

Abstract
TRPML1 (mucolipin-1/MCOLN1) is predicted to be an intracellular late endosomal and lysosomal ion channel protein belonging to the mucolipin subfamily of Transient Receptor Potential (TRP) proteins 1–3. Mutations in the human TRPML1 gene cause mucolipidosis type IV disease (ML4) 4, 5. ML4 patients exhibit motor impairment, mental retardation, retinal degeneration, and iron-deficiency anemia. Since aberrant iron metabolism may cause neural and retinal degeneration 6, 7, it may be a primary cause of ML4 phenotypes. In most mammalian cells, release of iron from endosomes and lysosomes following iron uptake via endocytosis of Fe3+-bound transferrin receptors 6, or following lysosomal degradation of ferritin-Fe complexes and autophagic ingestion of iron-containing macromolecules 6, 8, is the major source of cellular iron. The Divalent Metal Transporter protein (DMT1) is the only endosomal Fe2+ transporter currently known and is highly expressed in erythroid precursors 6, 9, but genetic studies suggest the existence of a DMT1-independent endosomal/lysosomal Fe2+ transport protein 9. Here, by measuring radiolabeled iron uptake, monitoring the levels of cytosolic and intra-lysosomal iron and directly patch-clamping the late endosomal/lysosomal membrane, we show that TRPML1 functions as a Fe2+ permeable channel in late endosomes and lysosomes. ML4 mutations are shown to impair TRPML1’s ability to permeate Fe2+ at varying degrees, which correlate well with the disease severity. A comparison of TRPML1−/− ML4 and control skin fibroblasts showed a reduction of cytosolic Fe2+ levels, an increase of intra-lysosomal Fe2+ levels, and an accumulation of lipofuscin-like molecules in TRPML1−/− cells. We propose that TRPML1 mediates a mechanism by which Fe2+ is released from late endosomes/lysosomes. Our results suggest that impaired iron transport may contribute to both hematological and degenerative symptoms of ML4 patients.
Keywords: iron transport, calcium channel, transient receptor potential (TRP) channel, iron-deficiency, anemia, patch clamp, whole-lysosome recording, lumenal-side-out patch, mucolipidosis, ML4, endosomes, lysosomes, lipofuscin, aging pigment, free radical oxide
Release of Fe2+ from late endosomes and lysosomes into the cytosol is essential for cellular iron metabolism (see Supplementary Fig. 1) 6. Since TRPML1 is ubiquitously expressed (in cells of every tissue) 5 and primarily localized in the late endosome/lysosome (LE/Ly) 1, 3, 10, we hypothesize that TRPML1 may serve as a novel endo-lysosomal Fe2+ release channel. The intracellular localization of wild-type (wt) TRPML1 1, 3, 10 (also see Supplementary Fig. 2), however, makes it difficult to assay TRPML1’s channel function. To overcome this problem, we recently developed a functional assay for TRPML channels 11. A spontaneous mouse mutation (A419P) 12 dramatically increases TRPML3-mediated currents at the plasma membrane without altering its permeation properties 11. Mice carrying this mutation (Varitint-Waddler, Va) are deaf and exhibit skin pigmentation defects. When the Va mutation (proline substitution) was introduced into a homologous position in TRPML1 (V432P), the mutant channel TRPML1Va was (mis-)localized in many cellular compartments including both LE/Ly and the plasma membrane (Supplementary Fig. 2). Importantly, TRPML1Va, but not wt TRPML1, generated a cationic whole-cell current that can be measured by patch clamp 11.
We studied TRPML proteins by transiently expressing them in HEK293T cell lines. To monitor expression, TRPML channels were fused to enhanced green fluorescent protein (EGFP) at their N-termini. In response to a voltage step protocol, TRPML1Va-expressing cells displayed strong inwardly rectifying step currents (Fig. 1a & Supplementary Fig. 3) in a standard extracellular bath solution (a modified Tyrode’s solution). TRPML1Va-mediated current (ITRPML1-Va) was 101 ± 8.6 pA/pF at −80 mV (mean ± SEM, n = 48). To mimic the acidic environments of lysosomes where the extracellular side (analogous to the intra-lysosomal lumenal side) of the wt TRPML1 protein is exposed to, extracellular solutions were adjusted to pH 4.6. No significant inward current was detected when NMDG+ was the only major cation in the bath (pH 4.6; Fig. 1b), indicating that ITRPML1-Va is a proton-impermeable cationic current 11. Bath perfusion of 30 mM [Fe2+]o (pH 4.6) to TRPML1Va-expressing cells induced large inwardly rectifying currents (Fig. 1c). The current density of [Fe2+]o–elicited ITRPML1-Va (IFeTRPML1-Va) at −80 mV was 74 ± 8.6 pA/pF (mean ± SEM, n = 24). To keep Fe2+ in a reduced state, ascorbic acid was used as the counter anion in the bath solution. The only other major cation in the 30 mM Fe2+ solution was NMDG+. Thus IFeTRPML1-Va is carried solely by Fe2+. Consistent with this, the amplitude of IFeTRPML1-Va was strongly dependent on [Fe2+]o (Supplementary Fig. 4).
