many survive to become resting memory CD8+ T cells (CD8M) that remain at higher frequencies than the original CD8N population (2). If a secondary infection occurs, the CD8M rapidly expand and become secondary CD8E. CD8M can contribute to reduce the severity of a secondary viral infection by achieving high numbers of effectors more rapidly than CD8N would. Moreover, the efficient generation of CD8M may be important for the effectiveness of some vaccines.
The genus Orthopoxvirus (OPV) comprises highly conserved DNA viruses that are antigenically highly cross-reactive. Vaccinia virus (VACV) is an OPV that can infect multiple species but is poorly pathogenic and highly immunogenic. Because of this, it was exploited as the vaccine that eliminated human smallpox, a highly lethal disease caused by the human-specific OPV variola virus (VARV). Thus, VACV remains as the gold standard of a highly effective vaccine, and VACV recombinants are currently being tested as vaccines for other infectious diseases and cancer (3, 4). In addition to preventing smallpox, VACV is also effective as a vaccine against lethal mousepox, a disease caused by the mouse-specific OPV ectromelia virus (ECTV) (5–9). Hence, VACV and ECTV can be paired as a unique model to understand the mechanisms of highly effective vaccination that is likely translatable to humans. Using this model, we have previously shown that in addition to antibodies (Abs), CD8M induced by VACV immunization can fully protect immunocompetent but susceptible mice from lethal mousepox (10, 11). Yet, how these protective CD8M are induced and maintained is not fully understood.
For some but not all infections, the transition of CD8N to CD8E requires CD4+ T cell help (TH) in the form of cytokines and/or costimulation (12). It has also been shown in several infectious models that TH is required for the conditioning and/or maintenance of the CD8M pool and/or their secondary expansion and differentiation into CD8E (13–16). In the case of OPVs, however, these issues remain controversial (17–25). Given that VACV is a mildly virulent virus used as a vaccine against more-pathogenic OPVs, we thought that it was important not only to perform additional experiments to confirm or exclude the need for TH for the establishment of anti-VACV CD8M but also, more importantly, to determine whether the absence of TH affects the ability of CD8M to become CD8E protective against a highly pathogenic OPV in its natural host. Thus, we used unattenuated VACV WR as the vaccine and ECTV as the pathogen to address the role of TH in the generation of protective CD8M. Our experiments measuring polyclonal rather than transgenic CD8+ T cell responses show that unhelped CD8M that expand and differentiate into CD8E are as effective as helped CD8M in their ability to protect from mousepox. Thus, TH is not essential for the generation and maintenance of memory CD8+ T cells capable of protecting against an OPV in its natural host.
MATERIALS AND METHODS
Ethics statement.
All animal experiments were conducted following the eighth edition of the Guide for the Care and Use of Laboratory Animals, National Research Council of the National Academy of Sciences, as mandated by the Office of Laboratory Animal Welfare (OLAW), NIH, and in accordance with protocols approved by the Fox Chase Cancer Center Institutional Animal Care and Use Committee.
Cells and virus.
The dendritic cell line DC2.4 was a gift from K. Rock (University of Massachusetts Medical Center, Worcester, MA). HeLa S3 and BSC-1 cells were obtained from the American Type Culture Collection. Initial stocks of VACV virus strain Western Reserve (WR) were obtained from B. Moss (National Institute of Allergy and Infectious Diseases, Bethesda, MD) and amplified in HeLa S3 cells as described previously (26). Initial stocks of the wild-type (WT) ECTV Moscow were obtained from ATCC (number VR-1374), and virus production and determination of virus titers were done as described previously (27).
Mice, immunizations, and infections.
Mice were bred at the Fox Chase Cancer Center Laboratory Animal Facility in specific-pathogen-free rooms from homozygous mice obtained from commercial vendors as follows: C57BL/6-Tg(Thy1-Sncg)HvP36Putt/J mice (B6.Thy1.1) and B6.D2-(D6Mit149-D6Mit15)/LusJ (B6.D2-D6) mice were originally purchased from Jackson Laboratories. B6 (CD45.2+) mice, B6.SJL (CD45.1+) mice, B6.129-H2-Ab1tm1GruN12 (major histocompatibility complex class II-deficient [MHC-II0/0]) mice, and B6.SJL (129)Ptprca/BoyAiTac H2-Ab1tm1Gru N7+N6 (CD45.1+-MHC-II0/0) mice were from Taconic Farms. Genotyping was according to the vendor's protocols. Before VACV immunization or ECTV infection, sex-matched animals 8 to 12 weeks old were transferred to a biosafety level 3 room. For VACV immunizations, mice were infected intraperitoneally (i.p.) with 500 μl phosphate-buffered saline (PBS) containing 5 × 106 PFU VACV WR. For ECTV infections, mice were infected in the left footpad with 3 × 103 PFU ECTV, approximately 9,000 LD50 (50% lethal dose) (28), in 25 μl PBS. All the infected mice were observed daily during the course of the experiments. In survival experiments, when death was imminent as evidenced by ruffled fur, hunched posture, lethargy, and unresponsiveness to touch, mice were euthanized according to the guidelines of the Institutional Animal Care and use Committee (IACUC) of the Fox Chase Cancer Center.
Adoptive transfers.
Adoptive transfers were performed as described previously (10, 11, 19). Briefly, lymph nodes (LN) and spleens of indicated donor mice were aseptically collected, and red blood cells (RBC) were lysed with 0.84% NH4Cl. The remaining cells were washed and labeled with rat anti-mouse CD8 magnetic beads according to the manufacturer's instructions (Miltenyi Biotec), and CD8+ cells were magnetically purified using an Automacs magnetic cell sorter (Miltenyi Biotec). The efficiency of the purification was monitored by fluorescence-activated cell sorter (FACS). Normally, the purity of CD8 T cells is >95%. In some experiments, purified cells were labeled with carboxyfluorescein succinimidyl ester (CFSE; Molecular Probes) according to published procedures (29). Indicated amounts of purified CD8+ T cells were resuspended in 0.5 ml PBS and inoculated intravenously (i.v.) into the recipient mice.
Flow cytometry.
