

Abstract
There have been a number of clinical trials testing the efficacy of FLT3 tyrosine kinase inhibitors (TKIs) in acute myeloid leukemia (AML). patients harboring a constitutively activating mutation in FLT3 However, there has been limited efficacy, most often due to inadequate achievement of FLT3 inhibition through a variety of mechanisms In a previous study, TTT-3002 was identified as a novel FLT3 inhibitor with the most potent activity to date against FLT3 internal tandem duplication (FLT3/ITD) mutations Here the activity of TTT-3002 is demonstrated against a broad spectrum of FLT3 activating point mutations (FLT3/PMs), including the most frequently occurring D835 mutations The compound is also active against a number of point mutations selected for in FLT3/ITD alleles that confer resistance to other TKIs, including the F691L gatekeeper mutation TTT-3002 maintains activity against relapsed AML patient samples that are resistant to sorafenib and AC220 Studies utilizing human plasma samples from healthy donors and AML patients indicate that TTT-3002 is only moderately protein bound compared to several other TKIs currently in clinical trials Tumor burden of mice in a FLT3 TKI-resistant transplant model is significantly improved by oral dosing of TTT-3002 Therefore, TTT-3002 has demonstrated preclinical potential as a promising new FLT3 TKI that may overcome some of the limitations of other TKIs in the treatment of FLT3-mutant AML
Keywords: FLT3 inhibitor, drug resistance, acute myeloid leukemia
Introduction
FMS-like tyrosine kinase-3 (FLT3) is a receptor tyrosine kinase that is commonly mutated in acute myeloid leukemia (AML). Approximately 23% of patients are found to harbor a constitutively activating FLT3/ITD mutation that confers poor prognosis (1). Kinase activating FLT3/PMs such as those reported in the tyrosine kinase domain are also frequently observed (7-10% of patients) (2) Thus, mutant FLT3 is an attractive therapeutic target in the treatment of this disease.
A number of clinical trials for AML patients with FLT3 mutations have been performed using novel tyrosine kinase inhibitors (TKIs) that target FLT3 (3). However, there have been limitations in the responses observed in patients on these trials, often related to insufficient achievement of thorough FLT3 inhibition An illustrative example is lestaurtinib (CEP-701), an indolocarbazole that showed activity in Phase 1 and 2 trials, though only in those patients in which a high level of inhibition of FLT3 signaling was achieved It failed in a randomized Phase 3 trial to improve remission and survival rates for relapsed FLT3 mutant patients for those randomized to chemotherapy plus CEP-701 vs those receiving chemotherapy alone in part due to failure to achieve inhibition in a large enough fraction of patients (4, 5). A potential reason for the failure of CEP-701 to effectively inhibit FLT3 signaling in vivo was a high level of human plasma protein binding (6). This shifts the IC50 against FLT3 from 2-3nM in assays conducted in media with 10% fetal bovine serum (FBS, typical for most in vitro culture conditions) to 700nM in 100% human plasma (reflective of in vivo binding in patients) (7).
A number of FLT3 TKI active against FLT3/ITD have little activity against FLT3/PMs, such as the most frequently occurring D835Y mutation. Sorafenib is a biaryl urea compound that targets multiple tyrosine kinases including FLT3/ITD (8). The results of clinical studies using sorafenib in combination with chemotherapy are promising, demonstrating reduction in bone marrow (BM) and/or peripheral blood (PB) blasts as well as increased CR rates in FLT3/ITD+ AML patients (9-11). Quizartinib (AC220) is another biaryl urea FLT3 inhibitor that has demonstrated significant responses in FLT3/ITD+ AML patients in recent trials (12, 13). However, both sorafenib and AC220 are inactive against many FLT3/PMs, including the D835Y mutation, and thus do not benefit patients harboring this mutation (14-16).
Finally treatment failure has also been observed due to the selection for resistance-conferring point mutations that have appeared in FLT3/ITD-expressing AML patients following TKI treatment. These mutations either occur in residues within the ATP binding pocket or to residues thought to affect protein structure in ways that also affect the binding of the drug allosterically (3). Midostaurin (PKC412) is a FLT3 TKI that demonstrated reduction of blasts in a Phase 2 trial of relapsed or refractory AML patients (17) and is moderately active against a number of known FLT3/PMs (14). However, in a trial of relapsed/refractory AML patients, PKC412 selected for a mutation at residue N676K in a FLT3/ITD patient that conferred drug resistance (18). A number of initially responsive patients on AC220 and sorafenib trials were also found to have selected for additional resistance-conferring point mutations in the FLT3/ITD allele. These frequently include a F691L mutation (analogous to the T315I mutation in BCR/ABL that confers resistance to Gleevec) or D835 mutations (Y/F/V/H) in the kinase domain (19, 20). Crenolanib is a next generation FLT3 inhibitor that is currently in Phase II trials of relapsed AML with FLT3/D835 activating mutations. This compound has demonstrated in vitro and in vivo activity against FLT3/D835 mutations (Y/F/V/H) and the dual FLT3-D835(Y/H)/ITD mutant receptors (21, 22). However, it is unable to target the F691L mutation, and therefore has the potential to select for this resistance mutation in trials. Currently, the most potent activity against the F691L mutation in vitro has been observed for the BCR-ABL inhibitor ponatinib. However, ponatinib still shows a nearly 20-fold shift in IC50 for the F691L mutation compared to FLT3/ITD alone, and plasma samples from patients show marginal levels of inhibition in the plasma inhibitory activity (PIA) assay against the F691L mutation and no activity against D835 mutations (23). Therefore, the search for novel FLT3 TKIs that overcome some of the mechanisms that result in persistent FLT3 activation is necessary to improve the cure rate for this disease.
We sought to explore the ability of a novel FLT3 inhibitor, TTT-3002, to overcome several mechanisms of drug resistance associated with current FLT3 TKIs. We have previously reported that TTT-3002 is the most potent FLT3 inhibitor discovered to date, with picomolar IC50 values against FLT3/ITD phosphorylation (24). Here we assess the activity of TTT-3002 against a broad spectrum of known FLT3/PMs, as well as a number of TKI resistance mutations within the FLT3/ITD allele. Finally, the protein binding characteristics of TTT-3002 in human plasma are explored to predict whether it is likely to maintain activity against FLT3/ITD and FLT3/PM in patients.
Materials and Methods
Compounds
TTT-3002 was a generous gift of Hanno Roder from TauTaTis, Inc. (San Diego, CA). CEP-701, AC220, sorafenib and PKC412 were purchased from LC Laboratories. Compounds were dissolved in 100% DMSO and prepared as 10μM stock solutions in RPMI with 0.1% DMSO and stored at -80°C.
Immunoprecipitation and Western Blotting
Cells were cultured in the presence of inhibitor for 1 hour at 37°C, and FLT3 and phospho-FLT3 expression was analyzed by performing immunoprecipitation of whole cell extracts for FLT3 (S-18), followed by SDS-PAGE and Western blotting as described previously (24). Other proteins were detected from whole cell lysates using the indicated antibodies and a horseradish peroxidase conjugated goat anti-rabbit secondary antibody followed by ECL.
