

Abstract
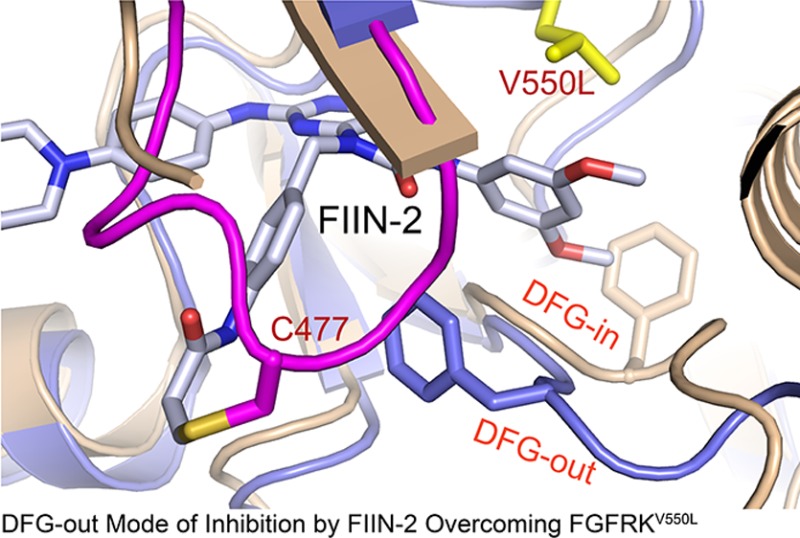
Drug-resistance acquisition through kinase gate-keeper mutations is a major hurdle in the clinic. Here, we determined the first crystal structures of the human FGFR4 kinase domain (FGFR4K) alone and complexed with ponatinib, a promiscuous type-2 (DFG-out) kinase inhibitor, and an oncogenic FGFR4K harboring the V550L gate-keeper mutation bound to FIIN-2, a new type-1 irreversible inhibitor. Remarkably, like ponatinib, FIIN-2 also binds in the DFG-out mode despite lacking a functional group necessary to occupy the pocket vacated upon the DFG-out flip. Structural analysis reveals that the covalent bond between FIIN-2 and a cysteine, uniquely present in the glycine-rich loop of FGFR kinases, facilitates the DFG-out conformation, which together with the internal flexibility of FIIN-2 enables FIIN-2 to avoid the steric clash with the gate-keeper mutation that causes the ponatinib resistance. The structural data provide a blueprint for the development of next generation anticancer inhibitors through combining the salient inhibitory mechanisms of ponatinib and FIIN-2.
The FGF family of ligands consists of 18 structurally related polypeptides that signal in paracrine or endocrine fashion through four FGFRs (FGFR1-FGFR4) and their alternatively spliced isoforms to regulate a myriad of biological processes in human development, metabolism, and tissue homeostasis.1,2 FGFs bind and dimerize the extracellular domains of FGFRs in concert with heparan sulfate glycosaminoglycans or single-pass Klotho coreceptor proteins positioning the cytoplasmic kinase domains in proper proximity/orientation for transphosphorylation on A-loop tyrosines.3,4 This event elevates the intrinsic kinase activity of FGFRs leading to subsequent autophosphorylation on tyrosines in the flanking juxtamembrane (JM) and C-tail regions that mediate recruitment and phosphorylation of a distinct set of intracellular effector proteins by the activated FGFR evoking activation of intracellular signaling pathways.4−6
Uncontrolled activation of FGF signaling due to gain-of-function mutations in FGFRs, FGFR gene fusions involving various dimerizing partners, or overexpression/misexpression of FGFs and FGFRs contributes to a number of developmental disorders and cancer.7−11 Gain-of-function mutations in FGFRs were initially discovered in human congenital craniosynostosis and dwarfism syndromes. Later studies showed that the very same mutations occur somatically in diverse cancers, including multiple myeloma,12 bladder cancer,13 endometrial cancer,14 glioblastoma,15 lung cancer,16 adenoid cystic carcinoma,17 and benign skin cancer.18 FGFR gene fusions, originally found in the 8p11 myeloproliferative syndrome (an aggressive atypical stem cell myeloproliferative disorder),7,19 have since been extended to glioblastoma, bladder, and lung cancers.20,21 Overexpression of FGFs and FGFRs has been documented in breast, prostate, and bladder cancers.22 Single nucleotide polymorphism in FGFR2 has been linked with susceptibility to breast cancer,23 and SNP in FGFR4 has been associated with resistance to chemotherapy.24 In light of these data, FGFRs are now considered major targets for cancer drug discovery.
Indeed, several small molecule ATP-competitive inhibitors are being pursued in the clinic for FGFR-associated cancers including endometrial and prostate cancer. These include dovitinib,25 ponatinib,26,27 brivanib,28 multitargeted RTK inhibitors with coverage of FGFRs, and AZD4547,29 which has a more restricted FGFR target specificity profile. In addition, there are historical FGFR inhibitors such as PD173074,30 SU5402,31 and FIIN-132 which have been extensively used as pharmacological probes. All of these inhibitors are reversible ATP-competitive inhibitors with the exception of FIIN-1, which covalently targets an unusual cysteine located in the glycine-rich loop of FGFR1–4. These inhibitors exhibit differential activity profiles with most acting primarily on the autoinhibited FGFRKs, while others also show activity against FGFR kinases carrying gain-of-function mutations. However, these inhibitors are ineffective against gate-keeper mutations,33,34 a mechanism that has been well documented to confer resistance in the clinic to many drugs targeting oncogenic kinases such as Bcr-Abl (T315I), EGFR (T790M), PDGFR (T674I), and c-Kit (T670I).
There is a major impetus to elucidate the structure–function relationships of FGFR kinases including the mechanisms of action of gain-of-function mutations and inhibitors as such data can provide crucial information to guide the development of inhibitors with improved selectivity and potency toward FGFR isoforms. To date, crystal structures of FGFR1–3 kinases in an autoinhibited state or in an activated state induced either by A-loop phosphorylation or by gain-of-function mutations have been determined.35−37 In addition, for FGFR1 and FGFR2 kinases, crystal structures exist of inhibitor bound forms.38−40 These structural data have guided the discovery of inhibitors with narrowed specificity toward FGFR kinases. Notably, the FGFR1K–PD173074 structure40 was used as template to develop FIIN-132 and FIIN-2, pyridopyrimidine-based irreversible inhibitors that exhibit greater specificity toward FGFRs. These inhibitors carry a reactive acrylamide group that is capable of forming a covalent bond with the thiol group of a cysteine uniquely present in the glycine-rich loop of FGFRs. Importantly, FIIN-2 shows activity against the FGFR kinase harboring gate-keeper mutation.
