

Abstract
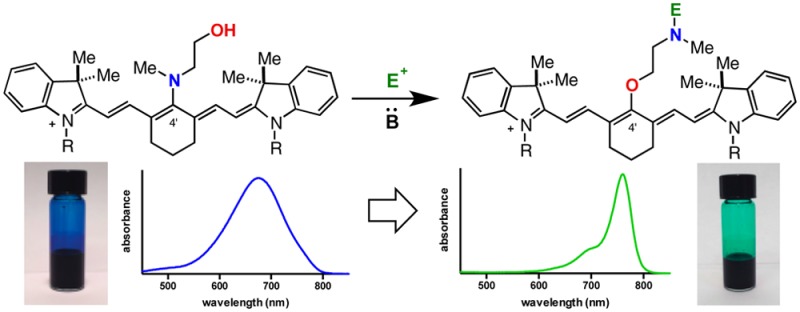
New synthetic methods to rapidly access useful fluorophores are needed to advance modern molecular imaging techniques. A new variant of the classical Smiles rearrangement is reported that enables the efficient synthesis of previously inaccessible C4′-O-alkyl heptamethine cyanines. The key reaction involves N- to O- transposition with selective electrophile incorporation on nitrogen. A representative fluorophore exhibits excellent resistance to thiol nucleophiles, undergoes productive bioconjugation, and can be used in near-IR fluorescence imaging applications.
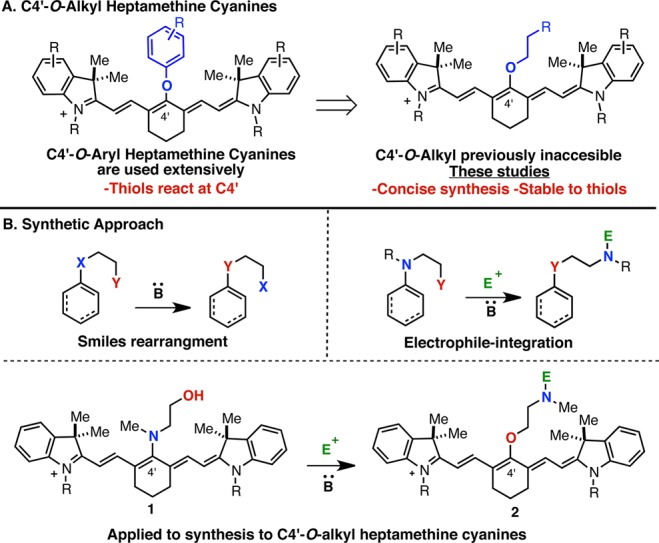
Given the central role of fluorescent small molecules in many modern biological techniques, it is surprising their preparation still often relies on inefficient condensation reactions requiring harsh reaction conditions with poor substrate scope.1,2 New synthetic methodologies are needed to enable the identification of optimal agents. This is particularly true for near-IR fluorophores, which find increasing use for a variety of techniques, including in clinical settings, due to the low autofluorescence and high tissue penetration of light in this range.3 Heptamethine cyanines represent a privileged scaffold, and various derivatives are the small molecule bioimaging agents of choice with emission maxima approaching 800 nm.4−7 Current state-of-the-art compounds are often substituted at the C4′ position with phenols (Figure 1A).8−11 These widely used molecules suffer from significant liabilities including poor chemical stability, likely arising from nucleophilic exchange reactions at the C4′ position.12−15 Cyanines modified at the C4′ position with an O-alkyl substituent are desirable because these are likely to be quite stable, while maintaining the excellent optical and physical properties associated with C4′-O-substitution. However, such molecules have only rarely been described16 and are unknown when functionalized for biomolecule conjugation.
Figure 1.
General considerations.
