

ABSTRACT
The first discovered and sequenced hepatitis C virus (HCV) genome and the first in vivo infectious HCV clones originated from the HCV prototype strains HCV-1 and H77, respectively, both widely used in research of this important human pathogen. In the present study, we developed efficient infectious cell culture systems for these genotype 1a strains by using the HCV-1/SF9_A and H77C in vivo infectious clones. We initially adapted a genome with the HCV-1 5′UTR-NS5A (where UTR stands for untranslated region) and the JFH1 NS5B-3′UTR (5-5A recombinant), including the genotype 2a-derived mutations F1464L/A1672S/D2979G (LSG), to grow efficiently in Huh7.5 cells, thus identifying the E2 mutation S399F. The combination of LSG/S399F and reported TNcc(1a)-adaptive mutations A1226G/Q1773H/N1927T/Y2981F/F2994S promoted adaptation of the full-length HCV-1 clone. An HCV-1 recombinant with 17 mutations (HCV1cc) replicated efficiently in Huh7.5 cells and produced supernatant infectivity titers of 104.0 focus-forming units (FFU)/ml. Eight of these mutations were identified from passaged HCV-1 viruses, and the A970T/I1312V/C2419R/A2919T mutations were essential for infectious particle production. Using CD81-deficient Huh7 cells, we further demonstrated the importance of A970T/I1312V/A2919T or A970T/C2419R/A2919T for virus assembly and that the I1312V/C2419R combination played a major role in virus release. Using a similar approach, we found that NS5B mutation F2994R, identified here from culture-adapted full-length TN viruses and a common NS3 helicase mutation (S1368P) derived from viable H77C and HCV-1 5-5A recombinants, initiated replication and culture adaptation of H77C containing LSG and TNcc(1a)-adaptive mutations. An H77C recombinant harboring 19 mutations (H77Ccc) replicated and spread efficiently after transfection and subsequent infection of naive Huh7.5 cells, reaching titers of 103.5 and 104.4 FFU/ml, respectively.
IMPORTANCE Hepatitis C virus (HCV) was discovered in 1989 with the cloning of the prototype strain HCV-1 genome. In 1997, two molecular clones of H77, the other HCV prototype strain, were shown to be infectious in chimpanzees, but not in vitro. HCV research was hampered by a lack of infectious cell culture systems, which became available only in 2005 with the discovery of JFH1 (genotype 2a), a genome that could establish infection in Huh7.5 cells. Recently, we developed in vitro infectious clones for genotype 1a (TN), 2a (J6), and 2b (J8, DH8, and DH10) strains by identifying key adaptive mutations. Globally, genotype 1 is the most prevalent. Studies using HCV-1 and H77 prototype sequences have generated important knowledge on HCV. Thus, the in vitro infectious clones developed here for these 1a strains will be of particular value in advancing HCV research. Moreover, our findings open new avenues for the culture adaptation of HCV isolates of different genotypes.
INTRODUCTION
Hepatitis C virus (HCV) has chronically infected over 130 million people worldwide and is a leading cause of liver fibrosis, cirrhosis, and hepatocellular carcinoma. More than 350,000 deaths annually are due to HCV-related liver diseases (World Health Organization website, 2014). HCV belongs to the Hepacivirus genus within the Flaviviridae family, and its genome is a positive-sense single-strand RNA of ∼9.6 kb consisting of a single open reading frame (ORF) and 5′ and 3′ untranslated regions (UTRs). The ORF encodes viral structural proteins (Core and envelope glycoproteins E1 and E2), a small membrane protein (p7), and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (1). HCV has been classified into 7 major genotypes differing in nucleotide and amino acid sequences by ∼30% and numerous subtypes with sequence heterogeneity of 15 to 20% (2–4). Genotype 1 accounts for the majority of HCV infections worldwide, and subtypes 1a and 1b are predominant. Furthermore, genotype 1 strains were found to be relatively resistant to alpha interferon/ribavirin therapy (1). Although incorporation of directly acting antivirals (DAA) improves the sustained virological response rate, the emergence of drug resistance is a concern and may influence the outcome of these new therapies (5).
Robust infectious HCV cell culture systems from isolates of different genotypes represent valuable tools for the in vitro study of HCV genetic heterogeneity, which plays a major role in disease progression and response to antiviral therapy and poses a significant challenge for vaccine development. Since the discovery of HCV-1 in 1989 (6), many attempts have been directed to adapt prototype strains of HCV to grow in cell culture. However, success did not come until 2005, when the cloned JFH1 (genotype 2a) full-length sequence was found to be able to spontaneously establish infection in hepatoma Huh7 cells and derivatives (7, 8). To date, JFH1 remains the only cloned HCV sequence reported with spontaneous growth in vitro. Recently, we identified three mutations in NS3, NS4A, and NS5B (F1464L, A1672S, and D2979G, respectively, referred to as the LSG mutations; the nucleotide and amino acid numbering throughout is according to the H77 [genotype 1a] reference sequence; GenBank accession number AF009606), which enabled adaptation of HCV full-length genomes in cell culture. These mutations led to the development of robust and highly infectious full-length cell culture systems for HCV genotype 1a (TNcc) (9), 2a (J6cc) (10), and 2b (J8cc, DH8cc, and DH10cc) (10, 11) strains; the TNcc represented the first effective cell culture system for genotype 1. Infectious culture systems were reported for only a few other genotype 1 and 2 strains (12–15).
The HCV-1 strain was the first HCV genome to be cloned (6) and has been a key tool commonly used by investigators in the HCV field. Studies using HCV-1 have led to important discoveries, such as the elucidation of the genetic organization of the HCV genome (16), identification of CD81 as an important viral receptor (17), and discovery of the frame-shifted F protein (18). An HCV-1 E1/E2-based vaccine was found to induce neutralizing antibody responses with cross-reactivity against various HCV genotypes in rodents (19), chimpanzees (20), and humans (21, 22), thus making it a promising vaccine candidate for further development. H77 is another genotype 1a strain that has significantly contributed to HCV research. H77 is the reference sequence for HCV genome numbering (23). The RNA transcripts of two H77 full-length cDNA clones, named H77C (24) and H77 (25), were the first HCV genomes found to be infectious, as demonstrated by intrahepatic transfections in chimpanzees. Patient serum-derived H77 virus was reported to be able to replicate in lymphoblastoid cell lines at low levels (26) and could be passaged to chimpanzees (27). Subsequently, studies using the sequences of H77 contributed greatly to HCV research, for example, in the study of viral entry using HCV-like particles (28), the development of HCV pseudoparticles (29), the selection of highly permissive Huh7.5 cells (30), and the discovery of the importance of microRNA miR-122 in HCV replication (31). Efforts to propagate H77 in cell culture have been reported, using different cell lines (27, 32–34), and H77C harboring mutations derived from its replicon (designated H77-S) could release infectious virus particles (12, 35). Given the historical importance of HCV-1 and H77 isolates in HCV research, efficient infectious cell culture systems for these isolates would be very valuable tools for studies on HCV.
In this study, we developed robust and efficient infectious cell culture systems for HCV-1 and H77C by using the LSG mutations and approaches recently discovered for the TNcc and J6cc culture systems. An HCV-1 full-length genome with 17 amino acid changes, named HCV1cc, produced infectious virus particles with titers of ∼104.0 focus-forming units per milliliter (FFU/ml). By using novel mutations identified in TN full-length viruses and in HCV-1 and H77C recombinants expressing the NS5B-3′UTR from JFH1 (5-5A recombinants), we finally succeeded in developing an infectious culture system for H77, designated H77Ccc. The H77Ccc virus, with 19 amino acid changes, replicated efficiently, spread to most culture cells within 3 to 5 days after transfection and subsequent infection of Huh7.5 cells, and produced infectivity titers of ∼104.0 FFU/ml. HCV1cc and H77Ccc represent robust infectious cell culture systems for these key prototype strains that will contribute to HCV basic research and the development of better antiviral therapies and vaccines.
MATERIALS AND METHODS
Plasmids.
The HCV-1 clone HCV-1/SF9_A (GenBank accession number AF271632), which has 12 amino acid (aa) differences from the first reported HCV-1 sequence (M62321) (6, 36), was shown to be infectious in chimpanzees (36) and thus was selected for this study. The HCV-1/SF9_A genome with the LSG (F1464L, A1672S, and D2979G) and TNcc-derived mutations (9) was synthesized (GenScript) and assembled into a pGEM-9zf(−) vector containing the T7 promoter for initiation of in vitro transcription immediately upstream of the 5′UTR and an XbaI cleavage site at the end of the HCV genome (Promega), which was previously used in pCV-H77C (24). Other mutations were introduced by fusion PCR or by site-directed mutagenesis using the QuikChange II XL kit (Agilent Technologies). The junction of the HCV-1 NS5A and JFH1 NS5B-3′UTR was synthesized (GenScript). For strain H77, we used the in vivo infectious clone pCV-H77C (GenBank accession number AF011751) (24). The LSG and other mutations were introduced into H77C by fusion PCR or site-directed mutagenesis using the QuikChange II XL kit. All final plasmid preparations were confirmed by sequence analysis spanning the T7 promoter and the entire HCV genome (Macrogen).
Transfection and infection of Huh7.5 cells.
The human hepatoma cell line Huh7.5 was maintained as described previously (37, 38). Cells were plated in 6-well plates (∼3.5 × 105 cells/well) ∼24 h before RNA transfection or viral infection, reaching 80 to 90% confluence at the time of inoculation. RNA transfection and viral infection were performed as previously described (10). The transfected or infected cultures were incubated for ∼16 h and subcultured every 2 to 3 days; culture supernatant was collected, filtered (0.45 μm), and stored at −80°C until analysis.