Rhabdomyosarcoma is the most common soft tissue sarcoma in children.8 FGFR4 activation due to overexpression or gain-of-function mutations in the FGFR4 kinase domain has been correlated with advanced-stage cancer and poor survival.8,41 FGFR4 inhibition has been shown to stop growth of rhabomyosarcoma cell lines and cause tumor shrinkage in xenograft studies,42,43 supporting the notion that these mutations play causal roles in tumorigenesis. To facilitate the ongoing drug discovery for rhabdomyosarcoma, we solved the first crystal structures of FGFR4K alone and in complex with ponatinib and FIIN-2. These structures provide the first examples for a DFG-out mode of inhibition of FGFRK by an ATP-competitive inhibitor. Remarkably, FIIN-2 also binds in the DFG-out mode despite not conforming to the pharmacophore required for this binding mode.44 In addition, the FIIN-2 gate-keeper mutant complex demonstrates how the internal rotational flexibility allows this compound to adapt to the bulkier side chains at the gate-keeper location, thus retaining its inhibitory activity. These findings have general implications for the structure-guided design of inhibitors that can overcome the gate-keeper mutation in FGFR and likely other kinases.
Results
Crystal Structure of Autoinhibited FGFR4 Kinase
As expected, the FGFR4 kinase domain (FGFR4K) adopts the canonical bilobate fold of protein kinases with the smaller N-terminal lobe exhibiting the characteristic twisted five-stranded β sheet and the αC helix and the larger C-terminal lobe consisting of mainly α helices (Figure 1A). Both the A-loop and the loop connecting αD and αE helices, referred to as the kinase insert, are fully ordered. The ordering of the A-loop is due to the intramolecular contacts between the loop and the rigid body of the C-lobe. By contrast, the observed conformation of the kinase insert is solely attributable to favorable crystal lattice contacts. The kinase insert region of FGFR4K bulges out of the main body of the kinase domain and, unlike FGFR1–3 kinases, lacks tyrosine autophosphorylation sites (Supporting Information Figure S1A).
Figure 1.

The crystal structure of wild-type FGFR4 kinase. (A) Ribbon diagram of the wild-type FGFR4K structure. β strands and α helices are colored cyan and green, respectively. The A-loop, catalytic loop, kinase insert, and kinase hinge are colored magenta, orange, wheat, and yellow, respectively. (B) Close-up view of the N-lobe showing formation of the catalytically critical salt bridge between K503 and E520. (C) Close-up view of the molecular brake at the kinase hinge region. (D) Close-up view of the active site of FGFR4KWT and activated FGFR2K (in slate) complexed with peptide (in yellow sticks) following superimposition of these two structures. Note that bidentate hydrogen bonds between R650 and D612 (the general base) block the access of substrate tyrosine into the active site of FGFR4K. The side chain of R650 in the FGFR4K structure occupies roughly the same space as the substrate tyrosine in the FGFR2K–peptide complex structure. In all figures, side chains of selected residues are shown as sticks, and atom colorings are as follows: oxygens in red, nitrogens in blue, and coloring of carbons follow the coloring scheme of the specific region of the kinase from which they derive. The hydrogen bonds are shown as black dashed lines.
The unphosphorylated FGFR4K is in an autoinhibited catalytically incompetent state, as evidenced by its comparison with the published crystal structures of unphosphorylated auoinhibited FGFR1–2 kinases, and activated FGFR1–3 kinases either by A-loop tyrosine phosphorylation or by pathogenic gain-of-function mutations (Supporting Information Figure S2). As in FGFR1–3 kinases, FGFR4K autoinhibition is principally governed by a network of inhibitory hydrogen bonds at the kinase hinge region termed the molecular brake (Figure 1C). This network, which is mediated by a triad of residues consisting of Asn-535, Glu-551, and Lys-627 in FGFR4K, restrains the N-lobe movement toward the C-lobe that accompanies kinase activation. Reminiscent of the unphosphorylated FGFR1K structure, an additional constraint is provided by the DFGLAR motif at the beginning of the A-loop, whose conformation physically interferes with N-lobe rotation.36 In fact, as in the unphosphorylated FGFR1K and FGFR2K structures, FGFR4K also contains the catalytically important salt bridge between Lys-503 and Glu-520 (αC), which is known to help orient Lys to coordinate α and β phosphates of ATP (Figure 1B). Last, as in FGFRK1,36 the FGFR-invariant Arg-650 at the C-terminal end of the A-loop makes bidentate hydrogen bonds with Asp-612 (the general base) from the catalytic loop, thereby directly blocking the access of substrate into the active site (Figure 1D).
The FGFR4K Gate-Keeper Mutations Confer Resistance to Ponatinib but Are Sensitive to FIIN-2
As alluded to previously, ponatinib (previously AP24534; Supporting Information Figure S3A) is a multitargeted RTK inhibitor with coverage of FGFR kinases that is currently being evaluated in clinical trials for several cancers including endometrial cancer and rhabdomyosarcoma.27,45 Importantly, we have recently shown that, in contrast to other FGFR inhibitors including dovitinib and PD173074, ponatinib is capable of effectively targeting not only the autoinhibited FGFR2 kinases but also FGFR2K that has undergone activation by gain-of-function mutations with the exception of gate-keeper mutation.33 Using the FGFR1K–PD173074 complex structure as a template, we have recently developed an irreversible covalently acting inhibitor, termed FIIN-2 (Supporting Information Figure S3B), that is capable of targeting FGFRKs harboring gate-keeper mutations.
To date, four oncogenic FGFR4 mutations have been identified in rhabdomyosarcoma tumors including N535K, N535D, V550L, and V550E.8 According to our structure, the N535K or N535D mutations confer gain-of-function by disengaging the autoinhibitory molecular brake at the kinase hinge region. By contrast, the mechanism by which the V550L and V550E mutations confer gain-of-function is not fully understood. These mutations affect the gate-keeper residue of FGFR4 kinase that is known to control the access of ATP-competitive inhibitors to the rear hydrophobic pocket in the ATP binding cleft. We first assessed the kinase activities of oncogenic FGFR4K variants harboring the V550E, V550L, N535D, or N535K rhabomyosarcoma mutations using an in vitro peptide phosphorylation assay. Consistent with the published cell-based data,8 all four mutants exhibited elevated kinase activity compared to wild-type kinase as evident by the rapid completion of substrate monophosphorylation and appearance of diphosphorylated substrate peaks in the mass spectra of the oncogenic variants (Figure 2).
Figure 2.
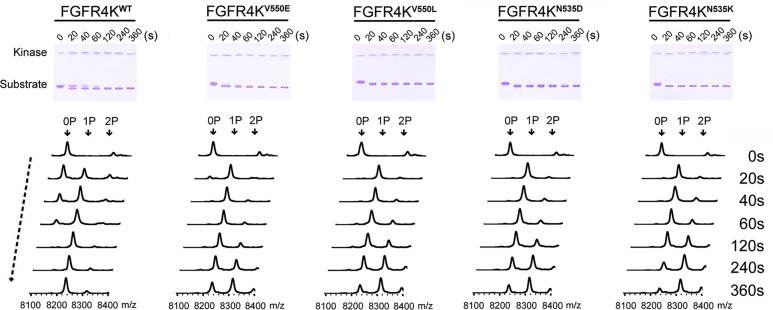
FGFR4K mutants harboring rhabdomyosarcoma mutations exhibiting elevated kinase activity. The substrate phosphorylation activities of FGFR4KWT, FGF4KN535K, FGFR4KN535D, FGFR4KV550L, and FGFR4KV550E were compared using native-PAGE (upper panel) coupled with time-resolved MALDI-TOF MS (lower panel). 0P, 1P, and 2P indicate the positions of the unphosphorylated, monophosphorylated, and dephosphorylated substrate peptide.