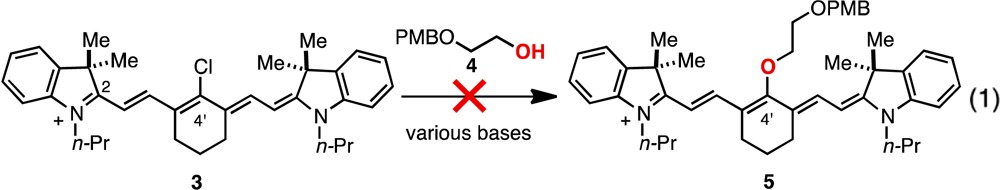
Here we report a general approach to access C4′-O-alkyl heptamethine cyanines through a new variant of the classical Smiles rearrangement. Traditionally, Smiles rearrangements employ a combination of strong bases and elevated temperatures to induce the α,δ-transposition of heteroatoms in reactions driven by formation of the thermodynamically preferred isomer (Figure 1B).17,18 A distinct alternative selectively incorporates an electrophile on the heteroatom originally affixed to the unsaturated carbon providing otherwise inaccessible products. Here we provide the first examples of this concept, a Smiles rearrangement with concurrent electrophile incorporation, to gain access to promising C4′-O-alkyl heptamethine cyanines (2) from easily synthesized C4′-N-methylethanolamine-substituted precursors (1). This method avoids the shortcomings of the more orthodox preparative approach for these compounds, intermolecular C4′-chloride substitution by direct addition of an alkoxide. Such chloride substitution reactions, which proceed rapidly in excellent yield with amine, phenol, and thiol nucleophiles, generally fail in this case due to the poor kinetics of alkoxides in electron transfer SRN1 pathways, the putative substitution mechanism.8a In one report, competitive addition of alkoxides to the imine-like C2 position has been suggested to intercede.19 In our own initial studies, which are consistent with these previous findings, we found that 5 was not observed when 3 and 4 were combined under a variety of basic conditions (eq 1).
 |
1 |
Using the Smiles rearrangement approach, we describe the synthesis of a diverse array of C4′-O-alkyl cyanines and detail several mechanistic experiments. The resulting fluorophores exhibit optical properties ideal for near-IR imaging and excellent chemical stability to thiol nucleophiles. Critically, access to C4′-O-alkyl substitution will provide an opportunity to develop agents with improved properties relative to those prepared through existing strategies.7,13,20 Toward these goals, we show that an antibody conjugate is suitable for both fluorescence microscopy and fluorescence-activated cell sorting (FACS).