Determination of cytokine production by intracellular staining was done as described previously (10, 11, 27, 30). Briefly, lymphocytes from spleens were obtained from mice at different days postinfection (days p.i.) and made into single-cell suspensions. Following osmotic lysis of RBC with 0.84% NH4Cl, lymphocytes were washed, and 106 cells were cultured at 37°C in 96-well plates in the presence of 10 U/ml interleukin-2 (IL-2) and 2 × 105 VACV-infected DC2.4 cells or uninfected DC2.4 cells as control. After 5 h, brefeldin A (Sigma-Aldrich) was added to block the secretory pathway and to allow for the accumulation of cytokines inside the cells. Following an additional 1.5 h of incubation, Ab 2.4G2 was added to block nonspecific binding of labeled Ab to FcR. The cells were then stained for cell surface molecules, fixed, permeabilized, and stained for intracellular molecules using the Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer's instructions. To stain for TSYKFESV epitope-specific T cells, H-2Kb:Ig recombinant fusion protein complexes (Mouse DimerX Kb dimers; BD) were incubated with synthetic TSYKFESV (GenScript) and used as recommended by the manufacturer. TSYKFESV is the dominant CD8+ T cell determinant of VACV and ECTV in the B6 background. At least 100,000 cells were analyzed by flow cytometry using the LSR II system (BD Biosciences).
In vivo cytotoxicity assay.
The in vivo cytotoxicity assay was done as described previously (10, 11, 19). Briefly, single-cell suspensions of lymphocytes from naive B6 mice were split into two populations. One population was labeled with a high concentration (4 μM) of CFSE (CFSEhigh) and pulsed with 10 μg/ml of TSYKFESV (31). The second population of lymphocytes was labeled with a low concentration (0.8 μM) of CFSE (CFSElow) and was not pulsed with peptide. The two cell populations were mixed together at a 1:1 ratio, and 2 × 107 total cells were injected i.v. into the indicated mice. Six hours later, recipient mice were euthanized, and the presence of CFSElow and CFSEhigh cells was determined by flow cytometry in cell suspensions of spleens. To calculate specific lysis, the following formula was used: % specific lysis = [1 − (ratio unprimed/ratio primed) × 100], where “ratio” is (% CFSElow/% CFSEhigh).
Data displayed and statistical analysis.
Unless indicated, all displayed data correspond to one representative experiment from at least three similar experiments with groups of three to five mice. Statistical analysis was performed using Prism software (GraphPad Software, Inc.). All statistical analyses were performed using an unpaired two-tailed Student's t test or the Mann-Whitney test as applicable. When applicable, data are displayed as means ± standard errors of the means (SEM).
RESULTS
TH plays a role, but is not critical, in generating anti-VACV CD8E and/or the maintenance of anti-VACV CD8M.
To determine whether TH is required for the primary endogenous anti-VACV CD8+ T cell response, we compared the endogenous responses of 6- to 12-week-old WT B6 and MHC-II-deficient (MHC-II0/0) mice to VACV strain WR at 7 days postpriming (dpp). The frequency of CD8+ T cells expressing granzyme B (GzB), which for CD8E does not require in vitro restimulation (19, 32), was similar for MHC-II0/0 and B6 mice, albeit the absolute numbers of GzB+ CD8+ T cells were slightly reduced in MHC-II0/0 mice. Yet, the frequency and absolute numbers of CD8+ T cells expressing IFN-γ, which requires ex vivo restimulation) or that stained with H-2Kb dimers (Dimer-X; BD) loaded with the immunodominant determinant TSYKFESV were similar in B6 and MHC-II0/0 mice (Fig. 1A). Thus, TH is not essential to generate large numbers of endogenous anti-VACV CD8E.
FIG 1.

TH plays a role but is not critical in generating anti-VACV CD8E and/or the maintenance of anti-VACV CD8M. B6 and MHC-II0/0 mice were primed i.p. with 5 × 106 PFU VACV strain WR. (A) CD8 T cell responses in spleens were determined at 7 dpp. Representative flow cytometry plots on the left show IFN-γ and GzB expression after in vitro restimulation with VACV-infected DC2.4 cells (upper row) and staining with Kb-TSYKFESV dimers (bottom row) by gated CD8+ cells. The summary graphs on the right show the frequency (top row) and number in millions (bottom row) of IFN-γ+, GzB+, and Kb-TSYKFESV+ CD8+ T cells for three mice/group. (B) Frequency of Kb-TSYKFESV+ CD8M at different dpp. NT, not tested because the data are from spleens pooled from 2 or 3 mice. (C) Frequency of Kb-TSYKFESV+ CD8M expressing the indicated cell surface molecules at 60 dpp. Data are representative of results from two or three similar experiments. n.s., not significant.
Despite being similar during the effector phase and as described for other models, the frequency of Kb-TSYKFESV-specific CD8M in MHC-II0/0 mice declined faster and was about one-half that of B6 mice at various times after immunization. Nevertheless, MHC-II0/0 mice still maintained a relatively large population of TSYKFESV-specific CD8M as late as 180 dpp (Fig. 1B). Side-by-side comparison of TSYKFESV-specific CD8M in B6 and MHC-II0/0 mice at 60 dpp showed that fewer CD8M cells expressed the IL-7 receptor alpha chain CD127 and the memory cell marker Gr-1(Ly6G/C) while more expressed the inhibitory molecule PD-1 in MHC-II0/0 mice. Yet, while statistically significant, it is arguable that these differences were minor because the vast majority of CD8M in MHC-II0/0 and B6 mice had similar mean fluorescence intensity (MFI) for these markers (not shown). Moreover, the expression of the activation and memory markers CD25, CD43, CD69, CD122, and KLRG1 were indistinguishable by frequency (Fig. 1C) and MFI (not shown). Thus, in the absence of TH, the frequency of anti-VACV CD8M is reduced compared to that of the wild type but still high compared to that of naive CD8M; and while some unhelped CD8M are phenotypically altered, the vast majority of the unhelped CD8M have a phenotype that is indistinguishable from that of helped CD8M.
Unhelped CD8M expand and generate secondary CD8M when maintained and boosted in an MHC-II-deficient environment.