Next, the abilities of ponatinib and FIIN-2 to inhibit wild-type FGFR4K and its four oncogenic variants were examined. The data showed that ponatinib inhibited not only the wild-type FGFR4K (Figure 3A, left panel) but also FGFR4Ks that have undergone activation either by A-loop phosphorylation (Supporting Information Figure S4) or by gain-of-function mutations (N535K or N535D; Supporting Information Figure S5). The V555L and V550E gate-keeper mutants were resistant to inhibition by ponatinib, however (Figure 3A and Supporting Information Figure S5). By contrast, FIIN-2 effectively inhibited the substrate phosphorylation ability of all mutants including the V550L gate-keeper mutant (Figure 3B). To understand the molecular basis for the differential sensitivity of the FGFR4K gate-keeper mutations to ponatinib and FIIN-2, we decided to determine the crystal structures of wild-type FGFR4K bound to ponatinib, the FGFR4KV550L gate-keeper mutant alone, and in complex with FIIN-2.
Figure 3.

The V550L gate-keeper mutation conferring resistance to ponatinib but not to FIIN-2. The abilities of ponatinib (A) and FIIN-2 (B) to inhibit substrate phosphorylation activities of FGFR4KWT and the FGFR4KV550L (gate-keeper mutant) were compared using native-PAGE (upper panel) and time-resolved MALDI-TOF MS (lower panel). 0P, 1P, and 2P indicate the positions of the unphosphorylated, monophosphorylated, and dephosphorylated substrate peptide. (C) Antiproliferative activity of ponatinib and FIIN-2 on transformed Ba/F3 cells.
Ponatinib Inhibits FGFR4K by Inducing a DFG-in → DFG-out Rearrangement
The FGFR4K–ponatinib complex crystallized under identical conditions to the free FGFR4K, yielding crystals that were isomorphous to the free FGFR4K crystals. As anticipated, ponatinib binds into the ATP binding pocket between the N and C lobes (Figure 4A). Drug binding does not induce any significant change in the interlobe angle. The C-alpha atoms of FGFR4K–ponatinib and apo-FGFR4K structures superimpose very well (RMSD of 0.2 Å) with the exception of the DFG motif at the beginning of the A-loop, which undergoes a dramatic DFG-in → DFG-out rearrangement in response to ponatinib binding (Figure 4A and Supporting Information Figure S6A).
Figure 4.

Structural basis for FGFR4KWT inhibition by ponatinib and resistance caused by the V550L gate-keeper mutation. (A) Ribbon diagram of the FGFR4KWT–ponatinib cocrystal structure. The middle panel shows the close-up view of the main interactions between FGFR4KWT and ponatinib. The hydrogen bonds are indicated as black dashed lines, and the hydrophobic interactions are shown as surface. The right panel shows a close-up view of the DFG motif in the FGFR4KWT (in orange) and FGFR4KWT–ponatinib (in cyan) following superimposition of the two structures. Note that ponatinib forces the DFG out of the ATP binding pocket. The phenylalaninein DFG region and K503 are rendered as orange and cyan sticks and labeled in black and cyan, respectively. (B) Ribbon diagrams of the FGFR4KV550L structure. Superimposition of the FGFR4KWT–ponatinib complex structure onto FGFR4KV550L reveals steric clashes between the added methyl group in Leu-550 and the imidazo[1,2-b]pyridazine scaffold of ponatinib which underlie the resistance of the FGFR4KV550L to ponatinib. The V550 in FGFR4KWT and L550 in FGFR4KV550L are shown in pink and yellow sticks, and labeled in pink and red, respectively. In all the structures, the ponatinib is rendered as sticks and labeled in black.
Ponatinib consists of an imidazo[1,2-b]pyridazine heterocyclic scaffold that is linked via an acetylene group to a methylphenyl ring that in turn is joined via an amide bond to a trifluoromethylphenyl aromatic ring (Figure 4A). A methylpiperazine ring has been appended via methyl linkage to the trifluoromethylphenyl aromatic ring to aid in penetration of the drug into cells. Ponatinib binds into the ATP binding cleft in an extended conformation. The methylphenyl and trifluoromethylphenyl rings are almost coplanar and have a 90° interplanar angle with the (imidazo[1,2-b]pyridazine) bicyclic aromatic ring. Ponatinib engages a vast area that stretches from the kinase hinge region at the back of the kinase all the way to the catalytic pocket at the front end of the kinase.
The aromatic rings of the drug engage three sites within the ATP binding cleft (Figure 4A). At site 1, the imidazo[1,2-b]pyridazine scaffold occupies approximately the same space as the adenine ring of ATP and makes a single hydrogen bond with the backbone amide nitrogen atom of Ala-553 in the kinase hinge region (Figure 4A, middle panel). Reminiscent of ATP binding, the imidazo[1,2-b]pyridazine aromatic ring is sandwiched between hydrophobic residues from N- and C-lobes of the kinase. The rigid acetylene linkage directs the remaining portion of the drug deep toward the rear corner of the ATP binding pocket where the methylphenyl and 3-trifluoromethylphenyl aromatic rings of the inhibitor engage sites 2 and 3, respectively (Figure 4A, middle panel). Importantly, these aromatic rings induce two significant conformational changes in the ATP binding cleft including (I) displacement of the catalytic Lys-503 side chain in site 2 and (II) an outward flipping of the DFG motif in site 3 (Figure 4A, right panel). These structural rearrangements are necessary to alleviate steric conflicts with the compound ultimately optimizing binding interactions of the inhibitor.
At site 2, the methylphenyl group pushes the side chain of catalytic Lys-503, which along with Val-550, the gate-keeper residue, and Met-524 from the αC helix form a hydrophobic pocket that binds the methylphenyl aromatic ring. The displacement of Lys-503 indirectly helps the catalytic Glu-520 from the αC helix to make a direct hydrogen bond with the amide linkage between aromatic rings of the drug (Figure 4A, middle panel). At site 3, the 3-trifluoromethylphenyl moiety expels the DFG phenylalanine out of the cleft and occupies the hydrophobic pit that becomes vacant upon outward movement of the phenylalanine (Figure 4A, right panel). The displaced phenylalanine side chain is now in position to engage in favorable hydrophobic contacts with the scaffold and the acetylene linker (Figure 4A, middle panel). As another important consequence of phenylalanine displacement, Asp-630 from DFG is also forced into a catalytically incompetent orientation where the backbone atoms of Asp-630 gain the ability to make hydrogen bonds with the amide linkage between the aromatic rings of the inhibitor. Interestingly, even the piperazine moiety of the drug contributes to drug binding affinity. The piperazine ring falls in the vicinity of the catalytic loop where it engages in hydrogen bonds with the loop (Figure 4A, middle panel). The overall binding mode of ponatinib in the FGFR4K–ponatinib structure resembles that observed in the Abl–Ponatinib complex structure where the DFG-out mode of inhibition was initially observed.46 Unlike Abl–Ponatinib, however, the glycine-rich loop of FGFR4 does not partake in ponatinib recognition.