Analysis of HCV in cultured cells.
To monitor HCV infection in the transfected and infected cultures, a combination of monoclonal anti-core antibody C7-50 (Enzo Life Sciences or Abcam) and the anti-NS5A antibody 9E10 (38) was used for immunostaining, as previously described (10, 37). The percentage of HCV antigen-positive cells in the culture was determined with fluorescence microscopy. Culture supernatants were collected when 80% of cells were HCV antigen positive (peak infection), and HCV infectivity titers were determined by an FFU assay using a combination of C7-50 and 9E10, as previously described (9, 10, 37). Full-length adapted HCV-1 and H77C viruses showed slightly lower intensity in staining than the positive-control virus, J65′UTR-NS2/JFH1 (39). The number of FFU was automatically determined with an ImmunoSpot Series 5 UV analyzer with customized software (CTL Europe GmbH) (40, 41). HCV RNA titers in the culture supernatant were determined using the real-time reverse transcription (RT)-PCR TaqMan method (37). Core antigen levels were determined by the Architect HCV antigen (Ag) detection system (Abbott) following the manufacturer's instructions. Whole ORF sequences of passaged viruses were determined using procedures previously described for the sequencing of H77C and JFH1 genomes (37, 42).
Determination of intracellular and extracellular HCV core levels and infectivity titers.
For single-cycle production assays, an Huh7-derived CD81-deficient cell line, S29 (43), was transfected with HCV RNA, and intracellular HCV core levels were measured 4 and 48 h posttransfection. Additionally, intracellular and extracellular HCV infectivity titers, as well as extracellular HCV core concentration, were determined 48 h posttransfection, as described previously (44, 45). Briefly, for this assay, S29 cells were seeded in 6-well plates (∼3.5 × 105 cells/well) 24 h prior to transfection. Plasmids of the different full-length clones were digested with XbaI (New England BioLabs [NEB]) for linearization and treated with mung bean nuclease (NEB). HCV RNA was generated by in vitro transcription using T7 RNA polymerase (Promega). In vitro transcripts were then digested with the RNase-free DNase set (Qiagen) and purified with the RNeasy MinElute Cleanup kit (Qiagen). RNA was quantified using spectrophotometry (NanoDrop), and 10 μg of RNA was used for transfection with Lipofectamine 2000 (Invitrogen). Transfection was performed in duplicate, and the transfection medium was replaced by complete Dulbecco's modified Eagle medium (DMEM) after 4 h in one of the replicate wells, while cells from the other replicate well were harvested for determination of intracellular HCV core levels. Briefly, cells were rinsed with phosphate-buffered saline (PBS) and resuspended in radioimmunoprecipitation assay (RIPA) buffer (Pierce) supplemented with protease inhibitors (Calbiochem). Samples were stored at −80°C until analysis. Prior to analysis, cell lysates were cleared by centrifugation at 14,000 rpm for 15 min and supernatants were transferred to a new tube. The same procedure was used to harvest cells at 48 h posttransfection. At this time point, supernatants were collected and filtered for determination of extracellular core levels and infectivity titrations, as described above. Both intracellular and extracellular core antigen levels were determined by the Architect HCV Ag detection system (Abbott) according to the manufacturer's instructions. For intracellular infectivity titers, cells were harvested and washed with PBS and then resuspended in complete DMEM and subjected to three cycles of freeze-thaw to release intracellular virus particles. Specific dilutions were analyzed in triplicate for HCV infectivity titers, and the FFU were determined using an ImmunoSpot Series 5 UV analyzer with customized software (CTL Europe GmbH) (40, 41) and confirmed by manual count.
Western blotting.
Intracellular HCV core in transfected S29 cells was also visualized by Western blotting. Briefly, S29 transfected cell lysates (the same sample as the one used for determination of core with the Architect detection system) were subjected to protein denaturation at 70°C for 10 min in the presence of NuPAGE sample reducing agent (Invitrogen) and NuPAGE LDS sample loading buffer (Invitrogen). Samples were run through 10% Bis-Tris SDS-polyacrylamide precasted gels (Invitrogen) for 1 h 30 min at 150 V. Afterwards, separated proteins were transferred to a Hybond-P polyvinylidene difluoride (PVDF) membrane (GE Healthcare Amersham) by wet electroblotting (XCell SureLock minicell; Invitrogen) at constant current for 1 h. Membranes were then washed with PBS plus 1% Tween 20 (PBS-T) and blocked with PBS plus 1% Tween 20 and 3% bovine serum albumin for 1 h. Blocked membranes were incubated overnight at 4°C with anti-HCV core C7-50 or anti-β-actin (Santa Cruz Biotechnology) with gentle rocking. Immunoblotting was followed by washes with PBS-T and 1 h of incubation with enhanced chemiluminescence (ECL) sheep anti-mouse IgG horseradish peroxidase-linked whole antibody (GE Healthcare Amersham). After washing, membranes were developed by chemiluminescence using Signal West Femto maximum-sensitivity substrate (Pierce) and visualized with the AutoChemi system (UVP Bio-Imaging Systems).
Nucleotide sequence accession numbers.
Sequences have been submitted to GenBank under accession numbers KP098532 (HCV1cc) and KP098533 (H77Ccc).
RESULTS
Adaptation of an HCV-1 5′UTR-NS5A (5-5A) recombinant leads to identification of the S399F mutation.
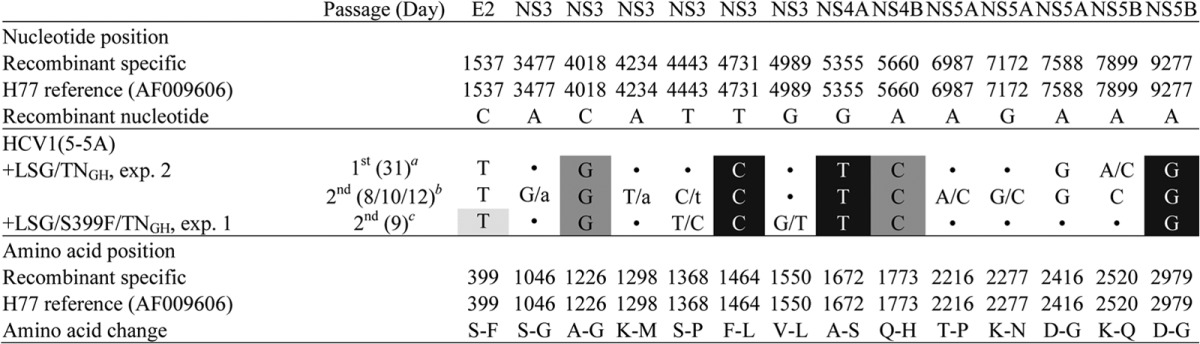
We previously identified the LSG mutations (F1464L, A1672S, and D2979G), which permitted development of full-length HCV infectious culture systems for genotypes 1a (TNcc) (9), 2a (J6cc) (10), and 2b (J8cc, DH8cc, and DH10cc) (10, 11), as well as 5-5A recombinants with JFH1 NS5B-3′UTR for genotypes 3a(S52), 4a(ED43), 5a(SA13), and 6a(HK6a) (46). In this study, we initially attempted to use the LSG mutations and a similar approach previously applied to J6cc and TNcc cultures to generate an HCV-1 infectious culture system. We selected the in vivo infectious clone HCV-1/SF9_A, a genome with 12 aa differences in comparison to the first reported HCV-1 sequence (M62321) (16, 36). The HCV-1/SF9_A clone shares nucleotide sequence identities of 96% and 95% with genotype 1a infectious clones H77C (24) and TN (47), respectively. Additionally, in our previously reported infectious J6cc and TNcc cell culture systems, we demonstrated that culture adaptation of 5-5A recombinants can lead to the identification of mutations critical for replication of full-length HCV genomes (9, 10). Based on these prior findings, we constructed an HCV-1 5-5A recombinant containing LSG substitutions, HCV1(5-5A)_LSG (Fig. 1A), and tested its viability by RNA transfection of Huh7.5 cells. In two independent transfections, HCV core and NS5A antigens were detected in <1% of cells at day 1, but spread of infection was not observed after 45 and 56 days of follow-up. Therefore, we concluded that the genome was viable but highly attenuated. We previously showed that combination of A1226G (NS3 helicase, NS3 aa position 200) and Q1773H (NS4B aa 62) could efficiently enhance the viability of TN and H77C 5-5A recombinants (9, 46), and they were both included in the TNcc recombinant (9). Thus, we added A1226G/Q1773H, designated TNGH, into HCV1(5-5A)_LSG (Fig. 1A). Culture with HCV1(5-5A)_LSG/TNGH showed 25% HCV-positive cells at day 1 in two transfection replicates, and the infection spread to ≥80% of the cultured cells (peak infection) at day 5 posttransfection (Table 1). However, titers of transfected cultures were below the detection limit (<102.4 FFU/ml). Transfection supernatants could be passaged to naive Huh7.5 cells, and in the first passage the infectivity titers reached 104.0 FFU/ml (Table 1). We continued passaging one of the viruses, and the second-passage recovered virus reached 104.5 FFU/ml (Table 1, experiment 2). Sequence analysis of the ORF of first- and second-passage viruses revealed that the engineered mutations were maintained and that two additional complete changes had emerged, S399F in the hypervariable region 1 (HVR1) of E2 and D2416G in the NS5A domain III (Table 2). Interestingly, F399 was also found in the originally published HCV-1 sequences (M62321 [16] and AF387806 [48]). To determine the effects of the adaptive S399F mutation, we engineered it into the HCV1(5-5A)_LSG/TNGH recombinant (Fig. 1A). HCV1(5-5A)_LSG/S399F/TNGH showed efficient viral replication with 60% of HCV-positive cells at day 1, viral spread to most cultured cells at day 4, and peak infectivity titers of 102.7 and 103.0 FFU/ml at days 8 and 12 in two transfections (Fig. 1B and Table 1), indicating that S399F could enhance virus spread and infectivity. Collected culture supernatant from the two transfections was passaged to naive Huh7.5 cells. In the first passage, the peak infectivity titers increased to 103.8 and 103.9 FFU/ml and in the second passage to 104.4 and 104.2 FFU/ml (Table 1). Sequence analysis of one of the second-passage viruses revealed that the engineered mutations were maintained and that two additional partial changes had emerged (Table 2). Taken together, these results indicate that the combination of LSG, S399F, and TN-derived A1226G/Q1773H mutations permitted the HCV-1 5-5A recombinant to efficiently grow in Huh7.5 cells.