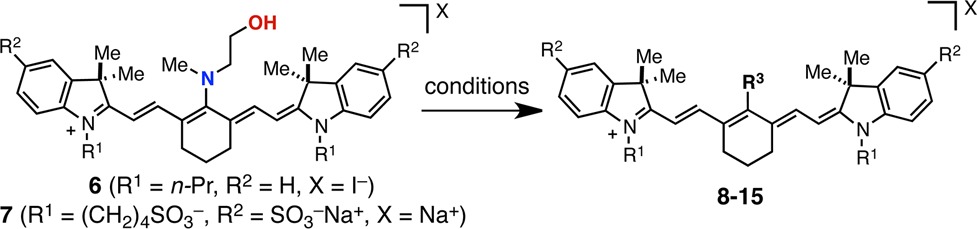
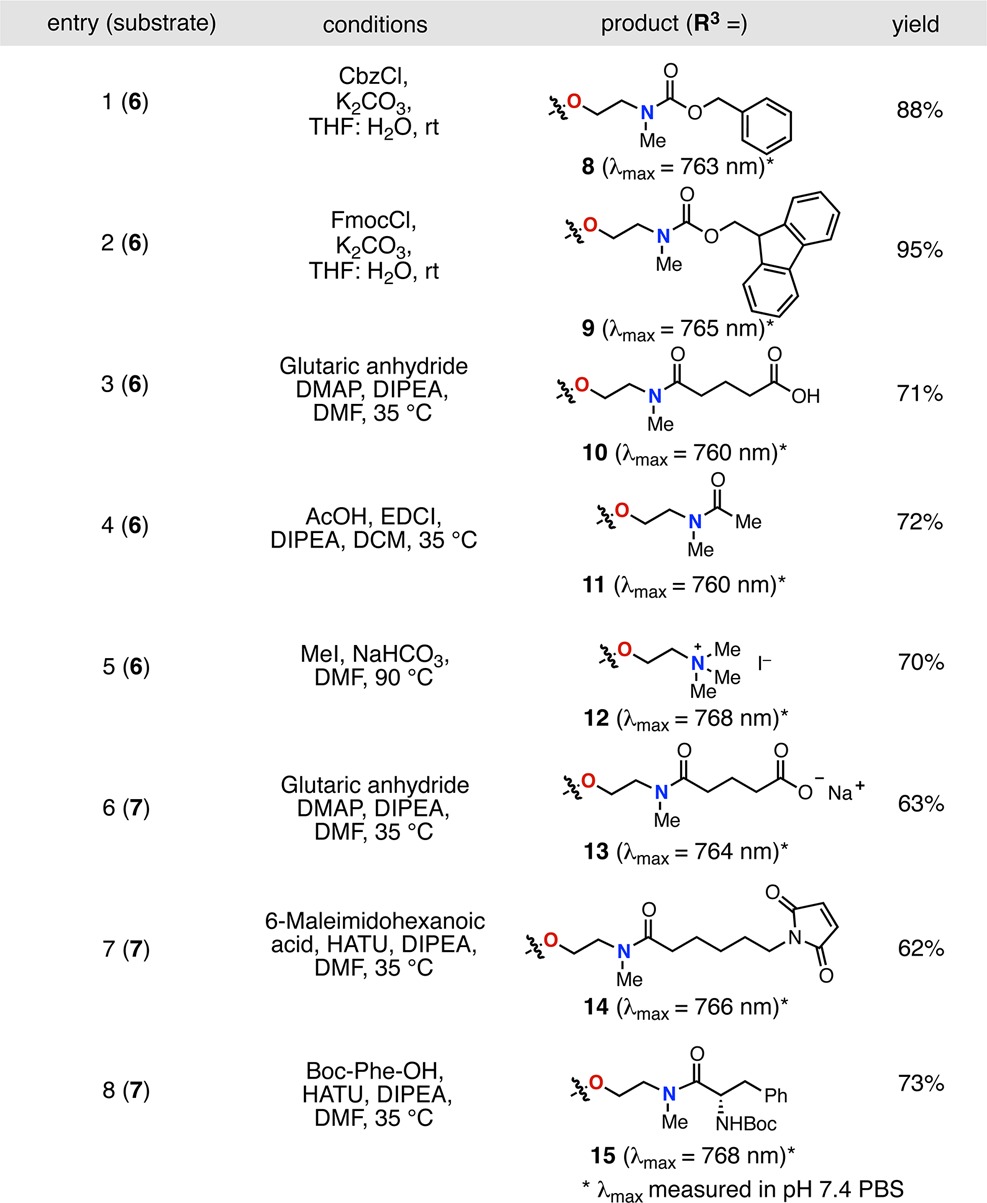
As shown in Table 1, the rearrangement process proceeds readily with a variety of substrates. The N-methylethanolamine-substituted 6 and 7 precursors were synthesized from known C4′-chloro intermediates as described previously.21,22 Characteristic of C4′-N-substitution, 6 and 7 exhibit a broad hypsochromic (blue-shifted) absorption, with maxima at 687 and 644 nm, respectively.23,24 With these two substrates in hand, we investigated a number of electrophiles and conditions to characterize the rearrangement process. Representative of productive rearrangement is the use of chloroformates to convert 6 to carbamates 8 and 9. These reactions undergo a dramatic blue to green color transition. Indicative of C4′-O-substitution, 8 exhibits an 76 nm bathochromic shift relative to N-linked 6, a small Stokes shift, and a high molar extinction coefficient.25 2-D NMR experiments were also employed to provide an unambiguous assignment. Similarly, we found that 6 reacted with cyclic anhydrides and activated carboxylic acids to provide amide products 10 and 11 in 71% and 72% yield, respectively. Methyl iodide also promotes N- to O- transposition to afford ammonium 12 in 70% yield. Demonstrating that the rearrangement is tolerant of sulfonate-bearing cyanines, tetrasulfonylated 7 is converted to carboxylate 13 in 63% yield with glutaric anhydride. Tetrasulfonate 7 also undergoes HATU coupling to produce maleimide and protected amino acid variants 14 and 15 in yields of 62% and 73% yield, respectively. In these examples, preactivation of the carboxylic acid with HATU, DIPEA was critical for efficient rearrangement. Notably, 13 and 14 bear suitable chemical handles for bioconjugation, and the four sulfonate groups impart excellent aqueous solubility and a reduced tendency toward aggregation—desirable traits for biological imaging.