Gate-Keeper Mutations Confer Resistance to Ponatinib Inhibition by Introducing a Steric Clash with the Inhibitor
As shown in Supporting Information Figure S5, ponatinib is capable of silencing all the FGFR4 pathogenic mutations with the exception of the V550L and V550E gate-keeper mutations. To understand the molecular basis for how these mutations render the kinase refractory to inhibition, we also solved the crystal structure of FGFR4K harboring the V550L gate-keeper mutation (Figure 4B). Superimposition of the FGFR4K–ponatinib complex structure onto FGFR4KV550L reveals steric clashes between the added methyl group in Leu-550 and the imidazo[1,2-b]pyridazine scaffold of ponatinib which underlie the resistance of the FGFR4KV550L to ponatinib (Figure 4B).
Crystal Structure of FGFR4KV550L Complexed with FIIN-2
As shown in Figure 3, unlike ponatinib, FIIN-2 is capable of inhibiting the gate-keeper V550L mutant. To understand how FIIN-2 is capable of overcoming gate-keeper mutations, the crystal structure of the FGFR4KV550L mutant in complex with FIIN-2 was solved. FIIN-2 is a PD173074-based compound in which the cyclic urea N of the pyridopyrimidine scaffold has been derivatized with an acrylamidobenzyl substituent possessing a reactive acrylamide in the para position (Figure 5A and Supporting Information Figure S3B). The benzyl moiety serves as a spacer to position the acrylamide, the electrophilic center of the compound in the vicinity of the thiol group of a unique cysteine in the glycine-rich loop of FGFRs (Supporting Information Figure S1C) allowing for formation a covalent bond via a Michael addition reaction. To this end, we first used mass spectrometry to demonstrate that FIIN-2 irreversibly reacts with Cys-477 (in the glycine-rich loop) in both FGFR4KWT and FGFR4KV550L. To do so, FGFR4KWT and FGFR4KV550L were incubated with FIIN-2 and digested with trypsin, and adduct formation between the tryptic peptide containing Cys-477 and FIIN-2 was analyzed by tandem mass spectrometry. As shown in Figure 5B, in the presence of FIIN-2, the mass of the tryptic peptide containing the Cys-477 increased by 634.3 Da, corresponding to the mass of the drug, confirming that FIIN-2 can irreversibly inhibit both FGFR4KWT and FGFR4KV550L.
Figure 5.

Structural basis for the inhibition of FGFR4KV550L gate-keeper mutant by FIIN-2. (A) The chemical structure of FIIN-2. (B) The LC-MS/MS spectra of the kinase peptide (Pro42-Arg53) from FGFR4KWT and FGFR4KV550L with and without FIIN-2. The reacting Cys477 from the glycine-rich loop of the kinase is highlighted in red color. (C) Ribbon diagram of the FGFR4KV550L–FIIN-2 cocrystal structure. (D) The close-up view of the main interactions between FGFR4KV550L and FIIN-2. The hydrogen bonds are indicated as black dashed lines, and the hydrophobic interactions are shown as surface. (E) Close-up view of the DFG motif conformation in the FGFR4KWT (in orange), FGFR4KV550L (in teal), and FGFR4KV550L–FIIN-2 (in blue) structures following superimposition of the three structures. Note that FIIN-2 also binds to the ATP-binding site of FGFR4K in DFG-out mode. The phenylalanines in the DFG region of FGFR4KWT, FGFR4KV550L, and the FGFR4KV550L–FIIN-2 complex are rendered as orange, teal, and blue sticks and labeled in black and blue, respectively. (F) Superimposition of the FGFR4KWT–FIIN-2 complex structure onto the FGFR4KV550L–FIIN-2 structure. Note that rotational freedom around the single bond linking the scaffold and dimethoxyphenyl of FIIN-2 allows for small structural adjustments to bypass any potential steric clash with the bulkier side chain of L550. The V550 in FGFR4KWT and L550 in FGFR4KV550L are shown in orange and yellow sticks, and labeled in orange and red, respectively. (G) The distances between L550 of the FGFR4KV550L gate-keeper mutant and FIIN-2 are shown as dashed lines and labeled in a black color. In all of the structures, the FIIN-2 is rendered as sticks and labeled in black.
The FGFR4KV550L–FIIN-2 complex structure shows that like ponatinib, FIIN-2 binding does not affect the kinase interlobe angle (Figure 5C and Supporting Information Figure S7A). The C-alpha atoms of FGFR4KV550L–FIIN-2 and apo-FGFR4KV550L match closely with the exceptions of the glycine-rich loop and the DFG motif, both of which undergo major conformational rearrangements in response to drug binding (Figure 5C,D,E and Supporting Information Figure S7B). Remarkably, the DFG motif undergoes an outward transition reminiscent of that seen in the FGFR4K–ponatinib structure (Figure 5E). This is rather surprising because FIIN-2, like its parent molecule PD173074, lacks a functional group necessary to actively force the DFG out of the ATP binding cleft. As detailed below, this unusual property of FIIN-2, a type-I inhibitor, to bind FGFR4K like a type-II inhibitor is a direct consequence of the covalent bonding between FIIN-2 to FGFR4K.
The pyridopyrimidine scaffold of FIIN-2 occupies roughly the same space as the bicyclic ring of ponatinib (site 1) and is sandwiched by hydrophobic residues from the N and C lobes of kinase (Figure 5C,D). Likewise, the dimethoxyphenyl ring, the key determinant of selectivity of PD173074 and likewise FIIN-2 for FGFRs, penetrates deep into the back pocket of the cleft engaging the same site as the methylphenyl ring of ponatinib (site 2). The acrylamidobenzyl group protrudes out of the ATP-binding pocket and places its a,b-unsaturated acrylamide, the electrophilic center of the drug, in the vicinity of nucleophilic thiol group of Cys-477, resulting in a covalent bond formation via a Michael addition reaction (Figure 5). This covalent bonding pulls the glycine-rich loop toward the compound enabling additional contacts between the compound and the glycine-rich loop to form. Specifically, the phenylalanine from the glycine-rich loop makes hydrophobic contacts with the acrylamidobenzyl ring of drug and backbone amid nitrogens of the glycine-rich loop form a hydrogen bond with the carbonyl group of acrylamide of the drug (Figure 5D). In addition to causing the observed conformational change of the glycine-rich loop, the covalent bond between FIIN-2 and the glycine-rich loop cysteine is ultimately responsible for the DFG-flip seen in the structure. Specifically, the altered conformation of the glycine-rich loop creates favorable p–p stacking contacts between the Phe-478 from the glycine-rich loop and the Phe-631 from the DFG motif (Figure 5D), which stabilizes the DFG-out conformation enabling the Phe-631 to also contribute to inhibitor binding.