Flow cytometry analysis
Flow cytometry analysis was performed using a BD FACSCalibur machine (BD Biosciences). The following anti-mouse monoclonal antibodies and dyes were used for the staining of cellular markers: CD135-PE, Annexin V-APC, 7-amino-actinomycin (7-AAD, BD Biosciences). For hematopoietic stem/progenitor staining, femur, tibia, hips, and spine were isolated from Ba/F3-F691L/ITD Luc+ transplant recipient mice. BM cells were isolated by crushing and stained with anti-mouse CD135-PE (5 μL) for 40 minutes. Apoptosis was measured by incubating treated cells with Annexin V-APC antibody and 7-AAD for 10 minutes according to the manufacturer’s instructions (BD Biosciences). All data were analyzed by FlowJo analysis software (Tree Star).
Macromolecular Modeling
To position TTT-3002, CEP-701, PKC412, AC220 and sorafenib on the FLT3 kinase structure (1RJB) (25), the kinase domains of the VEGFR2 kinase complexed with sorafenib (4ASD) (26) and the C-terminal Src kinase complexed with staurosporine (1BYG) (27) were superimposed on the FLT3 kinase structure. The program COOT (28) was used to generate the N676K, F691L, and G697R substitutions by replacing the native side chain with the most common rotamer of the resistance variant.
Transplantation experiments
Transplantation of Ba/F3-F691L/ITD luciferase expressing cells was performed as described previously (24, 29). For engraftment, 2 × 106 cells were injected by tail vein into female BALB/C mice (Day 0). Starting seven days later, engraftment was confirmed by bioluminescence imaging of luciferase-expressing cells and mice were then treated with 6 mg/kg of TTT-3002 hydrochloride suspended by brief sonication 1 h prior to dosing in 1mM HCl twice daily or 10 mg/kg of sorafenib suspended in 30% (w/v) Cremophor EL, 30% w/v PEG 400, 10% ethanol, and 10% glucose (all Sigma-Aldrich), as described previously, once daily by oral gavage for 2 weeks (30). Mice were imaged by injecting D-luciferin (3 mg intraperitoneally) and visualized on an IVIS Spectrum imager (Caliper LifeSciences) using Living Image software for analysis on day 7 post-inoculation in order to monitor engraftment. BM cells were harvested from the femur, tibia and hips by crushing, and spleens were harvested by mechanically filtering through a 40μm nylon cell strainer (BD).
Patient Samples
Human AML BM samples were collected under an institutionally-approved protocol with informed patient consent in accordance with the Declaration of Helsinki. Mononuclear cells were isolated by Ficoll centrifugation and cryopreserved until use in liquid nitrogen. All studies used freshly thawed cells. Plasma was purified by centrifugation (450 ×g) and stored at -20°C.
Statistical Analysis
Statistical analysis was performed with Student’s t test by use of the GraphPad software analysis program (Prism). P values of less than .05 were considered to be statistically significant. All data are presented as the mean ± standard deviation (SD).
Additional information on Materials and Methods can be found online in Supplemental Information.
Results
TTT-3002 activity against FLT3 activating point mutations
Many FLT3 activating point mutations have been observed in AML patients and the response of each FLT3/PM to each FLT3 TKI varies widely (2, 3, 31-34). Notably, AC220 has reduced potency against many FLT3/PMs, and is thus ineffective in treating patients with these mutations. To predict the ability of TTT-3002 to inhibit FLT3/PM autophosphorylation, we generated a panel of twelve stably transfected Ba/F3 cell lines in which FLT3/PMs known to transform the cells to IL-3 independence were introduced, and its activity was compared directly to the cytotoxic activity of AC220 (supplemental Table 1). This panel included the most frequently occurring D835 mutations (Y/F/V/H) and other D835 variants, as well as I836 mutations and mutations in the juxtamembrane domain (V579A and D593Δ). TTT-3002 is a potent inhibitor of all twelve mutants screened, with proliferation IC50 values in the low nanomolar range, and is thus active against many FLT3/PMs for which AC220 is completely ineffective (Fig. 1A and supplemental Table 1). Treatment with TTT-3002, but not AC220, results in a marked induction of apoptosis of FLT3/PM-expressing cells as determined by Annexin V binding, reaching the half maximal effective concentration (EC50) by 10-50nM for all mutants. In contrast, activity against FLT3/ITD-expressing cells is similar for the two compounds (Fig. 1B). TTT-3002 inhibits FLT3/PM phosphorylation in a dose-dependent manner, achieving greater than 50% inhibition of FLT3 signaling at a concentration of 0.5nM (Fig. 1C, D). In contrast, AC220 failed to achieve thorough FLT3/PM inhibition even at 10nM concentrations (Fig. 1C). Therefore, TTT-3002’s broad spectrum of activity against FLT3/PMs increases its therapeutic potential in patients with this class of non-ITD activating FLT3 mutations.
Figure 1.
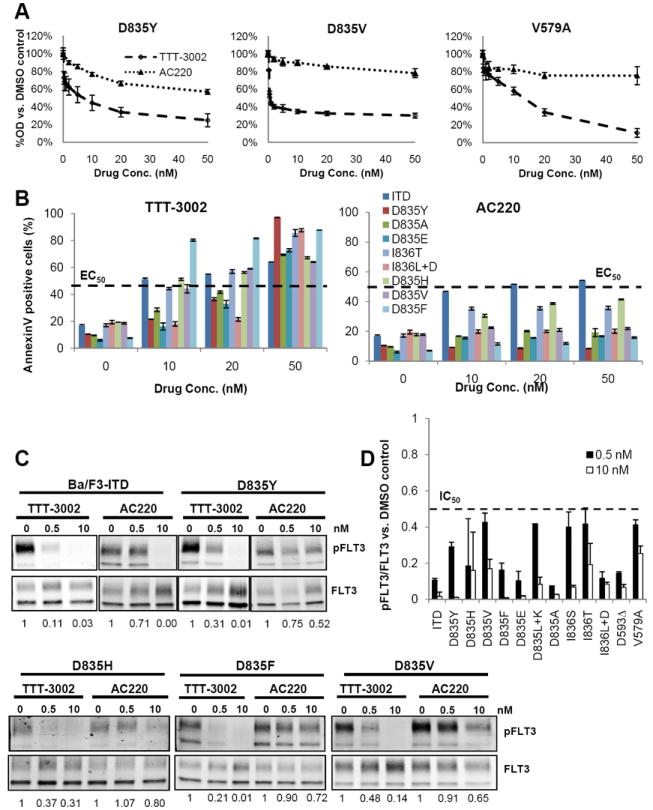
TTT-3002 is active against FLT3 activating point mutations. A, Ba/F3 cells stably transfected with the indicated FLT3 activating point mutations were treated for 48 h, and cell proliferation was measured by MTT assay. Error bars indicate average ± SD, data are representative of three independent experiments. B, Annexin V binding at 48 h following treatment with TTT-3002 or AC220, with error bars indicating average ± SD. The half maximal effective concentration (EC50) is indicated on the graph. C, Inhibition of pFLT3 in Ba/F3-FLT3/ITD and FLT3/PM cells. Fraction of pFLT3/FLT3 relative to DMSO treatment is indicated below each blot. D, Quantitation of (C), represented by fraction of pFLT3/FLT3 relative to DMSO control, at 0.5nM and 10nM TTT-3002. Data represent average of three independent experiments ± SD. The half maximal inhibitory concentration (IC50) is indicated on the graph.