Table 1. Scope of N- to O-Rearrangement.
We sought to provide initial insight regarding the various mechanistic possibilities. Divergent reaction pathways were observed between several methylation and acylation conditions (Table 2). While methyl iodide yields the rearrangement product, 12, trimethyloxonium tetrafluoroborate affords methyl ether 16. Similarly, unlike the peptide coupling conditions of entry 4 in Table 1, a combination of acetyl chloride and 2,6-lutidine provided only the O-acetylation product 17 in high yield. The latter result indicates that O-acylated compounds are unlikely to be reaction intermediates en route to N-acylated products (e.g., 11). We also have compared the rearrangement kinetics of N-methylethanolamine substituted 6, and the one-carbon N-methylpropanolamine homologue, 18 (Figure 2). Derivative 6 and 18 were exposed to identical conditions (4 equiv of EDCI, 4 equiv of AcOH, 1 equiv of DIPEA), and the reaction progress was monitored by HPLC. While 6 reacts relatively rapidly (t1/2 = 37 min) to form 11, 18 proceeds much more slowly to 19 (t1/2 = 720 min), though ultimately in good conversion and with satisfying isolated yield (72%). One reasonable mechanistic scenario involves initial nitrogen quaternerization and facile intramolecular displacement. This mechanism rationalizes the kinetic dependence on ring size (5 faster than 6) by suggesting that acyl-ammonium formation, which is likely reversible,26 precedes rate determining and, presumably, irreversible tetrahedral intermediate formation. An alternative mechanism consisting of N-selective electrophilic trapping of a minor equilibrating C4′-O-substituted component of 6 or 7 is also conceivable. However, the lack of a ∼780 nm peak or peak shoulder in the absorption spectra under relevant reaction conditions suggests that O-linked species are, at most, negligible components of 6 or 7. Nevertheless, further studies are required.
Table 2. Oxygen-Selective Electrophiles.
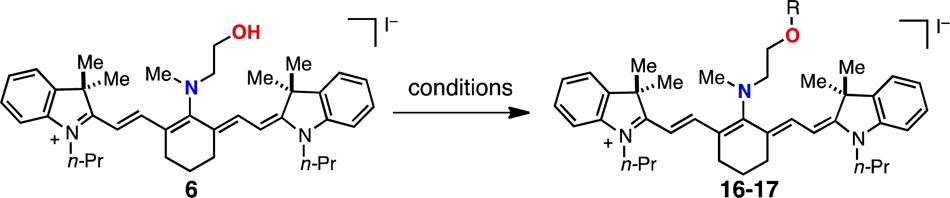
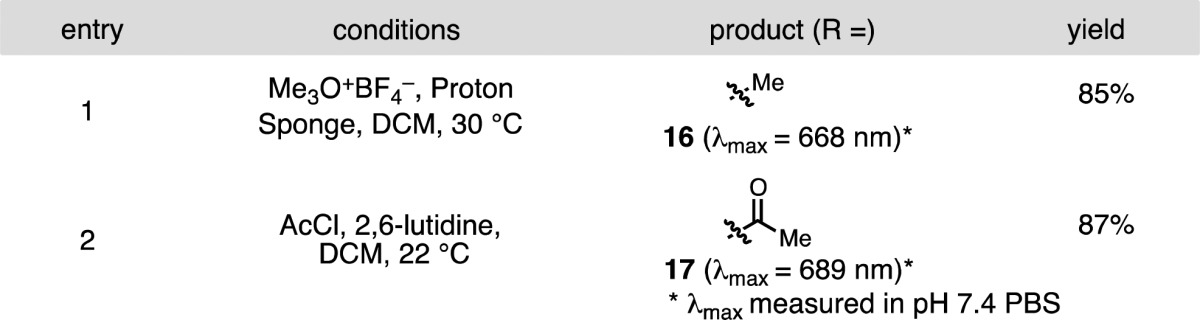
Figure 2.