Comparison of the FGFR4KV550L–FIIN-2 and FGFR4KWT–ponatinib structures explains the molecular basis for the differential sensitivity of these two inhibitors toward the V550L gate-keeper mutation. While the added methyl group in Leu-550 introduces steric clash with the imidazo[1,2-b]pyridazine scaffold of ponatinib (Figure 4B, right panel), Leu-550 is still able to make favorable hydrophobic contacts with the dimethoxyphenyl ring of FIIN-2 (Figure 5D). It is noteworthy that the rigidity of the acetylene linkage is disadvantageous as it precludes any structural adjustment of the methylphenyl moiety of ponatinib (Supporting Information Figure S3A) to alleviate this steric clash. In contrast, rotational freedom around the single bond linking the scaffold and dimethoxyphenyl of FIIN-2 (Supporting Information Figure S3B) would allow for small structural adjustments to bypass any potential steric clash with the bulkier side chain of L550. Indeed, an overlay of the crystal structure of the FGFR4KWT–FIIN-2 complex onto the FGFR4KV550L–FIIN-2 structure shows that the dimethoxyphenyl ring in the FGFR4KV550L–FIIN-2 complex undergoes slight rotation around the single bound linker to accommodate for the bulkier leucine side chain in the gate-keeper mutant (Figure 5F,G).
Discussion
In this report, we elucidated the molecular bases for FGFR4 kinase autoinhibition, inhibition by ponatinib and FIIN-2, and drug resistance caused by gate-keeper mutations. The FGFR4K–ponatinib and FGFR4K–FIIN-2, reported here, are the first examples of FGFR–inhibitor complexes featuring a DFG-out mode of inhibition. In fact, only a tiny fraction of published tyrosine kinase-inhibitor structures depict a DFG-out mode of inhibition. Among RTKs, KIT, TIE2, MET, and VEGFR2 are the only examples that have been shown so far to bind inhibitors in a DFG-out fashion.47−49
Previously, crystal structures of FGFR1 and FGFR2 kinases in complex with inhibitors bearing oxyindole and pyridopyrimidine and quinazolin as scaffolds have been solved.38,39,50 In none of these structures, however, does the inhibitor penetrate deep enough to access site 3, and accordingly the DFG motif remains in its original “in” conformation. In fact, the oxyindoles do not even make use of site 2, although they can induce conformational changes in the glycine-rich loop to create hydrophobic contacts between conserved phenylalanine from the glycine-rich loop and the drug contributing to drug affinity. The FGFR4K–ponatinib (Figure 4) and FGFR4K–FIIN-2 (Figure 5) complex structures elegantly demonstrate that these two inhibitors attain their superior inhibitory potency against FGFR kinases by binding the kinase via a DFG-out mechanism. In addition, the FGFR4K–FIIN-2 structure, the first structure of an FGFR kinase with a covalently acting inhibitor (Figure 5), shows how this compound takes advantage of a unique cysteine in the glycine-rich loop of FGFRs to achieve FGFR target specificity.
Interestingly, in contrast to FGFRs, where the DFG-out conformation has never been visualized in the previously published apo crystal structures, there are structures of unliganded KIT and Abl that display a DFG-out conformation in the absence of an inhibitor.51,52 These data imply that in FGFRs, the DFG motif rarely transitions into the out conformation, whereas the DFG-out conformation can occur with significant frequencies in these other RTKs. Since the relative distribution of the DFG-in/DFG-out states will dictate the energetics of drug binding, future efforts should be directed toward exploring the dynamics of this transition in solution. Taken together, comparison of FGFR4K–ponatinib and FGFR4K–FIIN-2 complex structures with previous FGFRK–inhibitor complexes showcases a substantial degree of conformational heterogeneity both in the DFG motif and glycine-rich loop that should be harnessed when tailoring more efficacious FGFRK inhibitors. The structural data provide roadmaps for the design of novel inhibitors for FGFRKs which incorporate the salient inhibitory features of ponatinib and FIIN-2. In the FGFR4K–FIIN-2 structure, the hydrophobic pocket (site 3) that becomes vacant upon outward flipping of the DFG phenylalanine remains unutilized. Hence, the inhibitory potency of FIIN-2 may be significantly improved by derivatizing it with an aromatic ring such that it gains the ability to engage this hydrophobic pocket as it occurs in the FGFR4K–ponatinib structure. Likewise, the structural data pinpoint two unique cysteines, one in the catalytic loop of all four FGFRKs35 and the other in the hinge region of FGFR4K (Supporting Information Figure 1B) that can be exploited for the design of more selective covalent inhibitors for FGFR4K and other FGFRs.
Methods
Please refer to the Supporting Information for full details.
Protein Expression and Purification
The human FGFR4 kinase domains FGFR3K445–753, including its mutated forms, and the C-terminal tail peptide of FGFR2 kinase (FGFR2K761–821) were all expressed using pET bacterial expression vectors with an N-terminal 6XHis-tag to aid in protein purification.
Crystallization and Structure Determination
All the crystals were grown by hanging drop vapor diffusion method either at 4 °C (FGFR4KWT, FGFR4KV550L, and FGFR4KWT–ponatinib) or 18 °C (FGFR4KV550L/Cys477–FIIN-2). FGFR4KWT crystallized in a buffer composed of 0.1 M MES (pH 5.5), 20% (w/v) PEG 4000, 0.2 M Li2SO4, and 0.01 M taurine. Crystals of the FGFR4KV550L were obtained using a crystallization buffer composed of 0.1 M Tris (pH 7.5), 20% (w/v) PEG 1500, and 0.2 M (NH4)2SO4. The FGFR4KWT–ponatinib complex was crystallized using a crystallization buffer composed of 0.1 M MES (pH 5.5), 25% (w/v) PEG 4000, 0.15 M (NH4)2SO4, and 4% (v/v) formamide. The FGFR4KV550L/C477–FIIN-2 complex was crystallized using a crystallization buffer composed of 0.1 M HEPES (pH 7.5), 1.0–1.2 M (NH4)2SO4, and 10 mM yttrium(III) chloride hexahydrate. All diffraction data were processed using the HKL2000 suite,53 and the crystal structures were solved using maximum likelihood molecular replacement program Phaser in the PHENIX software suite.54 The crystal structure of wild-type FGFR2 kinase (PDB ID: 2PSQ)35 was used as the search model. Model building was carried out using Coot,55 and refinements were done using phenix.refine in the PHENIX suite.54 Data collection and structure refinement statistics are listed in Table 1.