FIG 1.

Viability of HCV-1 5′UTR-NS5A (5-5A) and full-length recombinants in Huh7.5 cells. (A) Schematic diagrams of HCV genomes. LSG mutations (F1464L/A1672S/D2979G) previously identified from J6 and JFH1 recombinants and Y2981F (designated F) mutation are highlighted by black dots, additional TNcc-adaptive mutations (TNm) are indicated by gray dots, S399F from HCV-1 5-5A recombinant is indicated by a circle, and eight mutations (8m) identified from passaged full-length HCV-1 viruses are indicated by broken circles. (B to D) RNA transcripts of HCV-1 5-5A and full-length recombinants with the indicated mutations were transfected into Huh7.5 cells, and cultures were monitored for HCV core/NS5A antigens by immunostaining. HCV infectivity titers in culture supernatant collected at the days indicated, after ≥80% of the cells were found to be HCV antigen positive, were determined by FFU assays and are shown as the means from triplicate infections ± standard errors of the means (SEM). J65′UTR-NS2/JFH1 (39) was used as a positive control. LSGF, F1464L/A1672S/D2979G/Y2981F. TNGH, combination of two TNcc-adaptive mutations A1226G/Q1773H; TNm, combination of four TNcc-adaptive mutations A1226G/Q1773H/N1927T/F2994S; HCV1cc, HCV1_LSGF/S399F/TNm/8m, in which 8m indicates the mutations A970T/I1312V/I1326V/V2198A/I2268T/C2419R/E2622D/A2919T.
TABLE 1.
Characteristics of the HCV-1 5′UTR-NS5A (5-5A) recombinant and full-length viruses in Huh7.5 cell culturesa
| Virus and experiment no. | Transfection |
First passage |
Second passage |
||||
|---|---|---|---|---|---|---|---|
| Day with ≥80% of cells infected | Peak log10 FFU/ml (day) | Day with ≥80% of cells infected | Peak log10 FFU/ml (day) | Day with ≥80% of cells infected | Peak log10 FFU/ml (day) | Peak log10 IU/ml | |
| HCV1(5-5A) | |||||||
| +LSG/TNGH, exp. 1 | 5 | <2.4 | 38 | 4.0 (42) | ND | — | — |
| +LSG/TNGH, exp. 2b | 5 | <2.4 | 27 | 4.0 (31) | 8 | 4.5 (8/10/12)c | ND |
| +LSG/S399F/TNGH, exp. 1 | 4 | 2.7 (8) | 16 | 3.8 (18) | 5 | 4.4 (7) | ND |
| +LSG/S399F/TNGH, exp. 2 | 4 | 3.0 (12) | 16 | 3.9 (16) | 5 | 4.2 (7) | ND |
| Full-length HCV1 | |||||||
| +LSGF/S399F/TNmd | 26 | 3.3 (28) | 7 | 3.4 (11) | 7 | 4.3 (9) | 8.3 |
| +LSGF/S399F/TNm/8 m (HCV1cc) | 3 | 3.8 (5)e | 5 | 3.8 (7) | 7 | 3.8 (9) | 7.5 |
| +LSGF/S399F/TNm/4 m | 4 | 3.4 (13) | 11 | 3.5 (13) | 11 | 3.9 (15) | ND |
One milliliter of transfection- or first-passage-recovered supernatant was used for subsequent infection of cells grown in 6-well-plates. Abbreviations and symbols: TNGH, mutations A1226G/Q1773H; LSG, mutations F1464L/A1672S/D2979G; LSGF, mutations F1464L/A1672S/D2979G/Y2981F; TNm, mutations A1226G/Q1773H/N1927T/F2994S; 8 m, mutations A970T/I1312V/I1326V/V2198A/I2268T/C2419R/E2622D/A2919T; 4 m, mutations A970T/I1312V/C2419R/A2919T; ND, not done; exp., experiment; —, not applicable.
The first- and second-passage viruses were sequenced (Table 2).
The viruses collected at days 8, 10, and 12 were pooled and used for analysis.
The third-passage virus reached 4.2 log10 FFU/ml at day 6.
In two other independent transfections, HCV1cc produced 3.8 to 4.0 log10 FFU/ml at day 5.
TABLE 2.
The virus had a noncoding nucleotide change, T5750C.
The viruses collected at days 8, 10, and 12 were pooled and used for analysis; the recovered sequence contained noncoding nucleotide changes T4868C/t, T5750C, and T6251T/C.
The virus acquired a noncoding nucleotide change, C6131C/T.
Shadings indicate the engineered mutations; LSG mutations (F1464L/A1672S/D2979G) are indicated in white letters with black background, TN-derived mutations are in dark shading, and S399F identified in this study is in light shading. Coding changes are shown; the capital/capital letters indicate a 50/50 nucleotide quasispecies, while the capital/lowercase letters indicate a dominant/minor ratio. Dots indicate identity with the original plasmid sequence. TNGH, mutations A1226G/Q1773H; exp., experiment.
Development of an efficient full-length infectious culture system for HCV-1.
We previously demonstrated that LSG plus Y2981F (designated “F” mutation, NS5B aa 561 [49]) was important for in vitro viability of the TNcc (9) and full-length J6 (10) viruses. LSG plus S399F/TNGH could efficiently adapt the HCV-1 5-5A recombinant, in which S399F enhanced virus spread and infectivity (Fig. 1B and Table 1). Thus, here we attempted to combine these mutations and to test their adaptation potential in the full-length HCV-1 genome. For that purpose, we generated HCV-1 with LSGF/S399F/TNGH, LSGF/S399F/TNm (where TNm stands for the four TNcc adaptive mutations A1226G/Q1773H/N1927T/F2994S [9]), or LSGF/TNm (Fig. 1A). Replication was not observed for HCV1_LSGF in transfected cultures for up to 20 days of follow-up. Cultures transfected with HCV1_LSGF/S399F/TNGH showed HCV-positive cells beginning from day 4 but continued to have <1% of HCV-positive cells for up to 20 days of follow-up, with no evidence of viral spread. The HCV1_LSGF/TNm culture showed 1% HCV-positive cells at day 1 but no evidence of viral spread for up to 41 days. In contrast, the HCV1_LSGF/S399F/TNm culture showed 10% HCV-infected cells at day 1, reached the peak of infection after 26 days, and released HCV infectivity titers of 103.3 FFU/ml (Fig. 1C), indicating that S399F mediated the viral spread of full-length HCV1_LSGF/TNm and that N1927T and F2994S also contributed to a more efficient viral propagation. After passages to naive Huh7.5 cells, the first-, second-, and third-passage HCV1_LSGF/S399F/TNm showed peak infectivity titers of 103.4, 104.3, and 104.2 FFU/ml, respectively (Table 1). ORF sequence analysis of the second- and third-passage viruses revealed that the introduced mutations were all maintained and that eight additional amino acid changes (A970T, I1312V, I1326V, V2198A, I2268T, C2419R, E2622D, and A2919T [designated “8m”]) had emerged (Table 3).
TABLE 3.

The constructed plasmid contained a noncoding nucleotide change, C3210A, which was maintained in the passage-recovered viruses. The second- and third-passage viruses acquired noncoding nucleotide changes T2408C and C2765T.
No noncoding change was found in ORF sequence analysis.
Shadings indicate the engineered mutations; LSGF mutations are indicated in white letters with black background, TNcc-derived mutations (A1226G/Q1773H/N1927T/F2994S) (TNm) are in dark shading, and mutations identified in this study are in light shading. The in vivo viable HCV-1/SF9_A (GenBank accession number AF271632) (36) engineered with LSGF (F1464L/A1672S/D2979G/Y2981F), S399F identified from HCV-1 5-5A recombinant (Table 2), and TNm was adapted for growth in transfected Huh7.5 cells. Eight mutations (A970T/I1312V/I1326V/V2198A/I2268T/C2419R/E2622D/A2919T, designated 8m) were identified from passaged HCV1_LSGF/S399F/TNm viruses and were engineered back to the genome to make HCV1_LSGF/S399F/TNm/8m, designated HCV1cc. The HCV1cc genome, containing 17 mutations, showed efficient virus spread in transfection cultures and released infectious virus particles with HCV infectivity titers of 103.8 FFU/ml (Fig. 1). An HCV-1 full-length virus, HCV1_LSGF/S399F/TNm/4m (4m consists of mutations A970T/I1312V/C2419R/A2919T), replicated efficiently in the culture, with infectivity levels close to those of HCV1cc. The HCV-1 full-length viruses were passaged to naive Huh7.5 cells, the viruses spread efficiently, and the culture supernatant was collected at the indicated time points for sequence analysis.