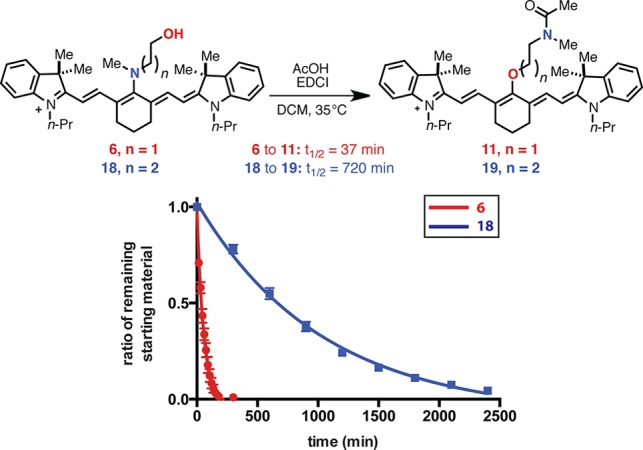
Relative rearrangement kinetics of 6 and 18.
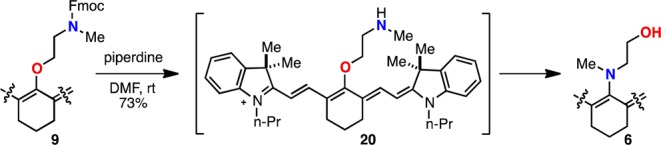
We also examined the reversibility of the rearrangement (Scheme 1). When Fmoc-protected 9 was exposed to piperidine in DMF, the C4′-N-methylethanolamine-substituted product 6, its synthetic precursor, was rapidly generated. This process likely occurs through the intermediacy of O-linked species 20 and further illustrates the thermodynamic preference for C4′-N-substitution in preference to O-substitution. It is noted that the facile conversion of 9 to 6 alters the absorption maximum by almost 100 nm, suggesting potential for various optical sensing applications.
Scheme 1. O- to N-Rearrangement of 9 to 6.
With access to a range of C4′-O-alkyl heptamethine cyanines, we explored their suitability for imaging purposes. Representative compounds 8 and 13 possess similar absorption properties and improved quantum yields relative to a standard heptamethine cyanine, indocyanine green (ICG) (Table 3). Previous studies have shown that widely used C4′ phenol- and thiol-substituted heptamethine cyanines can be rapidly exchanged by thiol nucleophiles under aqueous conditions, with problematic consequences during conjugation reactions of cysteine-containing peptides and macromolecules and during DNA sequencing applications.12,14 In addition, phenol-to-thiol exchange has been recently observed intracellularly in the context of a thiol-sensing platform.15 The thiol reactivity of 13 was compared with phenol-substituted 21 and to S-mercaptoethanol-substituted 22. Solutions of 13, 21, and 22 (10 μM in pH 7.4 PBS buffer with no intentional O2 exclusion) were exposed to 1 mM glutathione, and the reaction course was monitored by HPLC (Figure 3A). With 21 and 22, rapid conversion to the glutathione adduct 23 was observed (t1/2 = 95 and 40 min, respectively). By contrast, alkyl-ether 13 showed no decomposition over the same time period and >90% was present after 3 days. These experiments clearly demonstrate the superior chemical stability of these new alkyl-ether variants over conventional C4′ phenol- or thiol-substituted heptamethine cyanines. We also measured the near-IR fluorescence signal under identical conditions. As shown in Figure 3B, the C4′-O-linked compounds, 13 and 21, initially exhibit significantly greater signal than C4′-S-linked 22. Whereas 13 maintains the initial value, fluorescence from the mixture of 21 and forming 23 diminishes and approaches that of the mixture of 22 and 23. Thus, the loss of C4′-O-linkage is detrimental to the emissive properties of these molecules.