Next, we compared the abilities of helped and unhelped CD8M to expand and generate secondary CD8M after booster VACV immunization within their respective WT or MHC-II0/0 environments. B6 and MHC-II0/0 mice were immunized with VACV, rested for 5 weeks, and boosted with VACV. Sixty days postboost (dpb), the frequency of CD8M as determined by IFN-γ production and Kb-TSYKFESV dimer staining increased sharply in both B6 and MHC-II0/0 mice (Fig. 2A), demonstrating that unhelped CD8M can be recalled into secondary effectors. Still, the frequency of secondary CD8M cells in B6 mice was significantly higher than in MHC-II0/0 mice (P < 0.001 for IFN-γ production and P < 0.001 for Kb-TSYKFESV dimer staining). Yet, because MHC-II0/0 mice had a lower frequency of CD8M than B6 mice before boosting, it cannot be concluded that the differences in the frequency of secondary CD8M were due to intrinsic defects of the unhelped CD8M. Yet, consistent with results from Fuse et al. (20), the MFI for IFN-γ in the IFN-γ+ gates were significantly lower in MHC-II0/0 than in B6 mice (not shown), suggesting an altered effector function of recalled CD8M after VACV.
FIG 2.

Unhelped CD8M expand and generate secondary CD8M cells when maintained and boosted in an MHC-II-deficient environment. B6 mice and MHC-II0/0 mice were primed i.p. with 5 × 106 PFU VACV and, when applicable, similarly boosted 5 weeks later. (A) CD8M in spleen were determined at 60 dpp or dpb; the cells were gated on CD8+ T cells. Representative flow cytometry plots on the left show IFN-γ and GzB expression after in vitro restimulation with VACV-infected DC2.4 cells (top row) and staining with Kb-TSYKFESV+ dimers (bottom row). The summary graphs on the right show the frequency of IFN-γ+ and Kb-TSYKFESV+ CD8M. (B) Frequency of Kb-TSYKFESV+ CD8M expressing the indicated cell surface molecules at 60 dpp or dpb. (C) In vivo killing of target cells by CD8M. Splenocytes from naive B6 mice were labeled with CFSE at 3.0 μM (CFSEHigh) or 0.8 μM (CFSELow). CFSEHigh cells were pulsed with TSYKFESV, and CFSELow were not. Mixtures (1/1) of CFSEHigh and CFSELow cells were injected i.v. into primed or primed/boosted B6 and MHC-II0/0 mice at 60 dpp or dpb. Killing was determined 6 h later in spleen. Representative flow cytometry plots are shown on the left. Plots in the upper row are gated on CD8+ cells. The transferred cells can be distinguished by their CFSE expression. The histograms in the lower row show the peaks of CFSELow and CFSEHigh cells. The number inside the plot indicates the percentage of specific killing of CFSEHigh (TSYKFESV-pulsed) cells. Data correspond to groups of three to five mice and are representative of results from two or three experiments.
We also compared the phenotypes of secondary helped and unhelped CD8M. Compared with B6 mice, boosted MHC-II0/0 mice had significantly larger frequencies of CD8M that expressed PD-1 and lower frequencies that expressed KLRG1 (Fig. 2B). Nevertheless, in MHC-II0/0 and B6 mice, most secondary CD8M did not express PD-1 and 40 to 60% of CD8M were KLRG1+. Furthermore, all other markers analyzed were indistinguishable. Thus, as with primary unhelped CD8M, most unhelped secondary CD8M have a normal phenotype.
We next compared the effector function of helped and unhelped primary and secondary CD8M in in vivo cytotoxicity assays at 60 dpp or dpb (10, 19, 33). Similar to secondary CD8M in B6 mice, secondary CD8M killed splenocytes loaded with TSYKFESV with high efficiency in MHC-II mice (Fig. 2C).
Thus, when maintained in an MHC-II0/0 environment, unhelped primary CD8M proliferate after booster immunization to generate an expanded secondary CD8M pool. Most of these secondary CD8M maintain a normal phenotype and are effective killers in vivo. However, with this experimental setting, it is difficult to quantitatively compare the secondary response of helped and unhelped CD8M because they start from different numbers of primary CD8M precursors and are maintained in an immunodeficient environment.
TH is not essential during priming for the conditioning of anti-VACV CD8M.
The data above show that MHC-II0/0 mice have a reduction in their polyclonal anti-VACV primary CD8M pools and somewhat decreased expansion during the secondary response. Why unhelped CD8M are deficient remains controversial. Using tumor cells and lymphocytic choriomeningitis virus (LCMV), Janssen et al. (34) showed that in the absence of help, the responding polyclonal CD8+ T cells upregulated proapoptotic molecules such as TRAIL. As a consequence, when the unhelped CD8M reencountered antigen, they suffered antigen-induced cell death (AICD). This indicated that, in this system, the unhelped phenotype is programmed during priming. On the other hand, Sun et al. used adoptive transfer of T cell receptor (TCR) transgenic CD8+ T cells during LCMV and Listeria monocytogenes infections to show that the defect of the unhelped CD8M was acquired during the maintenance phase and not during priming (15). To test whether the quantitative differences that we observed in our model were acquired during priming, we immunized B6 and MHC-II0/0 mice (CD45.2) with VACV to prime in the presence or the absence of TH. At 7 dpp, 5 × 106 CD8E from these mice were transferred into naive B6-CD45.1 mice. Seven days later, both CD45.2 B6 and MHC-II0/0 donor cells represented 3 to 4% of the total CD8+ T cells and ∼2% of these were TSYKFESV specific (Fig. 3A). Hence, the early contractions of helped and unhelped CD8E were similar in a WT environment. At later time points, the VACV-specific donor CD8M could not be detected by flow cytometry, regardless of the donor strain (not shown). We hypothesized that if CD8M were still present, they would expand after VACV challenge and any differences would be apparent by alterations in the frequency of helped and unhelped donor CD8E. Thus, we performed experiments in which groups of 2 or 3 B6-CD45.1 mice received equal numbers of helped (B6) or unhelped (MHC-II0/0) CD45.2+ CD8E and 1.5 to 4 months later were infected in the footpad with the OPV ECTV, a natural pathogen of the mouse. Of note, ECTV and VACV share most of their CD8+ T cell determinants. At 8 days p.i., helped and unhelped donor CD8+ T cells represented 23 to 24% of the total CD8+ T cells, demonstrating extensive expansion. The vast majority of the donor cells were effectors, as ∼90% expressed GzB and 6 to 7% expressed IFN-γ. Fifteen to 20% were Kb-TSYKFESV specific, whether helped or unhelped during priming (Fig. 3B). Because the analysis was performed with pooled spleens, no statistics could be calculated. Yet, comparable results were obtained in two similar experiments, indicating that if there were any differences, these are minimal. Thus, during VACV infection, TH does not condition the polyclonal CD8M response at priming.