In order to generate an efficient HCV-1 full-length virus, we tested the importance of the mutations identified from passaged HCV1_LSGF/S399F/TNm viruses (see above). We introduced individual mutations or a combination of the 8m mutations into the HCV1_LSGF/S399F/TNm recombinant. At day 1 after transfection, the recombinants with single mutation A970T, I1312V, I1326V, V2198A, I2268T, C2419R, or E2622D showed a low number of HCV-positive cells, and in addition no evidence of viral spread was observed after 1 week of follow-up. In contrast, culture containing the recombinant with A2919T showed 40% HCV-positive cells at day 1 and reached 80% at day 4, remaining at this percentage for 2 weeks, albeit with low HCV infectivity titers (<102.4 FFU/ml). These results indicated that A2919T may play a greater role than the remaining seven mutations in adaptation of HCV1_LSGF/S399F/TNm. Culture containing the genome with all mutations combined, designated HCV1_LSGF/S399F/TNm/8m, showed 55% HCV-positive cells at day 1 in two transfection replicates and released peak HCV infectivity titers of 103.8 to 104.0 FFU/ml (Fig. 1D and Table 1). The transfection-derived virus showed efficient spread in the first and second passages, and both-passage-recovered viruses had peak infectivity titers of 103.8 FFU/ml (Table 1). ORF sequence analysis of the second-passage virus demonstrated that all the engineered mutations were maintained and that no additional mutations were present. Taken together, these results indicate that the combination of 8m efficiently enhances the replication and viral production of HCV-1, resulting in an efficient full-length HCV-1 infectious culture system. We therefore designated HCV1_LSGF/S399F/TNm/8m “HCV1cc” (for “HCV-1 cell culture-derived”).
Mutations important for the viability of HCV1cc.
As addition of the 8m mutations led to a robust HCV1_LSGF/S399F/TNm virus (Fig. 1C and D), we next examined which of the mutations primarily contributed to efficient viral viability. For this purpose, we mutated each of the 8m mutations individually back to the wild-type sequence and tested the effect on the viability of the virus after transfection of Huh7.5 cells (Fig. 2A and B). Compared to HCV1cc, viruses with −A970T (adaptive mutation A970T reverted to the wild type), −I1312V, and −C2419R were attenuated and did not produce detectable HCV infectivity titers until day 8 after transfection, at which time point their titers were approximately 7.9-, 1.7-, and 2.0-fold lower than that of HCV1cc, respectively. Moreover, the virus with −A2919T was highly attenuated, as HCV titers were first detected on day 13. Additionally, peak infectivity titers for both −A970T and −A2919T viruses were slightly lower than for the remaining viruses (Fig. 2A), specifically 2.8- and 3.2-fold lower than for HCV1cc. When analyzing secreted core antigen levels in the supernatants of transfected cells, we observed that the virus with mutation −A2919T, the most attenuated, had core levels that were 4.7- to 21-fold lower than those for HCV1cc at the same time points (Fig. 2B), whereas other viruses showed core levels that varied between 0.5- and 3-fold compared to those of HCV1cc at the same time points. Together, these results indicate that the absence of each of mutations A970T, I1312V, C2419R, and A2919T affects the viability of HCV1cc, with the absence of A2919T having the greatest effect.
FIG 2.

Effects of individual adaptive mutations on the viability of HCV1cc. RNA transcripts of HCV1cc and HCV1cc with each of eight putative adaptive mutations (named 8m) mutated back to the wild-type sequence were transfected into Huh7.5 cells. J65′UTR-NS2/JFH1 (39) was used as a positive control. Culture supernatants were collected at the days indicated. (A) HCV infectivity titers (FFU/ml) in supernatants from cultures with ≥80% of the cells found to be HCV antigen positive by immunostaining are shown as the means from triplicate infections ± SEM. *, not determined; #, FFU titers were below the detection limit of 102.4 FFU/ml. (B) HCV supernatant core antigen levels as determined by the Architect HCV Ag detection system (Abbott). HCV1cc, HCV1_LSGF/S399F/TNm/8m; LSGF, mutations F1464L/A1672S/D2979G/Y2981F; TNm, mutations A1226G/Q1773H/N1927T/F2994S; 8m, mutations A970T/I1312V/I1326V/V2198A/I2268T/C2419R/E2622D/A2919T.
Next, we explored whether the four mutations A970T, I1312V, C2419R, and A2919T (designated “4m”), singly or combined, were sufficient to adapt HCV1_LSGF/S399F/TNm to growth comparable to that of HCV1cc. Since A2919T played a major role in the viability of HCV1cc (Fig. 2A), in addition to the 4m, we tested HCV1_LSGF/S399F/TNm with A2919T plus any combinations of the other three mutations, namely, A970T/A2919T, I1312V/A2919T, C2419R/A2919T, A970T/I1312V/A2919T (designated “3m”), A970T/C2419R/A2919T, and I1312V/C2419R/A2919T. After transfection of Huh7.5 cells, only the viruses containing 3m and 4m spread to ≥80% of culture cells within 6 days (Fig. 3A); the viruses with other combinations did not spread. HCV1_LSGF/S399F/TNm/4m produced detectable infectivity titers from day 4, though the HCV peak titers were slightly lower than those of HCV1cc (Fig. 3A), whereas HCV1_LSGF/S399F/TNm/3m did not produce detectable HCV infectivity titers until day 13. However, supernatant core levels of both 3m and 4m viruses were similar to those of HCV1cc at each time point (Fig. 3B). From these results, we conclude that the 4m mutations are the minimum required for efficient production of infectious viruses of the HCV1_LSGF/S399F/TNm genome in vitro.
FIG 3.

Identification of adaptive mutations sufficient for the viability of HCV-1 full-length genomes. RNA transcripts from HCV1_LSGF/S399F/TNm recombinant with A970T/A2919T, I1312V/A2919T, C2419R/A2919T, A970T/I1312V/A2919T (designated 3m), A970T/C2419R/A2919T, I1312V/C2419R/A2919T, and A970T/I1312V/C2419R/A2919T (4m) were transfected into Huh7.5 cells. The recombinants with A970T/A2919T, I1312V/A2919T, C2419R/A2919T, A970T/C2419R/A2919T, and I1312V/C2419R/A2919T did not spread in the transfection cultures. In contrast, recombinants with 3m or 4m spread to ≥80% of the cells during the first week. J65′UTR-NS2/JFH1 and HCV1cc were used as controls. Culture supernatants were collected at the indicated days. (A) Supernatant HCV infectivity titers (FFU/ml), shown as means from triplicate infections ± SEM. *, not determined; #, FFU titers were below the detection limit of 102.4 FFU/ml. (B) HCV supernatant core antigen levels as determined by the Architect HCV Ag detection system (Abbott). LSGF, mutations F1464L/A1672S/D2979G/Y2981F; TNm, mutations A1226G/Q1773H/N1927T/F2994S; 3m, mutations A970T/I1312V/A2919T; 4m, mutations A970T/I1312V/C2419R/A2919T.
Effect of HCV-1 adaptive mutations on viral replication, assembly, and release.
To address the role of the identified adaptive mutations in replication, assembly, and release of HCV1cc, we performed a single-cycle production assay using Huh7-derived S29 cells, a cell line that is deficient for the HCV entry receptor CD81 (43). Since the A970T/I1312V/C2419R/A2919T (4m) mutations played a major role in the viability of HCV1cc, among which A970T and A2919T seemed to have a greater effect than the others (Fig. 2A), we tested the effect of A970T/A2919T with I1312V, C2419R, or I1312V/C2419R in the HCV1_LSGF/S399F/TNm backbone. After transfection of S29 cells, the intracellular and extracellular infectivity titers (Fig. 4A) and corresponding HCV core antigen levels (Fig. 4B and C) were determined. In addition, the intracellular HCV core levels were visualized by Western blotting (Fig. 4D), and the results agreed with the measurements obtained by using the Architect HCV Ag detection system (Fig. 4B and D).
FIG 4.

Functional analysis of the role of HCV1cc adaptive mutations in the HCV life cycle. RNA transcripts from the indicated recombinants were transfected into HCV entry-deficient S29 cells (43). All clones contained LSGF/S399F/TNm plus specific mutations as indicated under each bar graph. Cell lysates were collected at 4 and 48 h, and culture supernatants were collected at 48 h. Both intracellular and extracellular HCV infectivity titers and core levels were determined at 48 h after transfection. Intracellular core at 4 h was also determined as a measure of replication-independent genome translation following transfection and used to normalize the 48-h values. HCV1_LSGF and HCV1_LSGF/TNm were analyzed in a separate experiment (not shown); no intracellular and extracellular infectivity titers were detected, and intracellular and extracellular core levels were lower than for HCV1_LSGF/S399F/TNm. A replication-incompetent form of J6/JFH1 was included in each experiment (J6/JFH1-GND). (A) Intracellular and extracellular infectivity titers. #, no FFU detected by manual count. Values are expressed as log10 FFU/ml for extracellular titers and as log10 FFU/well for intracellular infectivity titers. (B) Intracellular core levels; the HCV core level at 48 h was normalized in percentage to the level at 4 h. (C) Extracellular core levels, expressed as log10 Fmol/liter. (D) Western blots. Cell lysates harvested 48 h posttransfection were separated through acrylamide gels, and proteins were transferred to PVDF membranes (see Materials and Methods). Immunoblotting was performed with anti-HCV core C7-50 for detection of HCV core and anti-β-actin for detection of host cell actin.