Table 3. Optical Properties of 8, 13, and ICG.
| compd | λabs (nm) | λem (nm) | ε (M–1 cm–1) | Φf |
|---|---|---|---|---|
| ICG | 785 | 822 | 204 000 | 0.078 |
| 8a | 774 | 797 | 187 000 | 0.22 |
| 13a | 774 | 798 | 214 000 | 0.23 |
Measured in methanol relative to ICG.
Figure 3.
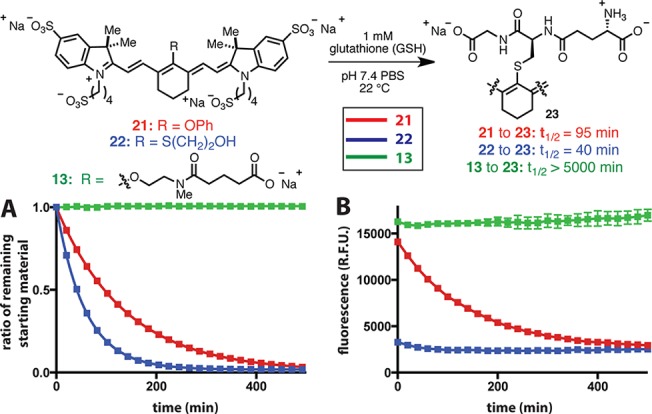
Stability of 21, 22, and 13 in the presence of 1 mM glutathione (GSH) in pH 7.4 PBS. (A) HPLC conversion of starting material (10 μM). These data were used to obtain the indicated half-lives. (B) Fluorescent signal over time (2 μM, λex = 740 nm, λem = 790 nm).
We have examined the use of these molecules as antibody labels (Figure 4).27 Carboxylate 13 was converted to its NHS-ester (TSTU, DMF, 35 °C) and then incubated with the anti-HER1 antibody, panitumumab, to provide the labeled antibody with a degree of labeling (DOL) of 2.1–2.2. The conjugate was incubated with HER1+ (MDA-MB-468) and HER1– (MCF-7) cells. Characteristic antibody labeling was only observed in HER1+ cells by fluorescence microscopy using a standard Cy7 filter set (λex = 710 nm, λem = 775 nm, Figure 4B). The efficient cellular labeling of the fluorophore–antibody conjugate was also confirmed using FACS (Figure 4C). These results suggest that 13 and other fluorophores that emerge from this new approach are likely to be suitable for a range of near-IR fluorescence applications.
Figure 4.