TTT-3002 activity against FLT3 TKI resistance mutations
The phenomenon of drug resistance caused by selection for FLT3 point mutations is occurring at increasing frequency as higher levels of inhibition by FLT3 TKIs are achieved in clinical trials of FLT3/ITD AML patients (19, 20, 35-37). To test the ability of TTT-3002 to successfully treat some of these resistance mutations, site-directed mutagenesis was used to generate a series of FLT3/ITD TKI resistant cell lines (29, 38). The activity of TTT-3002 was then compared to that of a number of other FLT3 TKIs currently in clinical trials. TTT-3002 is the most potent inhibitor against the six FLT3/ITD TKI resistance mutants screened, with proliferation IC50 values of approximately 1nM (with the single exception of Ba/F3-G697R/ITD, IC50=11nM) (Fig. 2A and Table 1). It effectively inhibits resistance mutations against which other TKIs are ineffective. The activities of CEP-701, sorafenib, AC220 and PKC412 against these resistance mutants is similar to what has been reported previously (21, 29). TTT-3002 inhibits FLT3 phosphorylation in a dose-dependent manner in FLT3/ITD TKI resistant cells, often with an IC50 of less than 250-500pM (Figure 2B). The extent of inhibition is also significantly greater than the other TKIs at a fixed concentration of 20nM, achieving 75-99% inhibition of FLT3 signaling (Fig. 2C, D). TTT-3002 also inhibits phosphorylation of downstream targets of FLT3 signaling, such as STAT5, AKT and MAPK (Fig. 2E, supplemental Fig. 1). Therefore, this compound’s broad spectrum of activity against FLT3/ITD TKI resistance mutations may enable it to successfully treat FLT3/ITD AML patients who have become resistant to other FLT3 TKIs.
Figure 2.
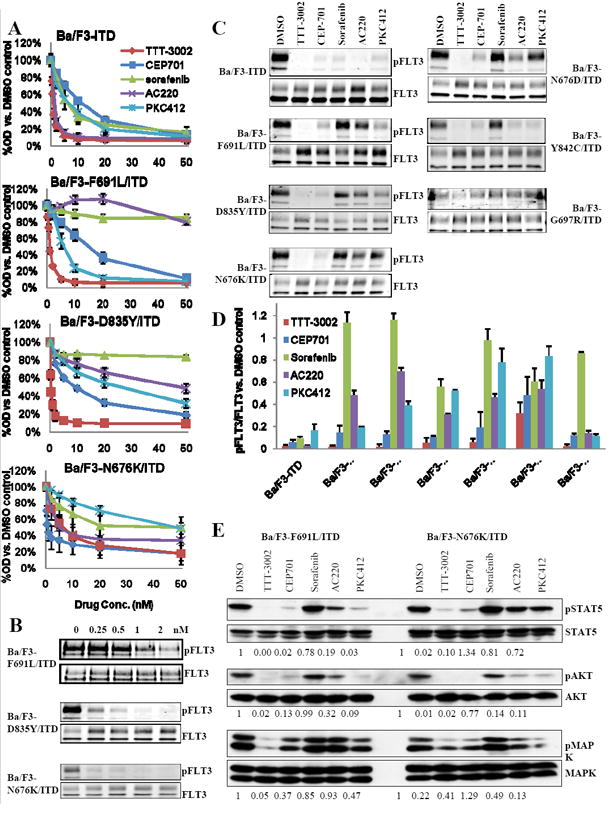
TTT-3002 is active against cells lines containing FLT3 TKI resistance mutations. A, Proliferation of Ba/F3-ITD and Ba/F3-F691L/ITD cells measured by MTT assay at 48 h following treatment with TTT-3002, CEP-701, sorafenib, AC220 or PKC412 (0-50nM). Error bars indicate average ± SD; data representative of three independent experiments. B, Ba/F3 cells transfected with TKI resistance mutations identified in patients (F691L/ITD, D835Y/ITD and N676K/ITD) were treated with TTT-3002, and inhibition of pFLT3 was assessed by Western blotting of cell extracts. C, Inhibition of pFLT3 with TKI resistance mutations by TTT-3002, CEP-701, sorafenib, AC220 or PKC412, each at 20nM. D, Quantitation of (C), represented by %pFLT3/FLT3 relative to DMSO control. Data represent average of three independent experiments ± SD. E, Inhibition of phospho-STAT5 (pSTAT5), phospho-AKT (pAKT) and phospho-MAPK (pMAPK) in Ba/F3-F691L/ITD and Ba/F3-N676K/ITD cells by TTT-3002, CEP-701, sorafenib, AC220 or PKC412, each at 20nM. Fraction of phospho-protein/total protein, relative to DMSO, is indicated below each blot.
Table 1.
Proliferation IC50 for FLT3 TKI treatment of FLT3 mutants
| Cell Line | TTT-3002 (nM) | CEP701 (nM) | Sorafenib (nM) | AC220 (nM) | PKC412 (nM) |
|---|---|---|---|---|---|
| Ba/F3-ITD | 0.6 | 10 | <2 | <2 | 5 |
| Ba/F3-F691L/ITD | 1 | 12 | >100 | >100 | 7 |
| Ba/F3-D835Y/ITD | 0.3 | 9 | >100 | 47 | 22 |
| Ba/F3-N676K/ITD | 1 | 6 | 36 | 8 | 44 |
| Ba/F3-N676D/ITD | 1 | 6 | 48 | 4 | >100 |
| Ba/F3-Y842C/ITD | 1.7 | 17 | >100 | 45 | 16 |
| Ba/F3-G697R/ITD | 11 | 55 | >100 | >100 | >100 |
| Ba/F3 parental | >100 | >100 | >100 | >100 | >100 |
Molecular modeling of the TKI-FLT3 binding interaction
To understand the patterns of drug resistance displayed by different FLT3 TKIs in this study, the positions of staurosporine-like (TTT-3002, CEP-701 and PKC412) and sorafenib-like inhibitors (sorafenib and AC220) bound to FLT3 were modeled (Fig. 3). Although crystal structures of the FLT3 kinase bound to these inhibitors have not been determined, crystal structures of the related VEGFR2 and C-terminal Src kinases bound to sorafenib (PDB code 4ASD) and staurosporine (PDB code 1BYG), respectively, have been determined (25, 26). Assuming that these drugs bind similarly to FLT3 kinase, we superimposed the kinase domains of the VEGFR2:sorafenib and C-terminal Src kinase:staurosporine complexes on the FLT3 kinase structure to generate a model for how sorafenib and staurosporine are likely to bind to FLT3. Inspecting these models to investigate the likely effects of amino-acid substitutions in FLT3 known to confer resistance to TKIs in clinical trials shows that substituting arginine for glycine at position 697 places side-chain atoms within 2.8 Å and 3.5 Å of atoms of staurosporine-like and sorafenib-like inhibitors, respectively, likely leading to steric clashes. Glycine 697 adopts main-chain dihedral angles (phi of 60°, psi of -170°) that are unfavorable for non-glycine amino-acids, and an arginine substitution at this site would also lead to at least local changes in main-chain conformation. It has been proposed that the F691 residue forms a stabilizing π-π stacking interaction with AC220, and thus a mutation to a Leu would result in reduced TKI binding affinity (20). TTT-3002 is predicted to bind FLT3 without making direct contact with the F691 residue, and thus is unaffected by mutations at this site. Likewise, the closer proximity of F691 and N676 to sorafenib compared to staurosporine atoms likely underlies the trend for substitutions at these sites to confer resistance to sorafenib-like but not staurosporine-like inhibitors.