FIGURE 1. TRPML1Va-expressing cells have a constitutively active H+-modulated Fe2+ current.
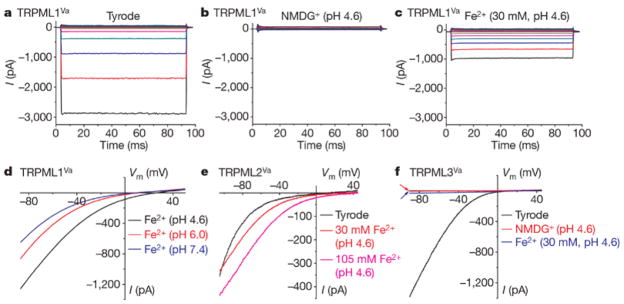
a, A TRPML1Va-expressing HEK293T cell showed a large inwardly rectifying whole-cell current elicited by voltage steps (from −140 mV to +80 mV in increments of 20 mV) in the standard extracellular (Tyrode’s) bath solution. Step duration, 90 ms; holding potential, 0 mV; Vm, membrane potential. b, No significant inward current was detected in NMDG+ (Na+-free, Ca2+-free, pH 4.6) solution. c, Inwardly rectifying step currents were evoked by 30 mM Fe2+ solution (pH 4.6) in the same cell as shown in a and b. d, pH-dependence of IFe/TRPML1Va. Whole-cell currents were elicited by repeated voltage ramps (−100 to +100 mV; 400 ms) with a 4-s interval between ramps. Only a portion of the voltage protocol is shown. Holding potential, 0 mV. e, Large IFe/TRPML2Va was seen in the presence of 30 and 105 mM Fe2+ (pH 4.6). f, Little or no IFe was seen in a TRPML3Va-expressing cell.
Both IFeTRPML1-Va (Fig. 1e) and ITRPML1-Va 11 were enhanced at low pH. Compared to experiments conducted at physiological pH (pH 7.4), IFeTRPML1-Va was enhanced ~2 fold at pH 4.6 (approximately the lumenal pH of lysosomes), respectively. In cells transfected with TRPML2Va (a short splice variant; see Methods), large current (23.0 ± 3.8 pA/pF at −80 mV, n = 7) was evoked by 30 mM Fe2+ (pH 4.6, Fig. 1e). Similar to ITRPML1-Va, ITRPML2-Va was also strongly potentiated by low pH (Supplementary Fig. 5). The ratio of IFe/ITyrode for TRPML2Va was 139 ± 22% (at −80 mV, n = 7), which was even larger than the ratio for TRPML1Va (72 ± 7 %, n = 11). In contrast with TRPML1Va and TRPML2Va, the ratio of IFe/ITyrode for TRPML3Va was only 2.3 ± 0.2% (n = 8; Fig. 1f). Thus Fe2+ permeability is specific for TRPML1 and TRPML2, but not for the closely-related TRPML3. Cationic selectivity analysis suggested that TRPML1Va was also permeable to most other divalent trace metals including Mn2+ and Zn2+, but not to Fe3+ (Supplementary Fig. 6). Detailed analyses of the permeation properties of TRPML1Va suggested that the conduction mechanism of Fe2+ resembled those of Na+ and Ca2+ (see Supplementary Figs. 7–9).