FIG 3.
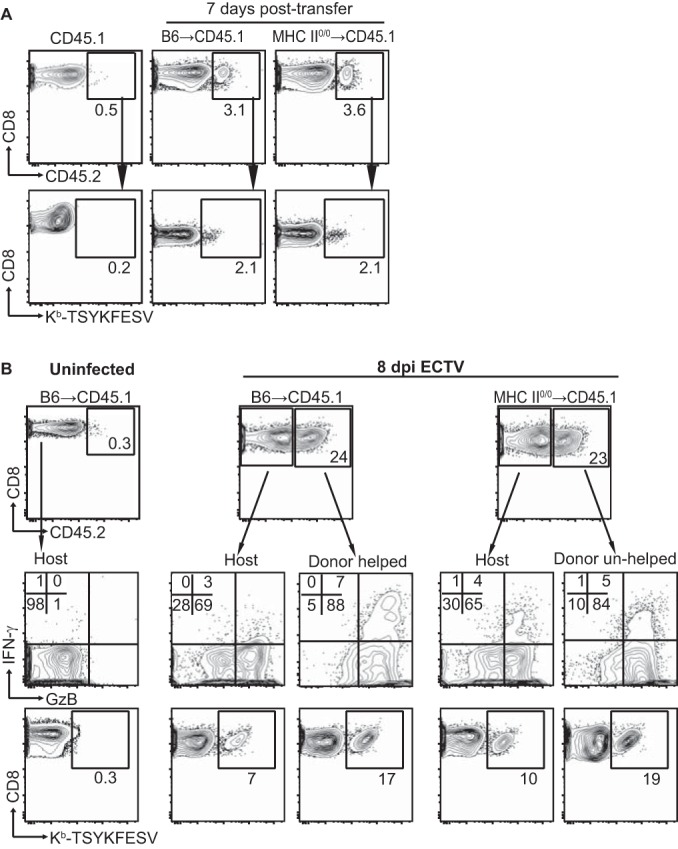
TH is not required during priming for the conditioning of anti-VACV CD8M. B6 and MHC-II0/0 mice were infected i.p. with 5 × 106 PFU VACV. At 7 days p.i., 5 × 106 magnetically purified CD8+ T cells from these mice were transferred i.v. into B6.CD45.1 mice. (A) Seven days after transfer, the donor cells were detected in the spleens of the recipient mice. Representative flow cytometry plots show the frequency of CD45.2+ transferred cells in the CD8+ gate (top row) and the frequency of Kb-TSYKFESV+ cells in the CD45.2 gate (bottom row). Data correspond to three pooled spleens, and the experiment was repeated three times. (B) Four months after transfer, the recipient mice were infected with 3 × 103 PFU ECTV in the footpad, and the CD8+ T cells responses were determined at 8 days p.i. Flow cytometry plots show the frequency of host and transferred cells in the CD8+ gate (top row), expression of IFN-γ and GzB after restimulation with VACV-infected DC2.4 cells (middle row), and the frequency of Kb-TSYKFESV+ cells in the CD45.2 gate (bottom row). The data correspond to two pools of two spleens and are representative of results from three experiments that were similar but had some differences in the methods (for example, less time between transfer and infection).
Quantitatively normal response to ECTV by unhelped secondary anti-VACV CD8M maintained in the absence of help.
As we show (illustrated in Fig. 2), it is difficult to quantitatively compare secondary CD8M responses in immunized WT and MHC-II-deficient hosts because they contain different numbers of CD8M. Also, most vaccines use prime/boost regimes. Thus, it is the secondary (or higher) CD8M that need to differentiate into tertiary (or higher) CD8E for vaccine-mediated protection. Therefore, we quantitatively compared the helped and unhelped CD8M responses primed, contracted, and maintained in their respective environments, after transfer into naive WT hosts. For this purpose, B6 and MHC-II0/0 mice were primed/boosted with VACV. At 60 dpb, their splenic CD8+ T cells were labeled with CFSE, adjusted to equal numbers of Kb-TSYKFESV+ CD8+ T cells, and transferred into B6.Thy1.1 mice. One day after transfer, the mice were infected in the footpad with ECTV, and the CD8+ T cell responses were determined in the draining lymph node (D-LN) at 5 days p.i. We found no differences in the abilities of helped and unhelped secondary CD8M in becoming CD8E as determined by frequency of the expanded donor cells (Fig. 4, first row), proliferation by CFSE dilution (Fig. 4, second row), or expression of the effector molecules GzB and IFN-γ (Fig. 4, third row). Thus, when transferred in equal numbers to WT hosts, unhelped anti-VACV CD8M primed, contracted, and maintained in a TH-deficient environment responded as efficiently as helped CD8M to pathogenic viral challenge.
FIG 4.