Figure 3.
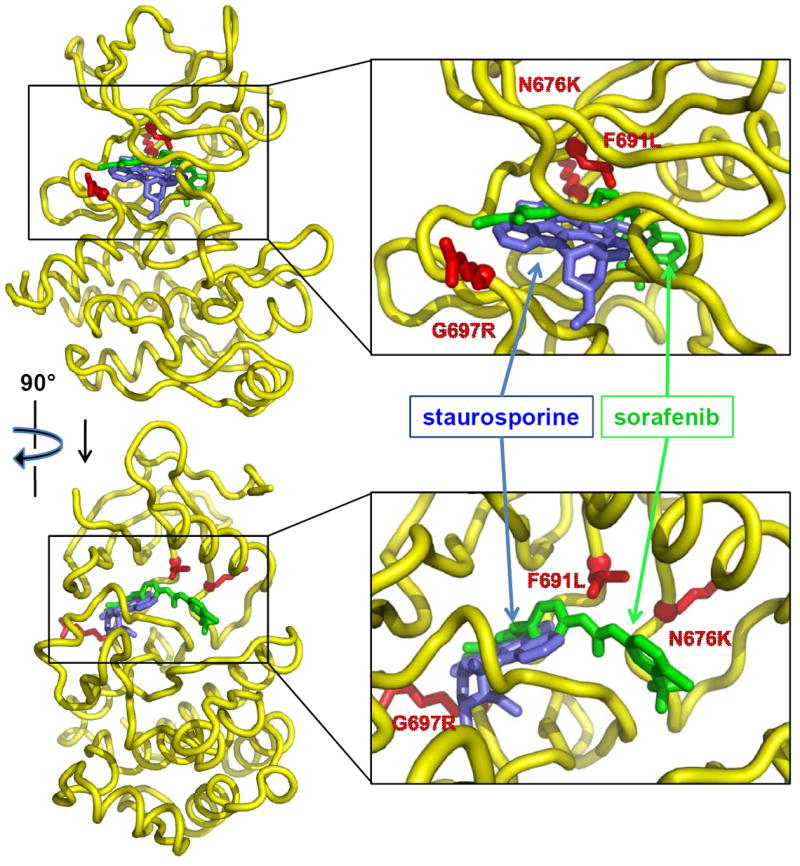
Molecular modeling of the FLT3-TKI binding interaction. Orthogonal views of the FLT3 kinase domain (yellow) are shown with the positions of staurosporine (blue, representing theoretical binding of TTT-3002, CEP-701 and PKC412) and sorafenib (green, representing binding of sorafenib and AC220). The side chains of Asn 676, Phe 691, and Gly 697 of FLT3 have been substituted with the side chains of Lys, Leu, and Arg, respectively, to mark the position and nature of common resistance variants and are shown in red.
TTT-3002 overcomes drug resistance in vivo
To assess the ability of TTT-3002 to overcome TKI resistance in vivo, a bioluminescent mouse model of leukemia was utilized in which luciferase expressing Ba/F3-F691L/ITD cells (Ba/F3-F691L/ITD Luc+) were transplanted into syngeneic BALB/c recipients. The Ba/F3-F691L/ITD Luc+ cell line is sensitive to TTT-3002 treatment in vitro with a proliferation IC50 of <250pM for TTT-3002 vs. >50nM for sorafenib (Fig. 4A). Engraftment was confirmed on Day 7 by bioluminescence imaging of luciferase-expressing cells following tail vein injection of 2 × 106 cells in recipient mice, which were then administered TTT-3002 and sorafenib at the maximum tolerated dose of 6 mg/kg b.i.d. and 10 mg/kg q.d., respectively, or vehicle control (1mM HCl b.i.d.) for 14 days (n=4 per group, Fig. 4B) (30). There was no significant effect on hemoglobin or platelet count with this drug administration schedule, with counts in the normal range at the end of the treatment period (11.0-15.1 g/dL and 592-2972 K/μL, respectively) (Figure 4C, D). Mice treated with vehicle or sorafenib succumbed to disease at 21 days post-transplant with splenomegaly and elevated CD135+ (FLT3) leukemic cells in the BM (Fig. 4E, F). In contrast, spleen weights and the number of leukemic cells (measured by their expression of CD135) in the BM of TTT-3002 treated mice were significantly reduced (P < 0.01) and these mice appeared healthy at the time of sacrifice (Fig. 4E, F). By comparison, both TTT-3002 (24) and sorafenib are effective against Ba/F3-ITD Luc+ cells in vivo under the same dosing schedule, indicating that the presence of the F691L point mutation confers resistance to sorafenib, but not TTT-3002.
Figure 4.