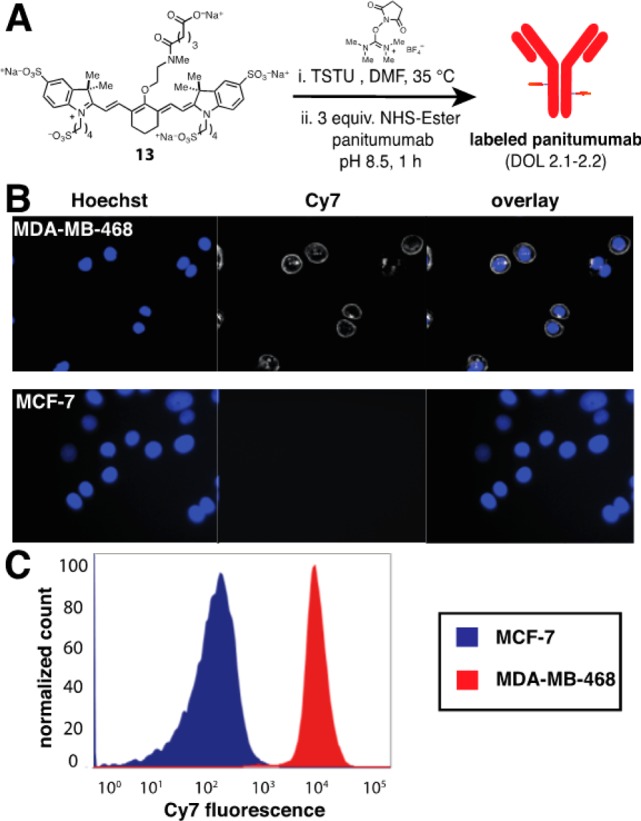
(A) NHS-ester formation and labeling of 13. (B) Fluorescence microscopy images of live MDA-MB-468 and MCF-7 cells treated with 100 nM labeled panitumumab and Hoechst 33342 (1 μM). (C) Flow cytometry of MDA-MB-468 and MCF-7 cells treated with 100 nM labeled panitumumab.
In conclusion, we have developed a synthetic approach to previously inaccessible C4′-O-alkyl heptamethine cyanines. This sequence provides concise and efficient access to a new class of useful heptamethine cyanine fluorophores resistant to thiol exchange reactions. As compared to conventional Smiles rearrangements, the alternative mode described here benefits from both lowering the barrier to productive reactivity and the additional structural complexity in the product acquired through electrophile incorporation. Current efforts are focused on extension of this Smiles rearrangement manifold and on further applications of molecules resulting from this approach.
Acknowledgments
We thank Dr. Joseph Barchi (NIH/NCI) for NMR assistance and Dr. James Kelley (NIH/NCI) for mass spectrometric analysis. Dr. Sibaprasad Bhattacharyya (Leidos) is gratefully acknowledged for advice regarding the antibody conjugation reactions. We also acknowledge the Frederick CCR Flow Cytometry Core (Cancer and Inflammation Program, NCI, Frederick) for flow cytometry analysis. This work was supported by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, and the National Cancer Institute, National Institutes of Health.
Supporting Information Available
Synthetic procedures, other experimental details, and 1H and 13C NMR spectra of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Lavis L. D.; Raines R. T. ACS Chem. Biol. 2014, 9, 855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie R. M.Colour Chemistry; Royal Society of Chemistry: Cambridge, U.K., 2001. [Google Scholar]
- Weissleder R. Nat. Biotechnol. 2001, 19, 316–317. [DOI] [PubMed] [Google Scholar]
- Frangioni J. V. Curr. Opin. Chem. Biol. 2003, 7, 626–634. [DOI] [PubMed] [Google Scholar]
- Kiyose K.; Kojima H.; Nagano T. Chem.—Asian J. 2008, 3, 506–515. [Google Scholar]
- Pansare V. J.; Hejazi S.; Faenza W. J.; Prud’homme R. K. Chem. Mater. 2012, 24, 812–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S. L.; Zhang E. L.; Su Y. P.; Cheng T. M.; Shi C. M. Biomaterials 2011, 32, 7127–7138. [DOI] [PubMed] [Google Scholar]
- a Strekowski L.; Lipowska M.; Patonay G. J. Org. Chem. 1992, 57, 4578–4580. [Google Scholar]; b Narayanan N.; Patonay G. J. Org. Chem. 1995, 60, 2391–2395. [Google Scholar]