Next, we tested whether TRPML1’s Fe2+ permeability is impaired by ML4 mutations. More than 15 ML4 mutations have been identified, most of which are located in the transmembrane regions of TRPML1 (Supplementary Fig. 10) 4, 5, 13, 14. We constructed six of these ML4 mutants into the TRPML1Va background and investigated the Fe2+ permeability of these combined mutations. We found that all ML4 mutant TRPML1Va channels had significantly smaller IFe than TRPML1Va. In particular, T232P, D362Y, and V446L mutations (combined with the Va mutation, below same) completely eliminated IFe, as well as ITyrode (Fig. 2a, Supplementary Fig. 11), although the protein expression levels and subcellular localization pattern of these mutants were similar to those of the parental TRPML1Va protein (Supplementary Fig. 12 & 13). Patients carrying these mutations are reported to have severe phenotypes 5, 15. On the other hand, a large IFe was detected in F408Δ-TRPML1Va-transfected cells. The average current density of this mutant, however, was still significantly smaller than IFeTRPML1-Va. Patients carrying the F408Δ mutation display unusually mild phenotypes 13, 15. Small but evident IFe was detected with expression of two other mutants (R403C and F465L). A patient carrying the R403C mutation was reported to have a relatively mild phenotype 14. Taken together, these results suggest that ML4 mutations significantly impair TRPML1’s Fe2+ permeability. The degree of the impaired Fe2+ permeability appears to correlate well with disease severity observed in ML4 patients.
FIGURE 2. Fe2+ permeability of the TRPML1 channel is impaired by ML4 mutations.

a, Current densities (mean s.e.m., n = 4–10) of IFe (30 mM Fe2+, pH 4.6) for TRPML1Va and ML4 mutant TRPML1Va channels. Asterisk indicates statistical difference (P < 0.01) compared to TRPML1Va. b, 55Fe2+ uptake (normalized) in HEK293T cells transfected with vector control, with TRPML1Va, with F408Δ-TRPML1Va and with T232P-TRPML1Va constructs. Error bars indicate the standard deviation on the basis of two independent triplicate experiments. c, [Fe2+]o-dependent quenching of Fura-2 fluorescence in TRPML1Va-transfected cells (arrows), but not in non-transfected control cells (arrowheads) or T232P-TRPML1Va-transfected cells (bottom row). The fluorescence intensity was measured at an excitation wavelength of 360 nm (F360). The original magnification used for all micrographs Was ×200. d, Average normalized responses of EGFP-positive TRPML1Va-transfected cells (typically n = 20–40 cells) to 1 or 10 mM Fe2+ (pH 4.6). e, No significant quenching was seen in T232P-TRPML1Va-transfected cells. f, Slightly less quenching was observed for the F408Δ-TRPML1Va-expressing cells. g, A small but significant quenching reaction was detected (with 10 mM Fe2+) in R403C-TRPML1Va-expressing cells.
To investigate TRPML1Va-mediated Fe2+ influx under less-invasive conditions in intact cells, we measured 55Fe2+ uptake at low pH (pH 5.8, 1μM 55Fe2+; Fig. 2b). Significant 55Fe2+ uptake was seen in HEK293T cells transfected with TRPML1Va, but not with T232P-TRPML1Va. An intermediate 55Fe2+ uptake was seen for F408Δ-TRPML1Va. These results suggest that TRPML1 Va can mediate significant Fe2+ entry even at micromolar [Fe2+]o. We also adopted a fluorescence-based Fe2+ quenching assay to measure TRPML1Va-mediated Fe2+ entry in HEK293T cells. Heavy metals such as Mn2+ and Fe2+ can bind Ca2+-sensitive dyes such as Fura-2 with higher affinity, resulting in strong fluorescence quenching effects 16. Substantial quenching of Fura-2 fluorescence was seen in TRPML1Va-expressing cells with addition of 1 mM Fe2+ (pH 4.6) (Fig. 2f, 2d). Increased quenching was observed with the addition of higher concentrations of Fe2+ (10 mM, pH 4.6). In contrast, no significant quenching was detected in neighboring non-transfected EGFP-negative cells, or TRPML3Va-transfected cells (1 mM Fe2+; Supplementary Fig. 14). ML4 mutant TRPML1Va–expressing HEK293T cells displayed an impairment of Fe2+ quenching. No significant Fe2+ quenching was seen in T232P-(Fig. 2e), D362Y-, or V446L-TRPML1Va-expressing cells (data not shown). Slightly less quenching was observed for F408Δ-TRPML1Va (Fig. 2f). In contrast, R403C-TRPML1Va (Fig. 2g) and F465L-TRPML1Va showed no response to addition of 1 mM Fe2+. However, a higher concentration of Fe2+ (10 mM) induced a slow but significant quenching. These results agree with the electrophysiological measurements of ML4 mutants, as well as the disease severity of ML4 patients carrying these mutations 5, 13–15. We conclude that ML4 phenotypes result from a loss-of-function of TRPML1 with respect to Fe2+ and/or Ca2+ permeability.