As expected, in the absence of 4m mutations, HCV1_LSGF/S399F/TNm failed to produce detectable intracellular and extracellular infectivity titers (Fig. 4A), and intracellular core levels were barely over those of the replication-deficient control, J6/JFH1-GND (Fig. 4B). In a separate experiment, we also tested HCV1_LSGF and HCV1_LSGF/TNm in parallel with HCV1cc. Likewise, HCV1_LSGF/S399F/TNm, HCV1_LSGF, and HCV1_LSGF/TNm failed to produce detectable levels of both intracellular and extracellular infectivity titers and showed a low level of intracellular and extracellular core antigen 48 h after transfection (data not shown). Addition of A970T/A2919T into the HCV1_LSGF/S399F/TNm genome had only a minor effect on core levels and no effect on infectivity titers. However, addition of I1312V or C2419R to A970T/A2919T mutations led to detectable intracellular infectious titers and an increase in extracellular core level; however, no extracellular infectious viruses were detected (Fig. 4A to C). These results suggest that both I1312V and C2419R played an important role in the assembly of infectious virus particles but had no or insufficient effect on virus release. Interestingly, when both I1312V and C2419R were combined with A970T/A2919T, thus making the genome with the 4m mutations, virus release of infectious viral particles was enhanced and the virus produced extracellular infectivity titers comparable to those of HCV1cc (Fig. 4A), with an increase in extracellular core levels (Fig. 4C). Based on the results of the single-cycle production assay, we concluded that the 4m mutations (A970T/I1312V/C2419R/A2919T), when added into the HCV1_LSGF/S399F/TNm genome, permit an efficient completion of the viral life cycle and that the combination of I1312V/C2419R apparently is required for efficient virus release of infectious viral particles. It should also be noted that although the 4m mutations were essential for the viability of HCV1_LSGF/S399F/TNm, the combination of the 8m mutations further increased intracellular core levels (Fig. 4B) and intracellular infectivity titers (Fig. 4A), thus suggesting that all 8m mutations further increased replication and virus assembly of HCV-1.
Development of an efficient full-length infectious culture system for H77.
After our success in adapting genotype 1a strains TN and HCV-1 for efficient growth in vitro, we wanted to determine whether the key adaptive mutations that we had uncovered could be used to adapt H77C, another important HCV prototype strain. We recently demonstrated that the LSGF and LSGF/TNm mutations were not sufficient to adapt the H77C genome after transfection in Huh7.5 cells (46). However, the LSG and TN(5-5A)-adaptive mutations A1226G/Q1773H could efficiently adapt an H77C 5-5A recombinant (designated “1a[H77]_LSG/A1226G/Q1773H” in reference 46). After passage, the virus acquired a partial change in the NS3 helicase, S1368P (46), which became dominant after the second passage. Interestingly, this mutation was also identified here in the two cell culture-adapted HCV-1 5-5A viruses (Table 2).
We previously found that a TN genome containing only LSGF, designated TN_LSGF, was nonviable in a single transfection (9). However, when we repeated transfections of the TN_LGSF for this study, we were able to obtain viral replication in one of three transfections. In that unique transfection, TN-LSGF showed a low number of HCV-positive cells at day 4, with infection spreading to ≥80% of cultured cells after 28 days of follow-up. The transfection-derived TN_LSGF culture supernatant had infectivity titers of 104.0 FFU/ml, and the titers reached 104.7 and 104.9 FFU/ml after the first and second passages, respectively. Among other mutations, viruses recovered from both passages acquired the change F2994R, which may be of importance since F2994 was changed to serine (F2994S) in our previously reported TNcc system (9).
Based on this new information, we hypothesized that combining mutations S1368P and F2994R might permit replication of an H77C genome with LSGF/TNm, and therefore we generated H77C_LSGF/TNmr/S1368P (TNmr indicates that F2994S of TNm was replaced with F2994R). After transfection of Huh7.5 cells, the H77C_LSGF/TNmr/S1368P showed a few HCV-positive cells for up to 3 months, but finally the infection spread to ≥80% of cells at day 96, producing HCV peak infectivity titers of 103.5 FFU/ml (Fig. 5A). First- and second-passage viruses spread to ≥80% of cells within 8 to 10 days and reached peak HCV infectivity titers of 103.5 and 104.2 FFU/ml (Fig. 5A). ORF sequence analysis revealed that viruses had acquired 10 complete amino acid changes, M345T, A828V, L864R, K1052R, V1663A, G1909S, M2105V, S2354G, V2417A, and V2431I, which we designated “10m” (Table 4). We therefore engineered the 10m into the H77C_LSGF/TNmr/S1368P clone. After transfection of Huh7.5 cells, H77C_LSGF/TNmr/S1368P/10m spread to ≥80% of culture cells within 3 days. The culture supernatant had a peak infectivity titer of 103.5 FFU/ml. The first- and second-passage viruses reached infectivity titers of 104.4 and 103.8 FFU/ml, respectively (Fig. 5B). In ORF sequencing analysis of the second-passage virus, all the engineered mutations were maintained, and no additional changes were found (Table 4). Hence, we had developed a robust infectious culture system for a full-length H77C clone, with efficient virus spread after transfection and subsequent infection, and we named the adapted recombinant “H77Ccc.”
FIG 5.

Viability of adapted H77C in Huh7.5 cells. RNA transcripts of H77C full-length recombinants with the indicated mutations were transfected into Huh7.5 cells, and cultures were monitored for HCV core/NS5A antigens by immunostaining. Cell-free transfection supernatants collected from peak infection were passaged to naive Huh7.5 cells (first passage), and after viral spread, the culture supernatant of the first passage was subsequently used to infect naive Huh7.5 cells (second passage). HCV infectivity titers in culture supernatant are shown as mean FFU/ml from triplicate infections ± SEM. (A) Transfection and passage of H77 full-length virus H77C_LSGF/TNmr/S1368P. (B) Transfection and passage of H77Ccc. J6/JFH1 was used as a positive control (38). H77Ccc, H77C_LSGF/TNmr/S1368P/10m; LSGF, mutations F1464L/A1672S/D2979G/Y2981F; TNmr, mutations A1226G/Q1773H/N1927T/F2994R; 10m, mutations M345T/A828V/L864R/K1052R/V1663A/G1909S/M2105V/S2354G/V2417A/V2431I.
TABLE 4.

Mutation S1368P was previously identified from H77C (46) and HCV-1 (Table 2) 5-5A recombinant viruses; the first- and second-passage H77C_LSGF/TNmr/S1368P viruses both acquired noncoding nucleotide changes A2558G, A3089G, G3860A, C4403T, T4904C, A6437G, A6713G, A7727G, A8804G, and T9227C.
Nucleotides at positions 9321 and 9322 are in the same codon for the F2994R change.
The in vivo-viable H77C genome (24) engineered with 10 nucleotide changes (resulting in nine amino acid changes, as nucleotides T9321 and T9322 are in the same codon; see footnote b above), named H77C_LSGF/TNmr/S1368P (see footnote a above), had low-level replication after transfection of Huh7.5 cells and spread to most culture cells at day 96. Ten mutations (10 m) were identified from H77C_LSGF/TNmr/S1368P and engineered into the genome to make H77Ccc. The H77Ccc genome, which had a total of 19 amino acid changes, showed efficient virus spread in transfection and infection cultures and released infectious virus particles with HCV infectivity titers of 103.5 to 104.4 FFU/ml (Fig. 5). The H77C full-length viruses were passaged to naive Huh7.5 cells, the viruses spread to ≥80% of culture cells within 3 days for H77Ccc and within 8 days for H77C_LSGF/TNmr/S1368P, and then the culture supernatants were collected for sequence analysis. Shadings indicate the engineered mutations; LSGF mutations (LSG, F1464L/A1672S/D2979G; F, Y2981F) are indicated in white letters with a black background, TNcc-adaptive mutations (TNm) are in dark shading, and mutations identified in this study are in light shading. H77Ccc, H77C_LSGF/TNmr/S1368P/10m; LSGF, mutations F1464L/A1672S/D2979G/Y2981F; TNmr, mutations A1226G/Q1773H/N1927T/F2994R (the F2994R mutation was identified from an LSGF-adapted TN full-length virus; see Results for details); 10m, mutations M345T/A828V/L864R/K1052R/V1663A/G1909S/M2105V/S2354G/V2417A/V2431I.
DISCUSSION
In this study, we developed highly efficient cell culture systems for full-length HCV prototype strains HCV-1 and H77 (both genotype 1a strains), named HCV1cc and H77Ccc, by using mutations and approaches previously developed for the J6cc (2a) and TNcc (1a) infectious clones (9, 10). HCV1cc and H77Ccc replicated efficiently following RNA transfection of human hepatoma Huh7.5 cells and produced HCV infectivity titers of ∼104 FFU/ml, showing no additional amino acid changes after a second-round viral passage. Given the clinical significance of genotype 1 and the uniquely important role of these prototype strains in HCV research, the HCV1cc and H77Ccc systems will be of particular value and will provide useful tools for future studies of HCV.
