

Abstract
Background
Seedless grapes are greatly appreciated for fresh and dry fruit consumption. Parthenocarpy and stenospermocarpy have been described as the main phenomena responsible for seedlessness in Vitis vinifera. However, the key genes underpinning molecular and cellular processes that play a significant role in seed development are not well characterized. To identify important regulators and mechanisms that may be altered in the seedless phenotype, we performed a comprehensive transcriptional analysis to compare the transcriptomes of a popular seeded wine cultivar (wild-type) and its seedless somatic variant (mutant) at three key developmental stages.
Results
The transcriptomes revealed by Illumina mRNA-Seq technology had approximately 98% of grapevine annotated transcripts and about 80% of them were commonly expressed in the two lines. Differential gene expression analysis revealed a total of 1075 differentially expressed genes (DE) in the pairwise comparison of developmental stages, which included DE genes specific to the wild-type background, DE genes specific to the mutant background and DE genes commonly shared in both backgrounds. The analysis of differential expression patterns and functional category enrichment of wild-type and mutant DE genes highlighted significant coordination and enrichment of pollen and ovule developmental pathways. The expression of some selected DE genes was further confirmed by real-time RT-PCR analysis.
Conclusions
This study represents the most comprehensive attempt to characterize the genetic bases of seed formation in grapevine. With a high throughput method, we have shown that a seeded wine grape and its seedless somatic variant are similar in several biological processes. Nevertheless, we could identify an inventory of genes with altered expression in the mutant compared to the wild-type, which may be responsible for the seedless phenotype. The genes located within known genomic regions regulating seed content may be used for the development of molecular tools to assist table grape breeding. Therefore the data reported here have provided a rich genomic resource for practical use and functional characterization of the genes that potentially underpin seedlessness in grapevine.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-1030) contains supplementary material, which is available to authorized users.
Keywords: Vitis vinifera, Seed content, mRNA-Seq, Differential gene expression, Functional category enrichment, Candidate gene
Background
Over the past decade there has been a sustained increase in the world production of table grapes, which reached 22.3 million tons [1]. This is largely due to consumer demand for seedless grape for fresh and dry fruit consumption. Nowadays, most breeding programs focus on the generation of new cultivars (cvs) combining seedlessness together with other traits such as large berry size, muscat flavor or crispiness.
Vitis vinifera L. is considered a good model for the study of seed development in fruit crops. Two different mechanisms are involved in grape seedlessness, namely parthenocarpy and stenospermocarpy. Usually, in parthenocarpic conditions fruit develops from the ovary in the absence of fertilization yielding small berries that completely lack seeds (e.g. cv Black Corinth), whereas in stenospermocarpy pollination and fertilization take place normally, but seed development aborts at an early stage (2–4 weeks) after fertilization and berry size at harvest is reduced (e.g. cv Sultanina) [2, 3]. Most cultivated seedless grapes exhibit stenospermocarpy. The major events that take place in grapevine normal seed development, parthenocarpy and stenospermocarpy are shown schematically in [4] and are described in detail by [5, 6].
In Arabidopsis, genetic studies have revealed several genes that participate in seed development like SHOOT MERISTEMLESS (STM), CUP-SHAPED COTYLEDON (CUC1 and CUC2), AINTEGUMENTA (ANT), SPATULA (SPT), AGAMOUS (AG) MADS box genes AG-SHATTERPROOF (SHP1 and SHP2), SEEDSTICK (STK, also known as AGL11), NOZZLE/SPOROCYTELESS (NZZ/SPL), EMBRYO-DEFECTIVE (EMB) and INO[7–9], including those that regulate endosperm formation such as CRINKLY4 and BET1[10, 11], embryo differentiation such as EMBRYO-DEFECTIVE (EMB) and LEAFY COTYLEDON (LEC) [12–14], and seed coat development such as APETALA 2 (AP2) and TRANSPARENT TESTA 16 (TT16) [15]. Also, molecular studies with Arabidopsis, tomatoes, and other plants have revealed cis-regulatory elements of several genes active during seed development, mostly the transcription factors (TFs) that play a role in their regulation, i.e. LEAFY COTYLEDON (LEC) genes and AGAMOUS-like 15 (AGL15) [16–18]. Nevertheless, in grapevine the identities of most regulators of seed development and their direct targets are largely unknown. To date, a number of studies have adopted QTL (Quantitative Trait Locus) analysis to dissect the genetic determinism of seedlessness [19–23]. A MADS-box ovule identity gene (VvAGL11) was proposed as the major positional and functional candidate gene for stenospermocarpy and tested for usefulness in marker-assisted selection [24, 25]. However, very few studies have looked for genes possibly responsible for seedlessness by comparison of gene expression profiles in seeded and seedless grapes. For instance, differential expression analysis in seeded and seedless clones of cv Sultanina by [26, 27] allowed the identification of a chloroplast chaperonin (ch-Cpn21) resulting in seed abortion when silenced in tobacco and tomato, and of a ubiquitin extension protein (S27a) having a probable general role in the control of organ development in grapevine. Recently, differential expression analysis during ovule development in seeded and seedless cultivars identified grape metacaspase genes, consistent with a role of programmed cell death in stenospermocarpy [28].
To identify regulators and processes required for seed development that may be altered in the seedless phenotype, somatic variants are invaluable resources. At the same time an analytical approach that provides a holistic view of the transcriptional landscape during seed development in both phenotypes is equally vital. In grapevine, somatic variation arises from mutation or epimutation events that first occur in a single cell belonging to a specific cell layer. Once at least one shoot apical meristem is colonized by the mutated cell in one or both cell layers, the mutation can be transmitted by bud propagation or eventually sexual reproduction [29]. However, identification of somatic variants in grapevine is a time and labor intensive task, which requires genetic and phenotypic characterization of large germplasm collections [30]. At the same time, the application of deep sequencing techniques to survey the total population of RNA within a tissue has made RNA-Seq a popular and comprehensive approach to deduce and quantify the transcriptome [31]. Its potential has been demonstrated in the de novo transcriptome characterization of Vitis vinifera cultivars [32] and gene expression profile of grape berry during key developmental stages [33–35].
In this paper, we exploited the availability of a spontaneous seedless somatic variant (hereafter mutant, MT) derived from Sangiovese (hereafter wild-type, WT), a widespread seeded wine cultivar in Italy [30]. This mutant has a gross morphology of vines identical to the wild-type except for absence of seeds, reduced berry and bunch size at harvest. With the aim of understanding the molecular mechanisms driving the seedless phenotype, we analyzed the transcriptional responses possibly related to seed development in the wild-type and the mutant using Illumina mRNA-Seq technology.
Methods
Sample collection
Samples were collected from wild-type and mutant plants in the germplasm collection of Grinzane Cavour maintained by CNR-Istituto di Virologia Vegetale di Grugliasco (Torino, Italy).
For molecular marker analysis young leaves were gathered.
To create inventories of gene expression at successive stages of seed formation, three key time points along grape berry development were selected corresponding to stages E-L 15 (single flowers in compact groups), E-L 27 (young berries enlarging) and E-L 38 (berries harvest-ripe) of the modified E-L system described by [36]. Samples were collected for both clones in the following dates: 12th May, 10th June and 16th September 2010. When matched to the number of days from bloom (DFB) shown in [4], these time points could be assigned to two main categories: “before” (E-L 15) and “after” (E-L 27 and 38) fertilization. A detailed description of how sampling dates were matched to DFB is reported in Additional file 1. For each developmental stage two independent samples (biological replicates) were collected. A biological replicate was composed of the whole inflorescence for stage E-L 15 and of the whole bunch for stages E-L 27 and 38.
Genomic DNA extraction and SSR genotyping of the wild-type and the mutant
Total genomic DNA was extracted from young immature leaves as described by [37]. Fifty-eight SSR (simple sequence repeat) markers, spread across the nineteen chromosomes of grapevine genome, were used to genotype the wild-type and the mutant (Additional file 2). Of this set, twenty SSR markers were previously described by [37], thirty-two SSR markers used by [23] and six SSR markers developed by [24].
PCR amplifications for multiplex panels were carried out in a final volume of 12.5 μl containing 10 ng of genomic DNA, 0.25 mM of each dNTP, 2 mM MgCl2, 1.5 U Taq DNA Polymerase (AmpliTaq Gold™, Applied Biosystems, Foster City, CA). The amplification protocol was as follows: 7 min at 95°C; 30 cycles of 45 sec at 95°C, 1 min at 54°C, 30 sec at 72°C; and 1 hour at 72°C. Primers failing to amplify at 54°C were further tested in single panel at different annealing temperatures.
PCR products (0.5 μl) were mixed with 9.3 μl of formamide and 0.2 μl of the GeneScan™ 500 ROX® Size Standard (Applied Biosystems) and 0.5 μl of this mix was subjected to capillary electrophoresis on an ABI PRISM 3130 Genetic Analyzer (Applied Biosystems) to separate DNA fragments. GeneMapper v3.5 (Applied Biosystems) was employed for the allele size estimation.
RNA extraction
For each sample total RNA extraction was performed from a lot of flowers/berries in triplicate (technical replicates), using the Spectrum™ Plant Total RNA kit (Sigma-Aldrich, St. Louis, MO) following the manufacturer’s protocol. RNA quality and quantity were determined using a Nanodrop 8000 (Thermo Scientific, Wilmington, DE) and a Bioanalyzer 2100 (Agilent, Santa Clara, CA).
Library preparation and sequencing
For transcriptomic analysis a single biological replicate was used due to economic constraints. Total RNA from the three technical replicates of each sample were pooled for a total six pools representing each developmental stage for the two genotypes.
Libraries were prepared using the TruSeq SBS v5 protocol (Illumina, San Diego, CA). In particular, 10 μg of total RNA were used to isolate poly(A) mRNA after double purification of transcripts using poly(T) oligos attached with magnetic beads. Subsequent mRNA quality control was carried out on a Bioanalyzer 2100 (Agilent). Purified mRNA was fragmented using Zn-catalyzed hydrolysis and converted into double-stranded cDNA by random priming. Following end repair, single “A” base addition to 3′-end, indexed adapters were ligated and cDNA fragments of 200 ± 25 bp were purified. Purified cDNA was amplified by PCR and quality control was done by TOPO cloning and capillary sequencing. The cDNA libraries were quantified and diluted to 10 nM, after which they were multiplexed and sequenced with an Illumina HiSeq 2000 sequencer at Fasteris (Fasteris SA, Switzerland). A hundred-bp paired-end sequences were generated. Image analysis, error estimation and base calling were carried out using Illumina Pipeline (version 1.4.5) to generate the sequence data. Indexed primers were used to identify the different reads from different samples in the sequence data. Some low-quality reads were removed using a custom algorithm. Illumina TruSeq adapter sequences were clipped and the remaining reads were considered suitable for further analysis after passing quality control at Fasteris.
cDNA sequence alignment and mapping to the reference genome
Short-read alignment and mapping of all the reads were carried on the 12x v1 annotation of the grapevine genome PN40024 [38] using BWA (Burrows Wheeler Aligner) software [39] with a maximum set of 2 mismatches in the first 32 bp sequences and a maximum of “n” mismatches in total (n from 2 to 9 depending on read length). The mapping results were processed with SAMtools [40] to extract for each transcript the number of mapped reads and determine, whether their mapping position is unique. Reads mapping to several positions on the reference sequence with the same “mapping quality” (i.e. number of mismatches and quality of the bases generating the mismatches) were attributed at random to one of them with a “0” mapping quality.
A Python script was developed to determine the distribution of mapped reads among genomic features for the wild-type and the mutant.
Gene expression analysis
Reads mapped to multiple locations and unmapped reads were excluded from gene expression analysis. Unique reads mapping to v1_mRNA annotated transcripts were summed for each gene model and normalized by million reads (RPM) because of read coverage bias towards 3′ end of transcripts. A lower limit of detection for expression estimate was designated to be an RPM of 0.5 or, if the RPM value was less than 0.5, at least five uniquely mapped reads with identity >98% over 100 bp, as previously described by [35]. The full raw expression dataset is available at GEO under the accession number GSE58061 [http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=ilkdqyqehtcrraz&acc=GSE58061].
We ranked the expression of all identified transcripts by order of magnitude. Briefly, p-values were computed to reflect the significance of the difference between two counts (n1 and n2 corresponding to any two library combination out of the six libraries) using a binominal model. The p-values were log-transformed in order to allow for greater numerical stability in comparing extreme values. Next all the p-values and the ratios of expression between the counts were considered to compute a ranking value for each transcript (Additional file 3).
Raw uniquely mapped read counts for the wild-type and the mutant were independently subjected to differential expression (DE) analysis in a pairwise comparison between developmental stages (E-L 15 vs E-L 27, E-L 27 vs E-L 38 and E-L 15 vs E-L 38) using the software DESeq [41] in R (parameters: false discovery rate (FDR) ≤5%, log2-fold change (FC) >1). Next, DE genes were compared between the wild-type and the mutant. This strategy was preferred to the direct comparison of the two clones at each developmental stage in order to minimize the eventual differences due to asynchronous sampling.