Table 1. X-ray Data Collection and Refinement Statistics.
| construct | FGFR4Kapo | FGFR4K-Ponatinib | FGFR4KV550L | FGFR4KV550L-FIIN-2 |
|---|---|---|---|---|
| data collection | ||||
| resolution (Å) | 50.0–1.50 (1.53–1.50) | 50–1.90 (1.93–1.90) | 50–1.68 (1.71–1.68) | 50–2.2 (2.24–2.20) |
| space group | P21 | P21 | P21 | R3 |
| unit cell parameters (Å, deg) | a = 42.384 | a = 42.722 | a = 42.671 | a = 139.599 |
| b = 61.336 | b = 61.593 | b = 61.472 | b = 139.599 | |
| c = 61.084 | c =60.311 | c = 61.819 | c = 49.660 | |
| α = 90.00 | α = 90.00 | α = 90.00 | α = 90.00 | |
| β = 99.01 | β = 97.94 | β = 99.43 | β = 90.00 | |
| γ = 90.00 | γ = 90.00 | γ = 90.00 | γ = 120.00 | |
| content of the asymmetric unit | 1 | 1 | 1 | 1 |
| measured reflections (#) | 348260 | 182746 | 243115 | 88774 |
| unique reflections (#) | 49375 | 24319 | 34654 | 18138 |
| data redundancy | 7.1 (6.0) | 7.5 (7.7) | 7.0 (5.4) | 4.9 (2.4) |
| data completeness (%) | 100 (100) | 99.2 (98.1) | 96.6 (94.2) | 99.1 (90.1) |
| Rsym (%) | 5.8 (19.5) | 7.5 (30.3) | 5.2 (16.7) | 10.2 (34.4) |
| I/sig | 53.6 (8.6) | 41.8 (7.4) | 50.2 (10.0) | 13.5 (1.7) |
| refinement | ||||
| R factor/R free | 22.2/24.8 | 17.8/21.2 | 23.5/26.9 | 19.3/23.4 |
| number of protein atoms | 2204 | 2317 | 2242 | 2139 |
| number of nonprotein/solvent atoms | 10 | 44 | 5 | 47 |
| number of solvent atoms | 0 | 49 | 0 | 0 |
| RMSD bond length (Å) | 0.005 | 0.015 | 0.006 | 0.008 |
| RMSD bond angle (deg) | 1.04 | 1.61 | 1.00 | 1.23 |
| PDB ID | 4QQT | 4QRC | 4QQJ | 4QQ5 |
Numbers in parentheses refer to the highest resolution shell.
Rsym = Σ|I – I|ΣI, where I is the observed intensity of a reflection, and I is the average intensity of all the symmetry related reflections.
Peptide Substrate Phosphorylation Assay by Native Gel and MALDI-TOF Mass Spectrometry
Peptide substrate phosphorylation activities of wild-type and pathogenic mutated FGFR4 kinases (FGFR4KWT, FGFR4KN535 K, FGFR4KN535D, FGFR4KV550L, FGFR4KV550E) and the inhibitory efficiency of ponatinib or FIIN-2 were analyzed by native gel electrophoresis and positive ion MALDI-TOF MS (Bruker Autoflex MALDI-TOF, Bruker Daltonics) in linear mode.
Inhibition of Kinase Autophosphorylation by Ponatinib
The autophosphorylation of the wild-type FGFR4 kinase and its pathogenic variants (FGFR4KWT, FGFR4KN535K, FGFR4KN535D, FGFR4KV550L, FGFR4KV550E) inhibited by ponatinib were analyzed by native gel electrophoresis and LTQ Orbitrap (Thermo Electron) LC-MS/MS.
BaF3 Cell Viability Assay
TEL-FGFR4-transformed BaF3 cells were seeded in a 96 well plate and treated with the indicated concentration of the compounds. After 72 h, cell viability was assessed by MTS assay. The IC50 values were calculated using GraphPad Prism version 5.0 (GraphPad Software Inc.). To generate the FGFR4V550L expressing BaF3 cell line, the V550L mutation was introduced into the Tel-FGFR4WT chimera, which had been subcloned into the retroviral expression vector, using site-directed mutagenesis (Agilent) and was tranduced into BaF3 cell line using retroviral infection.
Analysis of Covalent Bond Formation between FIIN-2 and FGFR4K by LC-MS/MS
To test if FIIN-2 can form a covalent bond with Cys477 in the glycine-rich loops, FGFR4KWT/C477 and FGFR4KV550L/C477 were incubated with FIIN-2 overnight at 4 °C, digested with trypsin, and analyzed by LC-MS/MS. The spectral region corresponding to the kinase tryptic peptide (Pro42–Arg53), which contains the reactive Cys477, was extracted from the raw data and was used to show the mass shift of the peptide in the presence of FIIN-2.
The coordinates and structure factors have been deposited in the RCSB Protein Data Bank under PDB IDs 4QQT, 4QRC, 4QQJ, and 4QQ5 and will be immediately released upon publication.
Acknowledgments
The authors are thankful to R. Goetz and A. Belov for critically reading the manuscript and making thoughtful suggestions. This work was supported by the U.S. National Institutes of Health NIDCR Grant DE13686 (to M.M.), CA130876-05 and DF/HCC Lung Spore Developmental Project (to N.S.G), CA016087 and National Institutes of Health Shared Instrumentation Grant S10RR027990 (to T.A.N.) and grants from Natural Science Foundation of China 31270789, 81102486, and 81273421 (to H.C., Z.H. and H.W.).