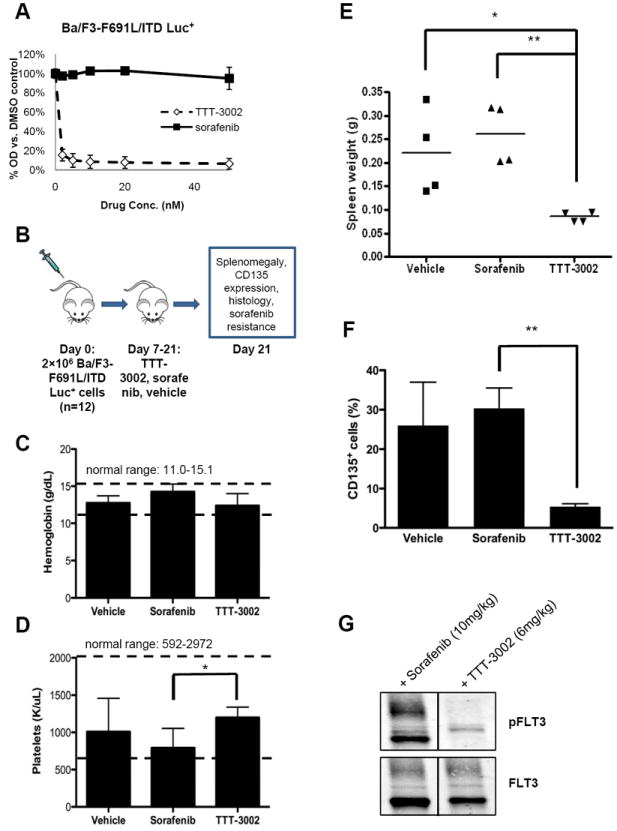
TTT-3002 is active against FLT3-F691L/ITD in vivo. A, Proliferation of Ba/F3-F691L/ITD Luc+ cells at 48 h by MTT assay (0-50nM TTT-3002 or sorafenib). Error bars indicate average ± SD. B, Schematic of, mouse xenograft study. Mice were treated with TTT-3002 (6 mg/kg b.i.d., n=4), sorafenib (10 mg/kg q.d., n=4) or vehicle control (1mM HCl, n=4) for 14 days. C, Hemoglobin (g/dL), D, platelet counts (K/μl), and E, spleen weights of TTT-3002, sorafenib and vehicle treated mice at Day 21 (*, P < 0.05; **, P < 0.01). F, CD135 expressing cells are decreased in TTT-3002 treated mouse BM at Day 21 (**, P < 0.01). G, TTT-3002 is active against sorafenib-resistant FLT3-F691L/ITD phosphorylation in vivo. Mice from the sorafenib cohort (n=2) following treatment with sorafenib for 2 weeks, received a single dose of sorafenib (10 mg/kg) or TTT-3002 (6 mg/kg). Spleens were harvested 2 hours later, and phosphorylation of FLT3 was assessed by Western blotting.
In order to further provide proof-of-principle that TTT-3002 is active against sorafenib-resistant cells in vivo, we utilized the Ba/F3-F691L/ITD transplant model to simulate a clinical setting in which a patient relapses while undergoing therapy with sorafenib with selection for a FLT3-F691L/ITD mutation and is treated with TTT-3002 instead. Leukemic mice that received sorafenib (10 mg/kg q.d.) for 2 weeks were administered a single dose of sorafenib or TTT-3002 (6 mg/kg). Leukemic Ba/F3-F691L/ITD cells from the enlarged spleens were harvested 2 hours later, and inhibition of pFLT3 was observed in vivo by TTT-3002, but not sorafenib (Fig.4G).
TTT-3002 activity against TKI-resistant relapsed AML patient samples
Primary leukemic BM samples were obtained from FLT3/ITD+ AML patients who had initially responded to sorafenib or AC220 on clinical trials but then had developed resistance. They had relapsed with a dual D835/ITD mutation or D835 point mutation alone while on a FLT3 TKI (Relapse AML 1-3, supplemental Table 2). Leukemic blasts from these patients were evaluated for their in vitro sensitivity to TTT-3002, sorafenib and AC220, along with a BM sample from a newly diagnosed FLT3/ITD AML patient sample (AML 1) for comparison. Significant effects on proliferation, induction of apoptosis, and cell signaling were observed when each of these samples was treated with TTT-3002 (Fig. 5A-C, supplemental Table 2). The signaling changes were especially prominent in pFLT3 (85-99% inhibition) and pSTAT5 (71-99% inhibition), with variable effects against pAKT (80-86% inhibition in 2/4 samples) and pMAPK (66-73% inhibition in 2/4 samples), dependent on their level and alternative sources of activation. The proliferation IC50 for TTT-3002 was 3-8nM for relapsed AML samples, and the diagnostic sample (AML1) had an IC50 of 19nM. The IC50 for AC220 against relapsed AML samples, however, ranged from 19nM to greater than 50nM, compared to its low nanomolar IC50 against blasts from the newly diagnosed FLT3/ITD patient (AML1). AC220 was moderately active against blasts from AML 1 and one relapsed patient sample with a D835/ITD mutation (Relapse AML 1) with regard to apoptosis, and was also able to inhibit FLT3 phosphorylation in AML 1 and Relapse AML 3. Sorafenib was active against only AML 1 cells and none of the three relapse samples, with proliferation IC50 values greater than 50nM, the highest concentration tested. Therefore, TTT-3002 maintains activity against human AML patient samples that have been selected for resistance to the TKIs sorafenib and AC220 in clinical trials.
Figure 5.
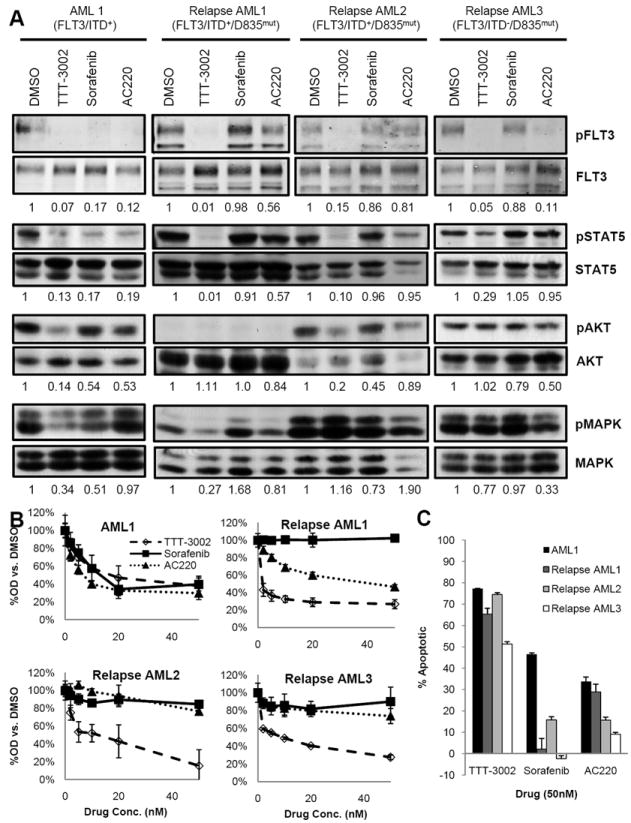
TTT-3002 is active against TKI-resistant patient samples with D835 mutations at relapse. A, Inhibition of pFLT3, pSTAT5, pAKT and pMAPK in FLT3/ITD+ (AML1) and relapsed AML patient samples with TKI resistance mutations at residue D835 (Relapsed AML1-3) by TTT-3002, sorafenib or AC220, each at 20nM. Fraction of phospho-protein/total protein, relative to DMSO treated samples, is indicated below each blot. B, Proliferation of AML patient blasts measured by MTT assay at 48 h following treatment with TTT-3002, sorafenib or AC220 (0-50nM). Error bars indicate average ± SD. C, Annexin V binding at 48 h following treatment with TTT-3002, sorafenib or AC220, with error bars indicating average of triplicate samples ± SD. Data represented as percentage Annexin V positive cells relative to DMSO control.