After development of the first HCV infectious culture system based on the genotype 2a JFH1 strain (7), tremendous efforts have been made to propagate HCV isolates of other genotypes in culture. Genotype 1 viruses account for ∼60% of HCV infections worldwide; thus, culture systems for genotype 1 have great interest for HCV research. Recently, we identified three mutations, designated LSG (F1464L/A1672S/D2979G), initially through studies of genotype 2a J6-JFH1 recombinants (10). The LSG mutations were essential for the development of full-length in vitro infectious clones of HCV genotypes 1a (TNcc) (9), 2a (J6cc) (10), and 2b (J8cc, DH8cc, and DH10cc) (10, 11). Replication of 2a and 2b recombinants could be initiated by LSG alone, thus permitting further adaptation that led to robust infectious culture systems. We also showed that the LSG mutations played an important role in the adaptation of JFH1-based 5-5A recombinants of genotypes 1 to 6 (46). In this study, we further demonstrated that LSG combined with NS5B mutation Y2981F (49) (interestingly, most genotype 2b isolates naturally have F2981 [11]) has the potential to initiate adaptation of additional genotypes, such as the genotype 1a strain TN. It would be worth exploring in future studies whether LSG or LSGF per se can promote cell culture viability for other HCV isolates, as a first step in the process of cell culture adaptation. Our experience with the TN_LSGF genome also suggests that multiple transfections may be required to start cell culture adaptation of isolates with low levels of viral replication and that negative results from a single experiment can be misleading.
Adaptation of HCV-1 and H77C required additional mutations other than LSGF, therefore strengthening the notion that cell culture adaptation is highly influenced by the nature of the genome sequence. In order to start replication of HCV-1 and H77C, we combined LSG or LSGF with other mutations, TNm or TNmr mutations from the efficient in vitro infectious full-length TN clones, S399F from HCV1(5-5A) recombinants, and S1368P from H77C(5-5A) or HCV1(5-5A) recombinants (Table 2 and reference 46). This broad set of adaptive mutations may represent a valid evolutionary path leading to the culture adaptation of several additional genotype 1a strains, including strains showing high genetic diversity to TN, H77, and HCV-1 (47), and thus permitting the generation of a wide panel of infectious clones, which could be relevant for studies of strain-related genetic variability in HCV genotype 1a.
Some positions in the HCV genome are under different selective pressure in vivo and in vitro. In this study, the HVR1 amino acid S399 was changed to F399, which matched the originally reported HCV-1 sequence (16, 48). This change may indicate that although an HCV-1 clone with S399 was found viable in vivo (36), F399 may be a better residue for in vitro viability. A recent study showed that cell culture-adaptive mutations in H77-S with replicon-adaptive mutations reverted to wild-type residues over time in persistently infected chimpanzees (50). In addition, culture replication-enhancing mutations were found to prevent productive in vivo replication of the Con1 genome (51). This discrepancy in requirements for in vitro and in vivo viability may explain the difficulties in propagating HCV recombinants in cell culture, even for those found viable in vivo, as was the case for the original genomes of HCV-1, H77, and TN (24, 25, 36, 47). In a recent study by Yamane et al. (52), the natural resistance to lipid-induced peroxidase stress of JFH1 was found to be correlated with its robust replication capacity in cell culture. Moreover, the authors demonstrated that our highly efficient cell culture-adapted TNcc (9) similarly had resistance to lipid peroxidation. It would be of interest for future studies to explore if this interesting finding is also applicable to other cell culture-adapted genomes, including genomes not depending on the specific TN-derived adaptive mutations.
In addition to the mutations used for promoting initial viral replication and adaptation of HCV-1 and H77C, most of which we had already described, the final HCV1cc and H77Ccc clones contained a number of additional mutations; for instance, HCV1cc contained a total of 17 amino acid changes compared to the original HCV1/SF_A clone (36). We demonstrated that 4 of the additional HCV1cc amino acid changes were critical for efficient adaptation (A970T, I1312V, C2419R, and A2919T). Among those, A970T (aa 161 in NS2) is a highly conserved position (Los Alamos HCV Sequence Databases), and only 1 of 4,306 sequences has a different residue. C2419R is another highly conserved residue located carboxy-terminally in NS5A. On the other hand, amino acid 1312, in the NS3 helicase, is a polymorphic site. I1312 is present in genotypes 1a, 1b, 2a, and 6a, while V1312 is primarily found in genotypes 3 and 5. The fact that V1312 can be found in natural viral sequences shows that some cell culture-adaptive changes can be found in circulating viruses, albeit from other genotypes. Likewise, amino acid 2919 in NS5B is a variable position, and A, V, and T can be found in various genotypes. Among the remaining cell culture-adaptive mutations present in HCV1cc, position 1326 (aa 300 in NS3, helicase domain) contains either I or V depending on the genotype. HCV isolates have different amino acids in NS5A position 2198 (aa 226 in NS5A), including L, V, E, M, or Q, similarly to position 2268 (296 in NS5A, in the protein kinase R [PKR] binding domain), where V, I, P, L, and M can be found. Finally, at NS5B position 2622 (aa 202 in NS5B), isolates from the HCV database contain only E or D.
In previous studies, it was assumed that certain cell culture-adaptive mutations identified in JFH1-based intergenotypic recombinants compensated for incompatibilities due to the chimeric nature of those genomes. However, since many of those changes also emerged in cell culture-adapted full-length genomes, they are most likely related to the cell culture adaptation process. One of these changes is the HCV-1 adaptive mutation I1312V, which was also identified in a passaged full-length DH10 (2b) virus (11), and which had been previously shown to adapt JFH1-based core-NS2 recombinants (53). Another HCV-1 change, C2419R, was identified in a DH8_LSG (2b) full-length genome (11) and was observed for adaptation of a J6/JFH1-based recombinant expressing the NS5A from H77 and with p7 mutations (54).
To understand the role of adaptive mutations in various steps of the HCV1cc life cycle, we performed single-cycle production assays using S29 cells. The S29 cells were derived from Huh7 cells but are deficient in CD81 and thus support only HCV RNA replication and virus particle assembly and release but not infection (43). HCV-1 genomes containing only LSGF or LSGF/S399F/TNm failed to efficiently replicate or to produce detectable infectious particles in the S29 cell assays (Fig. 4A). These results agreed with observations after transfection of Huh7.5 cells, in which both viruses started to spread only after 26 days, and this culture spread was related to the emergence of additional mutations (Table 3). In line with this finding, we had previously shown that LSG alone did not lead to high HCV infectivity titers of the J6 full-length genome (10). In contrast, addition of the 8m mutations in HCV-1 with LSGF/S399F/TNm resulted in efficient replication and virus production with augmented supernatant infectivity titers in S29 cells (Fig. 4). These results suggest that these mutations mediated efficient viral replication, assembly, and release, which was also in agreement with the rapid virus spread of genomes containing 8m mutations, after transfection of Huh7.5 cells (Fig. 1D). The 4m (A970T/I1312V/C2419R/A2919T) mutations were sufficient to achieve efficient virus spread and release, although with slightly lower levels than those of HCV1cc. Therefore, 4m mutations apparently were the minimal set of mutations required to complete an efficient viral life cycle of HCV-1 with LSGF/S399F/TNm. Of the 4m mutations, the A970T/A2919T combination was not sufficient to confer efficient viral replication and assembly in S29 cells (Fig. 4); however, when combined with either C2419R or I1312V, replication and assembly were significantly augmented, and therefore we concluded that they played a major role in these processes (Fig. 4A). Additionally, for our experiments in Huh7.5 cells, changing T2919 back to wild-type A2919 significantly reduced viability of HCV1cc (Fig. 2A). Amino acid 2919 (NS5B 499) is in the thumb domain of the polymerase and has been related to viral replication (55). This position shows a certain level of polymorphism, and amino acid changes at this site have different effects, depending on the strain. In genotype 2a replicon systems, the V2919A change had no effect on JFH1 (56), but A2919V increased the replication of J6 10-fold (57). In addition, in the context of full-length genotype 2a systems, A2919V was recently described as a cell culture-adaptive mutation in PR63cc, having an effect in replication of this strain (15). Interestingly, when both I1312V and C2419R were combined with A970T/A2919T, extracellular infectivity titers were detectable (Fig. 4A), but an increase in supernatant titers was not associated with an increase in replication, therefore indicating that the combination of I1312V and C2419R may have an important specific role in the release of infectious viral particles. It will be of interest to elucidate, in future studies, whether the rescue of release is mediated uniquely through the interaction of these two positions, located in NS3 and NS5A, or by the recruitment of other viral or host proteins.
Given the historical importance of H77 in HCV research, much effort has been invested in propagating this strain in cell culture, in particular since infectious clones were developed in 1997 (24, 25).
Strain H77Ccc, developed in this study, showed efficient replication after RNA transfection of Huh7.5 cells, with infectivity titers of 103.6 FFU/ml (Fig. 5B). Importantly, H77Ccc spread rapidly after passage to naive Huh7.5 cells, and the first- and second-passage viruses reached peak of infection within 5 to 8 days and produced infectivity titers of 104.4 and 103.8 FFU/ml, respectively (Fig. 5B and Table 4). Therefore, H77Ccc represents a robust and efficient infectious cell culture system for HCV strain H77, with high replication levels after transfection and rapid spread in viral passage cultures.
It was initially found by Yi et al. that an H77C genome carrying mutations derived from its replicon system replicated at a low level in transfected Huh7.5 cells (12). Subsequently, this genome was improved to yield higher infectivity titers by introducing an additional mutation in E2 (H77S.3 [35]), and during the preparation of this paper a further adapted genome (H77D) that replicated and spread efficiently in cell culture was reported (52). Similarly to the adaptation process described in this study, H77D was generated by introducing our previously described adaptive TNcc mutations (9) into the H77S.3 backbone. Replication of H77S.3/LSGF/TNm was inhibited, but removal of an adaptive mutation from the original H77S.3 (S2204I) permitted replication and further adaptation of this genome, which was passaged until high-titer viruses emerged. The cell culture-adapted emerging viruses, which showed significant replication enhancement, contained 3 additional amino acid changes, G1909S (NS4B), D2416G (NS5A), and G2963D (NS5B). Interestingly, our adapted H77Ccc also contains G1909S in NS4B, but with V2417A instead of D2416G in NS5A and V2431I instead of G2963D in NS5B. Both independent approaches for efficient adaptation of H77C thus depended on adaptive mutations from TN cultures, which was the first efficient genotype 1 culture system (9). Thus, the TNcc adaptive mutations might be valuable for adaptations of additional HCV strains to efficient growth in culture, as was also found for the HCV-1 strain in the present study.