An in-house R script was written to group DE genes with similar expression pattern based on the adjusted p-values. By indicating a significant up-regulation with “1”, a significant down-regulation with “-1” and a non-significant difference with “0”, the three comparisons between the developmental stages can be summarized with a triplet, e.g. “1, 0, 1”. This example indicates that there is a significant up-regulation going from the first to the second time point, no significant difference between the second and third time points, and a significant positive difference when comparing the first and last time points. Altogether, 27 different categories can be defined in this way, and 18 of these contain relevant patterns (for example the pattern “1, 1, −1” is impossible). These 18 groups are visualized in Figure 1. Each gene showing at least one significant difference between developmental stages was classified into one of these categories, for both the wild type and the mutant. The number of differentially expressed genes that fell to each pattern were compared between the wild-type and the mutant.
Figure 1.
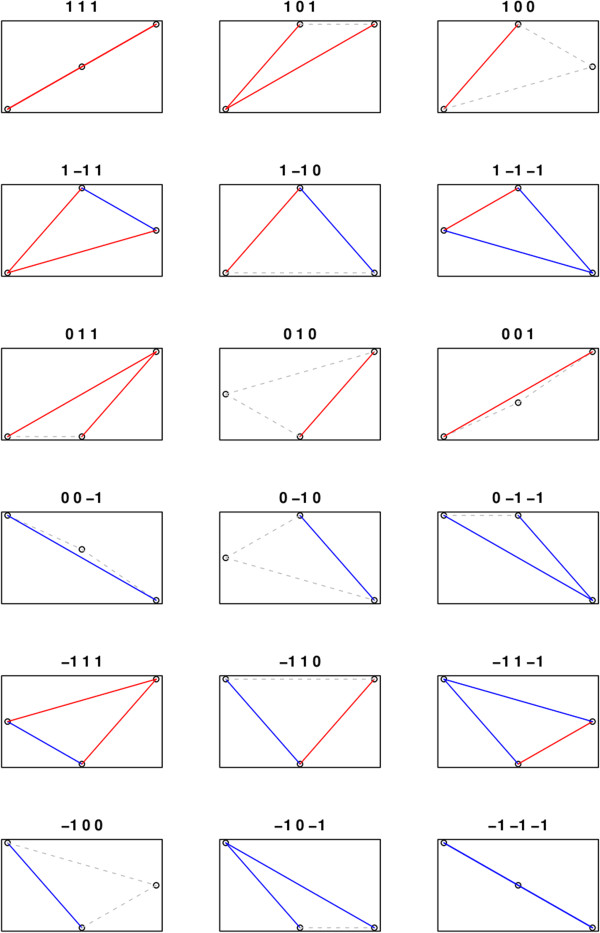
The eighteen relevant categories of triplets of significance. A red line indicates a significant up-regulation, a blue line a significant down-regulation, and a gray dashed line a non-significant difference between time points (the first position in the triplet corresponds to the shift from stage E-L 15 to E-L 27, the second position to the shift from stage E-L 27 to E-L 38 and the third position to the shift from stage E-L 15 to E-L 38). All DE genes for both the wild-type and the mutant formed 18 groups of differential expression patterns and the numbers of genes that fell within each group are shown in Table 3. Group 1 contains positively regulated genes along the whole time course; groups 2 and 3 consist of induced genes from stage E-L 15 to stage E-L 27, with no significant change afterwards; groups 4, 5 and 6 contain induced genes from stage E-L 15 to stage E-L 27, followed by a significant decrease in their expression; groups 7, 8 and 9 consist of stable genes from stage E-L 15 to stage E-L 27, with induced expression from stage E-L 15 or E-L 27 to stage E-L 38. Groups 10, 11 and 12 are made up of stable genes from stage E-L 15 to stage E-L 27 with reduced expression from stage E-L 15 or E-L 27 to stage E-L 38; groups 13, 14 and 15 contain repressed genes from stage E-L 15 to stage E-L 27, followed by a significant induction in their expression; groups 16 and 17 contain repressed genes from stage E-L 15 to stage E-L 27, with no significant subsequent change; group 18 contains negatively regulated genes along the whole time course.
Functional annotation and enrichment analysis
Wild-type and mutant genes were annotated against the v1 version of the 12x draft annotation of the grapevine genome using the CRIBI tools [42] combined with the grapevine molecular network VitisNet [43]. Next all DE genes for both genotypes were input into the AgriGO analysis tool [44]. This allowed us to identify significantly enriched gene ontology (GO) terms in the whole set of DE genes or within each group when compared with GO terms in the complete Vitis vinifera genome. Using a hypergeometric test, a GO term was considered significantly enriched, if the FDR was <0.05 and p-value <0.01 when compared to all gene transcripts annotated in the reference genome (supported in AgriGO). Further, the REVIGO web server [45] was used to summarize the processes represented in the lists of significantly enriched GO terms by removing redundant terms as described by [35].
Selection of candidate genes
Candidate genes were chosen belonging to the three following groups:
Wild-type and mutant specific not DE genes, i.e. the transcripts which are expressed in the wild-type but not in the mutant and vice versa, with no significant differences between developmental stages. These genes were tested for GO annotation enrichment using AgriGO. Ultimately, genes were selected, if they fulfilled the following criteria: significant GO enrichment, RPM values above the lower limit of detection (0.5) and putative function relevant to seed development;
Wild-type and mutant specific DE genes, chosen based on their expression profile, fold change value, functional category enrichment, and putative function relevant to seed development. In addition, candidates were selected among DE genes with different expression profile or level of fold change in the two clones;
Candidate genes affecting seed content, previously identified in QTL analyses [23, 46]. These genes were compared with DE genes in the wild-type and the mutant, and the overlapping candidates were evaluated, based on their expression profile and the level of fold change.
Real-Time PCR validation of RNA-Seq data
Quantitative real-time PCR was carried out on cDNA obtained from both biological replicates described above, one of which was used for RNA-Seq. First-strand cDNA synthesis was performed with 1 μg of total RNA in triplicate using SuperScript™ III Reverse Transcriptase (Invitrogen, Carlsbad, CA) and oligo-dT according to manufacturer’s protocol, after treatment with DNase I (Invitrogen). The transcriptional profiles of 14 genes were analyzed. SAND and GAPDH (glyceraldehyde 3-phosphate dehydrogenase) were chosen as constitutive genes for normalization after evaluation of a set of five genes with the geNorm software [47]. Their stable expression along development in the wild-type and the mutant was confirmed by RNA-Seq data. Details on gene IDs, gene annotations and primer sets are included in Additional file 4. Reactions were carried out with Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen) and specific primers using the LightCycler 480 (Roche Applied Science, Mannheim, Germany). The PCR conditions were: 95°C for 5 min as initial step, followed by 50 cycles of 95°C for 15 s, 68°C for 30 s and 72°C for 10 s. Finally, a post-PCR melting curve analysis was performed to verify the specificity of cDNA amplification. Each sample was examined in three technical replicates, and analyzed using the LightCycler 480 SV1.5.0 software (Roche Applied Science). REST 2009 software was used to calculate relative expression of each gene [48].
Results and discussion
SSR genotyping of the wild-type and the mutant
The two clones were found to have identical allele size at all the fifty-eight analyzed microsatellite loci (Additional file 2). This result further validates the data reported by Schneider et al. [30], confirming that the mutant, usually considered to be a different cultivar, is a synonym of the wild-type.
cDNA sequence alignment and mapping to the reference genome
Sequencing generated from 126 to 143 million and from 102 to 127 million 100-bp reads for the wild-type and the mutant, respectively (Additional file 5). After pre-processing and quality control, the majority of reads from wild-type (≈79-81%) and mutant (≈70-81%) were successfully aligned to v1_mRNA version of the 12x draft annotation of the grapevine genome [38]. A large fraction of mapped reads from each developmental stage for wild-type (≈87-89%) and mutant (≈85-87%) aligned to a single position. These uniquely mapped reads account on average for approximately 71% and 66% of the total number of sequenced reads for the wild-type and the mutant, respectively.
Distribution of mapped reads among genomic features showed that a high proportion (49% for both the wild-type and the mutant) mapped to protein coding regions indicating a high level of coverage of actual transcribed sequences (Additional file 6). The other reads mapped to splice junctions (27% and 26%), introns (14% and 16%) and untranslated regions (UTRs) (9% and 7%) for the wild-type and the mutant, respectively. The presence of intronic regions in RNA-Seq experiments is prevalent and has been attributed to various sources such as intron retention during splicing, DNA contamination during RNA-Seq preparation as well as alignment artifacts. Reads mapped to intronic regions in our data set are comparable to those obtained in similar experiments in grapevine [33]. Most of the intronic mapped reads in our data set show strand specificity hence we infer they are mainly due to unspliced mRNA in our samples and others may be due to alignment artifacts.
Gene expression analysis
The digital, count-based nature of RNA-Seq provided a number of potential advantages for downstream data analysis and interpretation. For every gene detected in wild-type and mutant samples, uniquely mapped reads were used to generate raw expression counts and normalized expression values. The normalized expression values were calculated as RPM since it provides a useful way to assess overall expression levels between samples. Following the normalization of read counts, we analyzed the most abundant transcripts within our samples by ranking them based on their p-value and ratio of expression. This in turn highlighted the top most highly expressed genes across all possible pairwise comparisons of the libraries (see Methods and Additional file 7).
Overall our data-set identified approximately 98% of grapevine annotated transcripts (representing 27,495 genes) expressed throughout the three developmental stages under study. We detected a gene expression gradient from “before flowering” to “after flowering”, i.e. for wild-type E-L 15 (25,785 expressed genes) > E-L 27 (25,706 expressed genes) > E-L 38 (24,822 expressed genes) and for mutant E-L 15 (25,848 expressed genes) > E-L 27 (25,197 expressed genes) > E-L 38 (24,089 expressed genes) (Additional file 8). To put these results into perspective, slightly more genes were expressed before fertilization in the mutant than in the wild-type and by far more genes were expressed after fertilization in the wild-type than in the mutant. In the wild-type and the mutant 23,640 and 23,072 genes were expressed in all three developmental stages, respectively (Figure 2). While it is not surprising the comparable number of genes shared by the three developmental stages in each clone, it is interesting to note that fewer genes were expressed specifically at each developmental stage: 586, 430 and 421 genes at stages E-L 15, E-L 27 and E-L 38 in the wild-type (Figure 2A) and 802, 337 and 351 genes at respective stages in the mutant (Figure 2B), which further highlights a reduction in gene expression in the mutant compared to the wild-type after fertilization. Thus we assessed what proportion of the expressed genes were common to both clones in the different stages and found that large number of expressed genes were shared among the wild-type and the mutant throughout development. In particular, 22,516 genes were commonly expressed in both clones in all three developmental stages (Table 1), 24,084 in the first two stages E-L 15 and E-L 27 (Additional file 9A) and 22,790 in the last two stages E-L 27 and E-L 38 (Additional file 9B). This was expected based on the phenotypic evaluation of the two clones that revealed similar berry development and ripening (they were at the same developmental stage in the same date). Nevertheless, a fewer number of genes were exclusively expressed in a particular developmental stage and clone (Table 1), suggesting they could be responsible for the specificity of each clone. Finally, a total of 565 genes were not expressed at all (Table 1). This set of genes could be genotype-specific and restricted to the grapevine clone PN40024 used for reference mapping.
Figure 2.
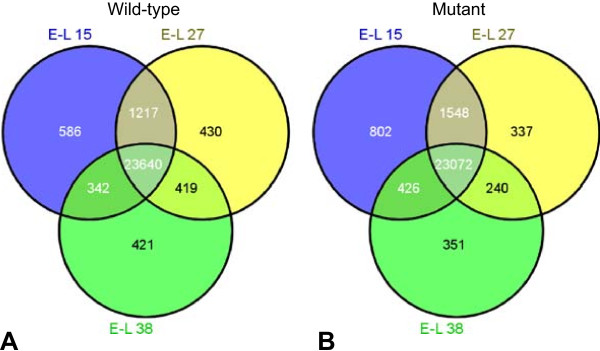
Gene expression overlap between the three key developmental stages in wild-type and mutant plants. Venn diagrams indicate the overlap of global gene expression signatures identified at stages E-L 15 (single flowers in compact groups), E-L 27 (young berries enlarging) and E-L 38 (berries harvest-ripe) of the wild-type (A) and mutant (B) genotypes.
Table 1.