To validate our use of TRPML1Va as a model to extrapolate potential intracellular functions of wt TRPML1, and more importantly, to confirm whether wt TRPML1 conducts Fe2+ from the lumen of LE/Ly into the cytosol, we performed patch clamp recordings directly on native LE/Ly membranes (Fig. 3a & Supplementary Fig. 15; see methods summary). In TRPML1Va-positive enlarged LE/Ly, large inwardly rectifying currents were seen under the lysosome-attached configuration (Fig. 3b, 3c). The currents became smaller when the patch was excised (lysosome lumenal-side-out) and exposed to Tyrode’s solution (Fig. 3c). These large inwardly rectifying currents were not seen in TRPML1Va-negative vacuoles, suggesting that these lysosomal currents are mediated by TRPML1Va (hence are referred to as lysosomal ITRPML1-Va). Note that inward lysosomal currents are actually currents that flow out of the lysosomes. Similar to ITRPML1-Va recorded at the plasma membrane, lysosomal ITRPML1-Va was impermeable to NMDG+ or H+, but potentiated by low pH or by removal of divalent cations in the bath solution (Nominal Divalent-free) (Fig. 3d, 3e). When the lumenal-side-out patches were exposed to 30 mM or 105 mM Fe2+ solutions (pH 4.6), smaller and less rectifying currents were seen, resembling IFe TRPML1-Va at the plasma membrane. These results suggest that TRPML1Va behave similarly at both plasma membrane and lysosomal membranes. Likewise, ML4 mutant TRPML1Va channels also behaved similarly as their plasma membrane counterparts (Supplementary Fig. 16), consistent with our conclusion that ML4 mutations primarily affect the channel function of TRPML1.
FIGURE 3. TRPML1 conducts Fe2+ in late endosomes and lysosomes.

a, Co-localization of mCherry–TRPML1 and EGFP–LAMP1 at the membrane of an isolated enlarged LEL (see Methods). The patch pipette was filled with red rhodamine B dye (shown in blue for the purpose of illustration). b, Cartoons of three distinct patch-clamp configurations of lysosomal recordings: lysosome-attached, lysosome luminal-side-out and whole-lysosome. In each configuration, the pink arrow indicates the direction of the inward (at negative potentials; flow out of the lysosomes) current mediated by TRPML1 (as shown in c–i). c, Lysosomal ITRPML1Va. Switching from lysosome-attached to (lysosome) luminal-side-out configuration significantly reduced the amplitude of the current. The luminal-side-out patch was exposed to the Tyrode’s solution. A Cs+-based solution (147 mM Cs-methanesulphonate (Cs-MSA)) was used as a pipette solution for both configurations. d, NMDG+-impermeable lysosomal ITRPML1Va was much larger in the absence of divalent cations (nominal divalent-free). e, Lowering pH potentiated lysosomal ITRPML1Va. f, IFe/TRPML1Va induced by 30 mM and 105 mM Fe2+. g, Whole-lysosome current in an enlarged lysosome expressing wild-type TRPML1. The pipette (lumen) solution contained nominal divalent-free Tyrode solution. A Cs+-based bath solution (147 mM Cs-MSA) was used. h, Whole-lysosome ITRPML1Va. i, Whole-lysosome IFe/TRPML1. The pipette (lumen) solution contained 105 mM Fe2+ (pH 4.6).
Next we performed whole-lysosome recordings on enlarged LE/Ly expressing wt TRPML1 and TRPML1Va. Significant inwardly rectifying currents were seen in wt TRPML1-positive LE/Ly (81.0 ± 9.0 pA/pF at −120 mV, n = 6; Fig. 3g) with Nominal Divalent-free solution in the lumenal side (i.e., pipette solution), although the current amplitude is still one order of magnitude smaller than the whole-lysosome current recorded from TRPML1Va-positive LE/Ly (1,713 ± 404 pA/pF at −120 mV, n = 4; Fig. 3h). Since we were not able to record any significant whole-cell current from wt TRPML1-expressing HEK293T cells 11, our results suggest that the Va mutation affects both channel gating and trafficking (between LE/Ly and the plasma membrane) of TRPML1. When isotonic (105 mM) Fe2+ solution was included in the lumenal side of wt TRPML1-positive enlarged LE/Ly, inwardly rectifying currents with very positive reversal potential were observed (18 ± 4 pA/pF at −120 mV, n = 4; Fig. 3i). Collectively, these results suggest that wt TRPML1 is a lysosomal Fe2+-permeable channel, and that the Va mutation does not alter the permeation properties of the TRPML1 channel.
