

Abstract
Receptor tyrosine kinases (RTKs) conduct biochemical signals via lateral dimerization in thwe plasma membrane, and their transmembrane (TM) domains play an important role in the dimerization process. Here we present two models of RTK-mediated signaling, and we discuss the role of the TM domains within the framework of these two models. We summarize findings of single amino acid mutations in RTK TM domains that induce unregulated signaling and, as a consequence, pathological phenotypes. We review current knowledge of pathology induction mechanisms due to these mutations, focusing on the structural and thermodynamic basis of pathogenic dimer stabilization.
RECEPTOR TYROSINE KINASES: ROLE IN BIOLOGY AND PATHOLOGY
Receptor tyrosine kinases (RTKs) are single-pass membrane proteins with an extracellular ligand-binding domain and an intracellular kinase domain. Members of this large group of membrane proteins have been classified based on their structural and ligand-affinity properties (1). The RTK family includes several sub-families, including the epidermal growth factor receptors (EGFRs or ErbBs), the fibroblast growth factor receptors (FGFRs), the insulin and the insulin-like growth factor receptors (IR and IGFR), the platelet-derived growth factor receptors (PDGFRs), the vascular endothelial growth factor receptors (VEGFRs), the hepatocyte growth factor receptors (HGFRs), and the nerve growth factor receptors (NGFRs) (2).
RTKs conduct biochemical signals via lateral dimerization in the plasma membrane (3). All the above RTKs, with the exception of the insulin and the insulin-like growth factor receptors, exist in a monomer-dimer equilibrium. The dimer, which is stabilized upon ligand binding, is the signaling competent structure. It is believed that the contact between the two cytoplasmic domains in the dimer stimulates catalytic activity, and results in the intermolecular autophosphorylation of the receptors. This activates the catalytic domains and triggers signaling cascades that lead to the phosphorylation of cytoplasmic substrates.
RTK-mediated signals play key roles in the regulation of various cellular processes, such as control of cell growth, differentiation, metabolism, and migration (3). The essential and diverse roles of RTKs are evident from the various developmental abnormalities and cancers that occur due to gain-of-function RTK signaling (4;5). In humans, signaling irregularities can be the result of (1) the generation of oncogenic fusion proteins (chromosomal translocation), (2) overactivation through mutations or deletions, and (3) overexpression as a result of gene amplification.
Below, we provide a short overview of some RTKs that are critical to cell life, and of their role in disease.
EGFRs
The subfamily of EGFRs consists of four members: EGFR (6), also designated as ErbB1, as well as ErbB2, ErbB3, and ErbB4. These receptors play important roles in cell growth and differentiation during embryonic development and in adult life. EGFR binds different ligands (including EGF, TGFα), while no ligand has been identified for ErbB2. Neuregulins bind to ErbB3 and ErbB4; however, ErbB3 has no intrinsic tyrosine activity. It has been shown that all the members of the ErbB family are capable of initializing signal cascades as heterodimers (7). ErbB3 is phosphorylated when it dimerizes with other EGFRs (8), and ErbB2 has been shown to be the preferred dimerization partner for the other EGFRs (9).
A variety of cancer cells have been shown to overexpress members of the EGFR subfamily (4;10). For instance, mammary carcinoma cells overexpress ErbB2 (11), while glioblastomas overexpress EGFR (12).
FGFRs
The four-member subfamily of the fibroblast growth factor receptors (FGFR1-4) mediates signaling cascades that induce cell growth, differentiation, migration and chemotaxis, angiogenesis, and cell survival. These receptors play a critical role in the development of the skeletal system. They bind at least 9 different FGFs; the binding is stabilized in the presence of heparan sulfate (13).
Targeted inactivation of FGFR genes can have dramatic consequences. FGFR1 gene disruption leads to embryonal death (14), FGFR2 inactivation causes abnormal development of the lungs (15), and FGFR3 inactivation results in bone overgrowth (16). In contrast, FGFR4 null mice exhibit no obvious phenotypes (17).
Mutations in FGFR1, 2 and 3 have been implicated in numerous skeletal and cranial disorders. More than 60 mutations, with very high frequency in FGFR2, are associated with a variety of craniosynostosis syndromes (premature ossification of the skull). Pfeiffer syndrome, characterized by additional abnormality in the fingers, has been observed in patients carrying mutations in FGFR1 and FGFR2. Mutations in FGFR2 have also been observed in Crouzon, Jackson-Weiss, Beare-Stevenson cutis gyrata, and Apert syndromes (18;19). Crouzon syndrome with acanthosis nigricans, commonly associated with mutations in FGFR2, has also been associated with a mutation in FGFR3 (20).
Deficiencies in long bone growth and development are often the result of mutations in FGFR3. Achondroplasia, the most common form of human dwarfism, is attributed often to a Gly380Arg mutation in the transmembrane domain of FGFR3 (21), and occasionally to a Gly375Cys mutation in the same region of the protein (22). Mutations in FGFR3 have also been found in patients with hypochondroplasia, a milder phenotype, and the much more severe thanatophoric (lethal) dysplasias TDI and TDII (23–27).
When overexpressed or mutated, FGFRs are also involved in carcinogenesis. Several FGFRs are overexpressed in human pancreatic cancers (28). In prostate cancer, expression of FGFR1 and FGFR4 is probably linked to tumor progression (29). FGFR subtypes and isoform expressions were shown to be altered in pituitive adenomas (30). Chromosomal translocations involving FGFRs are implicated in multiple myelomas and in leukemias and lymphomas (31–34). Jang et al. (35) has observed two mutations in FGFR2 in gastric cancer patients. An oncogenic role has been demonstrated for FGFR3 in human epithelial cancers and in multiple myelomas (36;37). Several of the mutations in FGFR3, causing skeletal and cranial dysplasias, have also been identified in bladder, cervical, and colorectal cancers (35–37).
VEGFRs
The development of new blood vessels, and the vascular system in general, is primarily regulated by the VEGFRs, VEGFR1-3. The physiological and pathological roles of these receptors were recently reviewed by Takahashi and Shibuya (38) and Ferrara et al. (39). The knockout of VEGFR1 and VEGFR2 genes in mice has been shown to be lethal. VEGFR1 is a negative regulator of angiogenesis, as evident from the overgrowth of endothelial cells and the disorganization of blood vessels in mice lacking the VEGFR1 gene. The opposite is observed in mice without the VEGFR2 gene; they die due to defects in endothelial cell growth (38).
IR and IGFR
The insulin receptor (IR) signals in response to insulin, and some of the outcomes are increased cellular glucose transport and increased glycogen synthase enzymatic activity. The IR further regulates metabolic processes such as protein or lipid synthesis, as well as growth. Defects in IR function can lead to diabetes and diabetes-related complications.
The IR receptor, unlike the RTKs described above, is constitutively dimeric. It is a dislfide-linked dimer of heterodimeric disulfide-linked proteins (αβ monomers). Upon ligand binding, a change is believed to occur from an inactive to active conformation that permits receptor autophosphorylation. In terms of structure and mechanism of activation, the IR is very similar to the the insulin-like growth factor receptor (IGFR), which regulates growth. Yet, one receptor cannot functionally compensate for the lack of the other (40).
STRUCTURAL DETERMINANTS OF RTK ACTIVATION
Extracellular domains
The solved crystal structures of RTK extracellulular domains have provided valuable information about the mechanism of signal transduction. The extracellular domain structures in the presence and absence of ligand have revealed that, at least for some RTKs, the extracellular domain is autoinhibited in the absence of ligand. For instance, EGFR, ErbB3, and ErbB4, which consist of four domains (I–IV), are autoinhibited in the absence of ligand when the dimerization-mediating sequence from domain II, the “dimerization arm”, is buried and in contact with domain IV from the same polypeptide chain (41–43). On the contrary, the ligand-bound EGFR extracellular domain exists in an “open” conformation that readily forms dimers. In the “open” EGFR structure, the ligand (EGF or TGF-a) is tightly bound to domains I and III (44;45) and the dimerization arm makes contacts with the neighboring chain (46). A similar open/closed switch mechanism has been proposed for FGFR extracellular domains, which are composed of three immunoglobulin- like domains (47). In FGFRs, domains II and III are the primary binding site for the ligands, FGF and heparin (18;47;48), while domain I in believed to play a role in autoinhibition.
In summary, the ligand is believed to control the equilibrium between the open and the closed state of the extracellular domains. (For general illustrations of the open and closed conformations of the extracellular domain, see Figure 1A.) By stabilizing the open state, the ligand shifts the equilibrium towards the active dimeric state. It should be noted, however, that ErbB2 extracellular domain does not require a ligand to dimerize and its crystal structure in the absence of ligand is open and resembles the ligand-bound conformation of EGFR, ErbB3 and ErbB4 (49). Therefore it can be expected that, unlike EGFR, ErbB3 and ErbB4, the unliganded ErbB2 extracellular domain exists primarily in the open configuration.
Figure 1.

Two models of RTK-mediated signaling. The difference between the two models is in the conformation and activity of the unliganded dimer. In model A, the unliganded dimer (3A) is active. The ligand (blue) stabilizes the dimer, but does not change the dimer structure (4A). In model B, the unliganded dimer (3B) is inactive. Ligand binding induces a structural change to an active dimer (4B). EC: extracellular domain. TM: transmembrane domain. IC: intracellular catalytic domain.