Plasma protein binding properties of TTT-3002
Only free, non-protein bound levels of TKI are available to bind to target and inhibit its kinase activity. In order to predict the impact of human plasma protein binding on TTT-3002 efficacy in patients, we utilized the PIA assay, which we developed previously to determine the extent of FLT3 inhibition being achieved in clinical trials of multiple FLT3 TKIs (7). The PIA assay is a valuable tool because human plasma contains the entire array of proteins capable of binding to and thus interfering with a TKI’s activity. Utilizing Ba/F3-ITD cells, we compared the IC50 of TTT-3002 with regard to inhibiting FLT3 phosphorylation (1 h treatment) in media supplemented with 10% FBS (standard culture conditions) to that achieved in 100% mouse or 100% human plasma from healthy donors. We found a 4- and 14-fold shift in IC50 values, respectively, in mouse and human plasma (Fig. 6A, B and supplemental Table 3). This shift corresponds to approximately 75 and 93% protein binding, which is significantly lower than the binding observed for many other TKIs (7, 39). For example, while the CEP-701 IC50 for FLT3 inhibition only shifts 6-fold from RPMI with 10% FBS to 100% mouse plasma, the IC50 shifts greater than 100-fold in 100% human plasma (from 2-3nM to an IC50 of 700nM), which indicates plasma protein binding of >99% (7). This was the major reason for the failure of CEP-701 to show improved CR and survival rates in a Phase 3 clinical trial (5).
Figure 6.
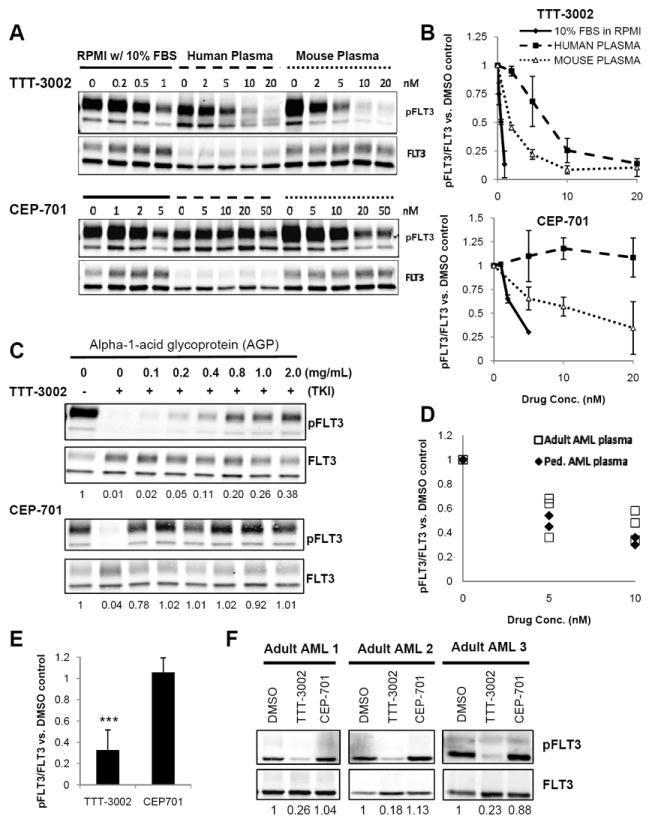
TTT-3002 is active in AML patient plasma and has lower affinity for human plasma proteins compared to CEP-701. A, Inhibition of phospho-FLT3 (pFLT3) by TTT-3002 or CEP-701 in Ba/F3-ITD cells cultured in RPMI-1640 with 10% FBS, 100% human plasma, or 100% BALB/C mouse plasma. B, Quantitation of (A), results of three independent experiments represented as %pFLT3/FLT3 relative to DMSO control ± SD. C, Inhibition of pFLT3 in Ba/F3-ITD cells cultured in RPMI-1640 supplemented with 10% FBS and AGP (0-2 mg/mL) by TTT-3002 (5nM, upper panel) or CEP-701 (20nM, lower panel). Fraction of pFLT3/FLT3 relative to DMSO control is indicated below each blot. Results representative of three independent experiments. D, Inhibition of pFLT3 in Ba/F3-ITD cells cultured in 100% plasma from pediatric (n=2) and adult (n=3) AML patients by TTT-3002 (0-10nM). Data represented as fraction of pFLT3/FLT3 relative to DMSO control. E, Ba/F3-ITD cells cultured in 100% plasma from 3 adults with AML and 3 healthy adults by TTT-3002 (20nM) or CEP-701 (100nM) for 1 h, and analyzed as in (D). Data represented as fraction of pFLT3/FLT3 relative to DMSO control. Error bars indicate average ± SD (n=6; ***, P < 0.0001; **, P = 0.01). F, Representative examples of adult AML plasma (n=3) from (E). Fraction of pFLT3/FLT3 relative to DMSO control is indicated below each blot.
To explore the disparate plasma protein binding of TTT-3002 and CEP-701, the impact of the two major human plasma proteins known to bind many TKIs, AGP and HSA, was evaluated. AGP is an acute phase plasma protein that is often elevated as much as 4-5 fold in leukemia patients (5). To ascertain whether TTT-3002 is significantly bound by this protein, we treated Ba/F3-ITD cells with TTT-3002 or CEP-701 at IC95 concentrations ascertained under typical culture conditions (5 and 20nM, respectively) in media supplemented with 0-2 mg/mL AGP and measured the percentage of phospho-FLT3 inhibition by Western blotting. While CEP-701 almost completely loses TKI activity with the addition of just 0.1 mg/mL AGP, the lowest concentration tested, TTT-3002 was still strongly active at physiological AGP concentrations, achieving greater than 60% FLT3 inhibition at 2 mg/mL AGP (Fig. 6C). The other major plasma protein capable of binding many drugs is human serum albumin (HSA). TTT-3002 and CEP-701 activities were not as greatly affected by physiological levels of HSA, the most abundant plasma protein (supplemental Fig. 2A) (40). Thus, TTT-3002 should require lower doses than CEP-701 to achieve sufficient concentrations of free drug in plasma due to lower protein binding to AGP.