Retention of Fe2+ in LE/Ly due to loss/inactivation of Fe2+ release mechanism may result in an insufficient supply of cytosolic Fe2+. We therefore measured the levels of free (chelatable) intracellular Fe2+ in skin fibroblasts from a ML4 patient (TRPML1−/−) and the non-diseased parent (TRPML1+/−) using a fluorescence-based iron de-quenching imaging method (see Methods) 17. Chelatable Fe2+ levels were significantly lower in TRPML1−/− cells compared to the control TRPML1+/−cells (Fig. 4a, 4b). TRPML1-deficient skin fibroblast cells exhibit auto-fluorescence (Fig. 4c) at excitation wavelengths between 440 nm and 500 nm, reminiscent of accumulated lipofuscin-like substances previously reported 18. The auto-fluorescence observed was primarily localized in Lamp1 (a marker for LE/Ly)-positive compartments (Fig. 4c), indicative of a lysosomal dysfunction in TRPML1−/− cells. Lipofuscin, also referred to as an “aging pigment”, is a non-degradable oxidized polymeric substance containing proteins, lipids, carbohydrates, and iron 19. The production of lipofuscin is facilitated by increased intra-lysosomal Fe2+ levels 19. Our results suggest that an accumulation of auto-fluorescent lipofuscin-like molecules in TRPML1−/− cells might result from impaired intra-lysosomal iron metabolism. Consistent with this, lysosomal iron staining experiments revealed that TRPML1−/− cells exhibited higher lysosomal iron content than control cells (Supplementary Fig. 17).
FIGURE 4. TRPML1-deficient cells have reduced free (chelatable) iron levels and lysosomal autofluorescence.

a, Cultured TRPML1−/ −(ML1−/ −) skin fibroblasts showed less de-quenching of the iron-sensitive fluorescence than ML1+/− cells did. De-quenching was achieved by preloading the fibroblasts with an iron-sensitive dye, Phen Green SK (PG SK), and then adding the membrane-permeable transition metal chelator, 2,2′-bipyridyl (BPD). 2,2′-BPD is predicted to chelate free cellular iron (also referred to as chelatable or labile iron), which subsequently increases PG SK fluorescence. The original magnification used was ×200. b, The average 2,2′-BPD induced normalized change of fluorescence (ΔF/F0) for ML4 cells (ML1−/−; n = 6 experiments) is significantly (asterisk, P < 0.01) lower than for the parental ML1+/− cells. Fibroblast cells (n = 10–20) were analysed for each individual experiment. 4,4′-BPD, a 2,2′-BPD analogue that cannot bind Fe2+, did not induce any significant restoration of PG SK fluorescence. Error bars, s.e.m. c, An ML1−/ − skin fibroblast cell showed autofluorescence in LAMP1-positive compartments. Autofluorescence (green) was detected within a range of excitation wavelengths (shown with excitation at 480 nm). No significant autofluorescence was observed for a ML1+/+ cell. Lysosomes were stained with a LAMP1 antibody (red). Differential interference contrast (DIC) images are shown for comparison.
We have demonstrated that TRPML1 plays a critical role in cellular iron homeostasis by showing the cytosolic Fe2+ deficiency with concurrent intra-lysosomal iron overload in ML4 cells. ML4 cells exhibit a defect in the late endocytic pathway 10, 20, 21, suggesting that TRPML1 may be required for Ca2+-dependent fusion/fission of LE/Ly. However, this defect in vesicular transport cannot explain the cytosolic Fe2+ deficiency that we observed since the internalization and recycling of the transferrin receptor is normal in ML4 cells 20, and lysosomal Fe2+ release (into the cytosol) is most likely to be mediated directly by an iron-conducting channel/transporter, as is the case for DMT122. Therefore, by far the simplest explanation of our results is that the Fe2+ release pathway between the cytosol and the lysosome lumen is blocked/inactivated in cells with ML4 mutations. While a ML4-induced-loss of Ca2+ permeability may result in a defect of lysosomal trafficking and subsequent accumulation of various lipids in LE/Ly, the degradation of these materials is normal 20, 21. Therefore, these accumulated substances would become most harmful only if they are oxidized to generate lipofuscin-like non-degradable materials in the presence of high intra-lysosomal Fe2+ 19. Lipofuscin-like molecules preferentially accumulate in post-mitotic cells such as muscle cells and neurons, which are primarily affected in ML4 patients 21. Our work suggests that lysosome-targeting iron chelators might alleviate the degenerative symptoms of ML4 patients. From the cell biology perspective, a key question to ask is how TRPML channels are regulated by various cytosolic and lumenal factors, and/or by proteins and lipids in the LE/Ly membrane, especially those of which are known to be involved in endo-lysosomal trafficking.