The view that EGFR extracellular domains in the absence of ligands inhibit dimerization was first proposed by Tanner and Kyte, who carried out dimerization studies of the extracellular domain (50). Tanner and Kyte argued that the role of the ligand is to eliminate the steric repulsion between the extracellular domains. However, work by Moriki et al. (51) has suggested a different role for the ligand in the activation of EGFR. Moriki et al. proposed that upon ligand binding, a conformational change occurs in the preformed dimers of EGFR extracellular domain, and this conformational change activates the receptor. The latter mechanism is similar to the activation mechanism of constitutive dimers, such as IR (52).
TM domains
The most direct answer as to whether the isolated TM domains have a propensity to dimerize in the absence of the extracellular domains and ligands comes from biophysical studies of the isolated TM domains. Using the Toxcat assay, Mendrola et al. (53) demonstrated that the isolated TM domains of EGFR and the three other members of the ErbB family, ErbB2-4, dimerize in bacterial membranes (53). Li et al. demonstrated that the isolated FGFR3 TM domain also dimerizes in the absence of extracellular domains and ligands (54). FGFR3 TM dimers were observed on SDS-PAGE gels and in lipid bilayers, and the free energy of dimerization in lipid bilayers was determined using FRET as −3 kcal/mole (54) with respect to a defined standard state (55). These results demonstrate that, at least for the TM domains of the EGFR family and for FGFR3, the isolated TM domains have a propensity to dimerize. Thus, the TM domains are not just passive membrane anchors.
It should be noted that the dimerization propensity of the studied RTK TM domains is low when compared to the dimerization propensity of the only well-characterized TM dimer, glycophorin A (GpA) (54;56). This finding is not surprising: unlike glycophorin A, which is a stable dimer in the erythrocyte membrane, RTKs exist in a monomer-dimer equilibrium under physiological conditions. Given the critical role that RTKs play in the control of vital biochemical processes, the ratio between monomers and dimers in the membrane needs to be tighly controlled, and a weak dimerization propensity allows for such a tight control (54).
A question arises about the relative contributions of the extracellular domain and the TM domain to the dimerization process in the absence of the ligand. Is dimerization in the absence of ligand driven by the conformational fluctuations of the extracellular domain (41;47), or by the TM domain? It is likely that both domains contribute, but their relative contributions are unknown. Furthermore, the catalytic domains also likely contribute to the dimerization process (57;58).
In one study, aimed at elucidating the relative contributions of the extracellular domain and the TM domain, Tanner and Kyte showed that the extracellular EGFR domains dimerize strongly only in the presence of the TM domains (50). The dissociation constant of the extracellular domains, when attached to the TM domains, was estimated to be least 10,000 fold smaller, as compared to the dissociation constant of the isolated extracellular domains. Furthermore, the interactions of the isolated TM domains can be detected, at least in membranes (53;54;59), while no interactions between the extracellular domains can be detected in solution in the absence of ligand (50;60). While these studies may seem to point to the importance of the TM domain contribution, caution needs to be exercised because the likelihood of dimer formation is predicted to increase 106-fold upon confining the proteins to a membrane (61). Therefore, the relative contributions of the three RTK domains in the formation of unliganded dimers should be investigated further.
Structural link between the TM and the catalytic domains
Bell et al. (62) have observed that the activation of an RTK occurs only for a specific TM dimer interface. The rotation of the dimer interface leads to periodic oscillations in kinase activity. Furthermore, inserting residues in the C-terminal TM flanking region, which causes the kinase domain to rotate with respect to the TM domain, restores the kinase activity of the inactive receptors. These experiments have demonstrated that the RTK TM dimer interface contains the critical structural information that positions the catalytic domains in such a way that they can phosphorylate each other. In view of this finding, it is not surprising that replacing the TM domain of a receptor with the TM domain from a different receptor sometimes does not lead to the expected activation. For instance, changing the TM domain of the insulin receptor with the highly dimerizing TM domain of GpA inhibited insulin signaling (63). Introducing the GpA TM in NeuT did not lead to transformation of COS-7 cells, even though the receptors were dimerizing (64). Constructs consisting of the extracellular and TM domain of EGFR, fused to the catalytic domain of NGFR, inhibited the differentiation of PC12 cells (65).
In other cases, substituting the TM domain of a receptor with another TM domain produced an active receptor. For instance, the insertion of the TM domain of NeuT in the PDGF β receptor led to constitutive activation of the chimeric receptor (66). The introduction of the FGFR3 TM domain in Neu did not affect receptor phosphorylation (67). Introducing the TM domain of NeuT in IR led to ligand-independent activation (68). It could be hypothesized that in the experiments described in this paragraph, the catalytic domain orientation was not affected by the TM domain substitution, thus yielding an active receptor.
Two models of RTK signaling
Based on the knowledge about changes in extracellular domains upon ligand binding, and the role of the TM domains in maintaining an active RTK dimer, two models can be proposed for the mode of activation of RTKs. The models, depicted as consecutive equilibrium processes (steps), are shown in Figures 1A and 1B.
Model A, Figure 1A
In the first step (IA), the closed autoinhibited conformation and the open conformation of the extracellular domain are in equilibrium. Ferguson et al. have estimated that for EGFR, 80–97% of EGFR extracellular domain is autoinhibited at equilibrium (41). It can be expected that this equilibrium will be different for different RTKs. For instance, unlike EGFR, ErbB2 extracellular domain is expected to be predominantly in the open conformation (49).
In the second step (IIA), equilibrium exists between monomers (with extracellular domain in the open configuration) and dimers. The dimer is expected to be not very stable in the absence of the ligand (structure 3A), but is greatly stabilized upon ligand binding (step IIIA, structure 4A).
In model A, the structure of the dimer is similar in the absence and presence of the ligand. The unliganded dimer (structure 3A) is active, because the TM domains interact via the correct dimerization motif, and the catalytic domains are oriented correctly. Upon ligand binding, the dimer is stabilized without major structural changes (structure 4A). Several lines of evidence appear to support this model. First, the structure of the open configuration of ErbB2 extracellular domain in the absence of ligand is very similar to the EGFR configuration in the presence of ligand, supporting the idea that no conformational change occurs in the “open” state upon ligand binding (49). Second, a basal level of autophosphorylation exists for RTKs, maybe due to active unliganded dimers, which ensures maximum sensitivity of the system and swift response (47). Third, ligand independent activation is observed for many pathogenic mutants (67;69), which further suggests that unliganded dimers can be active.
Model B, Figure 1B
In model B, the unliganded dimer (structure 3B) is inactive. The conformation of the extracellular domain in the absence of the ligand is different from the ligand-bound conformation. Furthermore, in the absence of ligand, the TM domains do not interact through the correct dimerization motif, and as a consequence the catalytic domains are inactive. A conformational change has been proposed to occur in the extracellular domain upon ligand binding (step IIIB), leading to the rotation of the whole receptor (51;57). Now, the correct TM domain dimerization motifs “lock” into place, thus positioning the catalytic domains in a signaling-competent orientation (structure 4B). Model B is similar to models proposed for receptors believed to be constitutively dimeric. It should be noted, however, that the activation of constitutively dimeric receptors (IR, IGFR, and non-RTK receptors) does not necessarily involve only rotation. In one model of IR activation, ligand binding brings the TM domains closer together, allowing a closer approach of the catalytic subunits (52). A model for the activation of the growth hormone receptor involves rotation of the subunits within the dimer, as well as a vertical movement (70). The erythropoetin receptor is believed to be activated by a combined scissor movement and subunit rotation (reviewed in (71)).
When considering the two models of activation depicted in Figure 1, it should be kept in mind that some TM domains are believed to have two dimerization motifs and are thus capable of forming active and inactive dimers, while other TM domains are characterized by a single dimerization motif (53;72). It can be hypothesized that the TM domains with a single dimerization motif follow model A, while the ones with two dimerization motifs follow model B. Future work should investigate this possibility.
A key question, pertaining to the exact mode of activation, is the question about the link between the extracellular domain and the TM domain, which, we believe, is still to be investigated in detail. Model B (Figure 1B) suggests a strong structural link between the extracellular domain and the TM domain, while the two domains could be structurally uncoupled in model A. The experiments of Moriki et al. (51) suggest that the ligand-induced rotation of EGFR extracellular domain is transmitted to the TM domain, thus supporting model B. On the other hand, activating cysteine mutations occur in consecutive residues close to the N-terminal flank of RTK TM domains, such as FGFR3 TM domain (see below). Therefore, the region between the extracellular domain and the TM domain may be flexible. Future experiments that shed light on the coupling between the extracellular domain and the TM domain will be therefore useful in understanding how RTKs signal, and in particular if their activation follows model A or model B.
ROLE OF RTK TM DOMAINS IN DISEASE
Pathogenic mutations in the TM domains of RTKs
The strongest supporting evidence that TM domains are critical in RTK signaling is the list of pathogenic mutations identified in the TM domains (see Table 1). Interest in RTK TM domain mutations first arose with the discovery of an oncogenic version of the rat homologue of ErbB2, NeuT. Overactivation of this oncogenic receptor was found to be the result of a Val664Glu mutation in its TM domain (73;74). Another mutation at the same residue (Val664Gln) was also found to lead to full oncogenic activation of the receptor (75). Later, it was demonstrated that the overactivation was due to an increase in receptor association (76).
Table 1.