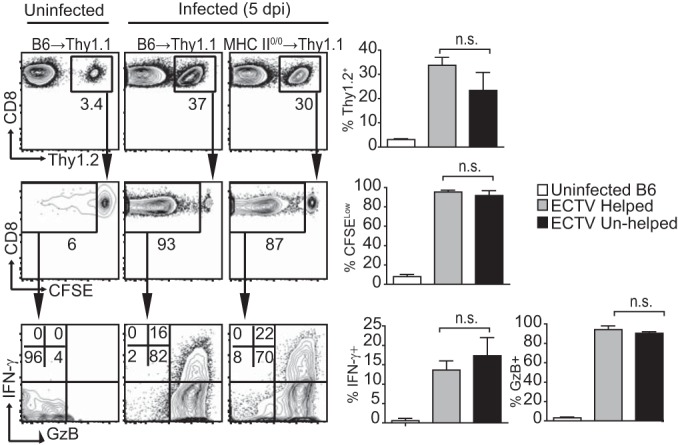
Quantitatively normal response to ECTV by unhelped secondary anti-VACV CD8M maintained in the absence of help. B6 mice and MHC-II0/0 mice were primed and boosted 5 weeks later with 5 × 106 PFU VACV i.v. At 60 dpp or dpb, the splenocytes were obtained and labeled with CFSE, and the CD8+ T cells were magnetically purified, equalized for similar numbers of Kb-TSYKFESV+ CD8+ T cells, and inoculated i.v. into B6.Thy1.1 mice. One day later, the mice were infected with 3 × 103 PFU ECTV in the footpad or left uninfected as a control. Cells in the draining popliteal lymph node were analyzed at 5 days p.i.. Representative flow cytometry plots on the left show the frequencies of transferred cells within the CD8+ gate (top row), the frequencies of cells within the transferred cells that diluted their CFSE (middle row), and the frequencies of cells that diluted CFSE and express GzB and IFN-γ. Summary graphs for the same parameters are shown on the right. Data correspond to three mice/group and are representative of results from three similar experiments. n.s., not significant.
Unhelped secondary CD8M protect susceptible mice from mousepox.
The results above show that CD8M generated by VACV immunization in the absence of TH can mount strong recall responses to secondary and tertiary ECTV challenge whether maintained in the presence or absence of TH. Because the goal of vaccination is to protect from disease, we next determined whether unhelped CD8M generated by VACV prime/boost immunization can protect susceptible mice from lethal mousepox.
B6.D2-D6 is a B6 congenic mouse strain that is susceptible to mousepox (35) but can be protected by CD8M after vaccination with dendritic cells pulsed with TSYKFESV (36) or adoptive transfer of CD8+ T cells from B6 mice primed/boosted with VACV (10). Helped and unhelped CD8M were generated by VACV prime/boost immunization of B6 and MHC-II0/0 mice, respectively. At 60 dpb, purified CD8+ T cells from these mice were adjusted to contain similar numbers of Kb-TSYKFESV+-specific CD8M and graded numbers were adoptively transferred into groups of five B6.D2-D6 mice, which were subsequently infected with ECTV. As shown in Table 1, all control (untransferred) mice succumbed to mousepox. However, all B6.D2-D6 mice transferred with as little as 4.5 × 104 Kb-TSYKFESV+ CD8M from VACV-primed/boosted B6 or MHC-II0/0 mice were fully protected. Consistently, mice receiving B6 or MHC-II0/0 CD8M did not have the splenic lymphopenia characteristic of lethal mousepox (Fig. 5A). Moreover, the virus titers at 7 days p.i. were significantly lower in mice that received helped or unhelped CD8M than in untransferred mice, while there were no significant differences in virus titers between B6 and MHC-II0/0 CD8M recipients (Fig. 5B). Because in these experiments we did not use mice with congenic markers, we were unable to analyze the CD8M response. Yet the fact that helped and unhelped CD8M are similarly responsive was already demonstrated, as illustrated in Fig. 4.
TABLE 1.
Unhelped secondary CD8M protect susceptible mice from mousepoxa
| No. of Kb-TSYKFESV+ cells transferred | No. dead/total no. of mice receiving: |
||
|---|---|---|---|
| Helped CD8Mb | Unhelped CD8Mc | No cells | |
| 1.8 × 105 | 0/5** | 0/5** | |
| 9 × 104 | 0/5** | 0/5** | |
| 4.5 × 104 | 0/5** | 0/5** | |
| 2.25 × 104 | 4/5 | 0/5** | |
| 0 | 5/5 | ||
B6 mice and MHC-II0/0 mice were primed and then boosted 5 weeks later with 5 × 106 PFU VACV i.v. Two months later, the splenocytes of these mice were obtained, the CD8+ T cells were magnetically purified, the frequency of Kb-TSYKFESV+ cells was determined by flow cytometry, and the indicated numbers of Kb-TSYKFESV+ cells from B6 (helped CD8M) and MHC II0/0 (unhelped CD8M) mice were inoculated i.v. into mousepox-susceptible B6.D2-D6 mice. One day later, the mice were infected with 3 × 103 PFU ECTV in the footpad and observed for lethality as detailed in Materials and Methods. **, P < 0.01 compared with mice receiving no cells (control).
Helped CD8M contained 2.25% Kb-TSYKFESV+ cells.
Unhelped CD8M contained 1% Kb-TSYKFESV+ cells.
FIG 5.
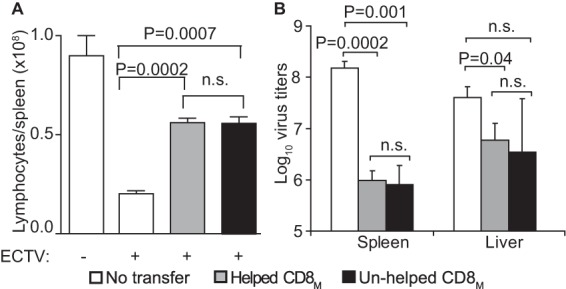
Unhelped secondary CD8M protect susceptible mice from mousepox. B6 mice and MHC-II0/0 mice were primed and then boosted 5 weeks later with 5 × 106 PFU VACV i.v. At 60 dpb, the splenocytes of these mice were obtained, the CD8+ T cells were magnetically purified, the frequency of Kb-TSYKFESV+ cells was determined by flow cytometry, and 4.5 × 104 Kb-TSYKFESV+ cells were inoculated i.v. into mousepox-susceptible B6 mice. One day later, the mice were infected with 3 × 103 PFU ECTV in the footpad. (A) Total number of lymphocytes in the spleen. (B) Virus titers in spleen and liver. n.s., not significant.