Comparison of gene expression between the wild-type and the mutant
| Wild-type | Mutant | |||||
|---|---|---|---|---|---|---|
| Developmental stage | E-L 15 | E-L 27 | E-L 38 | E-L 15 | E-L 27 | E-L 38 |
| Genes expressed in all developmental stages in the two clones (common genes) | 22516 | 22516 | 22516 | 22516 | 22516 | 22516 |
| Exclusively uniquely expressed genes for each developmental stage | 183 | 187 | 169 | 190 | 70 | 97 |
| Non-detected expression for each developmental stage | 1145 | 1224 | 2108 | 1082 | 1733 | 2841 |
| Constitutively non-expressed genes in the two clones | 565 | 565 | 565 | 565 | 565 | 565 |
The results of differential gene expression analysis of RNA-Seq data in the pairwise comparison between developmental stages are shown in Figure 3. In total 1075 genes were differentially expressed (DE) in both clones. With respect to the wild-type a total of 942 genes were found to be differentially expressed during development: 522 between stages E-L 15 and E-L 27, 354 between stages E-L 27 and E-L 38 and 393 between stages E-L 15 and E-L 38 (Figure 3A). For the mutant a total of 634 DE genes were identified: 458 between stages E-L 15 and E-L 27, 191 between stages E-L 27 and E-L 38 and 41 between stages E-L 15 and E-L 38 (Figure 3B). Analysis of data set overlap (Additional file 10) revealed that about 47% of the total DE genes (501/1075) were expressed in both the wild-type and the mutant (commonly shared expression), which supports the developmental alignment of the two clones. More strikingly, the percentage of DE genes specific to the wild-type with respect to all three developmental stages is 41% (441/1075), while for the mutant it is 12% (133/1075). We further evaluated the percentage of significantly up-regulated and down-regulated genes in each pairwise comparison in both the wild-type and the mutant. On average approximately 67% of DE genes in the wild-type and 75% of DE genes in the mutant were down-regulated along development, while 33% and 25% of DE genes were induced in the wild-type and the mutant, respectively (Table 2). Taken together these results suggest that most of the expressed genes were active in different contexts along the grape berry developmental gradient (Table 1). However, significant quantitative changes occurred in individual gene expression level that corresponds to a particular stage or switch in development during seed formation. Here the mutant exhibited the strongest reduction in gene expression after fertilization (Additional file 8). It is tempting to speculate that it might be due to shut down in transcriptional processes resulting from incomplete fertilization or failure of embryo development. However, further work will be necessary to test this hypothesis.
Figure 3.
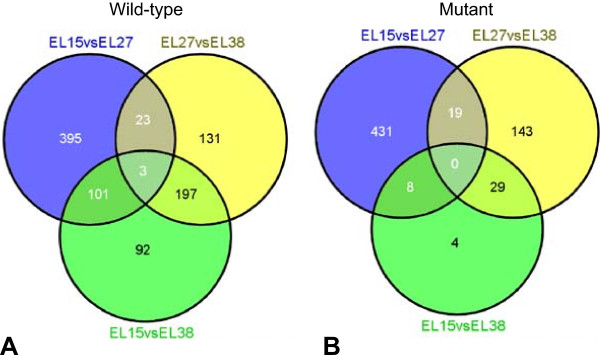
Comparison of differential gene expression in the pairwise comparison of developmental stages in wild-type and mutant plants. Venn diagrams indicate overlap of all differentially expressed genes obtained from each pairwise comparison between developmental stages (E-L 15 vs E-L 27, E-L 27 vs E-L 38 and E-L 15 vs E-L 38) in wild-type (A) and mutant (B).
Table 2.
Evaluation of significantly up- and down-regulated genes in each pairwise comparison between developmental stages
| Wild-type | Mutant | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pairwise comparison | E-L 27 vs E-L 15 | Percentage | E-L 38 vs E-L 27 | Percentage | E-L 38 vs E-L 15 | Percentage | E-L 27 vs E-L 15 | Percentage | E-L 38 vs E-L 27 | Percentage | E-L 38 vs E-L 15 | Percentage |
| Down-regulated genes | 332 | 63.6 | 256 | 72.3 | 256 | 65.1 | 327 | 71.4 | 136 | 71.2 | 34 | 82.9 |
| Up-regulated genes | 190 | 36.4 | 98 | 27.7 | 137 | 34.9 | 131 | 28.6 | 55 | 28.8 | 7 | 17.1 |
| Total | 522 | 354 | 393 | 458 | 191 | 41 | ||||||
Finally, we determined the expression pattern of all DE genes over the three developmental stages under investigation using the technique described in the Methods. This approach revealed transcripts from a pool of DE genes that exhibit the same patterns of expression over the three developmental stages. We present here 18 relevant groups (Figure 1). The wild-type and the mutant exhibited similar differential expression pattern except in groups 6, 10 and 18. Four main groups (3, 11, 12 and 16), accounted for about 67% of the DE genes along the three developmental stages of the wild-type. Similarly, groups 3, 11 and 16 accounted for 87% of DE genes in the mutant (Table 3). Additionally the analysis of expression pattern of all DE genes enabled us to identify relevant groups showing significant difference in the number of DE genes between the two clones, such as groups 2, 9, 10, 12 and 17 (Table 3).
Table 3.
Number of genes in each group of differential expression patterns for the wild-type and the mutant
| Number of groups | Gene pattern | Number of genes (Wild-type) | Number of genes (Mutant) |
|---|---|---|---|
| 1 | 111 | 0 | 0 |
| 2 | 101 | 13 | 4 |
| 3 | 100 | 155 | 112 |
| 4 | 1-11 | 0 | 0 |
| 5 | 1-10 | 21 | 15 |
| 6 | 1-1-1 | 1 | 0 |
| 7 | 011 | 66 | 26 |
| 8 | 010 | 30 | 25 |
| 9 | 001 | 58 | 4 |
| 10 | 00-1 | 34 | 0 |
| 11 | 0-10 | 101 | 118 |
| 12 | 0-1-1 | 131 | 3 |
| 13 | −111 | 0 | 0 |
| 14 | −110 | 2 | 4 |
| 15 | −11-1 | 0 | 0 |
| 16 | −100 | 240 | 319 |
| 17 | −101 | 88 | 4 |
| 18 | −1-1-1 | 2 | 0 |
Functional enrichment analysis
To assess the biological meaning of the wild-type and the mutant differential expression pattern, we examined representation of GO terms in the whole set of DE genes and within each of the eighteen groups.
When considering the whole set of DE genes the most striking difference between the two clones was the wild-type specific enrichment in GO terms related to reproduction, such as anther wall tapetum development, cell division and microsporogenesis (Additional file 11).
When considering the DE gene in each of the eighteen groups, for the wild-type we detected a number of significantly enriched GO terms in groups 3, 11, 12, 16 and 17, whereas in the mutant significantly enriched GO terms were found only in groups 11 and 16 (however, many of the GO terms in the wild-type group 17 were present in the mutant group 16) (Additional file 12). For example, we observed a specific significant enrichment of positively regulated (from stage E-L 15 to stage E-L 27) functional categories in the wild-type group 3, for which the genes were mainly related to cell wall modification. Here stage E-L 27 corresponded to “after fertilization”, a phase of berry development mainly characterized with extensive cell division. Perhaps it is likely that these genes were highly active in the wild-type and may have played important role in cell wall re-assembly to encourage cell division during seed formation and embryo development.
Real-time PCR validation of RNA-Seq data
To confirm the results obtained by RNA-Seq, relative expression profiles of 14 genes were analyzed by real-time PCR in the wild-type and the mutant. The tested genes encoded enzymes involved in cell wall metabolism, transcription factors from different families (MYB, MADS-box, PHD and AS2) and molecules playing a role in signaling, including hormone-mediated signaling. For both clones and all genes, the real-time PCR results were consistent with the expression profiles determined from RNA-Seq data. Seven genes had similar expression profiles in the wild-type and the mutant, while the expression of the remaining 7 genes ranged from slightly different to completely opposite which suggests that some pathways may be altered in the seedless phenotype (Figure 4). In most cases biological replicates showed a consistent expression profile.
Figure 4.
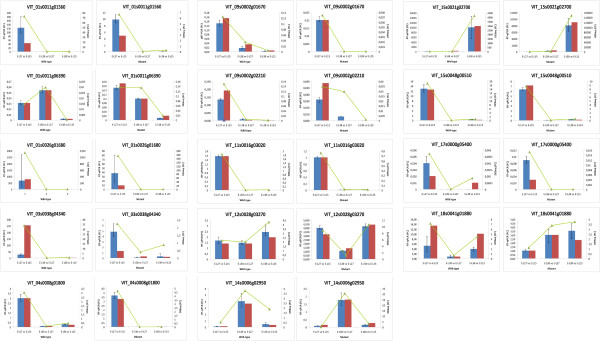
Quantitative real-time PCR validation of RNA-Seq data. Relative expression profile of 14 genes shows the expression fold change (FC) in the pairwise comparison between developmental stages for the wild-type and the mutant. Histograms represent expression fold changes as assessed by real-time PCR (by using REST), data are reported as means ± SE of three technical replicates (left axis). Green lines represent expression fold changes as assessed by RNA-Seq (by using DESeq, right axis). Blue column with error bar corresponds to the first biological replicate, while red column corresponds to the second biological replicate on which RNA sequencing was carried out.
Selection of candidate genes
In this work gene expression analysis highlighted several genes with common and contrasting expression profiles in the two clones, which may contribute to trait variation (seed content, and the resulting berry size, are the only phenotypic differences between the two somatic variants). Therefore, in order to narrow down to specific genes whose expression and effect were altered in the seedless phenotype, we have applied the criteria described in Methods. This allowed us to select a number of candidate genes for the seedless phenotype, which are listed in Table 4 and described hereafter. Among them are genes required for fertility, cell growth and development, transcription factors and signaling molecules.
Table 4.