Pathogenic RTK TM domain mutations
| FGFR1 | Tyr372Cys | Osteoglophonic dysplasia (82) |
| Cys379Arg | Osteoglophonic dysplasia (82) | |
| FGFR2 | Ser372Cys | Beare-Stevenson cutis gyrata syndrome (83) |
| Tyr375Cys | Beare-Stevenson cutis gyrata syndrome (83) | |
| FGFR3 | Gly370Cys | Thanatophoric dysplasia I (26) Bladder cancer (37) |
| Ser371Cys | Thanatophoric dysplasia I (25) | |
| Tyr373Cys | Thanatophoric dysplasia I (26) Bladder cancer (37) |
|
| Gly375Cys | Achondroplasia (22) | |
| Gly380Arg | Achondroplasia (21) | |
| Gly380Arg | Achondroplasia with acanthosis nigricans (112) | |
| Val381Glu | Hypochondroplasia (113) | |
| Gly382Asp | Multiple myeloma (78) | |
| Ala391Glu | Crouzon syndrome with acanthosis nigricans (20) | |
| Ala391Glu | Bladder cancer (37) | |
| FGFR4 | Gly388Arg | Tumor progresion (79) |
| ErbB2 | Ile654Val | Increased risk of breast cancer (77) |
The discovery of the oncogenic Val664Glu and Val664Gln mutations in NeuT prompted investigators to search for similar TM mutations in cancer patients. However, despite the fact that overexpression of ErbB receptors is frequently found in cancers, very few incidents of cancer-inducing TM mutations have been identified so far in ErbB receptors. One example is the Ile654Val mutation in ErbB2, associated with an elevated risk for breast cancer (77). More oncogenic mutations have been identified in the TM domains of the FGFR family. Otsuki et al. (78) identified a FGFR3 Gly382Asp mutation in a multiple myeloma cell line. The Ala391Glu mutation in FGFR3 has been implicated in bladder cancer (37). The mutations Gly370Cys, Gly380Arg and Tyr373Cys in FGFR3 have been associated with bladder carcinomas (37). In FGFR4, the Gly388Arg mutation has been shown to accelerate tumor progression in cancer patients (79).
The members of the FGFR receptor subfamily are critical for the development of the skeletal system, and therefore abnormal developments (dysplasias) of the bones have been linked to mutations in the TM domain of FGFRs. The disruption of endochondral ossification due to mutations in FGFR3 results in several forms of dwarfism, including achondroplasia (associated with the Gly380Arg and the G375Cys mutations), and thanatophoric dysplasia (due to the Gly370Cys, Ser371Cys, and Tyr373Cys mutations) (80;81). The Ala391Glu mutation in FGFR3 is linked to Crouzon syndrome with acanthosis nigricans (20). In FGFR1, several patients with osteoglophonic dysplasia have been shown to carry the Tyr372Cys and Cys379Arg mutations (82). The Tyr375Cys and Ser372Cys mutations in FGFR2 lead to Beare-Stevenson cutis gyrata syndrome, characterized by cranial abnormalities and wrinkled skin (83;84).
Mechanism of pathogenic TM dimer stabilization
Mutations in the TM domains are believed to cause receptor overactivation by stabilizing the RTK dimer (67;74–76;85). Wild-type RTKs and RTKs containing mutations in the TM domains, expressed at identical levels (i.e. identical rate of synthesis and subcellular localization), exhibit different mitogenic activities. The mutants, linked to disease, are overactivated, as determined by measuring the level of autophosphorylation and the ability to transform cells (67;85). Furthermore, at least in the case of the Val664Glu mutation in NeuT, the transforming activity has been directly linked to dimer stabilization (76).
Currently, it is accepted that TM dimer stabilization often occurs via specific, mutant amino acid-mediated contacts, discussed below. A second mechanism, specific to TM domains with two dimerization motifs, may be the stabilization of inactive TM dimers (discussed in (72) and not reviewed here).
Structural requirements for TM dimer stabilization due to pathogenic TM domain mutations
As discussed above, the structure of the TM domain dimer ensures that the catalytic domains are oriented correctly with respect to each other and that the whole receptor is signaling-competent. Li et al. (86) have therefore proposed that only TM mutations that stabilize the dimer without altering the orientation of the helices in the TM dimer can be overactivating. While some modest changes could be tolerated, a dramatic change in the TM dimer interface should render the receptor inactive. For overactivation to occur due to TM domain mutations, (1) the mutant TM dimer needs to be more stable, and (2) the wild-type and mutant TM dimer structures have to be similar, particularly at the C-terminal end (see also Figure 2). This hypothesis could explain why engineered (not naturally occurring) mutations, expected to be stabilizing, not always cause receptor overactivation (87;88). If this structural hypothesis is correct, RTK TM dimer structure determination would be key to understanding RTK signaling. Therefore, the development of routine structure determination techniques in the future will help us better understand how mutations in the TM domains of RTKs cause disease.
Figure 2.
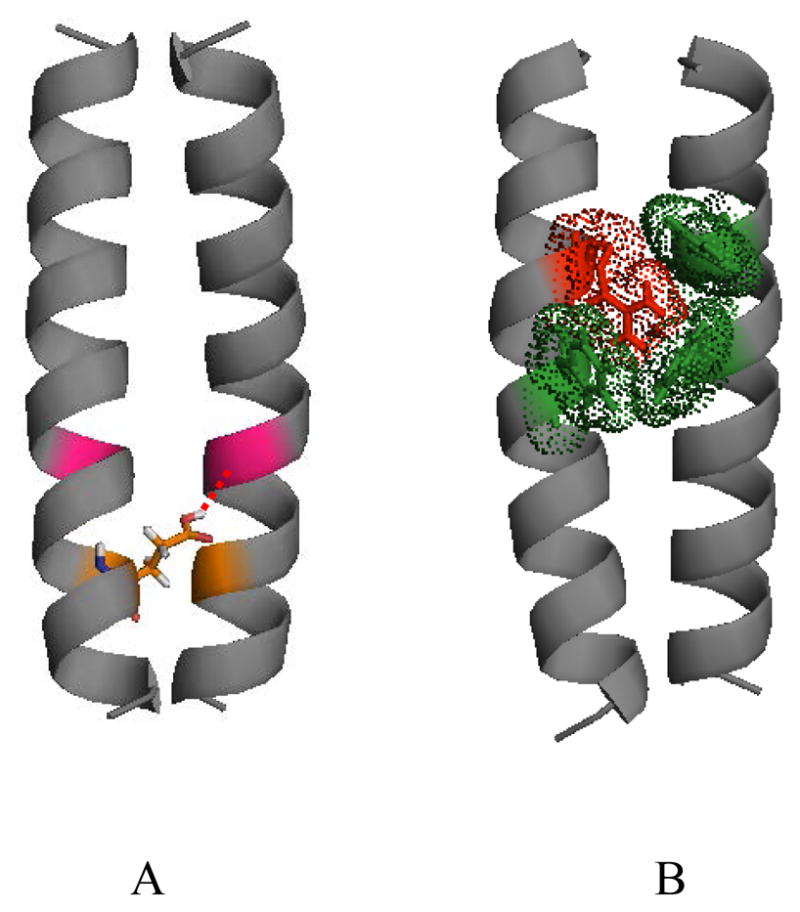
Pathogenic interactions, believed to contribute to the stability of mutant RTK TM domain dimers. (A) A putative hydrogen bond between Glu391 (orange) and Ile387 (magenta) in the pathogenic Ala391Glu FGFR3 mutant, linked to Crouzon syndrome with acanthosis nigricans and bladder cancer (86). (B) Putative cation-π interactions between Arg380 (red) and aromatic residues (green) in the achondroplasia-causing Gly380Arg FGFR3 mutant. These cation-π interactions likely compensate for the electrostatic repulsion between the two arginines in the mutant dimer. The comparison of these two mutant structures with the wild-type FGFR3 TM dimer structure (86) shows that the mutations induce modest changes in the relative orientation of the two helices close to the C-terminus (not discussed here in detail). Thus, the orientation of the catalytic domains is likely not perturbed due to the mutations, inaccordance with the structural hypothesis discussed here.
The only method that has so far produced a detailed structure of a TM dimer in detergent micelles is high-resolution NMR. Furthermore, a single structure has been solved, the structure of the GpA TM dimer (89). RTK TM domains have much weaker dimerization propensity than GpA, and it is not clear if their structure can be determined in micelles by high-resolution NMR. Solid state NMR, which interrogates the TM dimers in their native environment, the lipid bilayer, can be an alternative method that addresses specific structural questions (90;91). An approach combining mutagenesis and dimerization propensity studies has been used to determine the structure of the GpA dimer (92). The idea behind this approach is to (1) mutate residues believed to mediate helix-helix contacts and (2) assess the dimerization propensities of these mutants. If the mutated residues participate in the dimer interface, the dimer will be destabilized. Thus, critical residues that mediate dimerization can be identified. This “mutagenesis” method, however, is based on an assumption, which may be not always correct: a single TM dimer structure exists for all mutants, and a destabilizing mutation affects the monomer-dimer equilibrium, but does not alter the dimer structure. This approach has been successful for the stable GpA (93) and BNIP3 dimers (94), but it is not clear if it works for the weakly dimerizing RTK TM domains.
Hydrogen bonds
One possible mechanism of mutant TM dimer stabilization is hydrogen bonding. Indeed, some pathogenic mutations introduce amino acids with hydrogen bonding capabilities, such as Glu. The role of Glu/Gln hydrogen bonding in the stability of the oncogenic Neu mutant (NeuT) has been studied (91;95). Polarized FTIR and magic angle spinning NMR spectroscopy revealed that the side chain of Glu664 is protonated and participates in hydrogen bonding (95). Measurements of interhelical distances further demonstrated that Glu/Gln664 in one helix interacts with Gly665 in the other helix (91). Similarly to NeuT, the mutant Glu residue in the Ala391Glu FGFR3 mutant, linked to Crouzon syndrome with acanthosis nigricans and cancer, has been hypothesized to form hydrogen bonds (86). A structural model, created with the softwares CHI (96–98) and Insight II, shows the putative hydrogen bond in the mutant Ala391Glu FGFR3 TM domain in Figure 2A (see (86) for details).