Online Methods
Molecular biology and biochemistry
Full length mouse TRPML1 and TRPML3, and a short splicing variant of mouse TRPML2 (GeneBank # NP_001005846), were cloned into the EGFP-C2 vector (Clontech), the mCherry vector, or HA tag containing pcDNA3 as described previously 11. No significant functional difference was observed among EGFP-tagged, mCherry-tagged, and HA-tagged TRPMLVa constructs. ML4 mutants were constructed on a TRPML1Va background using a site-directed mutagenesis kit (Qiagen). All constructs were confirmed by sequencing analysis and protein expression was verified by Western blotting. HEK293T cells were transiently transfected with wt TRPML1, TRPML1Va, and ML4 mutants for electrophysiology, confocal imaging, 55Fe2+ uptake, and Fe2+ quenching assays. TRPML1 Western blot analyses were performed with an anti-EGFP monoclonal antibody (Covance).
Confocal imaging
All images were taken using a Leica (TCS SP5) confocal microscope. Lamp1 antibody was from the Iowa Hybridoma Bank and LysoTracker was from Molecular Probes (Invitrogen).
Mammalian cell electrophysiology
Recordings were performed in transiently transfected HEK293T cells. Cells were cultured at 37°C, transfected using Lipofectamine 2000 (Invitrogen), and plated onto glass coverslips. No significant difference was observed for EGFP-tagged-versus untagged TRPML1Va-transfected cells. Unless otherwise stated, pipette solution contained 147 mM Cs, 120 mM methane-sulfonate, 4 mM NaCl, 10 mM EGTA, 2 mM Na2-ATP, 2 mM MgCl2, 20 mM HEPES (pH 7.2; free [Ca2+]i < 10 nM). Standard extracellular bath solution (modified Tyrode’s solution) contained 153 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 20 mM HEPES, 10 mM glucose (pH 7.4). To reduce the background from an endogenous Cl− conductance activated by protons that was strongly outwardly rectifying 11, 25, gluconate− or methanesulfonic− (Mes−) was used to replace most of the Cl− (remaining [Cl−]o = 5–10 mM) for all low-pH bath solutions. Low-pH “Tyrode’s” solution contained 150 mM Na-Gluconate, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 10 mM HEPES, and 10 mM MES (pH 4.6). NMDG+ solution contained 160 mM N-methyl-D-glucamine (NMDG), 20 mM HEPES, 10 mM Glucose (pH 7.4). Low-pH NMDG+ solution contained 150 mM NMDG, 10 mM Glucose, 10 mM MES, 10 HEPES (pH adjusted to 4.6 using methanesulfonic acid). “Isotonic” metal solutions contained 105 mM metal ions, 30 mM Glucose, 10 mM HEPES, 10 mM MES, 0–30 mM NMDG+ (pH 4.6). Additional metal solutions (1, 3, 10, and 30 mM) derived from mixing “isotonic” solutions with low pH NMDG+ solutions at various ratios. Monovalent (Nominal Divalent-free) solutions contained 10 mM glucose, 20 mM HEPES, 160 mM NaCl, 5 mM KCl (pH 7.4; free Ca2+ <1–10 μM). All solutions were applied via a fast perfusion system to achieve a complete solution exchange within a few seconds. Data were collected using an Axopatch 2A patch clamp amplifier, Digidata 1440, and pClamp 10.0 software (Axon Instruments). Whole-cell currents and single channel recordings were digitized at 10 kHz and filtered at 2 kHz. Capacity current was reduced as much as possible using the amplifier circuitry. Series resistance compensation was 60–85%. All experiments were conducted at room temperature (~21–23°C) and all recordings were analyzed with pCLAMP10 (Axon Instruments, Union City, CA) and Origin 7.5 (Origin Lab, Northampton, MA).
Iron de-quenching imaging
Cultured skin fibroblast cells from a ML4 patient (TRPML1−/−, clone WG0909) and corresponding heterozygous parent (TRPML1+/−, clone WG0987) were obtained from the Repository for Mutant Human Cell Strains of Montreal Children’s Hospital (Montreal, Canada). Fibroblast cells were loaded with 20 μM Phen Green SK (Molecular Probes) in culture medium at 37°C for 20 min followed by a 15 min wash. Cellular fluorescence was excited at 488 nm on the PTI ERP system. Quenching of fluorescence was induced by the addition of the membrane permeable transition metal chelator 2, 2′-Bipyridyl (5 mM) 17. 4,4′-Bipyridyl, a 2, 2′-Bipyridyl analogue that cannot bind/chelate Fe2+, was used as a control. Since many variables such as dye loading may contribute to the variation of basal fluorescence (F) of PG SK, normalized change of fluorescence (ΔF/F) was used as readout to estimate the change of the cytosolic Fe2+ levels.