- Choi H. S.; Gibbs S. L.; Lee J. H.; Kim S. H.; Ashitate Y.; Liu F. B.; Hyun H.; Park G.; Xie Y.; Bae S.; Henary M.; Frangioni J. V. Nat. Biotechnol. 2013, 31, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shallal H. M.; Minn I.; Banerjee S. R.; Lisok A.; Mease R. C.; Pomper M. G. Bioconjugate. Chem. 2014, 25, 393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B. A.; Akers W. J.; Leevy W. M.; Lampkins A. J.; Xiao S. Z.; Wolter W.; Suckow M. A.; Achilefu S.; Smith B. D. J. Am. Chem. Soc. 2010, 132, 67–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaheer A.; Wheat T. E.; Frangioni J. V. Molecular Imaging 2002, 1, 354–364. [DOI] [PubMed] [Google Scholar]
- Lee H.; Mason J. C.; Achilefu S. J. Org. Chem. 2006, 71, 7862–7865. [DOI] [PubMed] [Google Scholar]
- Shealy D. B.; Lipowska M.; Lipowski J.; Narayanan N.; Sutter S.; Strekowski L.; Patonay G. Anal. Chem. 1995, 67, 247–251. [Google Scholar]
- Lim S. Y.; Hong K. H.; Kim D. I.; Kwon H.; Kim H. J. J. Am. Chem. Soc. 2014, 136, 7018–7025. [DOI] [PubMed] [Google Scholar]
- The only previous literature examples of C4′-O-alkyl heptamethine cyanines involve methanol adducts (first reported in ref (8a)) and one report of protected galactose derivatives, which were accessed in modest yield:Zhang Z. R.; Achilefu S. Org. Lett. 2004, 6, 2067–2070. [DOI] [PubMed] [Google Scholar]
- Bunnett J. F.; Zahler R. E. Chem. Rev. 1951, 49, 273–412. [Google Scholar]
- Truce W. E.; Kreider E. M.; Brand W. W. Organic Reactions 1970, 18, 99–215. [Google Scholar]
- Strekowski L.; Mason J. C.; Britton J. E.; Lee H.; Van Aken K.; Patonay G. Dyes Pigments 2000, 46, 163–168. [Google Scholar]
- Samanta A.; Vendrell M.; Yun S. W.; Guan Z. P.; Xu Q. H.; Chang Y. T. Chem.—Asian J. 2011, 6, 1353–1357. [DOI] [PubMed] [Google Scholar]
- Hilderbrand S. A.; Kelly K. A.; Weissleder R.; Tung C. H. Bioconjugate Chem. 2005, 16, 1275–1281. [DOI] [PubMed] [Google Scholar]
- Gorka A. P.; Nani R. R.; Zhu J.; Mackem S.; Schnermann M. J. J. Am. Chem. Soc. 2014, 136, 14153–14159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X. J.; Song F. L.; Lu E.; Wang Y. N.; Zhou W.; Fan J. L.; Gao Y. L. J. Am. Chem. Soc. 2005, 127, 4170–4171. [DOI] [PubMed] [Google Scholar]
- Myochin T.; Kiyose K.; Hanaoka K.; Kojima H.; Terai T.; Nagano T. J. Am. Chem. Soc. 2011, 133, 3401–3409. [DOI] [PubMed] [Google Scholar]
- Pascal S.; Haefele A.; Monnereau C.; Charaf-Eddin A.; Jacquemin D.; Le Guennic B.; Andraud C.; Maury O. J. Phys. Chem. A 2014, 118, 4038–4047. [DOI] [PubMed] [Google Scholar]
- Aquino E. C.; Brittain W. J.; Brunelle D. J. Macromolecules 1992, 25, 3827–3828. [Google Scholar]
- a Barrett T.; Koyama Y.; Hama Y.; Ravizzini G.; Shin I. S.; Jang B. S.; Paik C. H.; Urano Y.; Choyke P. L.; Kobayashi H. Clin. Cancer Res. 2007, 13, 6639–6648. [DOI] [PubMed] [Google Scholar]; b Bhattacharyya S.; Patel N. L.; Wei L.; Riffle L. A.; Kalen J. D.; Hill G. C.; Jacobs P. M.; Zinn K. R.; Rosenthal E. Med. Chem. Comm. 2014, 5, 1337–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.