Disulfide bonds
Not surprisingly, some of the pathogenic mutations in RTK TM domains are mutations to cysteines (81). The cysteine residues are expected to form disulfide bonds and stabilize the dimer, such that tyrosine kinase activity is stimulated, even in the absence of a ligand. While disulfide bonding has not been demonstrated directly, the effect of the pathogenic Gly370Cys, Ser371Cys, Tyr373Cys and Gly375Cys FGFR3 mutations on dimerization and downstream signaling has been studied. The extent of mutant receptor dimerization, phosphorylation, and downstream MAPK activation was found to depend on the position of the mutation, thus implying specific Cys-mediated stabilizing interactions (69).
The idea that two unpaired Cys residues from the two polypeptide chains form a disulfide bond and stabilize the dimer is intuitive and straight-forward. However, wild-type RTKs also have unpaired cysteines. For instance, wild-type VEGFR1, FGFR1, and FGFR2, have cysteines in their TM domains. Furthermore, FGFR3 TM domain has a wild-type Cys in position 396 that does not cause overactivation. Molecular modeling of the wild-type FGFR3 dimer suggest that the two wild-type cysteines face away from the interface (86). We reason that if the two wild-type 396 cysteines from the two polypeptide chains come into contact due to thermal fluctuation-induced helix unwinding and form a disulfide bond in the membrane cytoplasmic interface, then a large structural perturbation will be introduced that will uncouple the TM domain from the catalytic domain and inhibit signaling. Therefore, it can be expected that a putative Cys396-mediated disulfide bond will stabilize an inactive FGFR3 dimer. If a cysteine residue, however, occurs between the TM domain and the extracellular domain, as in the case of the pathogenic FGFR3 cysteine mutants, the cysteine residue may participate in disulfide bond formation without interfering with the orientation of the TM domain and the catalytic domains. Then, the signaling-competent dimer will be stabilized and the receptor will be overactivated.
It should be always kept in mind that disulfide bonds do not form in reducing environments, such as the cytoplasm. What about the membrane environment? The question whether disulfide bonds form between native cysteines in membranes has not been explored in detail. Furthermore, a question arises if the probability for disulfide bonding is different in the two interfacial regions (extracellular and cytoplasmic) of the plasma membrane.
Another interesting observation is that the mutation of any of four consecutive FGFR3 residues (370, 371, 372, and 373) to Cys can cause receptor overactivation (99). Based on this finding, we could hypothesize that this region is rather flexible and/or not α-helical. This hypothesis has implications for the mechanism of RTK signal transduction, as depicted in the two models in Figure 1. As discussed above, structural flexibility and a weak structural coupling between the extracellular domain and the catalytic domain would support model A of RTK activation.
Cation-π interactions
Recently, You et al. addressed the effect of the Gly380Arg mutation on FGFR3 TM dimer stability (100). This mutation is linked to achondroplasia, the most common form of human dwarfism (21). Previously, Webster and Donoghue have shown that this mutation increases the phosphorylation of FGFR3 and of a chimeric Neu/FGFR3 receptor, and have proposed that dimer stabilization occurs via Arg-mediated hydrogen bonding (67). You et al., however, have shown that the mutation does not alter the dimerization energetics of the isolated FGFR3 TM domain (100). This finding is surprising, since Arg-mediated dimer stabilization can be expected based the observed elevated autophosphorylation reported by Webster and Donoghue. More surprising, however, is probably the fact that the Gly380Arg mutation does not destabilize the FGFR3 TM dimer. Two previous reports have demonstrated that mutations to Arg in TM helix dimers are destabilizing (101;102). In this respect, the finding that Arg380 does not affect the stability of the FGFR3 TM dimer is unexpected: even if Arg380 faces away from the dimer interface (i.e. even if it is exposed to lipids), there should be electrostatic repulsion between the two guanidinium groups. Computer modeling of the mutant TM dimer structure using the program CHI (96–98) has produced a model in which Arg380 is surrounded by Phe384 on the same helix and Tyr379 and Phe383 on the neighboring helix (Figure 2B). In this geometry, no Arg-mediated hydrogen bonds can be identified. However, cation-π interactions, identified by the program CAPTURE (103), may play an important role in dimer stabilization and may compensate for the electrostatic repulsion between the two Arg residues, such that the dimer stability is not altered due to the mutation. The putative structure of the Arg380 FGFR3 mutant, stabilized by cation-π interactions, is shown in Figure 2B.
It should be noted that a direct observation of stabilizing interactions, such as hydrogen bonds, disulfide bonds, or cation-π interactions, is problematic. In soluble proteins, such interactions are usually inferred from the solved crystal structures. As TM dimer structures are very demanding to solve, direct identification of stabilizing interactions is currently a challenge.
The thermodynamics of pathogenic stabilization
Many questions of interest pertain to the thermodynamics of the dimerization process. These include: What is the “correct” dimerization energy for normal RTK signaling? How much excess dimerization energy is required for persistent activation of the receptors, and therefore, for induction of pathologies? The dimerization propensity of RTK TM domains has been probed using the TOXCAT and ToxR assays (53;59), FRET (54;104), and analytical ultracentrifugation (56). Recently, the change in free energy of dimerization of an RTK TM domain due to a pathogenic mutation was determined in lipid bilayers using FRET (55;86). In this study, the change in free energy of dimerization due to the pathogenic FGFR3 Ala391Glu mutation was measured as −1.3 kcal/mole (86). This seemingly modest value was shown to have a significant effect on the equilibrium between monomers and dimers, thus providing a plausible mechanism for pathogenesis due to the Ala391Glu mutation.
As discussed above, pathogenic TM domain mutations are not expected to significantly alter the structure of the TM domain dimer, and as a consequence, the structure of the whole receptor. Based on this assumption, it has been proposed that the measured −1.3 kcal/mole could be the mutation-induced change in dimerization free energy that transforms normal FGFR3 signaling into pathogenic in the context of the whole receptor (86). This hypothesis could be tested in the future by comparing the mutation-induced change in dimerization propensities of the isolated TM domains and of the whole receptors in cells.
Pathogenic RTK TM domain mutations are often dominant, and the cells of the affected heterozygotes express both wild-type and mutant proteins. For instance, the Ala391Glu mutation in the TM domain of FGFR3 is autosomal dominant. A method was recently developed to characterize the energetics of heterodimerization in lipid bilayers using FRET, and was used to determine the propensity for heterodimer formation between wild-type FGFR3 TM domain and the Ala391Glu mutant (105). The heterodimer was shown to be more stable than the wild-type homodimer by −0.6 kcal/mole. Furthermore, this method allows quantitative predictions of shifts in the over-all monomer-dimer equilibrium, thus shedding light on pathology induction mechanisms in heterozygotes.
Effects of receptor expression and ligand concentration
Li et al. (86) have proposed that mutation-induced changes in receptor dimer fraction in cells can be observed only for a certain range of receptor concentrations in the plasma membrane. This concentration range is determined by (1) the difference in dimerization propensities between wild-type and mutant receptors, and (2) the dimerization propensity of the wild-type receptor. Figure 3 illustrates the dependence of the relative increase in dimer fraction on the expression level and the dimerization propensities of the wild-type and mutant receptors (see figure caption for detail). The relative increase in dimer fraction due to TM mutations should also depend on the availability and the concentration of the ligand, since the dimerization propensity of the receptors depends on the ligand concentration (discussed in detail in (86)).
Figure 3.

The increase in receptor dimer fraction due to a pathogenic mutation depends of three parameters: (1) the difference in dimerization propensities between wild-type and mutant receptors, (2) the dimerization propensity of the wild-type receptor, which in turn depends on the ligand concentration, and (3) the receptor concentration in the plasma membrane. The functional dependence of the receptor dimer fraction on receptor concentration in the membrane is shown for an arbirary dimerization propensity of the wild-type receptor (solid line). The dashed line depicts a higher dimer fraction due to a pathogenic mutation (86). The figure illustrates that the relative increase in dimer fraction [ΔD]/[D], where [D] is the wild-type dimer fraction and [ΔD] is the increase due to the mutation, depends on the receptor concentration in the membrane. For example, the relative increase in dimer fraction [ΔD1]/[D1] for concentration “1” is [ΔD1]/[D1] ~ 2, such that the dimer fraction increases 3- fold due to the mutation. However, a 100-fold increase in expression level will result in only about 10% increase in dimer fraction ([ΔD2]/[D2] ~ 0.1 for concentration “2”) due to the same mutation. Note that the dimerization propensity of the wild-type receptors in the plasma membrane, determining the exact position of the solid line, is yet to be characterized (the difference between the solid and the dashed line correspond to −1.3 kcal/mole (86)). This is a challenging task since the dynamic monomer-dimer distribution in the plasma membrane is expected to depend on the concentration of the available ligand (86), but may be accomplished in the future using FRET. Until this is accomplished, the dependence of the receptor dimer fraction on the expression level, as well as the increase in receptor dimer fraction due to pathogenic mutations, cannot be predicted quantitatively.