Iron Uptake Assay
Iron uptake was measured as described previously 25. Briefly, 30hrs after transfection, HEK293T cells were washed twice with pre-warmed PBS, and then incubated with 1ml of pre-warmed uptake assay buffer (25mM Tris, 25 mM Mes, 140 mM NaCl, 5.4 mM KCl, 5 mM glucose, 1.8 mM CaCl2, 800 μM MgSO4, 50 μM ascorbic acid, pH 5.8) containing 55Fe-nitrilotriacetic acid (NTA, 1μM 55Fe) at 37°C for 5min. 55Fe-NTA was made by mixing 55FeCl3 (PerkinElmer) with 100mM NTA in a 1:50 molar ratio. Ascorbic acid (100 mM) was used to promote the formation and maintenance of ferrous (Fe2+) iron (the pH was adjusted to pH 5.8). All uptake assays were performed with 20 μM Fe2+ (with a 1:20 molar ratio for 55FeCl3 and FeSO4) at pH 5.8. Assays were stopped by the addition of 2ml ice-cold PBS. After washing three times with ice-cold PBS, the cells were detached and collected by adding 1ml 0.25% trypsin-EDTA. Parallel experiments were conducted at 0°C to measure the cell surface 55Fe binding, which was subtracted from the values at 37 °C to obtain the net iron uptake. Cell-associated radioactivity was measured with liquid scintillation spectrometry.
Lysosomal iron staining
Lysosome iron was stained using a modified sulfide-silver method (SSM) 19. Briefly, fibroblast cells were grown on coverslips and exposed to 50 μM Fe3+-Dextran overnight. Cells were then washed with PBS and fixed with 2% glutaraldehyde in 0.1 M Na-cacodylate buffer with 0.1 M sucrose (pH 7.2) for 1.5 h at room temperature (22°C). Next cells were sulfidated at pH ≈ 9 with 1% (w/v) ammonium sulfide in 70% (v/v) ethanol for 15 min. The development was then performed using a physical, colloid-protected developer containing silver-lactate and hydroquinone for 1–2h.
Reagents
2, 2′-BPD, 4, 4′-BPD, iron ascorbate, ferric chloride, iron dextran, and all other metal salts were from Sigma Chemicals (St. Louis, MO).
Data analysis
Most data are presented as the mean ± SEM. Statistical comparisons were made using analysis of variance (ANOVA). A P value < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This work is supported by startup funds to H.X. from the Department of MCDB and Biological Science Scholar Program, University of Michigan.
This work was also supported by the National Institutes of Health under award number: T32HL007572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We are grateful to Ulf Brunk, Saito Mitsu, Rich Hume, Cunming Duan, Mohammed Akaaboune, John Kuwada, Sean Low, Sukanya Punthambaker, and Shlomo Dellal for assistance, and David Clapham, Nancy Andrews, Lou DeFelice, Lixia Yue, Dejian Ren, Chun Jiang, and Shawn Xu for comments on an earlier version of the manuscript. We are also grateful to Kirill Kiselyov for sharing with us his unpublished results on lysosomal iron staining of ML4 cells. We appreciate the encouragement and helpful comments from other members of the Xu laboratory.