Candidate genes for seed content that have altered expression in the wild-type and the mutant
| Gene ID | Wild-type gene expression (RPM) | Mutant gene expression (RPM) | Wild-type fold change | Mutant fold change | Gene enrichment | Annotation | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E-L 15 | E-L 27 | E-L 38 | E-L 15 | E-L 27 | E-L 38 | E-L 27 vs E-L 15 | E-L 38 vs E-L 27 | E-L 38 vs E-L 15 | E-L 27 vs E-L 15 | E-L 38 vs E-L 27 | E-L 38 vs E-L 15 | |||
| Non-DE genes specific to the wild-type | ||||||||||||||
| VIT_09s0002g01980 | 0.5 | 0.7 | 0.2 | 0 | 0 | 0 | Myosin-like protein XIK | |||||||
| VIT_15s0048g01070 | 0.01 | 0.01 | 1.2 | 0 | 0 | 0 | Vacuolar iron transporter 1 | |||||||
| VIT_04s0044g01520 | 0 | 0.8 | 0 | 0 | 0 | 0 | GA 20-oxidase 2 | |||||||
| VIT_08s0058g01200 | 0 | 0.4 | 0 | 0 | 0 | 0 | sig | Alpha-expansin 2 | ||||||
| Non-DE genes specific to the mutant | ||||||||||||||
| VIT_13s0106g00290 | 0 | 0 | 0 | 0.01 | 0 | 0.1 | Histone deacetylase HDA14 | |||||||
| VIT_03s0088g00900 | 0 | 0 | 0 | 0 | 0.1 | 0.01 | Pathogenesis-related protein 1B | |||||||
| VIT_14s0006g00050 | 0 | 0 | 0 | 0.1 | 0 | 0 | Transposase, IS4 | |||||||
| Common genes differentially regulated in wild-type and mutant | ||||||||||||||
| VIT_01s0026g01680 | 0.02 | 24.6 | 0 | 0.01 | 1.9 | 0 | 1,133 | 0 | nd | 172.5 | nd | nd | sig | Pectate lyase |
| VIT_05s0020g04850 | 1.0 | 2.4 | 89.1 | 0.9 | 0.9 | 101.8 | nd | nd | 113.1 | nd | 153.3 | nd | sig | H1flk |
| VIT_15s0021g02700 | 0.3 | 7.4 | 1,103 | 0 | 2.0 | 1,396 | nd | nd | 4,154 | inf | nd | nd | sig | Beta-expansin (EXPB4) |
| VIT_15s0048g00510 | 9.1 | 170.5 | 4.4 | 8.8 | 74.9 | 2.4 | 18.8 | nd | nd | 8.7 | nd | nd | Pectinesterase family | |
| VIT_15s0021g02170 | 631.1 | 3.1 | 0.3 | 81.4 | 0.2 | 0.4 | 0.005 | nd | 0.0006 | 0.003 | nd | nd | Chalcone and stilbene synthase | |
| VIT_18s0089g00140 | 40.7 | 0.2 | 2.7 | 3.5 | 0 | 8.0 | 0.004 | nd | nd | 0 | nd | nd | 1,4-beta-mannan endohydrolase | |
| VIT_19s0015g00960 | 150.2 | 0.7 | 0 | 59.3 | 0.1 | 0.1 | 0.004 | nd | 0 | 0.002 | nd | nd | ABC transporter G member 4 | |
| VIT_18s0001g01760 | 969.1 | 8.1 | 0.04 | 854.3 | 3.2 | 1.0 | 0.008 | nd | 0.00005 | 0.004 | nd | nd | PISTILLATA (PI) floral homeotic protein | |
| VIT_18s0001g13460 | 107.1 | 13.8 | 0 | 84.4 | 7.9 | 0.1 | 0.1 | nd | 0 | 0.1 | nd | nd | MADS-box AP3 | |
| Differentially expressed genes specific to the wild-type | ||||||||||||||
| VIT_01s0011g06390 | 29.9 | 0.4 | 0.01 | 3.2 | 0.4 | 0.04 | 0.01 | nd | 0.0004 | nd | nd | nd | sig | MALE STERILITY 1 |
| VIT_08s0007g07100 | 20.5 | 0.8 | 0.02 | 3.7 | 0.06 | 0.04 | 0.04 | nd | nd | nd | nd | nd | sig | MALE STERILITY 2 |
| VIT_07s0005g05680 | 17.6 | 0.2 | 3.5 | 4.0 | 0.2 | 0.04 | 0.009 | nd | nd | nd | nd | nd | MALE STERILITY 5 | |
| VIT_07s0005g05720 | 29.3 | 1.6 | 2.3 | 14.5 | 2.6 | 1.2 | 0.06 | nd | nd | nd | nd | nd | MALE STERILITY 5 | |
| VIT_15s0107g00550 | 172.2 | 19.7 | 21.8 | 111.7 | 26.7 | 2.5 | 0.1 | nd | nd | nd | nd | nd | sig | MALE STERILITY 5 |
| VIT_19s0014g03940 | 7.7 | 0.2 | 0.06 | 4.5 | 0.3 | 0.06 | 0.02 | nd | nd | nd | nd | nd | SPOROCYTELESS | |
| VIT_12s0142g00040 | 49.6 | 2.6 | 0.07 | 12.5 | 0.7 | 0.08 | 0.05 | nd | 0.002 | nd | nd | nd | Glycerol-3-phosphate acyltransferase 1 | |
| VIT_00s1404g00010 | 17.5 | 22.6 | 0.04 | 18.1 | 14.6 | 0.04 | nd | 0.002 | nd | nd | nd | nd | sig | Calmodulin-binding |
| VIT_01s0026g01420 | 22.2 | 58.8 | 0.3 | 22.8 | 57.2 | 0.2 | nd | 0.007 | nd | nd | nd | nd | Wall-associated kinase 4 | |
| VIT_06s0061g00730 | 406.0 | 887.8 | 0.3 | 423.5 | 1,138 | 0.2 | nd | 0.0004 | 0.0009 | nd | nd | nd | Aquaporin GAMMA-TIP3/TIP1;3 | |
| VIT_18s0001g13200 | 31.7 | 143.9 | 0.3 | 38.7 | 110.9 | 0.5 | nd | 0.003 | nd | nd | nd | nd | Cytokinin dehydrogenase 5 precursor | |
| VIT_05s0094g00330 | 0.9 | 16.2 | 969.7 | 0.9 | 4.2 | 712.2 | 17.6 | nd | 1,332 | nd | nd | nd | Chitinase, class IV | |
| VIT_10s0003g03030 | 0.02 | 6.5 | 0.4 | 0.06 | 0.5 | 0.6 | 300.4 | nd | nd | nd | nd | nd | Cation/hydrogen exchanger (CHX15) | |
| VIT_01s0011g01560 | 0.2 | 14.2 | 0.07 | 0.4 | 2.4 | 0.07 | 72.6 | nd | nd | nd | nd | nd | TRANSPARENT TESTA16 | |
| VIT_18s0001g03010 | 0 | 1.5 | 0 | 0 | 0.2 | 0 | inf | nd | nd | nd | nd | nd | BZIP transcription factor | |
| VIT_18s0041g01880 | 9.5 | 67.4 | 33.2 | 9.8 | 10.7 | 26.7 | 7.1 | nd | nd | nd | nd | nd | sig | MADS-box protein SEEDSTICK |
| VIT_03s0038g04340 | 0.2 | 6.9 | 0.07 | 0.3 | 0.6 | 0.2 | 33.7 | nd | nd | nd | nd | nd | FERONIA receptor-like kinase | |
| VIT_17s0000g08110 | 1.1 | 23.1 | 0.01 | 0.2 | 1.4 | 0 | 20.1 | 0.0006 | nd | nd | nd | nd | Nodulin MtN3 | |
| VIT_17s0000g09000 | 0.8 | 0.4 | 36.3 | 0.5 | 0.009 | 0.04 | nd | 125.8 | nd | nd | nd | nd | Oleosin OLE-2 | |
| VIT_07s0151g00640 | 12.4 | 5.2 | 453.2 | 17.9 | 3.0 | 1.1 | nd | 109.3 | nd | nd | nd | nd | Globulin-1 S allele precursor | |
| VIT_14s0128g00200 | 0.04 | 0.1 | 42.5 | 0.09 | 0 | 0 | nd | 540.1 | 1,242 | nd | nd | nd | 7S globulin precursor | |
| VIT_13s0067g01250 | 0.06 | 0.1 | 26.0 | 0.02 | 0 | 0 | nd | 330.4 | 506.5 | nd | nd | nd | Em protein GEA6 (EM6) | |
| VIT_14s0108g00520 | 0 | 0.2 | 89.3 | 0.03 | 0.09 | 0.03 | nd | 637.6 | inf | nd | nd | nd | Protease inhibitor/seed storage/lipid transfer protein (LTP) | |
| VIT_16s0039g00220 | 0.4 | 0.2 | 25.2 | 0.3 | 0.1 | 0.7 | nd | 192.0 | nd | nd | nd | nd | Aquaporin BETA-TIP | |
| VIT_07s0005g05400 | 0 | 0.08 | 22.1 | 0.05 | 0 | 0 | nd | 360.2 | inf | nd | nd | nd | ABSCISIC ACID-INSENSITIVE protein 3 (ABI3) | |
| VIT_19s0014g04130 | 0.7 | 0.2 | 23.6 | 0.3 | 0.4 | 6.4 | nd | 179.8 | nd | nd | nd | nd | Serine/threonine-protein kinase receptor ARK3 | |
| VIT_18s0001g01570 | 0.2 | 0.5 | 265.1 | 0.3 | 0.07 | 0.3 | nd | 631.3 | 1,548 | nd | nd | nd | Seed maturation protein PM31 | |
| VIT_14s0128g00340 | 0.1 | 0.08 | 17.0 | 0.1 | 0 | 0 | nd | 277.3 | nd | nd | nd | nd | Seed maturation protein PM34 | |
| VIT_04s0008g01610 | 0 | 0.3 | 176.8 | 0 | 0 | 0 | nd | 776.9 | inf | nd | nd | nd | Heat shock protein 17.6 kDa class II | |
| Differentially expressed genes specific to the mutant | ||||||||||||||
| VIT_14s0219g00270 | 20.3 | 2.5 | 0.08 | 21.6 | 0.5 | 0.08 | nd | nd | nd | 0.03 | nd | nd | TEL1 (Terminal EAR1-like 1) | |
| VIT_12s0059g00560 | 14.6 | 1.1 | 1.1 | 17.8 | 0.6 | 0.4 | nd | nd | nd | 0.04 | nd | nd | Fimbrin 2 | |
| VIT_04s0008g04980 | 15.5 | 2.7 | 0.2 | 10.1 | 0.3 | 0.06 | nd | nd | nd | 0.03 | nd | nd | Boron transporter-like protein 4 | |
| VIT_09s0002g01670 | 9.0 | 1.1 | 0.03 | 11.8 | 0.09 | 0 | nd | nd | nd | 0.007 | nd | nd | Myb domain protein 26 | |
| VIT_09s0002g01370 | 13.2 | 1.3 | 0.2 | 14.0 | 0.3 | 0 | nd | nd | nd | 0.02 | nd | nd | AP2 AINTEGUMENTA | |
| VIT_14s0006g02950 | 25.4 | 12.6 | 37.4 | 53.1 | 5.4 | 123.3 | nd | nd | nd | 0.1 | nd | nd | LATERAL ORGAN BOUNDARIES protein 41 | |
| VIT_15s0046g03080 | 3.4 | 0.9 | 0.02 | 2.9 | 0 | 0 | nd | nd | nd | 0 | nd | nd | DTA2 (downstream target of AGL15 2) | |
| VIT_12s0134g00240 | 17.7 | 36.7 | 21.5 | 9.7 | 77.1 | 22.7 | nd | nd | nd | 8.1 | nd | nd | Avr9/Cf-9 rapidly elicited protein 20 | |
| VIT_12s0028g03270 | 14.2 | 30.5 | 48.0 | 7.3 | 62.5 | 55.7 | nd | nd | nd | 8.8 | nd | nd | Ethylene-responsive transcription factor 9 | |
| VIT_16s0013g00950 | 1.8 | 5.1 | 2.1 | 1.1 | 22.1 | 0.4 | nd | nd | nd | 20.5 | nd | nd | Ethylene-responsive transcription factor ERF105 | |
| VIT_16s0013g00990 | 2.4 | 5.0 | 0.7 | 1.2 | 18.6 | 0.4 | nd | nd | nd | 16.1 | nd | nd | Ethylene-responsive transcription factor ERF105 | |
| VIT_16s0013g01050 | 3.1 | 6.3 | 1.1 | 1.8 | 23.3 | 0.3 | nd | nd | nd | 13.2 | nd | nd | Ethylene-responsive transcription factor ERF105 | |
| VIT_16s0013g01120 | 3.2 | 13.2 | 63.7 | 3.1 | 29.5 | 49.1 | nd | nd | nd | 9.8 | nd | nd | Ethylene-responsive transcription factor ERF105 | |
Abbreviations : nd not detected in a pairwise comparison, inf infinity (when the mean of one stage in a pairwise comparison is the denominator with value 0), sig significant.
Non-differential transcriptional processes specific to the wild-type and mutant backgrounds
Non-DE genes specific to the wild-type
Within this category very few genes met the RPM selection criteria, however many genes were significantly enriched and some of them had a putative functional role relevant to seed development. We selected four genes that play roles in cellular process, transport and signaling.
Among cellular process genes, VIT_09s0002g01980 encodes the myosin-like protein XIK, which is involved in actin organization and biogenesis as well as actin-driven movement [43]. Among transporters, the gene VIT_15s0048g01070 encodes the vacuolar iron transporter 1 protein, implicated in iron transport and storage [43]. In seeds, iron has been demonstrated to be essential for Arabidopsis embryo development [49]. Among the signaling genes are VIT_04s0044g01520 and VIT_08s0058g01200. VIT_04s0044g01520 encodes GA 20-oxidase 2, which is involved in gibberellic acid biosynthesis, whereas VIT_08s0058g01200 codes for the alpha-expansin 2 protein that participates in auxin-mediated signaling pathway as well as regulating cell growth [43].
Non-DE genes specific to the mutant
All the genes that fell within this category did not meet the RPM selection criteria described in the Methods and did not have defined function when annotated; meaning that, many of them returned no hit upon functional annotation. Nevertheless, we noticed a few genes whose functional roles could be implicated in seed development. They included the histone deacetylase HDA14 gene (VIT_13s0106g00290), involved in chromatin organization through protein acetylation and deacetylation, a gene (VIT_03s0088g00900) coding for a pathogenesis-related protein 1B implicated in jasmonate-mediated signaling as well as in plant-pathogen interaction and a transposase IS4 gene (VIT_14s0006g00050) that encodes a transposable element protein [43].
Differential regulation of common transcriptional processes in the wild-type and the mutant
Significant number of expressed genes were common among wild-type and mutant growth stages, which suggests that the corresponding proteins may function in a common pathway to carry out a wide range of developmental processes. We reasoned that many of these shared genes will respond in both clones to the same signals that control the switch from one developmental phase (before fertilization) to another (after fertilization), and will have similar pattern of expression. Indeed, differential expression analysis revealed 501 DE genes common to the wild-type and the mutant (47% of the total 1075 DE genes) (Additional file 10).
Thirty-five of these genes showed different expression between the two clones along the time course. Among the 35 genes, six were significantly enriched and three of them had a functional annotation corresponding to seed development: pectate lyase, histone H1flk-like protein (H1flk), and beta-expansin (EXPB4). Pectate lyase is an enzyme involved in cell wall organization and biogenesis by catabolizing pectin. In tomato, two pectate lyases were found to be maximally expressed at the late stage of pollen development. It was suggested that the pollen expression of these genes might relate to a requirement for pectin degradation during pollen tube growth [50]. In the present study, the pectate lyase gene VIT_01s0026g01680 was up-regulated from stage E-L 15 to stage E-L 27 in both clones but the fold change was six times higher in the wild-type compared to the mutant. Based on its functional annotation, the histone H1flk-like gene VIT_05s0020g04850 plays a role in chromatin assembly. Its Arabidopsis homolog encodes a P-loop containing nucleoside triphosphate hydrolases superfamily protein that functions in ATP binding activity involved in cell killing [51]. In the mutant background, this gene was specifically up-regulated from stage E-L 27 to stage E-L 38 while in the wild-type a significant differential expression with a lower fold change was observed only between stages E-L 15 and E-L 38. The beta-expansin gene VIT_15s0021g02700 was not expressed at stage E-L 15 in the mutant. Differential expression analysis in the mutant showed specific up-regulation from stage E-L 15 to stage E-L 27, in contrast to a stable expression in the wild-type between the same stages. Based on its functional annotation, this gene encodes a protein involved in auxin-mediated signaling, which implies a late induction of auxin responsive genes in the mutant.