DISCUSSION
We have previously shown that CD8M induced by VACV immunization become CD8E and protect mice from mousepox, strongly suggesting that the establishment of a pool of CD8M cells is one of the mechanisms whereby the smallpox vaccine protects from pathogenic OPVs (10, 11, 36). Given the importance of CD8M induced by VACV in protection from lethal OPVs, we thought that it was important to determine the impact of TH in the establishment of this form of protective immunity. For this purpose, we used nonattenuated VACV WR as the vaccine and ECTV as the pathogen. Using this model, we showed that in MHC-II0/0 mice, which lack TH, CD8N generate potent CD8E that transition into long-lived CD8M. We also found that unhelped CD8M can be activated to become secondary CD8E in a WT as well as in an MHC-II-deficient environment and transition into secondary CD8M. Moreover, when transferred into mousepox-resistant WT mice, unhelped secondary CD8M expand and become effectors as efficiently as their helped counterparts in response to ECTV. More importantly, unhelped secondary CD8M are as effective as helped CD8M at protecting susceptible mice from lethal mousepox.
To date, the role of TH in primary and secondary CD8+ T cell responses to VACV remains controversial. While some have shown that TH is necessary for the induction of anti-VACV primary CD8+ T cell responses (21–24, 37), others, including us, have shown the opposite (17–20). Similarly, disparate results have been published regarding the role of TH in the generation, maintenance, and recall responses of anti-VACV CD8M. Some have shown that TH is required (14, 20–22, 37), and others have shown that it is not (24, 25). A possible explanation for these discrepancies may lie in the replicative capacity of the VACV strain used. In our experiments, we used VACV WR, which is not attenuated. Recently, Salek-Ardakani et al. (25) showed that MHC-II0/0 mice were protected by CD8M from intranasal VACV WR challenge when preimmunized with the nonattenuated VACV WR but were not protected after immunization with the attenuated VACV strain Lister. Moreover, several of the publications showing TH requirement were performed using VACV isolates with deletion of the thymidine kinase gene (14, 21–23, 37, 38), which results in attenuation (39). This suggests that the generation, maintenance, and recall response of CD8M to nonattenuated VACV may be less dependent on TH than strains with poorer replicative potential. Another reason for the differences might be that, similar to Salek-Ardakani et al. (25), we determined polyclonal responses against a natural epitope shared by VACV and ECTV (31, 36) rather than TCR transgenic responses to ectopic determinants (14, 21–23, 37, 38). In addition to attenuation, other factors that may affect the need for TH and protective capacity are the dose of VACV and the route of inoculation (20, 25).
It is also important to note that in our experiments we used MHC-II-deficient mice because they guarantee the absence of MHC-II-restricted TH. CD4+ T cells recognize antigen as peptides bound to MHC class II molecules. Peptide-MHC-II recognition is also required for their development in the thymus. Hence, mice deficient in MHC-II lack MHC-II-restricted cells. This is different for CD4-deficient mice, which have MHC-II-restricted CD4− CD8− helper T cells (40–42), and a large fraction of their CD8+ T cells are also restricted by MHC-II (43, 44), resulting in poorer overall MHC-I-restricted CD8+ responses (43). This can account for some of the deficits in the anti-VACV CD8M previously observed by others (38). MHC-II0/0 mice also lack possible complications arising from incomplete and/or prolonged CD4+ T cell depletion with the anti-CD4+ monoclonal antibody (MAb) GK1.5 (45), which we and others previously used (19, 38, 46, 47). Yet, experiments with MHC-II0/0 mice, which have a major immunodeficiency, may still have caveats. In our experiments, we have found significantly reduced numbers of CD8M in MHC-II0/0 mice and low but significant differences in the frequency of primary and/or secondary CD8M expressing CD127, Gr1, KLRG1, and PD-1. While it is possible that these differences are due to the lack of TH, it is also possible that they are a consequence of the imbalance of the immune system in MHC-II0/0 mice. For example, it is very likely that their interaction with the microbiome is different from that in immunocompetent mice, and this can affect their health and the quality of their immune responses, particularly as the mice age. In this regard, we have observed that naive aged MHC-II0/0 mice have poor health and tend to die earlier than age-matched B6 mice. Further, we have found that aged naive MHC-II0/0 mice have fewer cells with the CD8M phenotype, suggesting an overall dysregulation of the CD8+ T cell compartment with aging (our unpublished experiments). These defects may contribute to the faster decline in the frequency of CD8M in MHC-II0/0 than in the frequency of CD8M in B6 mice that we (Fig. 1) and others (16) have observed, an issue deserving of further exploration. To overcome this caveat, we have used adoptive transfer of polyclonal anti-VACV CD8M. Using this approach, we have shown that when cell numbers are adjusted, polyclonal anti-VACV CD8M generated in the absence or presence of help are similarly potent at protecting mice from a lethal viral disease.
In summary, our experiments confirm in the VACV model that CD8M in MHC-II0/0 mice are not completely normal, as they contracted more than in WT mice and a minority of them had an altered phenotype. Thus, our work does not dispute that TH contributes to the optimal generation and maintenance of CD8M. Yet, our work clearly shows that TH is not crucial for the establishment of CD8M or essential to confer CD8M the capacity to protect from a lethal infection. Because VACV is used as a vaccine in humans, our results may have implications for our understanding on how this vaccine induces protective immunity in this species.
ACKNOWLEDGMENTS
We thank Fox Chase Cancer Center (FCCC) Laboratory Animal, Flow Cytometry and Tissue Culture Facilities and Holly Gillin for assistance in the preparation of the manuscript.
This work was supported by grants R01AI048849, R01AI065544, and 5U19AI083008 to L.J.S. and P30CA006927 to the FCCC. M.F. was partially supported by FCCC's William J. Avery Fellowship. S.R. was supported by NIH grant T32 CA-009035035 to the FCCC.