References
- 1.Venkatachalam K, Hofmann T, Montell C. Lysosomal localization of TRPML3 depends on TRPML2 and the mucolipidosis-associated protein TRPML1. The Journal of biological chemistry. 2006;281:17517–17527. doi: 10.1074/jbc.M600807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 3.Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiological reviews. 2007;87:165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- 4.Bassi MT, et al. Cloning of the gene encoding a novel integral membrane protein, mucolipidin-and identification of the two major founder mutations causing mucolipidosis type IV. American journal of human genetics. 2000;67:1110–1120. doi: 10.1016/s0002-9297(07)62941-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun M, et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Human molecular genetics. 2000;9:2471–2478. doi: 10.1093/hmg/9.17.2471. [DOI] [PubMed] [Google Scholar]
- 6.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 7.Lee DW, Andersen JK, Kaur D. Iron dysregulation and neurodegeneration: the molecular connection. Molecular interventions. 2006;6:89–97. doi: 10.1124/mi.6.2.6. [DOI] [PubMed] [Google Scholar]
- 8.Kidane TZ, Sauble E, Linder MC. Release of iron from ferritin requires lysosomal activity. Am J Physiol Cell Physiol. 2006;291:C445–455. doi: 10.1152/ajpcell.00505.2005. [DOI] [PubMed] [Google Scholar]
- 9.Gunshin H, et al. Slc11a2 is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver. The Journal of clinical investigation. 2005;115:1258–1266. doi: 10.1172/JCI24356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pryor PR, Reimann F, Gribble FM, Luzio JP. Mucolipin-1 is a lysosomal membrane protein required for intracellular lactosylceramide traffic. Traffic (Copenhagen, Denmark) 2006;7:1388–1398. doi: 10.1111/j.1600-0854.2006.00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu H, Delling M, Li L, Dong X, Clapham DE. Activating mutation in a mucolipin transient receptor potential channel leads to melanocyte loss in varitint-waddler mice. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:18321–18326. doi: 10.1073/pnas.0709096104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Palma F, et al. Mutations in Mcoln3 associated with deafness and pigmentation defects in varitint-waddler (Va) mice. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:14994–14999. doi: 10.1073/pnas.222425399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Altarescu G, et al. The neurogenetics of mucolipidosis type IV. Neurology. 2002;59:306–313. doi: 10.1212/wnl.59.3.306. [DOI] [PubMed] [Google Scholar]
- 14.Goldin E, et al. Transfer of a mitochondrial DNA fragment to MCOLN1 causes an inherited case of mucolipidosis IV. Human mutation. 2004;24:460–465. doi: 10.1002/humu.20094. [DOI] [PubMed] [Google Scholar]
- 15.Bargal R, Goebel HH, Latta E, Bach G. Mucolipidosis IV: novel mutation and diverse ultrastructural spectrum in the skin. Neuropediatrics. 2002;33:199–202. doi: 10.1055/s-2002-34496. [DOI] [PubMed] [Google Scholar]
- 16.Kress GJ, Dineley KE, Reynolds IJ. The relationship between intracellular free iron and cell injury in cultured neurons, astrocytes, and oligodendrocytes. J Neurosci. 2002;22:5848–5855. doi: 10.1523/JNEUROSCI.22-14-05848.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petrat F, de Groot H, Rauen U. Determination of the chelatable iron pool of single intact cells by laser scanning microscopy. Archives of biochemistry and biophysics. 2000;376:74–81. doi: 10.1006/abbi.2000.1711. [DOI] [PubMed] [Google Scholar]
- 18.Goldin E, Blanchette-Mackie EJ, Dwyer NK, Pentchev PG, Brady RO. Cultured skin fibroblasts derived from patients with mucolipidosis 4 are autofluorescent. Pediatric research. 1995;37:687–692. doi: 10.1203/00006450-199506000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Kurz T, Terman A, Gustafsson B, Brunk UT. Lysosomes in iron metabolism, ageing and apoptosis. Histochemistry and cell biology. 2008;129:389–406. doi: 10.1007/s00418-008-0394-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen CS, Bach G, Pagano RE. Abnormal transport along the lysosomal pathway in mucolipidosis, type IV disease. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:6373–6378. doi: 10.1073/pnas.95.11.6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeevi DA, Frumkin A, Bach G. TRPML and lysosomal function. Biochim Biophys Acta. 2007 doi: 10.1016/j.bbadis.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 22.Andrews NC, Schmidt PJ. Iron homeostasis. Annual review of physiology. 2007;69:69–85. doi: 10.1146/annurev.physiol.69.031905.164337. [DOI] [PubMed] [Google Scholar]
- 23.Cerny J, et al. The small chemical vacuolin-1 inhibits Ca(2+)-dependent lysosomal exocytosis but not cell resealing. EMBO reports. 2004;5:883–888. doi: 10.1038/sj.embor.7400243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saito M, Hanson PI, Schlesinger P. Luminal chloride-dependent activation of endosome calcium channels: patch clamp study of enlarged endosomes. The Journal of biological chemistry. 2007;282:27327–27333. doi: 10.1074/jbc.M702557200. [DOI] [PubMed] [Google Scholar]
- 25.Xu H, Jin J, DeFelice LJ, Andrews NC, Clapham DE. A spontaneous, recurrent mutation in divalent metal transporter-1 exposes a calcium entry pathway. PLoS biology. 2004;2:E50. doi: 10.1371/journal.pbio.0020050. (online methods section) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.