TTT-3002 activity in human leukemia patient plasma
To further assess the likely activity of TTT-3002 in a clinical setting, human plasma samples were obtained from a panel of adult and pediatric AML patients at the time of diagnosis, when AGP levels are known to often be elevated. Ba/F3-ITD cells were then cultured in these plasma samples with the addition of 0-10nM TTT-3002 for 1 h (Fig. 6D). TTT-3002 potently inhibits phosphorylation of FLT3/ITD in the human AML plasma samples, with an average IC50 of 6.4 ± 2.6nM (n=5). There was no significant difference between phospho-FLT3 inhibition achieved in AML patient plasma samples relative to that achieved in normal adult plasma as determined in Fig. 6A, B (IC50 = 7.0 ± 0.5nM, n=3). Ba/F3-ITD cells cultured in AML plasma samples supplemented with a fixed concentration of TTT-3002 (20nM) or CEP-701 (100nM) showed that TTT-3002 achieved 75-85% inhibition of FLT3 phosphorylation whereas CEP-701 was inactive under these conditions (Fig. 6E, F). The CEP-701 result in plasma is in stark contrast to what was observed at the same concentrations in cell culture media with 10% FBS and remains the most likely explanation for its failure in clinical trials. We found that TTT-3002 was highly active across the AGP and HSA concentration ranges measured in the plasma samples (0.5-2 mg/mL AGP, 2.5-4.0 g/dL HSA) (supplemental Fig. 2B, C).
Discussion
TTT-3002 has been identified as a novel, potent small molecule inhibitor of mutant FLT3 phosphorylation (24). There are a number of mechanisms by which patients can develop resistance to FLT3 inhibition by current TKIs. One such clinical observation is the emergence of resistance mutations in FLT3/ITD expressing cells, which render the cells insensitive to TKI treatment. Other studies indicate that activating point mutations in FLT3, which are found in 7-10% of adult AML cases, provide a survival and proliferative advantage to cells, and respond to many FLT3 TKIs differently than do FLT3/ITD mutants. In the present study, the potent in vitro activity of TTT-3002 against a broad spectrum of FLT3/PM activating mutations, as well as a variety of TKI resistance mutations in the FLT3/ITD sequence was demonstrated. TTT-3002 reduced cell viability and inhibited FLT3 autophosphorylation in a dose-dependent manner at low nanomolar to picomolar concentrations. Although only the D835Y variant in the FLT3/ITD allele was tested, inhibition of FLT3/D835F, D835V, D835H and other activating FLT3 point mutations was also demonstrated. This implies that TTT-3002 would also likely target these D835 TKI resistant variants in a FLT3/ITD allele.
Another major factor that limits the efficacy of FLT3 TKIs in clinical trials, aside from lack of activity against FLT3/PMs and selection for TKI resistance mutations in FLT3/ITD, is the extensive protein binding for many TKIs that occurs in human plasma. In the case of CEP-701, the compound is >99.9% protein bound in plasma. Similar binding properties have also been reported for PKC412 (>99%), sorafenib (99.7%) and AC220 (99%) (7, 39, 41). This large amount of protein binding limits the amount of free drug available to inhibit target, reducing the efficacy of some of these TKIs in clinical trials. TTT-3002 shows more moderate protein binding in human plasma (93%). This predicts that the shift in the dose-response curve for TTT-3002 going from cell culture conditions with 10%FBS to human patients should not be as extreme as it is for some other TKIs. Thus, achieving a total level of ≥30nM should achieve >90% FLT3 inhibition, given the average IC90 of 2nM against FLT3/ITD phosphorylation by Western blotting. These are the approximate levels that one would aim to achieve at the trough level of the drug and thus would help to determine dose and frequency of administration in a future clinical trial.
The extensive preclinical studies highlighted both here and in a previous manuscript (24) support the advancement of TTT-3002 toward human clinical trials. Further studies will be required to assess the safety and toxicity in nonrodent animal models before moving this compound into human clinical trials, where assessment of absorption, bioavailability, pharmacokinetics and pharmacodynamics can be determined. Drug dosing schedule and tolerance in humans will also need to be determined in Phase 1 trials before moving to trials in relapsed/refractory/resistant FLT3 mutant AML patients.
Supplementary Material
Acknowledgments
We thank Ting Martin Ma and members of the D.S., P.B. and M.L. laboratories for help, critical comments, technical support and scientific discussions.
Financial support: The project described was supported by grants from the Alex’s Lemonade Stand Foundation for Childhood Cancer and NIH NCI (CA90668 and CA70970) as well as funds from the Giant Food Pediatric Cancer Research Program. D.S. is also supported by the Kyle Haydock Professorship.
Footnotes
Conflict-of-interest disclosure: The authors disclose no potential conflicts of interest.
References
- 1.Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–8. [PubMed] [Google Scholar]
- 2.Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97:2434–9. doi: 10.1182/blood.v97.8.2434. [DOI] [PubMed] [Google Scholar]
- 3.Weisberg E, Sattler M, Ray A, Griffin JD. Drug resistance in mutant FLT3-positive AML. Oncogene. 2010;29:5120–34. doi: 10.1038/onc.2010.273. [DOI] [PubMed] [Google Scholar]
- 4.Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103:3669–76. doi: 10.1182/blood-2003-11-3775. [DOI] [PubMed] [Google Scholar]
- 5.Levis M, Ravandi F, Wang ES, Baer MR, Perl A, Coutre S, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood. 2011;117:3294–301. doi: 10.1182/blood-2010-08-301796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gambacorti-Passerini C, Barni R, le Coutre P, Zucchetti M, Cabrita G, Cleris L, et al. Role of alpha1 acid glycoprotein in the in vivo resistance of human BCR-ABL(+) leukemic cells to the abl inhibitor STI571. J Natl Cancer Inst. 2000;92:1641–50. doi: 10.1093/jnci/92.20.1641. [DOI] [PubMed] [Google Scholar]
- 7.Levis M, Brown P, Smith BD, Stine A, Pham R, Stone R, et al. Plasma inhibitory activity (PIA): A pharmacodynamic assay reveals insights into the basis for cytotoxic response to FLT3 inhibitors. Blood. 2006;108:3477–83. doi: 10.1182/blood-2006-04-015743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Auclair D, Miller D, Yatsula V, Pickett W, Carter C, Chang Y, et al. Antitumor activity of sorafenib in FLT3-driven leukemic cells. Leukemia. 2007;21:439–45. doi: 10.1038/sj.leu.2404508. [DOI] [PubMed] [Google Scholar]
- 9.Borthakur G, Kantarjian H, Ravandi F, Zhang W, Konopleva M, Wright JJ, et al. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica. 2011;96:62–8. doi: 10.3324/haematol.2010.030452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang W, Konopleva M, Shi YX, McQueen T, Harris D, Ling X, et al. Mutant FLT3: A direct target of sorafenib in acute myelogenous leukemia. J Natl Cancer Inst. 2008;100:184–98. doi: 10.1093/jnci/djm328. [DOI] [PubMed] [Google Scholar]