Cellular studies: effects on downstream signaling
Usually pathogenic TM domain mutations result in enhanced receptor dimerization, in enhanced receptor phosphorylation, and in cell transformations. For instance, the Val664Glu mutation in NeuT increases the phosphorylation of the NeuT receptor and induces cell transformation (74;75). The Arg380 achondropalsia mutation increases the phosphorylation of FGFR3 and of a chimeric Neu/FGFR3 receptor (67).
Furthermore, FGFR3, in the presence of the pathogenic Gly370Cys, Ser371Cys, Tyr373Cys and Gly375Cys mutations, dimerizes in 293T cells in a ligand-independent way, while the wild-type dimerizes only on FGF stimulation. Receptor phosphorylation (in 293T cells) and MAPK activation levels (in L8 myoblasts) show a strong correlation with the severity of the corresponding phenotypes. They are the highest for Gly370Cys and Ser371Cys, two mutants causing lethal phenotypes (69).
The outcomes of such cellular studies, however, are often cell-specific. For instance, the Gly380Arg mutation in FGFR3 results in constitutive activation of the receptor in the absence of ligands in 3T3 (fibroblast), L6 (myoblast) and C2-7 (myoblast) cells (67;106), but not in 293 (kidney), PC12 (adrenal), and RCJ (chondrocyte) cells (106–108). An increase in phosphorylation due to the Gly375Cys FGFR3 mutation was observed in 293 cells, but not in PC12 cells (69;109). Measurements of MAPK activation showed that pathogenic cysteine mutants in FGFR3 induce ligand independent activation in L8 myoblasts, but not in RCJ chondrocytes (69).
One obvious explanation for the observed cell-specific responses is differences in downstream signaling. Differences in response have also been attributed to the potential interactions of overexpressed receptors with endogenous ones (69;107). The outcomes could be further affected by the particular concentrations of receptors in the plasma membrane (see Figure 3), as determined by receptor expression level, trafficking and downregulation.
Cellular studies: downregulation of the mutant receptors
The activity of RTKs must be tightly regulated and thus different mechanisms have evolved for the attenuation and termination of RTK-mediated signals. Such mechanisms include (1) autoinhibition mediated by the extracellular domains or the catalytic domains, (2) antagonistic ligands, (3) hetero-oligomerization with kinase-inactive mutant receptors, (4) inhibition by phosphatases, and (5) receptor endocytosis and degradation (3;47). Mutations that interfere with these regulatory mechanisms can lead to persistent activation of the receptors and to pathologies.
Pathogenic mutations in the TM domains of RTKs have been shown to interfere with at least one of the above inhibitory mechanisms, namely downregulation and internalization of the activated dimeric receptors. Monsonego-Ornan and colleagues (108) have demonstrated that the mutant Gly380Arg receptors are downregulated slowly, such that they accumulate in the plasma membrane. Cho et. al. (110) have shown that the same mutant escapes lysosomal degradation and is recycled back to the plasma membrane, thus amplifying FGFR3-mediated signaling. Yamada et al. (111) have measured the rate of ligand-induced receptor internalization of IR and IR mutants. Replacing the TM domain of IR with that of non-transforming Neu reduced the rate of internalization, while the introduction of the double mutation G933P934→A933A934 increased it.
How can a TM domain mutation, supposedly hidden inside the membrane, affect downregulation? One mechanism could be a mutation-induced change in protein topology. Some of the pathogenic mutations are mutations to hydrophilic residues, and might affect the disposition of the receptor in the bilayer. The achondroplasia Arg380 mutation was shown to induce a shift in the FGFR3 TM domain in model bilayers (X. Han and K. Hristova, manuscript in preparation). A TM domain shift could in turn affect the ubiquitination of FGFR3, and its subsequent downregulation. Yamada et al. (111) has attributed the differences in IR internalization to different mobilities of the wild-type IR and the mutants from microvilli to nonvillous domains prior to internalization. Future work should explore in greater detail the molecular mechanism behind the slow RTK downregulation due to TM domain mutations.
A LOOK AHEAD
Above all, this review underscores the important questions that are yet to be answered in order to arrive at a better understanding of the mechanism of RTK signaling in health and disease. While changes in the conformation of the extracellular domains upon ligand binding have been elucidated from recent crystal structures, the exact mechanism of signal transduction from the extracellular space to the cytoplasm is yet to be determined. No doubt, the TM domains will be shown to play an important role in the process.
The debate about the mechanism of RTK signal transduction in health and disease will continue in the future, at least until crystal structures of whole receptors, wild-type and mutants in various signaling states, become available. In the nearer future, the development of routine methods for determining structures of isolated wild-type and mutant TM domain dimers will help us better understand the structural determinants of TM mutation-induced pathologies. A method, based on FRET, has been developed to quantify the dimerization propensities of the isolated TM domains (wild-type and mutant homodimers and heterodimers) in the native lipid bilayer (55;105). Thus, the dimerization energetics of various RTK TM domains can be now determined in the membrane environment. Next, a question arises if quantitative measurements of dimerization propensities can be carried out for the whole receptors in cells. If this is accomplished, it will allow us to directly correlate receptor dimerization in the plasma membrane with receptor autophosporylation levels. This line of research will eventually allow us to develop a comprehensive model of unregulated cell signaling by RTKs.
Acknowledgments
Supported by NIH grant GM068619 and Research Scholar Grant # RSG-04-201-01 from the American Cancer Society to K.H.
Studies of RTK-mediated signaling and the roles of RTK TM domains in pathology induction in our laboratory are supported by the American Cancer Society and the National Institute of General Medical Sciences. We thank Drs. Graham Carpenter and Anne K. Kenworthy for reading the manuscript prior to publication, and for their useful comments. We are grateful to Drs. William C. Wimley, Stephen H. White, and Michael Edidin for many enjoyable and helpful discussions and to Dr. Paul Adams for providing us with the CHI software. We extend special thanks to Dr. Jay Gargus, who introduced us to the genetics of FGFR3-mediated skeletal disorders. We acknowledge the members of our laboratory, Dr. Min You, Dr. Mikhail Merzlyakov, Xue Han, Lijuan He, Dr. Ricky Soong, Jamie Spangler, Shannon O’Conner, William Tsao, Rachel Casas, Lirong Chen, Jesse Placone, and Dr. Vesselin Nikolov for their hard work and support.
ABBREVIATIONS
- RTK
receptor tyrosine kinase
- TM
transmembrane
- EGFR/ErbB
epidermal growth factor receptor
- FGFR
fibroblast growth factor receptor
- IR
insulin receptor
- IGFR
insulin-like growth factor receptor
- PDGFR
platelet-derived growth factor receptor
- VEGFR
vascular endothelial growth factor receptor
- HGFR
hepatocyte growth factor receptor
- NGFR
nerve growth factor receptor
- GpA
glycophorin A
- FRET
Förster resonance energy transfer
References
- 1.Fantl WJ, Johnson DE, Williams LT. Signaling by Receptor Tyrosine Kinases. Annu Rev Biochem. 1993;62:453–481. doi: 10.1146/annurev.bi.62.070193.002321. [DOI] [PubMed] [Google Scholar]
- 2.van der Geer P, Hunter T, Lindberg RA. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu Rev Cell Biol. 1994;10:251–337. doi: 10.1146/annurev.cb.10.110194.001343. [DOI] [PubMed] [Google Scholar]
- 3.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 4.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 5.Robertson SC, Tynan JA, Donoghue DJ. RTK mutations and human syndromes - when good receptors turn bad. Trends Genet. 2000;16:265–271. doi: 10.1016/s0168-9525(00)02077-1. [DOI] [PubMed] [Google Scholar]
- 6.Carpenter G, King L, Cohen S. Epidermal Growth-Factor Stimulates Phosphorylation in Membrane Preparations Invitro. Nature. 1978;276:409–410. doi: 10.1038/276409a0. [DOI] [PubMed] [Google Scholar]