As expected, 466 out of the 501 common DE genes shared the same group or expression profile in both the wild-type and the mutant. Functional annotation and GO term enrichment uncovered many biological processes, which included cell wall metabolism, cell cycling, primary and secondary metabolism, signaling and regulation of gene expression, water transport and abiotic stress responses. Within this set the following four genes are of interest. VIT_15s0048g00510 encodes a protein that belongs to the pectinesterase family, up-regulated from stage E-L 15 to stage E-L 27 with double fold change in the wild-type compared to the mutant. Functional annotation revealed the protein involvement in cell wall modification through pectin degradation. In Arabidopsis, it has been shown that cell type-specific pectin degradation is required to separate microspores during pollen development [52]. VIT_15s0021g02170, VIT_18s0089g00140 and VIT_19s0015g00960 showed a similar behavior: they were down-regulated from stage E-L 15 to stage E-L 27 in both clones, but much more expressed in the wild-type than in the mutant. VIT_15s0021g02170 encodes chalcone and stilbene synthase. Its Arabidopsis homolog is involved in phenylpropanoid biosynthetic process and pollen exine formation [51]. VIT_18s0089g00140 encodes 1,4-beta-mannan endohydrolase, which is implicated in fructose and mannose metabolic pathways [43]. Description of biological processes associated to its Arabidopsis homolog revealed a role in seed germination [51]. The Arabidopsis homolog of VIT_19s0015g00960 is required for male fertility and pollen exine formation as it encodes an ATP-binding cassette transporter involved in tapetal cell and pollen development [51].
Finally, within this category we identified two genes already proposed to affect seed and/or berry development [46]. They code for the PISTILLATA (PI) floral homeotic protein (VIT_18s0001g01760) and the MADS-box AP3 transcription factor (VIT_18s0001g13460). The latter co-localizes with the stable QTL for berry weight, seed number and fresh weight identified by [46].
Differentially expressed genes specific to the wild-type background
The 441 genes specifically modulated among the wild-type developmental stages represented 12 groups and included a range of functional categories. A large number (approximately 64%) of these genes were observed among nine groups, down-regulated from stage E-L 15 to stage E-L 27 and not differentially expressed from stage E-L 27 to stage E-L 38 or vice versa. The remaining 36% were observed in three groups, and were up-regulated in the same manner (Additional file 10).
Down-regulated genes specific to wild-type (from stage E-L15 to stage E-L 27)
Within this category we observed several interesting genes that showed significant enrichment of GO terms and very high negative fold change. They include five genes, three of which encode similar proteins: MALE STERILITY 1 (MS1, VIT_01s0011g06390), MALE STERILITY 2 (MS2, VIT_08s0007g07100) and MALE STERILITY 5 (MS5, VIT_07s0005g05680, VIT_07s0005g05720 and VIT_15s0107g00550). The gene coding for MS1 protein belongs to the PHD family of transcription factors. The Arabidopsis MS1 gene was described to be a sporophytic factor controlling anther and pollen development. It plays a critical role in the induction of pollen wall and pollen coat materials in the tapetum and, ultimately, the production of viable pollen. Indeed, mutants show a semi-sterile phenotype, as their pollen degenerates after microspore release. In addition their tapetum appears abnormally vacuolated [51, 53–55]. The MS2 gene has an unclear function in Vitis vinifera, however its Arabidopsis best match was described as a fatty acid reductase gene, involved in oxidation-reduction process and pollen exine formation [56]. The function of the MS5 gene in Vitis vinifera is unknown, however in Arabidopsis it was suggested to be similar to POLLENNESS3 gene [57]. Mutants of this gene in Arabidopsis were shown to have defects in functional microspore production that lead to the degeneration of cells within the anther locules [53]. One of the three MS5 gene predictions co-located with a minor QTL for mean seed fresh weight on chromosome 15 [23]. The significant down-regulation of these genes from stage E-L 15 to stage E-L 27 in the wild-type implies that they were highly induced at stage E-L 15, where they exhibited maximum expression levels, perhaps to ensure viable and functional pollen development for complete fertilization. On the other hand, in the mutant, these genes were not differentially expressed. Further analysis of their RPM values in the mutant revealed very low level of expression at stage E-L 15, when compared to the wild-type. This observation might suggest abnormal pollen development in the mutant resulting in non-functional or partially sterile pollen. However, it needs to be tested and confirmed experimentally.
Within this category we found two additional genes with a putative role in ovule and pollen differentiation: SPOROCYTELESS (VIT_19s0014g03940) and glycerol-3-phosphate acyltransferase 1 (VIT_12s0142g00040). The SPOROCYTELESS gene of Arabidopsis was described to encode a transcription factor that is required for the initiation of both micro- and megagametogenesis and is expressed in the sporogenous tissue of the anther and the ovule. It is involved in establishing the prospective chalaza of the ovule, plays a central role in patterning both the proximal-distal and the adaxial-abaxial axes in the ovule and regulates the anther cell differentiation. Mutant is defective in the differentiation of primary sporogenous cells into microsporocytes, and does not properly form the anther wall [51, 58, 59]. The Arabidopsis homolog of glycerol-3-phosphate acyltransferase 1 gene was shown to be expressed in flower buds and siliques. Its protein is involved in metabolic processes such as phosphatidylglycerol biosynthetic process, pollen sperm cell differentiation, and response to karrikin. Interestingly, the homozygous mutant plants are male sterile [51, 60].
Down-regulated genes specific to wild-type (from stage E-L 27 to stage E-L 38)
Within this category we observed about 30 genes with high negative fold change, the majority of which belong to the functional categories of cellular process and signaling. The most relevant for seed development appeared the genes encoding a calmodulin-binding protein (VIT_00s1404g00010), the wall-associated kinase 4 (WAK4, VIT_01s0026g01420), the aquaporin GAMMA-TIP3/TIP1;3 (VIT_06s0061g00730) and a precursor of cytokinin dehydrogenase (VIT_18s0001g13200). Indeed, in rice a calmodulin-binding protein was found to be essential to pollen development [61], the silencing of a member of the WAK family led to sterility due to anther indehiscence [62], while the aquaporin GAMMA-TIP3/TIP1;3 in Arabidopsis was reported to be a pollen-specific water transporter contributing to male sterility in the double knockout mutant tip1;3/tip5;1[63], and cytokinins were demonstrated to regulate seed yield [64].
Up-regulated genes specific to the wild-type (from stage E-L 15 to stage E-L 27)
Amongst this group we noticed a number of genes with high positive fold change value. Besides genes encoding proteins involved in cell wall organization and biogenesis, the most relevant for seed development were found in the categories: metabolism, transport, regulation overview and signaling. For instance, we identified a chitinase class IV gene (VIT_05s0094g00330), whose best Arabidopsis match was described to be expressed during somatic embryogenesis in nursing cells surrounding the embryos and additionally in mature pollen and growing pollen tubes until they enter the receptive synergid [51, 65]. Among transporters, a cation/hydrogen exchanger (VIT_10s0003g03030) showed its best match with an Arabidopsis protein involved in pollen tube growth [51].
Of particular interest were a set of genes encoding transcription factors and signaling molecules. Among the transcription factors were TRANSPARENT TESTA 16 TT16 or AGL32 (VIT_01s0011g01560), BZIP family protein (VIT_18s0001g03010) and the MADS-box protein SEEDSTICK (VIT_18s0041g01880). The TT16 gene encodes a MADS-box family transcription factor [43, 51]. In Arabidopsis it was reported to determine the identity of the endothelial layer within the ovule, to play a maternal role in fertilization and seed development and to regulate proanthocyanidin biosynthesis and cell shape of the inner-most cell layer of the seed coat [51, 66]. In canola (Brassica napus) it was further demonstrated that the tt16 deficiency affects pollen tube guidance, resulting in reduced fertility and negatively impacting embryo and seed development due to the altered expression of genes involved in gynoecium and embryo development, lipid metabolism, auxin transport, and signal transduction [67]. In addition, the TT16 gene was reported among the functional candidates potentially involved in seed and/or berry development that did not co-localize with QTLs detected for the same traits [46].
The BZIP gene was previously described by [68] to be expressed in pollen and other flower parts.
Although the MADS-box SEEDSTICK gene did not show high positive fold change, it was significantly enriched in our data. In Arabidopsis and rice, this gene was described to encode a MADS-box transcription factor expressed in the carpel and ovules and to play a maternal role in fertilization and seed development. Mutants indeed exhibited reduced ovule fertilization and high seed abortion [51, 69–71]. Interestingly, this gene was among those that co-localized with the stable QTLs for seed-related traits [23, 46].
The signaling molecules included FERONIA receptor-like kinase (VIT_03s0038g04340). In Arabidopsis, it was shown to mediate male–female interactions during pollen tube reception [72]. Feronia mutant had impaired fertilization because pollen tube failed to arrest by continue growth inside the female gametophyte [73]. This study concluded that female control of pollen tube reception is based on a FERONIA-dependent signaling pathway. In our investigation, we observed low expression level (0.6 RPM) of FERONIA receptor-like kinase gene in the mutant, compared to higher expression (6.9 RPM) in the wild-type.
Finally, within this category we identified a gene coding for a nodulin (VIT_17s0000g08110), which was up-regulated from stage E-L 15 to stage E-L 27 and down-regulated from stage E-L 27 to stage E-L 38. The Arabidopsis best match for this gene encodes a protein containing three domains, one of which is MtN3/saliva-related trans-membrane protein, and has function in sugar trans-membrane transporter activity [51]. In rice the genes Xa13/Os8N3/OsSWEET11 and Os11N3/OsSWEET14 encode proteins with two MtN3/saliva domains similar to that of Arabidopsis, and were identified to play important role in regulating reproductive development through promotion of fertilization. These genes were reported to have a very high expression level in rice panicles and anthers compared to other tissues. Suppressed plants showed reduced fertility or were sterile due to blockage of microspore development at the unicellular pollen grain stage. This resulted in the gradual degeneration of the immature pollen suggesting the proteins are required for pollen development in rice. In addition knockout mutants showed reduced seed size and delayed growth [74]. The significant up-regulation of the nodulin MtN3 gene from stage E-L 15 to stage E-L 27 in the wild-type compared to the mutant could imply an active role in promoting fertilization. In contrast, down-regulation of this gene from stage E-L 27 to stage E-L 38, which corresponds to a period of seed maturation (after fertilization), seems to support the notion that genes participating or promoting seed formation are tightly regulated.
Up-regulated genes specific to the wild-type (from stage E-L 27 to stage E-L 38)
Within this category we found a gene coding for oleosin OLE-2 protein (VIT_17s0000g09000), with a putative role in oil body organization and biogenesis as well as in reproduction and seed development. Functional studies in Arabidopsis showed that the double mutant ole1/ole2 had irregular enlarged oil-containing structures throughout the seed cells which led to defects in germination or seed mortality [75].
Three different genes encoded enzymes involved in primary metabolism, namely globulin-1 S allele precursor (GLB1, VIT_07s0151g00640), 7S globulin precursor (VIT_14s0128g00200) and Em protein GEA6 (EM6, VIT_13s0067g01250). Functional annotation revealed that the three genes participate in generation of metabolite precursors and serve as energy storage proteins. The maize GLB1 gene was found to be expressed throughout embryo development specifically in seed tissues [76]. Similarly, 7S globulin precursor was described as a major storage protein in legume species [77]. In our study, the expression of the 7S globulin precursor gene was highest at wild-type stage E-L 38 while it was almost abolished in the mutant. This suggests that induction of these genes may be required to complete seed development. The best Arabidopsis match for the EM6 gene was described to be the LATE EMBRYOGENESIS ABUNDANT 6 gene, involved in response to abscisic acid, required for normal seed development, and regulating the timing of desiccation tolerance and the rate of water loss during seed maturation [51, 78].
Other interesting genes are those involved in lipid and water transport, e.g. the genes coding for a protease inhibitor/seed storage/lipid transfer protein (VIT_14s0108g00520) and aquaporin BETA-TIP (VIT_16s0039g00220).
Equally worth mentioning are two genes coding for signaling molecules, namely the abscisic acid-insensitive protein 3 ABI3 (VIT_07s0005g05400) and the serine/threonine-protein kinase receptor ARK3 (VIT_19s0014g04130). The expression of ABI3 gene was completely abolished in the mutant from stage E-L 27 to stage E-L 38. ABI3 is a putative seed-specific transcriptional activator acting as a central regulator in ABA signaling. In different species it was described to play a major role in seed maturation and to regulate the transition between embryo maturation and early seedling development [51, 79, 80]. In Arabidopsis the ARK3 gene was proposed to participate in recognition of pollen [51, 81].
Four stress response genes were also present and specifically induced, including those coding for the seed maturation proteins PM31 (VIT_18s0001g01570) and PM34 (VIT_14s0128g00340). Finally, the gene prediction for the heat shock protein 17.6 kDa class II with a putative role in protein folding (VIT_04s0008g01610) was not expressed in the mutant in all three developmental stages.