REFERENCES
- 1.Harty JT, Tvinnereim AR, White DW. 2000. CD8+ T cell effector mechanisms in resistance to infection. Annu Rev Immunol 18:275–308. doi: 10.1146/annurev.immunol.18.1.275. [DOI] [PubMed] [Google Scholar]
- 2.Smith-Garvin JE, Sigal LJ. 2013. Immunology: memory cells sound the alarm. Nature 497:194–196. doi: 10.1038/497194b. [DOI] [PubMed] [Google Scholar]
- 3.Gomez CE, Najera JL, Krupa M, Perdiguero B, Esteban M. 2011. MVA and NYVAC as vaccines against emergent infectious diseases and cancer. Curr Gene Ther 11:189–217. doi: 10.2174/156652311795684731. [DOI] [PubMed] [Google Scholar]
- 4.Walsh SR, Dolin R. 2011. Vaccinia viruses: vaccines against smallpox and vectors against infectious diseases and tumors. Expert Rev Vaccines 10:1221–1240. doi: 10.1586/erv.11.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fields BN, Knipe DM, Howley PM. 2007. Fields virology, 5th ed. Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 6.Verardi PH, Titong A, Hagen CJ. 2012. A vaccinia virus renaissance: new vaccine and immunotherapeutic uses after smallpox eradication. Hum Vaccin Immunother 8:961–970. doi: 10.4161/hv.21080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobs BL, Langland JO, Kibler KV, Denzler KL, White SD, Holechek SA, Wong S, Huynh T, Baskin CR. 2009. Vaccinia virus vaccines: past, present and future. Antiviral Res 84:1–13. doi: 10.1016/j.antiviral.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kennedy RB, Ovsyannikova IG, Jacobson RM, Poland GA. 2009. The immunology of smallpox vaccines. Curr Opin Immunol 21:314–320. doi: 10.1016/j.coi.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moss B. 2011. Smallpox vaccines: targets of protective immunity. Immunol Rev 239:8–26. doi: 10.1111/j.1600-065X.2010.00975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Remakus S, Rubio D, Lev A, Ma X, Fang M, Xu RH, Sigal LJ. 2013. Memory CD8 T cells can outsource IFN-gamma production but not cytolytic killing for antiviral protection. Cell Host Microbe 13:546–557. doi: 10.1016/j.chom.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu RH, Fang M, Klein-Szanto A, Sigal LJ. 2007. Memory CD8+ T cells are gatekeepers of the lymph node draining the site of viral infection. Proc Natl Acad Sci U S A 104:10992–10997. doi: 10.1073/pnas.0701822104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiesel M, Oxenius A. 2012. From crucial to negligible: functional CD8(+) T-cell responses and their dependence on CD4(+) T-cell help. Eur J Immunol 42:1080–1088. doi: 10.1002/eji.201142205. [DOI] [PubMed] [Google Scholar]
- 13.Bachmann MF, Wolint P, Schwarz K, Oxenius A. 2005. Recall proliferation potential of memory CD8+ T cells and antiviral protection. J Immunol 175:4677–4685. doi: 10.4049/jimmunol.175.7.4677. [DOI] [PubMed] [Google Scholar]
- 14.Agnellini P, Wiesel M, Schwarz K, Wolint P, Bachmann MF, Oxenius A. 2008. Kinetic and mechanistic requirements for helping CD8 T cells. J Immunol 180:1517–1525. doi: 10.4049/jimmunol.180.3.1517. [DOI] [PubMed] [Google Scholar]
- 15.Sun JC, Williams MA, Bevan MJ. 2004. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol 5:927–933. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun JC, Bevan MJ. 2003. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goulding J, Bogue R, Tahiliani V, Croft M, Salek-Ardakani S. 2012. CD8 T cells are essential for recovery from a respiratory vaccinia virus infection. J Immunol 189:2432–2440. doi: 10.4049/jimmunol.1200799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salek-Ardakani S, Arens R, Flynn R, Sette A, Schoenberger SP, Croft M. 2009. Preferential use of B7.2 and not B7.1 in priming of vaccinia virus-specific CD8 T cells. J Immunol 182:2909–2918. doi: 10.4049/jimmunol.0803545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang M, Sigal LJ. 2006. Direct CD28 costimulation is required for CD8+ T cell-mediated resistance to an acute viral disease in a natural host. J Immunol 177:8027–8036. doi: 10.4049/jimmunol.177.11.8027. [DOI] [PubMed] [Google Scholar]
- 20.Fuse S, Tsai CY, Molloy MJ, Allie SR, Zhang W, Yagita H, Usherwood EJ. 2009. Recall responses by helpless memory CD8+ T cells are restricted by the up-regulation of PD-1. J Immunol 182:4244–4254. doi: 10.4049/jimmunol.0802041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Novy P, Huang X, Leonard WJ, Yang Y. 2011. Intrinsic IL-21 signaling is critical for CD8 T cell survival and memory formation in response to vaccinia viral infection. J Immunol 186:2729–2738. doi: 10.4049/jimmunol.1003009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Novy P, Quigley M, Huang X, Yang Y. 2007. CD4 T cells are required for CD8 T cell survival during both primary and memory recall responses. J Immunol 179:8243–8251. doi: 10.4049/jimmunol.179.12.8243. [DOI] [PubMed] [Google Scholar]
- 23.Wiesel M, Joller N, Ehlert AK, Crouse J, Sporri R, Bachmann MF, Oxenius A. 2010. Th cells act via two synergistic pathways to promote antiviral CD8+ T cell responses. J Immunol 185:5188–5197. doi: 10.4049/jimmunol.1001990. [DOI] [PubMed] [Google Scholar]
- 24.Obar JJ, Molloy MJ, Jellison ER, Stoklasek TA, Zhang W, Usherwood EJ, Lefrancois L. 2010. CD4+ T cell regulation of CD25 expression controls development of short-lived effector CD8+ T cells in primary and secondary responses. Proc Natl Acad Sci U S A 107:193–198. doi: 10.1073/pnas.0909945107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salek-Ardakani S, Flynn R, Arens R, Yagita H, Smith GL, Borst J, Schoenberger SP, Croft M. 2011. The TNFR family members OX40 and CD27 link viral virulence to protective T cell vaccines in mice. J Clin Invest 121:296–307. doi: 10.1172/JCI42056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elroy-Stein O, Moss B. 2001. Gene expression using the vaccinia virus/T7 RNA polymerase hybrid system. Curr Protoc Protein Sci Chapter 5:Unit5.15. doi: 10.1002/0471140864.ps0515s14. [DOI] [PubMed] [Google Scholar]