Supporting Information Available
Supporting figures and methods as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ These authors contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Beenken A.; Mohammadi M. (2009) The FGF family: biology, pathophysiology and therapy. Nat. Rev. Drug Discovery 8, 235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh N.; Ornitz D. M. (2011) Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J. Biochem. (Tokyo) 149, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz R.; Mohammadi M. (2013) Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 14, 166–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon M. A.; Schlessinger J. (2010) Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T. (2004) Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 116, 191–203. [DOI] [PubMed] [Google Scholar]
- Schlessinger J.; Lemmon M. A. (2003) SH2 and PTB domains in tyrosine kinase signaling. Sci. STKE 2003, RE12. [DOI] [PubMed] [Google Scholar]
- Roumiantsev S.; Krause D. S.; Neumann C. A.; Dimitri C. A.; Asiedu F.; Cross N. C.; Van Etten R. A. (2004) Distinct stem cell myeloproliferative/T lymphoma syndromes induced by ZNF198-FGFR1 and BCR-FGFR1 fusion genes from 8p11 translocations. Cancer Cell 5, 287–298. [DOI] [PubMed] [Google Scholar]
- Taylor J. G. t.; Cheuk A. T.; Tsang P. S.; Chung J. Y.; Song Y. K.; Desai K.; Yu Y.; Chen Q. R.; Shah K.; Youngblood V.; Fang J.; Kim S. Y.; Yeung C.; Helman L. J.; Mendoza A.; Ngo V.; Staudt L. M.; Wei J. S.; Khanna C.; Catchpoole D.; Qualman S. J.; Hewitt S. M.; Merlino G.; Chanock S. J.; Khan J. (2009) Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Invest. 119, 3395–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Huang Z.; Dutta K.; Blais S.; Neubert T. A.; Li X.; Cowburn D.; Traaseth N. J.; Mohammadi M. (2013) Cracking the molecular origin of intrinsic tyrosine kinase activity through analysis of pathogenic gain-of-function mutations. Cell Rep. 4, 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster M. K.; Donoghue D. J. (1997) FGFR activation in skeletal disorders: too much of a good thing. Trends Genet. 13, 178–182. [DOI] [PubMed] [Google Scholar]
- Wilkie A. O. (2005) Bad bones, absent smell, selfish testes: the pleiotropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev. 16, 187–203. [DOI] [PubMed] [Google Scholar]
- Chesi M.; Nardini E.; Brents L. A.; Schrock E.; Ried T.; Kuehl W. M.; Bergsagel P. L. (1997) Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat. Genet. 16, 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappellen D.; De Oliveira C.; Ricol D.; de Medina S.; Bourdin J.; Sastre-Garau X.; Chopin D.; Thiery J. P.; Radvanyi F. (1999) Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat. Genet. 23, 18–20. [DOI] [PubMed] [Google Scholar]
- Pollock P. M.; Gartside M. G.; Dejeza L. C.; Powell M. A.; Mallon M. A.; Davies H.; Mohammadi M.; Futreal P. A.; Stratton M. R.; Trent J. M.; Goodfellow P. J. (2007) Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene 26, 7158–7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand V.; Huang J.; Stockwell T.; Ferriera S.; Buzko O.; Levy S.; Busam D.; Li K.; Edwards J. B.; Eberhart C.; Murphy K. M.; Tsiamouri A.; Beeson K.; Simpson A. J.; Venter J. C.; Riggins G. J.; Strausberg R. L. (2005) Sequence survey of receptor tyrosine kinases reveals mutations in glioblastomas. Proc. Natl. Acad. Sci. U. S. A. 102, 14344–14349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grose R.; Dickson C. (2005) Fibroblast growth factor signaling in tumorigenesis. Cytokine Growth Factor Rev. 16, 179–186. [DOI] [PubMed] [Google Scholar]
- Frierson H. F. Jr.; Moskaluk C. A. (2013) Mutation signature of adenoid cystic carcinoma: evidence for transcriptional and epigenetic reprogramming. J. Clin. Invest. 123, 2783–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logie A.; Dunois-Larde C.; Rosty C.; Levrel O.; Blanche M.; Ribeiro A.; Gasc J. M.; Jorcano J.; Werner S.; Sastre-Garau X.; Thiery J. P.; Radvanyi F. (2005) Activating mutations of the tyrosine kinase receptor FGFR3 are associated with benign skin tumors in mice and humans. Hum. Mol. Genet. 14, 1153–1160. [DOI] [PubMed] [Google Scholar]
- Reiter A.; Sohal J.; Kulkarni S.; Chase A.; Macdonald D. H.; Aguiar R. C.; Goncalves C.; Hernandez J. M.; Jennings B. A.; Goldman J. M.; Cross N. C. (1998) Consistent fusion of ZNF198 to the fibroblast growth factor receptor-1 in the t(8;13)(p11;q12) myeloproliferative syndrome. Blood 92, 1735–1742. [PubMed] [Google Scholar]
- Wu Y.-M.; Su F.; Kalyana-Sundaram S.; Khazanov N.; Ateeq B.; Cao X.; Lonigro R. J.; Vats P.; Wang R.; Lin S.-F. (2013) Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discovery 3, 636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D.; Chan J. M.; Zoppoli P.; Niola F.; Sullivan R.; Castano A.; Liu E. M.; Reichel J.; Porrati P.; Pellegatta S. (2012) Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 337, 1231–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer C. L.; Dopheide B.; Savagner P.; Thompson E. W.; Williams E. D. (2007) Aberrant fibroblast growth factor receptor signaling in bladder and other cancers. Differentiation 75, 831–842. [DOI] [PubMed] [Google Scholar]
- Fletcher M. N.; Castro M. A.; Wang X.; de Santiago I.; O’Reilly M.; Chin S. F.; Rueda O. M.; Caldas C.; Ponder B. A.; Markowetz F.; Meyer K. B. (2013) Master regulators of FGFR2 signalling and breast cancer risk. Nat. Commun. 4, 2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama N.; Varjosalo M.; Meller P.; Lohi J.; Hyytiainen M.; Kilpinen S.; Kallioniemi O.; Ingvarsen S.; Engelholm L. H.; Taipale J.; Alitalo K.; Keski-Oja J.; Lehti K. (2010) Fibroblast growth factor receptor 4 regulates tumor invasion by coupling fibroblast growth factor signaling to extracellular matrix degradation. Cancer Res. 70, 7851–7861. [DOI] [PubMed] [Google Scholar]
- Andre F.; Bachelot T.; Campone M.; Dalenc F.; Perez-Garcia J. M.; Hurvitz S. A.; Turner N.; Rugo H.; Smith J. W.; Deudon S.; Shi M.; Zhang Y.; Kay A.; Porta D. G.; Yovine A.; Baselga J. (2013) Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin. Cancer Res. 19, 3693–3702. [DOI] [PubMed] [Google Scholar]
- Cortes J. E.; Talpaz M.; Kantarjian H. (2014) Ponatinib in Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 370, 577. [DOI] [PubMed] [Google Scholar]
- Gozgit J. M.; Wong M. J.; Moran L.; Wardwell S.; Mohemmad Q. K.; Narasimhan N. I.; Shakespeare W. C.; Wang F.; Clackson T.; Rivera V. M. (2012) Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol. Cancer Ther. 11, 690–699. [DOI] [PubMed] [Google Scholar]
- Llovet J. M.; Decaens T.; Raoul J. L.; Boucher E.; Kudo M.; Chang C.; Kang Y. K.; Assenat E.; Lim H. Y.; Boige V.; Mathurin P.; Fartoux L.; Lin D. Y.; Bruix J.; Poon R. T.; Sherman M.; Blanc J. F.; Finn R. S.; Tak W. Y.; Chao Y.; Ezzeddine R.; Liu D.; Walters I.; Park J. W. (2013) Brivanib in patients with advanced hepatocellular carcinoma who were intolerant to sorafenib or for whom sorafenib failed: results from the randomized phase III BRISK-PS study. J. Clin. Oncol. 31, 3509–3516. [DOI] [PubMed] [Google Scholar]