- 11.Ravandi F, Cortes JE, Jones D, Faderl S, Garcia-Manero G, Konopleva MY, et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol. 2010;28:1856–62. doi: 10.1200/JCO.2009.25.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fathi AT, Chabner BA. FLT3 inhibition as therapy in acute myeloid leukemia: A record of trials and tribulations. Oncologist. 2011;16:1162–74. doi: 10.1634/theoncologist.2011-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischer T, Stone RM, Deangelo DJ, Galinsky I, Estey E, Lanza C, et al. Phase IIB trial of oral midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28:4339–45. doi: 10.1200/JCO.2010.28.9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barry EV, Clark JJ, Cools J, Roesel J, Gilliland DG. Uniform sensitivity of FLT3 activation loop mutants to the tyrosine kinase inhibitor midostaurin. Blood. 2007;110:4476–9. doi: 10.1182/blood-2007-07-101238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clark JJ, Cools J, Curley DP, Yu JC, Lokker NA, Giese NA, et al. Variable sensitivity of FLT3 activation loop mutations to the small molecule tyrosine kinase inhibitor MLN518. Blood. 2004;104:2867–72. doi: 10.1182/blood-2003-12-4446. [DOI] [PubMed] [Google Scholar]
- 16.Kancha RK, Grundler R, Peschel C, Duyster J. Sensitivity toward sorafenib and sunitinib varies between different activating and drug-resistant FLT3-ITD mutations. Exp Hematol. 2007;35:1522–6. doi: 10.1016/j.exphem.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 17.Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105:54–60. doi: 10.1182/blood-2004-03-0891. [DOI] [PubMed] [Google Scholar]
- 18.Heidel F, Solem FK, Breitenbuecher F, Lipka DB, Kasper S, Thiede MH, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107:293–300. doi: 10.1182/blood-2005-06-2469. [DOI] [PubMed] [Google Scholar]
- 19.Moore AS, Faisal A, Gonzalez de Castro D, Bavetsias V, Sun C, Atrash B, et al. Selective FLT3 inhibition of FLT3-ITD+ acute myeloid leukaemia resulting in secondary D835Y mutation: A model for emerging clinical resistance patterns. Leukemia. 2012;26:1462–70. doi: 10.1038/leu.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260–3. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galanis A, Ma H, Rajkhowa T, Ramachandran A, Small D, Cortes J, et al. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood. 2014;123:94–100. doi: 10.1182/blood-2013-10-529313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zimmerman EI, Turner DC, Buaboonnam J, Hu S, Orwick S, Roberts MS, et al. Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood. 2013;122:3607–15. doi: 10.1182/blood-2013-07-513044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith CC, Lasater EA, Zhu X, Lin KC, Stewart WK, Damon LE, et al. Activity of ponatinib against clinically-relevant AC220-resistant kinase domain mutants of FLT3-ITD. Blood. 2013;121:3165–71. doi: 10.1182/blood-2012-07-442871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma H, Nguyen B, Li L, Greenblatt S, Williams A, Zhao M, et al. TTT-3002 is a novel FLT3 tyrosine kinase inhibitor with activity against FLT3-associated leukemias in vitro and in vivo. Blood. 2014;123:1525–34. doi: 10.1182/blood-2013-08-523035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F, et al. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell. 2004;13:169–78. doi: 10.1016/s1097-2765(03)00505-7. [DOI] [PubMed] [Google Scholar]
- 26.McTigue M, Murray BW, Chen JH, Deng YL, Solowiej J, Kania RS. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc Natl Acad Sci U S A. 2012;109:18281–9. doi: 10.1073/pnas.1207759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lamers MB, Antson AA, Hubbard RE, Scott RK, Williams DH. Structure of the protein tyrosine kinase domain of C-terminal src kinase (CSK) in complex with staurosporine. J Mol Biol. 1999;285:713–25. doi: 10.1006/jmbi.1998.2369. [DOI] [PubMed] [Google Scholar]
- 28.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams AB, Nguyen B, Li L, Brown P, Levis M, Leahy D, et al. Mutations of FLT3/ITD confer resistance to multiple tyrosine kinase inhibitors. Leukemia. 2013;27:48–55. doi: 10.1038/leu.2012.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L, Zhang L, Fan J, Greenberg K, Desiderio S, Rassool FV, et al. Defective nonhomologous end joining blocks B-cell development in FLT3/ITD mice. Blood. 2011;117:3131–9. doi: 10.1182/blood-2010-05-286070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thiede C, Steudel C, Mohr B, Schaich M, Schakel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: Association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–35. doi: 10.1182/blood.v99.12.4326. [DOI] [PubMed] [Google Scholar]
- 32.Armstrong SA, Mabon ME, Silverman LB, Li A, Gribben JG, Fox EA, et al. FLT3 mutations in childhood acute lymphoblastic leukemia. Blood. 2004;103:3544–6. doi: 10.1182/blood-2003-07-2441. [DOI] [PubMed] [Google Scholar]
- 33.Frohling S, Schlenk RF, Breitruck J, Benner A, Kreitmeier S, Tobis K, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: A study of the AML study group ulm. Blood. 2002;100:4372–80. doi: 10.1182/blood-2002-05-1440. [DOI] [PubMed] [Google Scholar]
- 34.Stirewalt DL, Meshinchi S, Kussick SJ, Sheets KM, Pogosova-Agadjanyan E, Willman CL, et al. Novel FLT3 point mutations within exon 14 found in patients with acute myeloid leukaemia. Br J Haematol. 2004;124:481–4. doi: 10.1111/j.1365-2141.2004.04808.x. [DOI] [PubMed] [Google Scholar]
- 35.Man CH, Fung TK, Ho C, Han HH, Chow HC, Ma AC, et al. Sorafenib treatment of FLT3-ITD(+) acute myeloid leukemia: Favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood. 2012;119:5133–43. doi: 10.1182/blood-2011-06-363960. [DOI] [PubMed] [Google Scholar]
- 36.Alvarado T, Kantarjian H, Ravandi F, Luthra R, Borthakur G, Manero GG, et al. FLT3 inhibitor treatment in FLT3-mutated AML is associated with development of secondary FLT3-TKD mutations. Blood. 2008;118:1493. [Google Scholar]
- 37.Zhang W, Konopleva M, Jacamo RO, Borthakur G, Chen W, Cortes J, et al. Acquired point mutations of TKD are responsible for sorafenib resistance in FLT3-ITD mutant AML. Blood. 2011;118 [Google Scholar]
- 38.Cools J, Mentens N, Furet P, Fabbro D, Clark JJ, Griffin JD, et al. Prediction of resistance to small molecule FLT3 inhibitors: Implications for molecularly targeted therapy of acute leukemia. Cancer Res. 2004;64:6385–9. doi: 10.1158/0008-5472.CAN-04-2148. [DOI] [PubMed] [Google Scholar]
- 39.Villarroel MC, Pratz KW, Xu L, Wright JJ, Smith BD, Rudek MA. Plasma protein binding of sorafenib, a multi kinase inhibitor: In vitro and in cancer patients. Invest New Drugs. 2012;30:2096–102. doi: 10.1007/s10637-011-9767-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kremer JM, Wilting J, Janssen LH. Drug binding to human alpha-1-acid glycoprotein in health and disease. Pharmacol Rev. 1988;40:1–47. [PubMed] [Google Scholar]
- 41.Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML) Blood. 2009;114:2984–92. doi: 10.1182/blood-2009-05-222034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.