- 27.Fang M, Cheng H, Dai Z, Bu Z, Sigal LJ. 2006. Immunization with a single extracellular enveloped virus protein produced in bacteria provides partial protection from a lethal orthopoxvirus infection in a natural host. Virology 345:231–243. doi: 10.1016/j.virol.2005.09.056. [DOI] [PubMed] [Google Scholar]
- 28.Xu RH, Cohen M, Tang Y, Lazear E, Whitbeck JC, Eisenberg RJ, Cohen GH, Sigal LJ. 2008. The orthopoxvirus type I IFN binding protein is essential for virulence and an effective target for vaccination. J Exp Med 205:981–992. doi: 10.1084/jem.20071854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurts C, Heath WR, Carbone FR, Kosaka H, Miller JF. 1998. Cross-presentation of self antigens to CD8+ T cells: the balance between tolerance and autoimmunity. Novartis Found Symp 215:172–181, discussion 181–190. [DOI] [PubMed] [Google Scholar]
- 30.Fang M, Sigal LJ. 2005. Antibodies and CD8+ T cells are complementary and essential for natural resistance to a highly lethal cytopathic virus. J Immunol 175:6829–6836. doi: 10.4049/jimmunol.175.10.6829. [DOI] [PubMed] [Google Scholar]
- 31.Tscharke DC, Karupiah G, Zhou J, Palmore T, Irvine KR, Haeryfar SM, Williams S, Sidney J, Sette A, Bennink JR, Yewdell JW. 2005. Identification of poxvirus CD8+ T cell determinants to enable rational design and characterization of smallpox vaccines. J Exp Med 201:95–104. doi: 10.1084/jem.20041912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuen TJ, Flesch IE, Hollett NA, Dobson BM, Russell TA, Fahrer AM, Tscharke DC. 2010. Analysis of A47, an immunoprevalent protein of vaccinia virus, leads to a reevaluation of the total antiviral CD8+ T cell response. J Virol 84:10220–10229. doi: 10.1128/JVI.01281-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barber DL, Wherry EJ, Ahmed R. 2003. Cutting edge: rapid in vivo killing by memory CD8 T cells. J Immunol 171:27–31. doi: 10.4049/jimmunol.171.1.27. [DOI] [PubMed] [Google Scholar]
- 34.Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD, Griffith TS, Green DR, Schoenberger SP. 2005. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature 434:88–93. doi: 10.1038/nature03337. [DOI] [PubMed] [Google Scholar]
- 35.Fang M, Orr MT, Spee P, Egebjerg T, Lanier LL, Sigal LJ. 2011. CD94 is essential for NK cell-mediated resistance to a lethal viral disease. Immunity 34:579–589. doi: 10.1016/j.immuni.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Remakus S, Rubio D, Ma X, Sette A, Sigal LJ. 2012. Memory CD8+ T cells specific for a single immunodominant or subdominant determinant induced by peptide-dendritic cell immunization protect from an acute lethal viral disease. J Virol 86:9748–9759. doi: 10.1128/JVI.00981-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bedenikovic G, Crouse J, Oxenius A. 2014. T-cell help dependence of memory CD8 T-cell expansion upon vaccinia virus challenge relies on CD40 signaling. Eur J Immunol 44:115–126. doi: 10.1002/eji.201343805. [DOI] [PubMed] [Google Scholar]
- 38.Shedlock DJ, Shen H. 2003. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 39.Lee MS, Roos JM, McGuigan LC, Smith KA, Cormier N, Cohen LK, Roberts BE, Payne LG. 1992. Molecular attenuation of vaccinia virus: mutant generation and animal characterization. J Virol 66:2617–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Locksley RM, Reiner SL, Hatam F, Littman DR, Killeen N. 1993. Helper T cells without CD4: control of leishmaniasis in CD4-deficient mice. Science 261:1448–1451. doi: 10.1126/science.8367726. [DOI] [PubMed] [Google Scholar]
- 41.Killeen N, Sawada S, Littman DR. 1993. Regulated expression of human CD4 rescues helper T cell development in mice lacking expression of endogenous CD4. EMBO J 12:1547–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rahemtulla A, Fung-Leung WP, Schilham MW, Kundig TM, Sambhara SR, Narendran A, Arabian A, Wakeham A, Paige CJ, Zinkernagel RM, Miller RG, Mak TW. 1991. Normal development and function of CD8+ cells but markedly decreased helper cell activity in mice lacking CD4. Nature 353:180–184. doi: 10.1038/353180a0. [DOI] [PubMed] [Google Scholar]
- 43.Tyznik AJ, Sun JC, Bevan MJ. 2004. The CD8 population in CD4-deficient mice is heavily contaminated with MHC class II-restricted T cells. J Exp Med 199:559–565. doi: 10.1084/jem.20031961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pearce EL, Shedlock DJ, Shen H. 2004. Functional characterization of MHC class II-restricted CD8+CD4- and CD8-CD4- T cell responses to infection in CD4−/− mice. J Immunol 173:2494–2499. doi: 10.4049/jimmunol.173.4.2494. [DOI] [PubMed] [Google Scholar]
- 45.Dialynas DP, Quan ZS, Wall KA, Pierres A, Quintans J, Loken MR, Pierres M, Fitch FW. 1983. Characterization of the murine T cell surface molecule, designated L3T4, identified by monoclonal antibody GK1.5: similarity of L3T4 to the human Leu-3/T4 molecule. J Immunol 131:2445–2451. [PubMed] [Google Scholar]
- 46.Shedlock DJ, Whitmire JK, Tan J, MacDonald AS, Ahmed R, Shen H. 2003. Role of CD4 T cell help and costimulation in CD8 T cell responses during Listeria monocytogenes infection. J Immunol 170:2053–2063. doi: 10.4049/jimmunol.170.4.2053. [DOI] [PubMed] [Google Scholar]
- 47.Wofsy D, Mayes DC, Woodcock J, Seaman WE. 1985. Inhibition of humoral immunity in vivo by monoclonal antibody to L3T4: studies with soluble antigens in intact mice. J Immunol 135:1698–1701. [PubMed] [Google Scholar]