Differentially expressed genes specific to the mutant background
The 133 DE genes, which were peculiar to the mutant, fell within 4 groups (3, 8, 11 and 17) and were all stage specifically induced. The majority of these genes (63%) were either down-regulated from stage E-L 15 to stage E-L 27 or from stage E-L 27 to stage E-L 38, whereas 37% of them were up-regulated in the same manner (Additional file 10). The genes related to seed development showed differential expression between stages E-L 15 and E-L 27.
Down-regulated genes specific to the mutant (from stage E-L 15 to stage E-L 27)
In this category we identified genes with high negative fold change encoding proteins with a role in cellular processes, transport and regulation of gene expression.
Among the genes involved in cellular processes we selected TERMINAL EAR1-like 1 (TEL1, VIT_14s0219g00270) and Fimbrin 2 (VIT_12s0059g00560). The TEL1 gene encodes an RNA binding protein with a function in shoot development, conserved among land and vascular plants [51, 82]. The Arabidopsis best match of TEL1 is a member of the MEI2-like gene family, which plays a role in meiosis. Specific multiple mutant combinations were reported to display sterility and a range of defects in meiotic chromosome behavior [83]. The Fimbrin 2 gene is involved in actin organization and biogenesis; its Arabidopsis homolog is FIMBRIN5, an actin bundling factor required for pollen germination and pollen tube growth [84]. The same function was reported in lily [85]. We observed high expression of the TEL1 and Fimbrin 2 genes at stage E-L 15 in both clones, however as development progressed towards stage E-L 27 a significant repression of both genes in the mutant was evident in their very low RPM values as compared to a stable expression of these genes in the wild-type. In addition the Fimbrin2 gene in grape fell within a stable QTL for mean seed fresh weight reported by [46].
In the transport category we identified a gene encoding the boron transporter-like protein 4 (VIT_04s0008g04980). Previously, boron deficiency has been associated with the occurrence of parthenocarpic seedless grapes in some varieties of Vitis vinifera L [86].
We also noticed a set of genes coding for transcription factors, which included the MYB domain protein 26 MYB26 (VIT_09s0002g01670), AP2 AINTEGUMENTA (VIT_09s0002g01370) and LATERAL ORGAN BOUNDARIES protein 41 LBD41 (VIT_14s0006g02950). The Arabidopsis MYB26 protein was described to be involved in anther dehiscence, response to gibberellin stimulus and secondary cell wall biogenesis. Mutants for this gene produced fertile pollen but plants were sterile because anthers did not dehisce. When compared to wild type, no cellulosic secondary wall thickening was seen in the anther endothecium of the mutant [87]. The AP2 AINTEGUMENTA gene belongs to the AP2 (APETALA2)/EREBP (ethylene-responsive element binding protein) family of transcription factors, known to be key regulators of several developmental processes [88]. The Arabidopsis homolog was reported to have a role in ovule development among other functions. Mutants exhibited female-sterility as integuments did not develop and megasporogenesis was blocked at the tetrad stage [89]. The LBD41 gene encodes a protein containing the conserved domain AS2/LOB. The Arabidopsis homolog of the LOB gene ASYMMETRIC LEAVES2 (AS2) was demonstrated to function in the repression of KNOX genes and in the specification of adaxial/abaxial organ polarity [90]. The maize ortholog was also reported to be required to prevent KNOX gene expression in lateral organs and, in addition, to promote the switch from proliferation to differentiation in the embryo sac. The failure to limit proliferation in mutant embryo sacs was shown to lead to a variety of structural defects, including the production of extra gametes and synergids. Moreover, the fertilization process was frequently abnormal, producing seeds with haploid embryos and embryos and endosperms derived from fertilization by different pollen tubes [91]. Although the role of these regulatory genes in growth and development is well documented in model species, in Vitis vinifera L. their specific functions are not well characterized and can only be inferred. However, we observed a general pattern in the mutant, in which expression of these genes was almost abolished at stage E-L 27 when compared to their stable expression in the wild-type.
Finally, a gene DTA2 was observed (VIT_15s0046g03080, downstream target of AGL15). In Arabidopsis DTA2 was reported to encode an unknown protein with no significant similarity to any known protein and to be expressed in developing seeds and in roots [92]. In our data, the DTA2 gene from the mutant was expressed at stage E-L 15, and the expression was abolished at stages E-L 27 and E-L 38 (in contrast to the stable expression in the wild-type).
Up-regulated genes specific to the mutant (from stage E-L 15 to stage E-L 27)
Within this category we selected six genes, one of which (VIT_12s0134g00240) encodes a signaling molecule involved in stress response. This Avr9/Cf-9 rapidly elicited protein 20 was shown to function in the initial development of the defense response in tomato [93]. The remaining five genes encode proteins involved in the ethylene-mediated signaling pathway. These are ethylene-responsive transcription factor ERF9 (VIT_12s0028g03270) and ethylene-responsive transcription factor ERF105 (VIT_16s0013g00950, VIT_16s0013g00990, VIT_16s0013g01050 and VIT_16s0013g01120). The ERF9 gene was shown to take part in repressing the activation of pathogen related genes in Arabidopsis[94]. The Arabidopsis homolog of ERF105 encodes a member of the ERF (ethylene response factor) subfamily B-3 of ERF/AP2 transcription factor family that is involved in processes such as regulation of transcription, respiratory burst involved in defense responses, as well as responses to mechanical stimulus and wounding [51, 94, 95]. We noticed that the expression levels of these genes were always higher at stage E-L 27 in the mutant compared to the wild-type.
It might be worthy of mention that a substantial proportion of our strongest candidate genes (that are the genes expressed specifically in either clone) were physically clustered in the vicinity of some previously identified QTLs [23, 46], mainly the loci on chromosomes 2 and 12 (Additional file 13). While there may be no causal link between their expression and trait variation, they might provide a valuable starting point for developing DNA markers linked to the target trait, as discussed in [96].
Conclusions
At the best of our knowledge, the present study represents the first transcriptomic analysis by mRNA-Seq technology in a seeded grapevine variety and its seedless somatic variant. The examination of absolute expression count for every gene has enabled us to carry out a global investigation of gene expression at three key time-points during seed formation covering from before anthesis to after fertilization. This has in turn allowed a comprehensive description of distinguishing transcriptional events in the two lines, based on the analysis of differentially expressed genes, gene patterns and enriched functional categories. We have given a detailed account of the expression profiles of the genes potentially required to initiate and to complete seed development, including genes involved in gametophyte development, cell division, cell wall organization, as well as signaling molecules and transcription factors. Reduction in the number of transcripts observed in the mutant after fertilization seems consistent with shut down in transcriptional processes resulting from incomplete fertilization or failure of embryo development. Here the significant low expression profile of male sterility genes in the mutant and the high induction of the same genes in the wild-type suggests non-functional or partially sterile pollen in the mutant, which is in agreement with preliminary observations from the phenotypic characterization underway and encourages further investigation. We surmise that some of the candidate genes derived from this study could be useful for the development of molecular markers to assist breeding programs, especially if these genes are located in a genomic region of a known QTL for seed content. In conclusion, the data reported here have provided a rich genomic resource for functional characterization of the genes that potentially underpin seedlessness in grapevine.
Supporting data
The data sets supporting the results of this article are included within the article and its additional files.
Electronic supplementary material
Additional file 1: Figure S1: Sample collection. Three key time points along grape berry development corresponding to stages E-L 15 (single flowers in compact groups), E-L 27 (young berries enlarging) and E-L 38 (berries harvest-ripe) of the modified E-L system 36 were matched to the number of days from bloom (DFB) and could be assigned to two main categories: “before” (E-L 15) and “after” (E-L 27 and 38) fertilization. (A) diagram showing the match of sampling dates (expressed as E-L stages) to days from bloom in reference 4. (B) picture of the materials collected from the two lines at each sampling date. (DOCX 233 KB)
Additional file 2: Table S1: Genotypic characterization of the wild-type and the mutant. Fifty-eight SSR (simple sequence repeat) markers, spread across the nineteen chromosomes of grapevine genome, were used to genotype the wild-type and the mutant. Marker details and PCR conditions are described in Methods. Symbols: * SSR markers commonly used to discriminate grapevine varieties, - indicates homozygous or null allele. (XLSX 35 KB)
Additional file 3: Text S1: Description of the procedure adopted to rank the transcripts by order of magnitude. P-values were computed to reflect the significance of the difference between 2 counts (n1 and n2 corresponding to any two library combination out of the six libraries, independently of the genotype) using a binominal model. (DOCX 117 KB)
Additional file 4: Table S2: List of the genes analyzed in real-time PCR and primers used for their amplification. The table reports the gene IDs, gene annotations and sequences of the primer sets used to analyze the transcriptional profile of 14 genes in addition to the two constitutive genes used as reference for normalization. (XLSX 23 KB)
Additional file 5: Table S3: Summary of read mapping to v1_mRNA version of 12x grapevine genome draft annotation. Short-read alignment and mapping of all the reads were carried on the 12x v1 annotation of the grapevine genome PN40024 38 using BWA (Burrows Wheeler Aligner) software 39. The mapping results were processed with SAMtools 40 to extract for each transcript the number of mapped reads and determine, whether their mapping position is unique. Reads mapping to several positions on the reference sequence with the same “mapping quality” (i.e. number of mismatches and quality of the bases generating the mismatches) were attributed at random to one of them with a “0” mapping quality. (XLSX 11 KB)
Additional file 6: Table S4: Distribution of mapped reads among genomic features. A Python script was developed to determine the distribution of mapped reads among genomic features in the wild-type and the mutant. (XLSX 13 KB)
Additional file 7: Table S5: List of transcripts ranked by order of magnitude. P-values (or scores) were computed to reflect the significance of the difference between 2 counts (n1 and n2 corresponding to any two library combination out of the six libraries, independently of the genotype) using a binomial model. The model is described in Additional file 3. The p-values were log-transformed in order to allow for greater numerical stability in comparing extreme values. The sign of the p-value reflects the direction of the comparison (whether n1 is greater or lesser than n2). The smaller is the absolute p-value, the more significant is the difference between the counts. Next all the p-values and the ratios of expression between the counts were considered to compute a ranking value for each transcript. Afterwards the ranking values were used to sort the transcripts and show on top the biggest differences in expressions between two of the libraries. (XLSX 9 MB)
Additional file 8: Table S6: Transcript abundance measurement at each developmental stage. A lower limit of detection for expression estimate was designated to be an RPM of 0.5 or, if the RPM value was less than 0.5, at least five uniquely mapped reads with identity > 98% over 100 bp. (XLSX 11 KB)
Additional file 9: Figure S2: Gene overlap between the wild-type and the mutant in the first two and last two developmental stages. (A) Venn diagram showing shared and unique expressed genes between the wild-type and the mutant during the first two developmental stages E-L 15 and E-L 27. (B) Venn diagram showing shared and unique expressed genes between the wild-type and the mutant during the last two developmental stages E-L 27 and E-L 38. Abbreviations: WT = wild-type, MT = mutant. (PDF 146 KB)
Additional file 10: Table S7: List of all differentially expressed genes in the pairwise comparison of developmental stages in the wild-type and the mutant. Differential expression (DE) analysis was carried out on the raw uniquely mapped reads using the software DESeq with the following parameters: false discovery rate (FDR) ≤ 0.05, log2-fold change (FC) > 1. An in-house R script was written to group DE genes with similar expression pattern based on the adjusted p-values, such that transcripts displaying significant negative differences, significant positive differences and insignificant differences between time points were fitted to -1, 1 and 0, respectively. The first position in each triplet corresponds to the shift from stage E-L 15 to E-L 27, the second position to the shift from stage E-L 27 to E-L 38 and the third position to the shift from stage E-L 15 to E-L 38. The different sheets report the genes differentially expressed in both the wild-type and the mutant, specific to the wild-type and the mutant, respectively. Abbreviations: nd = not detected in a pairwise comparison; imp (impossible to divide) = when the basemean of one stage in a pairwise comparison is the denominator with value 0; hyphen (-) = when the basemean of one stage in a pairwise comparison is the numerator with value 0 (the corresponding logarithm doesn’t exist). (XLS 524 KB)
Additional file 11: Table S8: Functional enrichment analysis in the whole set of differentially expressed genes in the wild-type and the mutant. Using a hypergeometric test in the AgriGO toolkit, a gene ontology (GO) term was considered significantly enriched, if the false discovery rate (FDR) was < 0.05 and p-value < 0.01 when compared to all gene transcripts annotated in the reference genome (supported in AgriGO). The two sheets report the results of functional enrichment analysis for the differentially expressed genes in the wild-type and the mutant, respectively. Abbreviations: P = biological process, F = molecular function, C = cellular component, BG = background, Ref = reference. Asterisks indicate terms commonly enriched in the wild-type and the mutant. (XLSX 61 KB)
Additional file 12: Table S9: Functional enrichment analysis within groups of differentially expressed genes in the wild-type and the mutant. Using a hypergeometric test in the AgriGO toolkit, a gene ontology (GO) term was considered significantly enriched, if the false discovery rate (FDR) was < 0.05 and p-value < 0.01 when compared to all gene transcripts annotated in the reference genome (supported in AgriGO). The different sheets report the results of functional enrichment analysis within specific clusters of differentially expressed genes in the wild-type and the mutant. Abbreviations: P = biological process, F = molecular function, C = cellular component, BG = background, Ref = reference. Asterisks indicate terms commonly enriched in the wild-type and the mutant clusters 11 and 16. (XLSX 182 KB)
Additional file 13: Table S10: Proportion of RNA-Seq-derived candidate genes in the physical proximity of seed-related QTLs. Genes that were specifically expressed in either clone (with or without significant modulation between developmental stages) were mapped to each of eight chromosomes carrying seed-related QTLs, identified previously by [23, 46]. The proportion of genes falling within a 10-Mb region centered on each QTL peak was calculated, as described in [96]. Abbreviations: chr = chromosome. (XLSX 9 KB)
Acknowledgements
We are grateful to Stefano Raimondi, Silvia Lorenzi, and Paula Moreno-Sanz for technical assistance and contributing in collection of plant materials. We thank Alessandro Cestaro and Emanuela Kerschbamer for assessment of read coverage and distribution of reads among genomic features. Lukasz Grzeskowiak is acknowledged for critical reading of the manuscript. This study was carried out with full financial support of Fondazione Edmund Mach, DGBPF-GAV-AdP 2010–2013.