In conclusion, we have developed two efficient high-titer culture systems for the globally prevalent HCV genotype 1. HCV1cc and H77Ccc represent efficient in vitro infectious systems for two historically important strains that have been the foundation for the development of diagnostic tests and key research material in the field, including the discovery of HCV. Both cell culture systems, together with other infectious full-length HCV genomes, will permit genotype- and isolate-specific functional studies of the viral life cycle and of specific viral proteins and their interactions with cellular components. This knowledge will then contribute to basic research on different aspects of HCV and help improve antiviral therapy and future vaccine development.
ACKNOWLEDGMENTS
We thank A. L. Sørensen and L. Ghanem for general laboratory support, D. Humes for his input and proofreading of the manuscript, and J. O. Nielsen, B. Ørskov Lindhardt, and O. Andersen for providing valuable support (all from Copenhagen University Hospital, Hvidovre, Denmark). We thank C. M. Rice (Rockefeller University, New York, NY), S. Emerson and R. Purcell (National Institutes of Health, Bethesda, MD), and T. Wakita (National Institute of Infectious Diseases, Tokyo, Japan) for providing reagents and H. Krarup (Aalborg University Hospital, Aalborg, Denmark) for performing the Abbott HCV Core antigen analyses.
This study was supported by research grants from the Lundbeck Foundation (J.B.), the Danish Cancer Society (J.B.), the Novo Nordisk Foundation (Y.-P.L. and J.B.), the A. P. Møller og Hustru Chastine Mc-Kinney Møllers Foundation (J.B.), and the Danish Council for Independent Research–Medical Sciences (Y.-P.L., S.R., and J.B.). S.R. was the recipient of an individual postdoctoral stipend from the Danish Council for Independent Research–Medical Sciences. J.B. is the 2014 recipient of an advanced-top researcher grant from the Danish Council for Independent Research.
We have no conflicts of interest to report.
REFERENCES
- 1.Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat Rev Microbiol 5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 2.Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton J T, Simmonds P. 2014. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology 59:318–327. doi: 10.1002/hep.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simmonds P, Bukh J, Combet C, Deleage G, Enomoto N, Feinstone S, Halfon P, Inchauspe G, Kuiken C, Maertens G, Mizokami M, Murphy DG, Okamoto H, Pawlotsky J M, Penin F, Sablon E, Shin I, Stuyver LJ, Thiel HJ, Viazov S, Weiner AJ, Widell A. 2005. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 42:962–973. doi: 10.1002/hep.20819. [DOI] [PubMed] [Google Scholar]
- 4.Bukh J, Purcell RH, Miller RH. 1993. At least 12 genotypes of hepatitis C virus predicted by sequence analysis of the putative E1 gene of isolates collected worldwide. Proc Natl Acad Sci U S A 90:8234–8238. doi: 10.1073/pnas.90.17.8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarrazin C, Hezode C, Zeuzem S, Pawlotsky JM. 2012. Antiviral strategies in hepatitis C virus infection. J Hepatol 56(Suppl 1):S88–S100. [DOI] [PubMed] [Google Scholar]
- 6.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 7.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li YP, Ramirez S, Jensen SB, Purcell RH, Gottwein J M, Bukh J. 2012. Highly efficient full-length hepatitis C virus genotype 1 (strain TN) infectious culture system. Proc Natl Acad Sci U S A 109:19757–19762. doi: 10.1073/pnas.1218260109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li YP, Ramirez S, Gottwein J M, Scheel TK, Mikkelsen L, Purcell RH, Bukh J. 2012. Robust full-length hepatitis C virus genotype 2a and 2b infectious cultures using mutations identified by a systematic approach applicable to patient strains. Proc Natl Acad Sci U S A 109:E1101–E1110. doi: 10.1073/pnas.1203829109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramirez S, Li YP, Jensen SB, Pedersen J, Gottwein J M, Bukh J. 2014. Highly efficient infectious cell culture of three hepatitis C virus genotype 2b strains and sensitivity to lead protease, nonstructural protein 5A, and polymerase inhibitors. Hepatology 59:395–407. doi: 10.1002/hep.26660. [DOI] [PubMed] [Google Scholar]
- 12.Yi M, Villanueva RA, Thomas DL, Wakita T, Lemon SM. 2006. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc Natl Acad Sci U S A 103:2310–2315. doi: 10.1073/pnas.0510727103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pietschmann T, Zayas M, Meuleman P, Long G, Appel N, Koutsoudakis G, Kallis S, Leroux-Roels G, Lohmann V, Bartenschlager R. 2009. Production of infectious genotype 1b virus particles in cell culture and impairment by replication enhancing mutations. PLoS Pathog 5:e1000475. doi: 10.1371/journal.ppat.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Date T, Kato T, Kato J, Takahashi H, Morikawa K, Akazawa D, Murayama A, Tanaka-Kaneko K, Sata T, Tanaka Y, Mizokami M, Wakita T. 2012. Novel cell culture-adapted genotype 2a hepatitis C virus infectious clone. J Virol 86:10805–10820. doi: 10.1128/JVI.07235-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu J, Xiang Y, Tao W, Li Q, Wang N, Gao Y, Xiang X, Xie Q, Zhong J. 2014. A novel strategy to develop a robust infectious hepatitis C virus cell culture system directly from a clinical isolate. J Virol 88:1484–1491. doi: 10.1128/JVI.02929-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choo QL, Richman KH, Han J H, Berger K, Lee C, Dong C, Gallegos C, Coit D, Medina-Selby R, Barr PJ. 1991. Genetic organization and diversity of the hepatitis C virus. Proc Natl Acad Sci U S A 88:2451–2455. doi: 10.1073/pnas.88.6.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S. 1998. Binding of hepatitis C virus to CD81. Science 282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 18.Xu Z, Choi J, Yen TS, Lu W, Strohecker A, Govindarajan S, Chien D, Selby MJ, Ou J. 2001. Synthesis of a novel hepatitis C virus protein by ribosomal frameshift. EMBO J 20:3840–3848. doi: 10.1093/emboj/20.14.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stamataki Z, Coates S, Evans MJ, Wininger M, Crawford K, Dong C, Fong YL, Chien D, Abrignani S, Balfe P, Rice CM, McKeating JA, Houghton M. 2007. Hepatitis C virus envelope glycoprotein immunization of rodents elicits cross-reactive neutralizing antibodies. Vaccine 25:7773–7784. doi: 10.1016/j.vaccine.2007.08.053. [DOI] [PubMed] [Google Scholar]
- 20.Meunier JC, Gottwein JM, Houghton M, Russell RS, Emerson SU, Bukh J, Purcell RH. 2011. Vaccine-induced cross-genotype reactive neutralizing antibodies against hepatitis C virus. J Infect Dis 204:1186–1190. doi: 10.1093/infdis/jir511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stamataki Z, Coates S, Abrignani S, Houghton M, McKeating JA. 2011. Immunization of human volunteers with hepatitis C virus envelope glycoproteins elicits antibodies that cross-neutralize heterologous virus strains. J Infect Dis 204:811–813. doi: 10.1093/infdis/jir399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Law JL, Chen C, Wong J, Hockman D, Santer DM, Frey SE, Belshe RB, Wakita T, Bukh J, Jones CT, Rice CM, Abrignani S, Tyrrell DL, Houghton M. 2013. A hepatitis C virus (HCV) vaccine comprising envelope glycoproteins gpE1/gpE2 derived from a single isolate elicits broad cross-genotype neutralizing antibodies in humans. PLoS One 8:e59776. doi: 10.1371/journal.pone.0059776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuiken C, Combet C, Bukh J, Shin I, Deleage G, Mizokami M, Richardson R, Sablon E, Yusim K, Pawlotsky JM, Simmonds P. 2006. A comprehensive system for consistent numbering of HCV sequences, proteins and epitopes. Hepatology 44:1355–1361. doi: 10.1002/hep.21377. [DOI] [PubMed] [Google Scholar]
- 24.Yanagi M, Purcell RH, Emerson SU, Bukh J. 1997. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc Natl Acad Sci U S A 94:8738–8743. doi: 10.1073/pnas.94.16.8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kolykhalov AA, Agapov EV, Blight KJ, Mihalik K, Feinstone SM, Rice CM. 1997. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 277:570–574. doi: 10.1126/science.277.5325.570. [DOI] [PubMed] [Google Scholar]
- 26.Nakajima N, Hijikata M, Yoshikura H, Shimizu YK. 1996. Characterization of long-term cultures of hepatitis C virus. J Virol 70:3325–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimizu YK, Igarashi H, Kiyohara T, Shapiro M, Wong DC, Purcell RH, Yoshikura H. 1998. Infection of a chimpanzee with hepatitis C virus grown in cell culture. J Gen Virol 79:1383–1386. [DOI] [PubMed] [Google Scholar]