- 7.Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim HH, Vijapurkar U, Hellyer NJ, Bravo D, Koland JG. Signal transduction by epidermal growth factor and heregulin via the kinase-deficient ErbB3 protein. Biochem J. 1998;334:189–195. doi: 10.1042/bj3340189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beerli RR, Grausporta D, Woodscook K, Chen XM, Yarden Y, Hynes NE. Neu Differentiation Factor Activation of Erbb-3 and Erbb-4 Is Cell-Specific and Displays A Differential Requirement for Erbb-2. Mol Cell Biol. 1995;15:6496–6505. doi: 10.1128/mcb.15.12.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holbro T, Civenni G, Hynes NE. The ErbB receptors and their role in cancer progression. Experim Cell Res. 2003;284:99–110. doi: 10.1016/s0014-4827(02)00099-x. [DOI] [PubMed] [Google Scholar]
- 11.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A, Press MF. Studies of the Her-2/Neu Proto-Oncogene in Human-Breast and Ovarian-Cancer. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 12.Barker FG, Simmons ML, Chang SM, Prados MD, Larson DA, Sneed PK, Wara WM, Berger MS, Chen PC, Israel MA, Aldape KD. EGFR overexpression and radiation response in glioblastoma multiforme. International Journal of Radiation Oncology Biology Physics. 2001;51:410–418. doi: 10.1016/s0360-3016(01)01609-1. [DOI] [PubMed] [Google Scholar]
- 13.Wilkie AOM, Morriss-Kay GM, Jones EY, Heath JK. Functions of fibroblast growth factors and their receptors. Curr Biol. 1995;5:500–507. doi: 10.1016/s0960-9822(95)00102-3. [DOI] [PubMed] [Google Scholar]
- 14.Deng CX, Wynshaw-Boris A, Shen MM, Daugherty C, Ornitz DM, Leder P. Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev. 1994;8:3045–3057. doi: 10.1101/gad.8.24.3045. [DOI] [PubMed] [Google Scholar]
- 15.Peters K, Werner S, Liao X, Wert S, Whitsett J, Williams L. Targeted Expression of A Dominant-Negative Fgf Receptor Blocks Branching Morphogenesis and Epithelial Differentiation of the Mouse Lung. EMBO J. 1994;13:3296–3301. doi: 10.1002/j.1460-2075.1994.tb06631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- 17.Weinstein M, Xu XL, Ohyama K, Deng CX. FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development. 1998;125:3615–3623. doi: 10.1242/dev.125.18.3615. [DOI] [PubMed] [Google Scholar]
- 18.Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 19.Webster MK, Donoghue DJ. FGFR activation in skeletal disorders: Too much of a good thing. Trends Genet. 1997;13:178–182. doi: 10.1016/s0168-9525(97)01131-1. [DOI] [PubMed] [Google Scholar]
- 20.Meyers GA, Orlow SJ, Munro IR, Przylepa KA, Jabs EW. Fibroblast-Growth-Factor-Receptor-3 (Fgfr3) Transmembrane Mutation in Crouzon-Syndrome with Acanthosis Nigricans. Nat Genet. 1995;11:462–464. doi: 10.1038/ng1295-462. [DOI] [PubMed] [Google Scholar]
- 21.Shiang R, Thompson LM, Zhu Y-Z, Church DM, Fielder TJ, Bocian M, Winokur ST, Wasmuth JJ. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994;78:335–342. doi: 10.1016/0092-8674(94)90302-6. [DOI] [PubMed] [Google Scholar]
- 22.Ikegawa S, Fukushima Y, Isomura M, Takada F, Nakamura Y. Mutations of the Fibroblast Growth-Factor Receptor-3 Gene in One Familial and 6 Sporadic Cases of Achondroplasia in Japanese Patients. Human Genetics. 1995;96:309–311. doi: 10.1007/BF00210413. [DOI] [PubMed] [Google Scholar]
- 23.Grigelioniene G, Hagenas L, Eklof O, Neumeyer L, Haereid PE, Anvret M. A novel missense mutation Ile538Val in the fibroblast growth factor receptor 3 in hypochondroplasia. Human Mutation. 1998;11:333. doi: 10.1002/(SICI)1098-1004(1998)11:4<333::AID-HUMU18>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 24.Bellus GA, Mcintosh I, Smith EA, Aylsworth AS, Kaitila I, Horton WA, Greenhaw GA, Hecht JT, Francomano CA. A Recurrent Mutation in the Tyrosine Kinase Domain of Fibroblast Growth-Factor Receptor-3 Causes Hypochondroplasia. Nat Genet. 1995;10:357–359. doi: 10.1038/ng0795-357. [DOI] [PubMed] [Google Scholar]
- 25.Tavormina PL, Shiang R, Thompson LM, Zhu YZ, Wilkin DJ, Lachman RS, Wilcox WR, Rimoin DL, Cohn DH, Wasmuth JJ. Thanatophoric Dysplasia (Type-I and Type-Ii) Caused by Distinct Mutations in Fibroblast Growth-Factor Receptor-3. Nat Genet. 1995;9:321–328. doi: 10.1038/ng0395-321. [DOI] [PubMed] [Google Scholar]
- 26.Rousseau F, el G, Delezoide VAL, Legeai-Mallet L, Le Merrer M, Munnich A, Bonaventure J. Missense FGFR3 mutations create cysteine residues in thanatophoric dwarfism type I (TD1) Hum Mol Genet. 1996;5:509–512. doi: 10.1093/hmg/5.4.509. [DOI] [PubMed] [Google Scholar]
- 27.Iwata T, Chen L, Li CI, Ovchinnikov DA, Behringer RR, Francomano CA, Deng CX. A neonatal lethal mutation in FGFR3 uncouples proliferation and differentiation of growth plate chondrocytes in embryos. Human Molecular Genetics. 2000;9:1603–1613. doi: 10.1093/hmg/9.11.1603. [DOI] [PubMed] [Google Scholar]
- 28.Kornmann M, Beger HG, Korc M. Role of fibroblast growth factors and their receptors in pancreatic cancer and chronic pancreatitis. Pancreas. 1998;17:169–175. doi: 10.1097/00006676-199808000-00010. [DOI] [PubMed] [Google Scholar]
- 29.Kwabi-Addo B, Wang JH, Erdem H, Vaid A, Castro P, Ayala G, Ittmann M. The expression of sprouty1, an inhibitor of fibroblast growth factor signal transduction, is decreased in human prostate cancer. Cancer Research. 2004;64:4728–4735. doi: 10.1158/0008-5472.CAN-03-3759. [DOI] [PubMed] [Google Scholar]
- 30.Abbass SAA, Asa SL, Ezzat S. Altered expression of fibroblast growth factor receptors in human pituitary adenomas. Journal of Clinical Endocrinology and Metabolism. 1997;82:1160–1166. doi: 10.1210/jcem.82.4.3896. [DOI] [PubMed] [Google Scholar]
- 31.Chesi M, Nardini E, Lim RSC, Smith KD, Kuehl WM, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood. 1998;92:3025–3034. [PubMed] [Google Scholar]
- 32.Chesi M, Nardini E, Brents LA, Schrock E, Ried T, Kuehl WM, Bergsagel PL. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet. 1997;16:260–264. doi: 10.1038/ng0797-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richelda R, Ronchetti D, Baldini L, Cro L, Viggiano L, Marzella R, Rocchi M, Otsuki T, Lombardi L, Maiolo AT, Neri A. A novel chromosomal translocation t(4;14)(p16.3;q32) in multiple myeloma involves the fibroblast growth-factor receptor 3 gene. Blood. 1997;90:4062–4070. [PubMed] [Google Scholar]
- 34.Chesi M, Brents LA, Fly SA, Bais C, Robbiani DF, Mesri E, Kuehl WM, Bergsagel PL. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Blood. 2001;97:729–736. doi: 10.1182/blood.v97.3.729. [DOI] [PubMed] [Google Scholar]
- 35.Jang JH, Shin KH, Park JG. Mutations in fibroblast growth factor receptor 2 and fibroblast growth factor receptor 3 genes associated with human gastric and colorectal cancers. Cancer Research. 2001;61:3541–3543. [PubMed] [Google Scholar]
- 36.Cappellen D, de Oliveira C, Ricol D, Diez de Medina SG, Bourdin J, Sastre-Garau X, Chopin D, Thiery JP, Radvanyi F. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18–20. doi: 10.1038/12615. [DOI] [PubMed] [Google Scholar]
- 37.van Rhijin B, van Tilborg A, Lurkin I, Bonaventure J, de Vries A, Thiery JP, van der Kwast TH, Zwarthoff E, Radvanyi F. Novel fibroblast growth factor receptor 3 (FGFR3) mutations in bladder cancer previously identified in non-lethal skeletal disorders. European Journal of Human Genetics. 2002;10:819–824. doi: 10.1038/sj.ejhg.5200883. [DOI] [PubMed] [Google Scholar]
- 38.Takahashi H, Shibuya M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clinical Science. 2005;109:227–241. doi: 10.1042/CS20040370. [DOI] [PubMed] [Google Scholar]
- 39.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nature Medicine. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 40.Entingh-Pearsall A, Kahn CR. Differential roles of the insulin and insulin-like growth factor-I (IGF-I) receptors in response to insulin and IGF-I. J Biol Chem. 2004;279:38016–38024. doi: 10.1074/jbc.M313201200. [DOI] [PubMed] [Google Scholar]
- 41.Ferguson KM, Berger MB, Mendrola JM, Cho HS, Leahy DJ, Lemmon MA. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Molecular Cell. 2003;11:507–517. doi: 10.1016/s1097-2765(03)00047-9. [DOI] [PubMed] [Google Scholar]
- 42.Cho HS, Leahy DJ. Structure of the extracellular region of HER3 reveals an interdomain tether. Science. 2002;297:1330–1333. doi: 10.1126/science.1074611. [DOI] [PubMed] [Google Scholar]
- 43.Bouyain S, Longo PA, Li SQ, Ferguson KM, Leahy DJ. The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15024–15029. doi: 10.1073/pnas.0507591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garrett TPJ, Mckern NM, Lou MZ, Elleman TC, Adams TE, Lovrecz GO, Zhu HJ, Walker F, Frenkel MJ, Hoyne PA, Jorissen RN, Nice EC, Burgess AW, Ward CW. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell. 2002;110:763–773. doi: 10.1016/s0092-8674(02)00940-6. [DOI] [PubMed] [Google Scholar]
- 45.Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, Kim JH, Saito K, Sakamoto A, Inoue M, Shirouzu M, Yokoyama S. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110:775–787. doi: 10.1016/s0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]