- Gavine P. R.; Mooney L.; Kilgour E.; Thomas A. P.; Al-Kadhimi K.; Beck S.; Rooney C.; Coleman T.; Baker D.; Mellor M. J.; Brooks A. N.; Klinowska T. (2012) AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 72, 2045–2056. [DOI] [PubMed] [Google Scholar]
- Nguyen P. T.; Tsunematsu T.; Yanagisawa S.; Kudo Y.; Miyauchi M.; Kamata N.; Takata T. (2013) The FGFR1 inhibitor PD173074 induces mesenchymal-epithelial transition through the transcription factor AP-1. Br. J. Cancer 109, 2248–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grand E. K.; Chase A. J.; Heath C.; Rahemtulla A.; Cross N. C. (2004) Targeting FGFR3 in multiple myeloma: inhibition of t(4;14)-positive cells by SU5402 and PD173074. Leukemia 18, 962–966. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Hur W.; McDermott U.; Dutt A.; Xian W.; Ficarro S. B.; Zhang J.; Sharma S. V.; Brugge J.; Meyerson M.; Settleman J.; Gray N. S. (2010) A structure-guided approach to creating covalent FGFR inhibitors. Chem. Biol. 17, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byron S. A.; Chen H.; Wortmann A.; Loch D.; Gartside M. G.; Dehkhoda F.; Blais S. P.; Neubert T. A.; Mohammadi M.; Pollock P. M. (2013) The N550K/H mutations in FGFR2 confer differential resistance to PD173074, dovitinib, and ponatinib ATP-competitive inhibitors. Neoplasia 15, 975–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blencke S.; Zech B.; Engkvist O.; Greff Z.; Orfi L.; Horvath Z.; Keri G.; Ullrich A.; Daub H. (2004) Characterization of a conserved structural determinant controlling protein kinase sensitivity to selective inhibitors. Chem. Biol. 11, 691–701. [DOI] [PubMed] [Google Scholar]
- Chen H.; Ma J.; Li W.; Eliseenkova A. V.; Xu C.; Neubert T. A.; Miller W. T.; Mohammadi M. (2007) A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol. Cell 27, 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae J. H.; Boggon T. J.; Tome F.; Mandiyan V.; Lax I.; Schlessinger J. (2010) Asymmetric receptor contact is required for tyrosine autophosphorylation of fibroblast growth factor receptor in living cells. Proc. Natl. Acad. Sci. U. S. A. 107, 2866–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.; Chen H.; Blais S.; Neubert T. A.; Li X.; Mohammadi M. (2013) Structural mimicry of a-loop tyrosine phosphorylation by a pathogenic FGF receptor 3 mutation. Structure 21, 1889–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M.; McMahon G.; Sun L.; Tang C.; Hirth P.; Yeh B. K.; Hubbard S. R.; Schlessinger J. (1997) Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science 276, 955–960. [DOI] [PubMed] [Google Scholar]
- Eathiraj S.; Palma R.; Hirschi M.; Volckova E.; Nakuci E.; Castro J.; Chen C. R.; Chan T. C.; France D. S.; Ashwell M. A. (2011) A novel mode of protein kinase inhibition exploiting hydrophobic motifs of autoinhibited kinases: discovery of ATP-independent inhibitors of fibroblast growth factor receptor. J. Biol. Chem. 286, 20677–20687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M.; Froum S.; Hamby J. M.; Schroeder M. C.; Panek R. L.; Lu G. H.; Eliseenkova A. V.; Green D.; Schlessinger J.; Hubbard S. R. (1998) Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain. EMBO J. 17, 5896–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzat S.; Zheng L.; Zhu X. F.; Wu G. E.; Asa S. L. (2002) Targeted expression of a human pituitary tumor-derived isoform of FGF receptor-4 recapitulates pituitary tumorigenesis. J. Clin. Invest. 109, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ye Y. W.; Hu S.; Shi Y. Q.; Zhang X. F.; Zhou Y.; Zhao C. L.; Wang G. J.; Wen J. G.; Zong H. (2013) Combination of the FGFR4 inhibitor PD173074 and 5-fluorouracil reduces proliferation and promotes apoptosis in gastric cancer. Oncol. Rep. 30, 2777–2784. [DOI] [PubMed] [Google Scholar]
- Ho H. K.; Nemeth G.; Ng Y. R.; Pang E.; Szantai-Kis C.; Zsakai L.; Breza N.; Greff Z.; Horvath Z.; Pato J.; Szabadkai I.; Szokol B.; Baska F.; Orfi L.; Ullrich A.; Keri G.; Chua B. T. (2013) Developing FGFR4 inhibitors as potential anti-cancer agents via in silico design, supported by in vitro and cell-based testing. Curr. Med. Chem. 20, 1203–1217. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Gray N. S. (2006) Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2, 358–364. [DOI] [PubMed] [Google Scholar]
- Li S. Q.; Cheuk A. T.; Shern J. F.; Song Y. K.; Hurd L.; Liao H.; Wei J. S.; Khan J. (2013) Targeting wild-type and mutationally activated FGFR4 in rhabdomyosarcoma with the inhibitor ponatinib (AP24534). PLoS One 8, e76551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T.; Commodore L.; Huang W. S.; Wang Y.; Thomas M.; Keats J.; Xu Q.; Rivera V. M.; Shakespeare W. C.; Clackson T.; Dalgarno D. C.; Zhu X. (2011) Structural mechanism of the Pan-BCR-ABL inhibitor ponatinib (AP24534): lessons for overcoming kinase inhibitor resistance. Chem. Biol. Drug Des. 77, 1–11. [DOI] [PubMed] [Google Scholar]
- Leproult E.; Barluenga S.; Moras D.; Wurtz J. M.; Winssinger N. (2011) Cysteine mapping in conformationally distinct kinase nucleotide binding sites: application to the design of selective covalent inhibitors. J. Med. Chem. 54, 1347–1355. [DOI] [PubMed] [Google Scholar]
- Han S.; Mistry A.; Chang J. S.; Cunningham D.; Griffor M.; Bonnette P. C.; Wang H.; Chrunyk B. A.; Aspnes G. E.; Walker D. P.; Brosius A. D.; Buckbinder L. (2009) Structural characterization of proline-rich tyrosine kinase 2 (PYK2) reveals a unique (DFG-out) conformation and enables inhibitor design. J. Biol. Chem. 284, 13193–13201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard J. R.; Getlik M.; Grutter C.; Schneider R.; Wulfert S.; Rauh D. (2010) Fluorophore labeling of the glycine-rich loop as a method of identifying inhibitors that bind to active and inactive kinase conformations. J. Am. Chem. Soc. 132, 4152–4160. [DOI] [PubMed] [Google Scholar]
- Liang G.; Liu Z.; Wu J.; Cai Y.; Li X. (2012) Anticancer molecules targeting fibroblast growth factor receptors. Trends Pharmacol. Sci. 33, 531–541. [DOI] [PubMed] [Google Scholar]
- Mol C. D.; Dougan D. R.; Schneider T. R.; Skene R. J.; Kraus M. L.; Scheibe D. N.; Snell G. P.; Zou H.; Sang B. C.; Wilson K. P. (2004) Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J. Biol. Chem. 279, 31655–31663. [DOI] [PubMed] [Google Scholar]
- Levinson N. M.; Kuchment O.; Shen K.; Young M. A.; Koldobskiy M.; Karplus M.; Cole P. A.; Kuriyan J. (2006) A Src-like inactive conformation in the abl tyrosine kinase domain. PLoS Biol. 4, e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Grosse-Kunstleve R. W.; Hung L. W.; Ioerger T. R.; McCoy A. J.; Moriarty N. W.; Read R. J.; Sacchettini J. C.; Sauter N. K.; Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.