- 28.Triyatni M, Saunier B, Maruvada P, Davis AR, Ulianich L, Heller T, Patel A, Kohn LD, Liang TJ. 2002. Interaction of hepatitis C virus-like particles and cells: a model system for studying viral binding and entry. J Virol 76:9335–9344. doi: 10.1128/JVI.76.18.9335-9344.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bartosch B, Dubuisson J, Cosset FL. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med 197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blight KJ, McKeating JA, Marcotrigiano J, Rice CM. 2003. Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J Virol 77:3181–3190. doi: 10.1128/JVI.77.5.3181-3190.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 32.Kanda T, Basu A, Steele R, Wakita T, Ryerse JS, Ray R, Ray RB. 2006. Generation of infectious hepatitis C virus in immortalized human hepatocytes. J Virol 80:4633–4639. doi: 10.1128/JVI.80.9.4633-4639.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kato T, Matsumura T, Heller T, Saito S, Sapp RK, Murthy K, Wakita T, Liang TJ. 2007. Production of infectious hepatitis C virus of various genotypes in cell cultures. J Virol 81:4405–4411. doi: 10.1128/JVI.02334-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heller T, Saito S, Auerbach J, Williams T, Moreen TR, Jazwinski A, Cruz B, Jeurkar N, Sapp R, Luo G, Liang TJ. 2005. An in vitro model of hepatitis C virion production. Proc Natl Acad Sci U S A 102:2579–2583. doi: 10.1073/pnas.0409666102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimakami T, Welsch C, Yamane D, McGivern DR, Yi M, Zeuzem S, Lemon SM. 2011. Protease inhibitor-resistant hepatitis C virus mutants with reduced fitness from impaired production of infectious virus. Gastroenterology 140:667–675. doi: 10.1053/j.gastro.2010.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lanford RE, Lee H, Chavez D, Guerra B, Brasky KM. 2001. Infectious cDNA clone of the hepatitis C virus genotype 1 prototype sequence. J Gen Virol 82:1291–1297. [DOI] [PubMed] [Google Scholar]
- 37.Gottwein JM, Scheel TK, Hoegh AM, Lademann JB, Eugen-Olsen J, Lisby G, Bukh J. 2007. Robust hepatitis C genotype 3a cell culture releasing adapted intergenotypic 3a/2a (S52/JFH1) viruses. Gastroenterology 133:1614–1626. doi: 10.1053/j.gastro.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 38.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 39.Li YP, Gottwein JM, Scheel TK, Jensen TB, Bukh J. 2011. MicroRNA-122 antagonism against hepatitis C virus genotypes 1-6 and reduced efficacy by host RNA insertion or mutations in the HCV 5′ UTR. Proc Natl Acad Sci U S A 108:4991–4996. doi: 10.1073/pnas.1016606108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scheel TK, Gottwein JM, Mikkelsen LS, Jensen TB, Bukh J. 2011. Recombinant HCV variants with NS5A from genotypes 1-7 have different sensitivities to an NS5A inhibitor but not interferon-alpha. Gastroenterology 140:1032–1042. doi: 10.1053/j.gastro.2010.11.036. [DOI] [PubMed] [Google Scholar]
- 41.Gottwein JM, Scheel TK, Callendret B, Li YP, Eccleston HB, Engle RE, Govindarajan S, Satterfield W, Purcell RH, Walker CM, Bukh J. 2010. Novel infectious cDNA clones of hepatitis C virus genotype 3a (strain S52) and 4a (strain ED43): genetic analyses and in vivo pathogenesis studies. J Virol 84:5277–5293. doi: 10.1128/JVI.02667-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bukh J, Thimme R, Meunier JC, Faulk K, Spangenberg HC, Chang KM, Satterfield W, Chisari FV, Purcell RH. 2008. Previously infected chimpanzees are not consistently protected against reinfection or persistent infection after reexposure to the identical hepatitis C virus strain. J Virol 82:8183–8195. doi: 10.1128/JVI.00142-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Russell RS, Meunier JC, Takikawa S, Faulk K, Engle RE, Bukh J, Purcell RH, Emerson SU. 2008. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc Natl Acad Sci U S A 105:4370–4375. doi: 10.1073/pnas.0800422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gottwein JM, Jensen SB, Serre SB, Ghanem L, Scheel TK, Jensen TB, Krarup H, Uzcategui N, Mikkelsen LS, Bukh J. 2013. Adapted J6/JFH1-based hepatitis C virus recombinants with genotype-specific NS4A show similar efficacy to lead protease inhibitors, interferon-alpha and a putative NS4A inhibitor. Antimicrob Agents Chemother 57:6034–6049. doi: 10.1128/AAC.01176-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prentoe J, Serre SB, Ramirez S, Nicosia A, Gottwein JM, Bukh J. 2014. Hypervariable region 1 deletion and required adaptive envelope mutations confer decreased dependency on scavenger receptor class B type I and low-density lipoprotein receptor for hepatitis C virus. J Virol 88:1725–1739. doi: 10.1128/JVI.02017-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li YP, Ramirez S, Humes D, Jensen SB, Gottwein JM, Bukh J. 2014. Differential sensitivity of 5′UTR-NS5A recombinants of hepatitis C virus genotypes 1-6 to protease and NS5A inhibitors. Gastroenterology 146:812–821. doi: 10.1053/j.gastro.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 47.Sakai A, Takikawa S, Thimme R, Meunier JC, Spangenberg HC, Govindarajan S, Farci P, Emerson SU, Chisari FV, Purcell RH, Bukh J. 2007. In vivo study of the HC-TN strain of hepatitis C virus recovered from a patient with fulminant hepatitis: RNA transcripts of a molecular clone (pHC-TN) are infectious in chimpanzees but not in Huh7.5 cells. J Virol 81:7208–7219. doi: 10.1128/JVI.01774-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weiner AJ, Paliard X, Selby MJ, Medina-Selby A, Coit D, Nguyen S, Kansopon J, Arian CL, Ng P, Tucker J, Lee CT, Polakos NK, Han J, Wong S, Lu HH, Rosenberg S, Brasky KM, Chien D, Kuo G, Houghton M. 2001. Intrahepatic genetic inoculation of hepatitis C virus RNA confers cross-protective immunity. J Virol 75:7142–7148. doi: 10.1128/JVI.75.15.7142-7148.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murayama A, Weng L, Date T, Akazawa D, Tian X, Suzuki T, Kato T, Tanaka Y, Mizokami M, Wakita T, Toyoda T. 2010. RNA polymerase activity and specific RNA structure are required for efficient HCV replication in cultured cells. PLoS Pathog 6:e1000885. doi: 10.1371/journal.ppat.1000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yi M, Hu F, Joyce M, Saxena V, Welsch C, Chavez D, Guerra B, Yamane D, Veselenak R, Pyles R, Walker CM, Tyrrell L, Bourne N, Lanford RE, Lemon SM. 2014. Evolution of a cell culture-derived genotype 1a hepatitis C virus (H77S.2) during persistent infection with chronic hepatitis in a chimpanzee. J Virol 88:3678–3694. doi: 10.1128/JVI.03540-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bukh J, Pietschmann T, Lohmann V, Krieger N, Faulk K, Engle RE, Govindarajan S, Shapiro M, St Claire M, Bartenschlager R. 2002. Mutations that permit efficient replication of hepatitis C virus RNA in Huh-7 cells prevent productive replication in chimpanzees. Proc Natl Acad Sci U S A 99:14416–14421. doi: 10.1073/pnas.212532699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamane D, McGivern DR, Wauthier E, Yi M, Madden VJ, Welsch C, Antes I, Wen Y, Chugh PE, McGee CE, Widman DG, Misumi I, Bandyopadhyay S, Kim S, Shimakami T, Oikawa T, Whitmire JK, Heise MT, Dittmer DP, Kao CC, Pitson SM, Merrill AH Jr, Reid LM, Lemon SM. 2014. Regulation of the hepatitis C virus RNA replicase by endogenous lipid peroxidation. Nat Med 20:927–935. doi: 10.1038/nm.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scheel TK, Gottwein JM, Carlsen TH, Li YP, Jensen TB, Spengler U, Weis N, Bukh J. 2011. Efficient culture adaptation of hepatitis C virus recombinants with genotype-specific core-NS2 by using previously identified mutations. J Virol 85:2891–2906. doi: 10.1128/JVI.01605-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scheel TK, Prentoe J, Carlsen TH, Mikkelsen LS, Gottwein JM, Bukh J. 2012. Analysis of functional differences between hepatitis C virus NS5A of genotypes 1-7 in infectious cell culture systems. PLoS Pathog 8:e1002696. doi: 10.1371/journal.ppat.1002696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bressanelli S, Tomei L, Rey FA, De Francesco R. 2002. Structural analysis of the hepatitis C virus RNA polymerase in complex with ribonucleotides. J Virol 76:3482–3492. doi: 10.1128/JVI.76.7.3482-3492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai Z, Yi M, Zhang C, Luo G. 2005. Mutagenesis analysis of the rGTP-specific binding site of hepatitis C virus RNA-dependent RNA polymerase. J Virol 79:11607–11617. doi: 10.1128/JVI.79.18.11607-11617.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmitt M, Scrima N, Radujkovic D, Caillet-Saguy C, Simister PC, Friebe P, Wicht O, Klein R, Bartenschlager R, Lohmann V, Bressanelli S. 2011. A comprehensive structure-function comparison of hepatitis C virus strain JFH1 and J6 polymerases reveals a key residue stimulating replication in cell culture across genotypes. J Virol 85:2565–2581. doi: 10.1128/JVI.02177-10. [DOI] [PMC free article] [PubMed] [Google Scholar]