Abbreviations
- bp
Base pair
- cDNA
Complementary DNA
- cv
Cultivar
- DE
Differentially expressed
- DFB
Days from bloom
- DNA
Deoxyribonucleic acid
- dNTP
Deoxyribonucleotide triphosphate
- FC
Fold change
- FDR
False discovery rate
- GO
Gene ontology
- mRNA-Seq
Messenger ribonucleic acid sequencing
- MT
Mutant
- QTL
Quantitative trait locus
- RPM
Reads per million
- qRT-PCR
Real time reverse transcription polymerase chain reaction
- SE
Standard error
- SSR
Simple sequence repeat
- UTR
Untranslated region
- v1
Version 1
- WT
Wild-type.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CCN carried out SSR genotyping, transcriptome data analysis, qPCR validation of RNA-Seq results and wrote the manuscript. IG and AS participated in the design and coordination of the study and took care of the plant materials. RW carried out statistical analysis of gene expression patterns and contributed to the discussion of results. MSG conceived the study, participated in its design and coordination, contributed to the discussion of results and revised the manuscript. LC conceived the study, participated in its design and coordination, assisted lab experiments, contributed to the discussion of results and revised the manuscript. All authors read and approved the final manuscript.
Contributor Information
Chinedu Charles Nwafor, Email: chinedu.nwafor@fmach.it.
Ivana Gribaudo, Email: i.gribaudo@ivv.cnr.it.
Anna Schneider, Email: a.schneider@ivv.cnr.it.
Ron Wehrens, Email: ron.wehrens@fmach.it.
Maria Stella Grando, Email: stella.grando@fmach.it.
Laura Costantini, Email: laura.costantini@fmach.it.
References
- 1.Statistical report on world vitiviniculture . International Organization of Vine and Wine (OIV) 2011. [Google Scholar]
- 2.Pratt C. Reproductive anatomy in cultivated grapes - a review. Am J Enol Vitic. 1971;22:92–109. [Google Scholar]
- 3.Ledbetter CA, Ramming DW. Seedlessness in grapes. Hort Rev. 1989;11:159–184. [Google Scholar]
- 4.Seed development flow-chart in grape berrieshttp://michaelstriem.com/files/Seed_Development_Flow-Chart_in_Grape_Berries.pdf
- 5.Striem MJ, Spiegel-Roy P, Baron I, Sahar N. The degrees of development of the seed-coat and endosperm as separate subtraits of stenospermocarpic seedlessness in grapes. Vitis. 1992;31:149–155. [Google Scholar]
- 6.Varoquaux F, Blanvillain R, Delseny M, Gallois P. Less is better: new approaches for seedless fruit production. Trends Biotechnol. 2000;18:233–242. doi: 10.1016/S0167-7799(00)01448-7. [DOI] [PubMed] [Google Scholar]
- 7.Skinner DJ, Hill TA, Gasser CS. Regulation of ovule development. Plant Cell. 2004;16:S32–S45. doi: 10.1105/tpc.015933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jenik PD, Gillmor CS, Lukowitz W. Embryonic patterning in Arabidopsis thaliana. Annu Rev Cell Dev Bi. 2007;23:207–236. doi: 10.1146/annurev.cellbio.22.011105.102609. [DOI] [PubMed] [Google Scholar]
- 9.Devic M. The importance of being essential: EMBRYO-DEFECTIVE genes in Arabidopsis. C R Biol. 2008;331:726–736. doi: 10.1016/j.crvi.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 10.Berger F. Endosperm development. Curr Opin Plant Biol. 1999;2:28–32. doi: 10.1016/S1369-5266(99)80006-5. [DOI] [PubMed] [Google Scholar]
- 11.Huh JH, Bauer MJ, Hsieh TF, Fischer RL. Cellular programming of plant gene imprinting. Cell. 2008;132:735–744. doi: 10.1016/j.cell.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 12.Braybrook SA, Harada JJ. LECs go crazy in embryo development. Trends Plant Sci. 2008;139:624–630. doi: 10.1016/j.tplants.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 13.Breuninger H, Rikirsch E, Hermann M, Ueda M, Laux T. Differential expression of WOX genes mediates apical-basal axis formation in the Arabidopsis embryo. Dev Cell. 2008;14:867–876. doi: 10.1016/j.devcel.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 14.Yin T, Pan G, Liu H, Wu J, Li Y, Zhao Z, Fu T, Zhou Y. The chloroplast ribosomal protein L21 gene is essential for plastid development and embryogenesis in Arabidopsis. Planta. 2012;235:907–921. doi: 10.1007/s00425-011-1547-0. [DOI] [PubMed] [Google Scholar]
- 15.Dean G, Cao Y, Xiang D, Provart NJ, Ramsay L, Ahad A, White R, Selvaraj G, Datla R, Haughn G. Analysis of gene expression patterns during seed coat development in Arabidopsis. Mol Plant. 2011;4:1074–1091. doi: 10.1093/mp/ssr040. [DOI] [PubMed] [Google Scholar]
- 16.Dutt M, Dhekney SA, Soriano L, Kandel R, Grosser JW. Temporal and spatial control of gene expression in horticultural crops. Horticulture Res. 2014;1:14047. doi: 10.1038/hortres.2014.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le BH, Cheng C, Bui AQ, Wagmaister JA, Henry KF, Pelletier J, Kwong L, Belmonte M, Kirkbride R, Horvath S, Drews GN, Fischer RL, Okamuro JK, Harada JJ, Goldberg RB. Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc Natl Acad Sci. 2010;107:8063–8070. doi: 10.1073/pnas.1003530107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruan YL, Patrick JW, Bouzayen M, Osorio S, Fernie AR. Molecular regulation of seed and fruit set. Trends Plant Sci. 2012;17:1360–1385. doi: 10.1016/j.tplants.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 19.Doligez A, Bouquet A, Danglot Y, Lahogue F, Riaz S, Meredith P, Edwards J, This P. Genetic mapping of grapevine (Vitis vinifera L.) applied to the detection of QTLs for seedlessness and berry weight. Theor Appl Genet. 2002;105:780–795. doi: 10.1007/s00122-002-0951-z. [DOI] [PubMed] [Google Scholar]
- 20.Fanizza G, Lamaj F, Costantini L, Chaabane R, Grando MS. QTL analysis for fruit yield components in table grapes (Vitis vinifera L) Theor Appl Genet. 2005;111:658–664. doi: 10.1007/s00122-005-2016-6. [DOI] [PubMed] [Google Scholar]
- 21.Cabezas JA, Cervera MT, Ruiz-Garcia L, Carreno J, Martinez-Zapater JM. A genetic analysis of seed and berry weight in grapevine. Genome. 2006;49:1572–1585. doi: 10.1139/g06-122. [DOI] [PubMed] [Google Scholar]
- 22.Mejía N, Gebauer M, Muñoz L, Hewstone N, Muñoz C, Hinrichsen P. Identification of QTLs for seedlessness, berry size, and ripening date in a seedless × seedless progeny. Am J Enol Viticult. 2007;58:499–507. [Google Scholar]
- 23.Costantini L, Battilana J, Lamaj F, Fanizza G, Grando MS. Berry and phenology-related traits in grapevine (Vitis vinifera L.): From Quantitative Trait Loci to underlying genes. BMC Plant Biol. 2008;8:38. doi: 10.1186/1471-2229-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mejía N, Soto B, Guerrero M, Casanueva X, Houel C, Miccono MA, Ramos R, Le Cunff L, Boursiquot JM, Hinrichsen P, Adam-Blondon AF. Molecular, genetic and transcriptional evidence for a role of VvAGL11 in stenospermocarpic seedlessness in grapevine. BMC Plant Biol. 2011;11:57. doi: 10.1186/1471-2229-11-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bergamini C, Cardone MF, Anaclerio A, Perniola R, Pichierri A, Genghi R, Alba V, Forleo LR, Caputo AR, Montemurro C, Blanco A, Antonacci D. Validation assay of p3_VvAGL11 marker in a wide range of genetic background for early selection of stenospermocarpy in Vitis vinifera L. Mol Biotechnol. 2013;54:1021–1030. doi: 10.1007/s12033-013-9654-8. [DOI] [PubMed] [Google Scholar]
- 26.Hanania U, Velcheva M, Or E, Flaishman M, Sahar N, Perl A. Silencing of chaperonin 21, that was differentially expressed in inflorescence of seedless and seeded grapes, promoted seed abortion in tobacco and tomato fruits. Transgenic Res. 2007;16:515–525. doi: 10.1007/s11248-006-9044-0. [DOI] [PubMed] [Google Scholar]
- 27.Hanania U, Velcheva M, Sahar N, Flaishman M, Or E, Degani O, Perl A. The ubiquitin extension protein S27a is differentially expressed in developing flower organs of Thompson seedless versus Thompson seeded grape isogenic clones. Plant Cell Rep. 2009;28:1033–1042. doi: 10.1007/s00299-009-0715-1. [DOI] [PubMed] [Google Scholar]
- 28.Zhang C, Gong P, Wei R, Li S, Zhang X, Yu Y, Wang Y. The metacaspase gene family of Vitis vinifera L.: characterization and differential expression during ovule abortion in stenospermocarpic seedless grapes. Gene. 2013;528:267–276. doi: 10.1016/j.gene.2013.06.062. [DOI] [PubMed] [Google Scholar]
- 29.Torregrosa L, Fernandez L, Bouquet A, Boursiquot JM, Pelsy F, Martínez-Zapater JM. Origins and Consequences of Somatic Variation in Grapevine. In: Zapater JM, Blondon AM, Kole C, editors. Genetics, genomics, and breeding of grapes. New Hampshire: Science Publishers; 2011. pp. 68–92. [Google Scholar]
- 30.Schneider A, Raimondi S, Moreira FM, De Santis D, Zappia R, Marinoni D, Librandi N, Grando MS. Contribution to the identification of the main varieties of Calabria. Frutticoltura. 2009;71:46–55. [Google Scholar]
- 31.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Venturini L, Ferrarini A, Zenoni S, Tornielli GB, Fasoli M, Dal Santo S, Minio A, Buson G, Tononi P, Zago ED, Zamperin G, Bellin D, Pezzotti M, Delledonne M. De novo transcriptome characterization of Vitis vinifera cv. Corvina unveils varietal diversity. BMC Genomics. 2013;14:41. doi: 10.1186/1471-2164-14-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zenoni S, Ferrarini A, Giacomelli E, Xumerle L, Fasoli M, Malerba G, Bellin D, Pezzotti M, Delledonne M. Characterization of transcriptional complexity during berry development in Vitis vinifera Using RNA-Seq. Plant Physiol. 2010;152:1787–1795. doi: 10.1104/pp.109.149716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fasoli M, Dal Santo S, Zenoni S, Tornielli GB, Farina L, Zamboni A, Porceddu A, Venturini L, Bicego M, Murino V, Ferrarini A, Delledonne M, Pezzotti M. The grapevine expression atlas reveals a deep transcriptome shift driving the entire plant into a maturation program. Plant Cell. 2012;24:3489–3505. doi: 10.1105/tpc.112.100230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sweetman C, Wong DC, Ford CM, Drew DP. Transcriptome analysis at four developmental stages of grape berry (Vitis vinifera cv. Shiraz) provides insights into regulated and coordinated gene expression. BMC Genomics. 2012;13:691. doi: 10.1186/1471-2164-13-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coombe BG. Growth stages of the grapevine: adoption of a system for identifying grapevine growth stages. Aust J Grape Wine R. 1995;1:100–110. doi: 10.1111/j.1755-0238.1995.tb00086.x. [DOI] [Google Scholar]
- 37.Emanuelli F, Lorenzi S, Grzeskowiak L, Catalano V, Stefanini M, Troggio M, Myles S, Martinez-Zapater JM, Zyprian E, Moreira FM, Grando MS. Genetic diversity and population structure assessed by SSR and SNP markers in a large germplasm collection of grape. BMC Plant Biol. 2013;13:39. doi: 10.1186/1471-2229-13-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grape Genomehttp://genomes.cribi.unipd.it/
- 39.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler Transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.SAMtoolshttp://samtools.sourceforge.net/
- 41.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grape Genome toolshttp://genomes.cribi.unipd.it/grape/tools.php