- 46.Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TPJ, Leahy DJ, Lemmon MA, Sliwkowski MX, Ward CW, Yokoyama S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Molecular Cell. 2003;12:541–552. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 47.Schlessinger J. Autoinhibition control. Science. 2003;300:750–752. doi: 10.1126/science.1082024. [DOI] [PubMed] [Google Scholar]
- 48.Olsen SK, Ibrahimi OA, Raucci A, Zhang FM, Eliseenkova AV, Yayon A, Basilico C, Linhardt RJ, Schlessinger J, Mohammadi M. Insights into the molecular basis for fibroblast growth factor receptor autoinhibition and ligand-binding promiscuity. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:935–940. doi: 10.1073/pnas.0307287101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW, Leahy DJ. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 50.Tanner KG, Kyte J. Dimerization of the extracellular domain of the receptor for epidermal growth factor containing the membrane-spanning segment in response to treatment with epidermal growth factor. J Biol Chem. 1999;274:35985–35990. doi: 10.1074/jbc.274.50.35985. [DOI] [PubMed] [Google Scholar]
- 51.Moriki T, Maruyama H, Maruyama IN. Activation of preformed EGF receptor dimers by ligand-induced rotation of the transmembrane domain. J Mol Biol. 2001;311:1011–1026. doi: 10.1006/jmbi.2001.4923. [DOI] [PubMed] [Google Scholar]
- 52.Ottensmeyer FP, Beniac DR, Luo RZT, Yip CC. Mechanism of transmembrane signaling: Insulin binding and the insulin receptor. Biochemistry. 2000;39:12103–12112. doi: 10.1021/bi0015921. [DOI] [PubMed] [Google Scholar]
- 53.Mendrola JM, Berger MB, King MC, Lemmon MA. The single transmembrane domains of ErbB receptors self-associate in cell membranes. J Biol Chem. 2002;277:4704–4712. doi: 10.1074/jbc.M108681200. [DOI] [PubMed] [Google Scholar]
- 54.Li E, You M, Hristova K. SDS-PAGE and FRET suggest weak interactions between FGFR3 TM domains in the absence of extracellular domains and ligands. Biochemistry. 2005;44:352–360. doi: 10.1021/bi048480k. [DOI] [PubMed] [Google Scholar]
- 55.You M, Li E, Wimley WC, Hristova K. FRET in liposomes: measurements of TM helix dimerization in the native bilayer environment. Analytical Biochemistry. 2005;340:154–164. doi: 10.1016/j.ab.2005.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stanley AM, Fleming KG. The transmembrane domains of ErbB receptors do not dimerize strongly in micelles. J Mol Biol. 2005;347:759–772. doi: 10.1016/j.jmb.2005.01.059. [DOI] [PubMed] [Google Scholar]
- 57.Yu XC, Sharma KD, Takahashi T, Iwamoto R, Mekada E. Ligand-independent dimer formation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Mol Biol Cell. 2002;13:2547–2557. doi: 10.1091/mbc.01-08-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Linggi B, Cheng QC, Rao AR, Carpenter G. The ErbB-4 s80 intracellular domain is a constitutively active tyrosine kinase. Oncogene. 2006;25:160–163. doi: 10.1038/sj.onc.1209003. [DOI] [PubMed] [Google Scholar]
- 59.Gerber D, Sal-Man N, Shai Y. Two motifs within a transmembrane domain, one for homodimerization and the other for heterodimerization. J Biol Chem. 2004;279:21177–21182. doi: 10.1074/jbc.M400847200. [DOI] [PubMed] [Google Scholar]
- 60.Lemmon MA, Bu ZM, Ladbury JE, Zhou M, Pinchasi D, Lax I, Engelman DM, Schlessinger J. Two EGF molecules contribute additively to stabilization of the EGFR dimer. EMBO J. 1997;16:281–294. doi: 10.1093/emboj/16.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grasberger B, Minton AP, DeLisi C, Metzger H. Interaction Between Proteins Localized in Membranes. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:6258–6262. doi: 10.1073/pnas.83.17.6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bell CA, Tynan JA, Hart KC, Meyer AN, Robertson SC, Donoghue DJ. Rotational coupling of the transmembrane and kinase domains of the Neu receptor tyrosine kinase. Mol Biol Cell. 2000;11:3589–3599. doi: 10.1091/mbc.11.10.3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gardin A, Auzan C, Clauser E, Malherbe T, Aunis D, Cremel G, Hubert P. Substitution of the insulin receptor transmembrane domain with that of glycophorin A inhibits insulin action. FASEB J. 1999;13:1347–1357. doi: 10.1096/fasebj.13.11.1347. [DOI] [PubMed] [Google Scholar]
- 64.Burke CL, Lemmon MA, Coren BA, Engelman DM, Stern DF. Dimerization of the p185(neu) transmembrane domain is necessary but not sufficient for transformation. Oncogene. 1997;14:687–696. doi: 10.1038/sj.onc.1200873. [DOI] [PubMed] [Google Scholar]
- 65.Yan H, Schlessinger J, Chao MV. Chimeric Ngf-Egf Receptors Define Domains Responsible for Neuronal Differentiation. Science. 1991;252:561–563. doi: 10.1126/science.1850551. [DOI] [PubMed] [Google Scholar]
- 66.Petti LM, Irusta PM, DiMaio D. Oncogenic activation of the PDGF beta receptor by the transmembrane domain of p185(neu) Oncogene. 1998;16:843–851. doi: 10.1038/sj.onc.1201590. [DOI] [PubMed] [Google Scholar]
- 67.Webster MK, Donoghue DJ. Constitutive activation of fibroblast growth factor receptor 3 by the transmembrane domain point mutation found in achondroplasia. EMBO J. 1996;15:520–527. [PMC free article] [PubMed] [Google Scholar]
- 68.Cheatham B, Shoelson SE, Yamada K, Goncalves E, Kahn CR. Substitution of the Erbb-2 Oncoprotein Transmembrane Domain Activates the Insulin-Receptor and Modulates the Action of Insulin and Insulin-Receptor Substrate-1. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:7336–7340. doi: 10.1073/pnas.90.15.7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adar R, Monsonego-Ornan E, David P, Yayon A. Differential activation of cysteine-substitution mutants of fibroblast growth factor receptor 3 is determined by cysteine localization. Journal of Bone and Mineral Research. 2002;17:860–868. doi: 10.1359/jbmr.2002.17.5.860. [DOI] [PubMed] [Google Scholar]
- 70.Brown RJ, Adams JJ, Pelekanos RA, Wan Y, McKinstry WJ, Palethorpe K, Seeber RM, Monks TA, Eidne KA, Parker MW, Waters MJ. Model for growth hormone receptor activation based on subunit rotation within a receptor dimer. Nature Structural & Molecular Biology. 2005;12:814–821. doi: 10.1038/nsmb977. [DOI] [PubMed] [Google Scholar]
- 71.Jiang GQ, Hunter T. Receptor signaling: When dimerization is not enough. Cur Biol. 1999;9:R568–R571. doi: 10.1016/s0960-9822(99)80357-1. [DOI] [PubMed] [Google Scholar]
- 72.Fleishman SJ, Schlessinger J, Ben-Tal N. A putative molecular-activation switch in the transmembrane domain of erbB2. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:15937–15940. doi: 10.1073/pnas.252640799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schechter AL, Stern DF, Vaidyanathan L, Decker SJ, Drebin JA, Greene MI, Weinberg RA. The Neu Oncogene - An Erb-B-Related Gene Encoding A 185,000-Mr Tumor-Antigen. Nature. 1984;312:513–516. doi: 10.1038/312513a0. [DOI] [PubMed] [Google Scholar]
- 74.Bargmann CI, Hung MC, Weinberg RA. Multiple Independent Activations of the Neu Oncogene by A Point Mutation Altering the Transmembrane Domain of P185. Cell. 1986;45:649–657. doi: 10.1016/0092-8674(86)90779-8. [DOI] [PubMed] [Google Scholar]
- 75.Bargmann CI, Weinberg RA. Oncogenic Activation of the Neu-Encoded Receptor Protein by Point Mutation and Deletion. EMBO J. 1988;7:2043–2052. doi: 10.1002/j.1460-2075.1988.tb03044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weiner DB, Liu J, Cohen JA, Williams WV, Greene MI. A Point Mutation in the Neu Oncogene Mimics Ligand Induction of Receptor Aggregation. Nature. 1989;339:230–231. doi: 10.1038/339230a0. [DOI] [PubMed] [Google Scholar]
- 77.Frank B, Hemminki K, Wirtenberger M, Bermejo JL, Bugert P, Klaes R, Schmutzler RK, Wappenschmidt B, Bartram CR, Burwinkel B. The rare ERBB2 variant Ile654Val is associated with an increased familial breast cancer risk. Carcinogenesis. 2005;26:643–647. doi: 10.1093/carcin/bgh342. [DOI] [PubMed] [Google Scholar]
- 78.Otsuki T, Nakazawa N, Taniwaki M, Yamada O, Sakaguchi H, Wada H, Yawata Y, Ueki A. Establishment of a new human myeloma cell line, KMS-18, having t(4;14)(p16.3;q32.3) derived from a case phenotypically transformed from Ig A-lambda to BJP-lambda, and associated with hyperammonemia. International Journal of Oncology. 1998;12:545–552. doi: 10.3892/ijo.12.3.545. [DOI] [PubMed] [Google Scholar]
- 79.Bange J, Prechtl D, Cheburkin Y, Specht K, Harbeck N, Schmitt M, Knyazeva T, Muller S, Gartner S, Sures I, Wang HY, Imyanitov E, Haring HU, Knayzev P, Iacobelli S, Hofler H, Ullrich A. Cancer progression and tumor cell motility are associated with the FGFR4 Arg(388) allele. Cancer Research. 2002;62:840–847. [PubMed] [Google Scholar]
- 80.Vajo Z, Francomano CA, Wilkin DJ. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: The achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocrine Reviews. 2000;21:23–39. doi: 10.1210/edrv.21.1.0387. [DOI] [PubMed] [Google Scholar]
- 81.Passos-Bueno MR, Wilcox WR, Jabs EW, Sertié AL, Alonso LG, Kitoh H. Clinical spectrum of fibroblast growth factor receptor mutations. Human Mutation. 1999;14:115–125. doi: 10.1002/(SICI)1098-1004(1999)14:2<115::AID-HUMU3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 82.White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, Fields J, Yu XJ, Shaw NJ, McLellan NJ, McKeown C, FitzPatrick D, Yu K, Ornitz DM, Econs MJ. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. American Journal of Human Genetics. 2005;76:361–367. doi: 10.1086/427956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Przylepa KA, Paznekas W, Zhang M, Golabi M, Bias W, Bamshad MJ, Carey JC, Hall BD, Stevenson R, Orlow S, Cohen MM, Jr, Jabs EW. Fibroblast growth factor receptor 2 mutations in Beare-Stevenson cutis gyrata syndrome. Nat Genet. 1996;13:492–494. doi: 10.1038/ng0896-492. [DOI] [PubMed] [Google Scholar]
- 84.Andrea R, Vargas P, Maegawa GHB, Taucher SC, Leite JCL, Sanz P, Cifuentes J, Parra M, Munoz H, Maranduba CM, Passos-Bueno MR. Beare-Stevenson syndrome: Two South American patients with FGFR2 analysis. American Journal of Medical Genetics Part A. 2003;121A:41–46. doi: 10.1002/ajmg.a.20101. [DOI] [PubMed] [Google Scholar]
- 85.Segatto O, King CR, Pierce JH, di Fiore PP, Aaronson SA. Different structural alterations upregulate in vitro tyrosine kinase activity and transforming potency of the erbB-2 gene. Mol Cell Biol. 1988;8:5570–5574. doi: 10.1128/mcb.8.12.5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li E, You M, Hristova K. FGFR3 dimer stabilization due to a single amino acid pathogenic mutation. J Mol Biol. 2006;356:600–612. doi: 10.1016/j.jmb.2005.11.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kashles O, Szapary D, Bellot F, Ullrich A, Schlessinger J, Schmidt A. Ligand-Induced Stimulation of Epidermal Growth-Factor Receptor Mutants with Altered Transmembrane Regions. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:9567–9571. doi: 10.1073/pnas.85.24.9567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Carpenter CD, Ingraham HA, Cochet C, Walton GM, Lazar CS, Sowadski JM, Rosenfeld MG, Gill GN. Structural-Analysis of the Transmembrane Domain of the Epidermal Growth-Factor Receptor. J Biol Chem. 1991;266:5750–5755. [PubMed] [Google Scholar]
- 89.MacKenzie KR, Prestegard JH, Engelman DM. A transmembrane helix dimer: Structure and implications. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 90.Smith SO, Song D, Shekar S, Groesbeek M, Ziliox M, Aimoto S. Structure of the transmembrane dimer interface of glycophorin A in membrane bilayers. Biochemistry. 2001;40:6553–6558. doi: 10.1021/bi010357v. [DOI] [PubMed] [Google Scholar]
- 91.Smith SO, Smith C, Shekar S, Peersen O, Ziliox M, Aimoto S. Transmembrane interactions in the activation of the Neu receptor tyrosine kinase. Biochemistry. 2002;41:9321–9332. doi: 10.1021/bi012117l. [DOI] [PubMed] [Google Scholar]
- 92.Lemmon MA, Flanagan JM, Hunt JF, Adair BD, Bormann BJ, Dempsey CE, Engelman DM. Glycophorin-A dimerization is driven by specific interactions between transmembrane α-helices. J Biol Chem. 1992;267:7683–7689. [PubMed] [Google Scholar]
- 93.Lemmon MA, Treutlein HR, Adams PD, Brunger AT, Engelman DM. A Dimerization Motif for Transmembrane Alpha-Helices. Nature Struct Biol. 1994;1:157–163. doi: 10.1038/nsb0394-157. [DOI] [PubMed] [Google Scholar]
- 94.Sulistijo ES, Jaszewski TM, MacKenzie KR. Sequence-specific dimerization of the transmembrane domain of the “BH3-only” protein BNIP3 in membranes and detergent. J Biol Chem. 2003;278:51950–51956. doi: 10.1074/jbc.M308429200. [DOI] [PubMed] [Google Scholar]
- 95.Smith SO, Smith CS, Bormann BJ. Strong hydrogen bonding interactions involving a buried glutamic acid in the transmembrane sequence of the neu/erbB-2 receptor. Nature Struct Biol. 1996;3:252–258. doi: 10.1038/nsb0396-252. [DOI] [PubMed] [Google Scholar]
- 96.Adams PD, Engelman DM, Brünger AT. Improved prediction for the structure of the dimeric transmembrane domain of glycophorin A obtained through global searching. Proteins. 1996;26:257–261. doi: 10.1002/(SICI)1097-0134(199611)26:3<257::AID-PROT2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 97.Adams PD, Arkin IT, Engelman DM, Brünger AT. Computational searching and mutagenesis suggest a structure for the pentameric transmembrane domain of phospholamban. Nature Struct Biol. 1995;2:154–162. doi: 10.1038/nsb0295-154. [DOI] [PubMed] [Google Scholar]
- 98.Adams PD, Brunger AT. In: Membrane protein assembly. von Heijne G, editor. R.G. Landes Co; Austin, TX: 1997. p. 97. [Google Scholar]
- 99.Adar R, Monsonego-Ornan E, David P, Yayon A. Differential activation of cysteine-substitution mutants of fibroblast growth factor receptor 3 is determined by cysteine localization. J Bone Miner Res. 2002;17:860–868. doi: 10.1359/jbmr.2002.17.5.860. [DOI] [PubMed] [Google Scholar]
- 100.You M, Li E, Hristova K. The achondroplasia mutation does not alter the dimerization energetics of FGFR3 transmembrane domain. Biochemistry. 2006 doi: 10.1021/bi060113g. in press. [DOI] [PubMed] [Google Scholar]
- 101.Sal-Man N, Shai Y. Arginine mutations within a transmembrane domain of Tar, an Escherichia coli aspartate receptor, can drive homodimer dissociation and heterodimer association in vivo. Biochemical Journal. 2005;385:29–36. doi: 10.1042/BJ20041022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Therien AG, Deber CM. Oligomerization of a peptide derived from the transmembrane region of the sodium pump gamma subunit: Effect of the pathological mutation G41R. J Mol Biol. 2002;322:583–590. doi: 10.1016/s0022-2836(02)00781-7. [DOI] [PubMed] [Google Scholar]
- 103.Gallivan JP, Dougherty DA. Cation-π interactions in structural biology. Proc Natl Acad Sci USA. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li E, Hristova K. Imaging FRET Measurements of Transmembrane Helix Interactions in Lipid Bilayers on a Solid Support. Langmuir. 2004;20:9053–9060. doi: 10.1021/la048676l. [DOI] [PubMed] [Google Scholar]
- 105.Merzlyakov M, You M, Li E, Hristova K. Transmembrane helix heterodimerization in lipids bilayers: probing the energetics behind autosomal dominant growth disorders. J Mol Biol. 2006;358:1–7. doi: 10.1016/j.jmb.2006.01.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li Y, Mangasarian K, Mansukhani A, Basilico C. Activation of FGF receptors by mutations in the transmembrane domain. Oncogene. 1997;14:1397–1406. doi: 10.1038/sj.onc.1200983. [DOI] [PubMed] [Google Scholar]
- 107.Raffioni S, Zhu YZ, Bradshaw RA, Thompson LM. Effect of transmembrane and kinase domain mutations on fibroblast growth factor receptor 3 chimera signaling in PC12 cells. A model for the control of receptor tyrosine kinase activation. J Biol Chem. 1998;273:35250–35259. doi: 10.1074/jbc.273.52.35250. [DOI] [PubMed] [Google Scholar]
- 108.Monsonego-Ornan E, Adar R, Feferman T, Segev O, Yayon A. The transmembrane mutation G380R in fibroblast growth factor receptor 3 uncouples ligand-mediated receptor activation from down-regulation. Mol Cell Biol. 2000;20:516–522. doi: 10.1128/mcb.20.2.516-522.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Thompson LM, Raffioni S, Wasmuth JJ, Bradshaw RA. Chimeras of the native form or achondroplasia mutant (G375C) of human fibroblast growth factor receptor 3 induce ligand-dependent differentiation of PC12 cells. Mol Cell Biol. 1997;17:4169–4177. doi: 10.1128/mcb.17.7.4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cho JY, Guo CS, Torello M, Lunstrum GP, Iwata T, Deng CX, Horton WA. Defective lysosomal targeting of activated fibroblast growth factor receptor 3 in achondroplasia. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:609–614. doi: 10.1073/pnas.2237184100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yamada K, Carpentier JL, Cheatham B, Goncalves E, Shoelson SE, Kahn CR. Role of the Transmembrane Domain and Flanking Amino-Acids in Internalization and Down-Regulation of the Insulin-Receptor. J Biol Chem. 1995;270:3115–3122. doi: 10.1074/jbc.270.7.3115. [DOI] [PubMed] [Google Scholar]
- 112.Van Esch H, Fryns JP. Acanthosis nigricans in a boy with achondroplasia due to the classical Gly380Arg mutation in FGFR3. Genetic Counseling. 2004;15:375–377. [PubMed] [Google Scholar]
- 113.Heuertz S, Le Merrer M, Cormier-Daire V, Legeai-Mallet L, Flori E, Munnich A, Bonaventure J. A novel missense mutation in the FGFR3 gene causing hypochondroplasia. American Journal of Human Genetics. 2003;73:262. [Google Scholar]