- 43.Grimplet J, Van Hemert J, Carbonell-Bejerano P, Díaz-Riquelme J, Dickerson J, Fennell A, Pezzotti M, Martínez-Zapater JM. Comparative analysis of grapevine whole-genome gene predictions, functional annotation, categorization and integration of the predicted gene sequences. BMC Res Notes. 2012;5:213. doi: 10.1186/1756-0500-5-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Du Z, Zhou X, Ling Y, Zhang Z, Su Z. AgriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010;38(Web Server issue):W64–W70. doi: 10.1093/nar/gkq310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.REVIGO: reduce + visualize Gene Ontologyhttp://revigo.irb.hr/
- 46.Doligez A, Bertrand Y, Farnos M, Grolier M, Romieu C, Esnault F, Dias S, Berger G, François P, Pons T, Ortigosa P, Roux C, Houel C, Laucou V, Bacilieri R, Péros JP, This P. New stable QTLs for berry weight do not colocalize with QTLs for seed traits in cultivated grapevine (Vitis vinifera L.) BMC Plant Biol. 2013;13:217. doi: 10.1186/1471-2229-13-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:7. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pfaffl MW, Horgan GW, Dempfle L. Relative Expression Software Tool (REST©) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stacey MG, Patel A, McClain WE, Mathieu M, Remley M, Rogers EE, Gassmann W, Blevins DG, Stacey G. The Arabidopsis AtOPT3 protein functions in metal homeostasis and movement of iron to developing seeds. Plant Physiol. 2008;146:589–601. doi: 10.1104/pp.107.108183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wing RA, Yamaguchi J, Larabell SK, Ursin VM, McCormick S. Molecular and genetic characterization of two pollen-expressed genes that have sequence similarity to pectate lyases of the plant pathogen Erwinia. Plant Mol Biol. 1990;14:17–28. doi: 10.1007/BF00015651. [DOI] [PubMed] [Google Scholar]
- 51.The Arabidopsis Information Resource (TAIR)http://www.arabidopsis.org/
- 52.Rhee SY, Somerville CR. Tetrad pollen formation in quartet mutants of Arabidopsis thaliana is associated with persistence of pectic polysaccharides of the pollen mother cell wall. Plant J. 1998;15:79–88. doi: 10.1046/j.1365-313X.1998.00183.x. [DOI] [PubMed] [Google Scholar]
- 53.Sanders PM, Anhthu QB, Weterings K, McIntire KN, Hsu Y, Lee PY, Troung MT, Beals TP, Goldberg RB. Anther developmental defects in Arabidopsis thaliana male-sterile mutants. Sex Plant Reprod. 1999;11:297–322. doi: 10.1007/s004970050158. [DOI] [Google Scholar]
- 54.Ito T, Nagata N, Yoshiba Y, Ohme-Takagi M, Ma H, Shinozaki K. Arabidopsis MALE STERILITY1 encodes a PHD-type transcription factor and regulates pollen and tapetum development. Plant Cell. 2007;19:3549–3562. doi: 10.1105/tpc.107.054536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang C, Vizcay-Barrena G, Conner K, Wilson ZA. MALE STERILITY1 is required for tapetal development and pollen wall biosynthesis. Plant Cell. 2007;19:3530–3548. doi: 10.1105/tpc.107.054981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen W, Yu XH, Zhang K, Shi J, De Oliveira S, Schreiber L, Shanklin J, Zhang D. Male Sterile2 encodes a plastid-localized fatty acyl carrier protein reductase required for pollen exine development in Arabidopsis. Plant Physiol. 2011;157:842–853. doi: 10.1104/pp.111.181693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uniprothttp://www.uniprot.org/
- 58.Yang WC, Ye D, Xu J, Sundaresan V. The SPOROCYTELESS gene of Arabidopsis is required for initiation of sporogenesis and encodes a novel nuclear protein. Genes Dev. 1999;13:2108–2117. doi: 10.1101/gad.13.16.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu X, Huang J, Parameswaran S, Ito T, Seubert B, Auer M, Rymaszewski A, Jia G, Owen HA, Zhao D. The SPOROCYTELESS/NOZZLE gene is involved in controlling stamen identity in Arabidopsis. Plant Physiol. 2009;151:1401–1411. doi: 10.1104/pp.109.145896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li XC, Zhu J, Yang J, Zhang GR, Xing WF, Zhang S, Yang ZN. Glycerol-3-phosphate acyltransferase 6 (GPAT6) is important for tapetum development in Arabidopsis and plays multiple roles in plant fertility. Mol Plant. 2012;5:131–142. doi: 10.1093/mp/ssr057. [DOI] [PubMed] [Google Scholar]
- 61.Zhang Q, Li Z, Yang J, Li S, Yang D, Zhu Y. A calmodulin-binding protein from rice is essential to pollen development. J Plant Biol. 2012;1:8–14. doi: 10.1007/s12374-011-9184-5. [DOI] [Google Scholar]
- 62.Kanneganti V, Gupta AK. Wall associated kinases from plants - an overview. Physiol Mol Biol Plants. 2008;14:109–118. doi: 10.1007/s12298-008-0010-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wudick MM, Luu DT, Tournaire-Roux C, Sakamoto W, Maurel C. Vegetative and sperm cell-specific aquaporins of Arabidopsis highlight the vacuolar equipment of pollen and contribute to plant reproduction. Plant Physiol. 2014;164:1697–1706. doi: 10.1104/pp.113.228700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bartrina I, Otto E, Strnad M, Werner T, Schmülling T. Cytokinin regulates the activity of reproductive meristems, flower organ size, ovule formation, and thus seed yield in Arabidopsis thaliana. Plant Cell. 2011;23:69–80. doi: 10.1105/tpc.110.079079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Passarinho PA, Van Hengel AJ, Fransz PF, de Vries SC. Expression pattern of the Arabidopsis thaliana AtEP3/AtchitIV endochitinase gene. Planta. 2001;212:556–567. doi: 10.1007/s004250000464. [DOI] [PubMed] [Google Scholar]
- 66.Nesi N, Debeaujon I, Jond C, Stewart AJ, Jenkins GI, Caboche M, Lepiniec L. The TRANSPARENT TESTA16 locus encodes the ARABIDOPSIS BSISTER MADS domain protein and is required for proper development and pigmentation of the seed coat. Plant Cell. 2002;14:2463–2479. doi: 10.1105/tpc.004127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deng W, Chen G, Peng F, Truksa M, Snyder CL, Weselake RJ. Transparent Testa16 plays multiple roles in plant development and is involved in lipid synthesis and embryo development in canola. Plant Physiol. 2012;160:978–989. doi: 10.1104/pp.112.198713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu J, Chen N, Chen F, Cai B, Dal Santo S, Tornielli GB, Pezzotti M, Cheng ZM. Genome-wide analysis and expression profile of the bZIP transcription factor gene family in grapevine (Vitis vinifera) BMC Genomics. 2014;15:281. doi: 10.1186/1471-2164-15-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Favaro R, Pinyopich A, Battaglia R, Kooiker M, Borghi L, Ditta G, Yanofsky MF, Kater MM, Colombo L. MADS-box protein complexes control carpel and ovule development in Arabidopsis. Plant Cell. 2003;15:2603–2611. doi: 10.1105/tpc.015123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mizzotti C, Mendes MA, Caporali E, Schnittger A, Kater MM, Battaglia R, Colombo L. The MADS box genes SEEDSTICK and ARABIDOPSIS Bsister play a maternal role in fertilization and seed development. Plant J. 2012;70:409–420. doi: 10.1111/j.1365-313X.2011.04878.x. [DOI] [PubMed] [Google Scholar]
- 71.Dreni L, Pilatone A, Yun D, Erreni S, Pajoro A, Caporali E, Zhang D, Kater MM. Functional analysis of all AGAMOUS subfamily members in rice reveals their roles in reproductive organ identity determination and meristem determinacy. Plant Cell. 2011;23:2850–2863. doi: 10.1105/tpc.111.087007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Escobar-Restrepo JM, Huck N, Kessler S, Gagliardini V, Gheyselinck J, Yang WC, Grossniklaus U. The FERONIA receptor-like kinase mediates male–female interactions during pollen tube reception. Science. 2007;317:656–660. doi: 10.1126/science.1143562. [DOI] [PubMed] [Google Scholar]
- 73.Zou Y, Aggarwal M, Zheng WG, Wu HM, Cheung AY. Receptor-like kinases as surface regulators for RAC/ROP-mediated pollen tube growth and interaction with the pistil. AoB Plants. 2011;2011:plr017. doi: 10.1093/aobpla/plr017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yuan M, Wang S. Rice MtN3/saliva/SWEET family genes and their homologs in cellular organisms. Mol Plant. 2013;6:665–674. doi: 10.1093/mp/sst035. [DOI] [PubMed] [Google Scholar]
- 75.Shimada TL, Shimada T, Takahashi H, Fukao Y, Hara-Nishimura I. A novel role for oleosins in freezing tolerance of oilseeds in Arabidopsis thaliana. Plant J. 2008;55:798–809. doi: 10.1111/j.1365-313X.2008.03553.x. [DOI] [PubMed] [Google Scholar]
- 76.Belanger FC, Kriz AL. Molecular characterization of the major maize embryo globulin encoded by the Glb1 gene. Plant Physiol. 1989;91:636–643. doi: 10.1104/pp.91.2.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kagawa H, Yamauchi F, Hirano H. Soybean basic 7S globulin represents a protein widely distributed in legume species. FEBS Lett. 1987;226:145–149. doi: 10.1016/0014-5793(87)80568-9. [DOI] [Google Scholar]
- 78.Gaubier P, Raynal M, Hull G, Huestis GM, Grellet F, Arenas C, Pages M, Delseny M. Two different Em-like genes are expressed in Arabidopsis thaliana seeds during maturation. Mol Gen Genet. 1993;238:409–418. doi: 10.1007/BF00292000. [DOI] [PubMed] [Google Scholar]
- 79.Zeng Y, Zhao T, Kermode AR. A conifer ABI3-interacting protein plays important roles during key transitions of the plant life cycle. Plant Physiol. 2013;161:179–195. doi: 10.1104/pp.112.206946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Delmas F, Sankaranarayanan S, Deb S, Widdup E, Bournonville C, Bollier N, Northey JG, McCourt P, Samuel MA. ABI3 controls embryo degreening through Mendel’s I locus. Proc Natl Acad Sci. 2013;110:E3888–E3894. doi: 10.1073/pnas.1308114110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pastuglia M, Swarup R, Rocher A, Saindrenan P, Roby D, Dumas C, Cock JM. Comparison of the expression patterns of two small gene families of S gene family receptor kinase genes during the defence response in Brassica oleracea and Arabidopsis thaliana. Gene. 2002;282:215–225. doi: 10.1016/S0378-1119(01)00821-6. [DOI] [PubMed] [Google Scholar]
- 82.Vivancos J, Spinner L, Mazubert C, Charlot F, Paquet N, Thareau V, Dron M, Nogué F, Charon C. The function of the RNA-binding protein TEL1 in moss reveals ancient regulatory mechanisms of shoot development. Plant Mol Biol. 2012;78:323–336. doi: 10.1007/s11103-011-9867-9. [DOI] [PubMed] [Google Scholar]
- 83.Kaur J, Sebastian J, Siddiqi I. The Arabidopsis-mei2-lLike genes play a role in meiosis and vegetative growth in Arabidopsis. Plant Cell. 2006;18:545–559. doi: 10.1105/tpc.105.039156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu Y, Yan J, Zhang R, Qu X, Ren S, Chen N, Huang S. Arabidopsis FIMBRIN5, an actin bundling factor, is required for pollen germination and pollen tube growth. Plant Cell. 2010;22:3745–3763. doi: 10.1105/tpc.110.080283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Su H, Zhu J, Cai C, Pei W, Wang J, Dong H, Ren H. FIMBRIN1 is involved in lily pollen tube growth by stabilizing the actin fringe. Plant Cell. 2012;24:4539–4554. doi: 10.1105/tpc.112.099358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pérez-Castro R, Kasai K, Gainza-Cortés F, Ruiz-Lara S, Casaretto JA, Peña-Cortés H, Tapia J, Fujiwara T, González E. VvBOR1, the grapevine ortholog of AtBOR1, encodes an efflux boron transporter that is differentially expressed throughout reproductive development of Vitis vinifera L. Plant Cell Physiol. 2012;53:485–494. doi: 10.1093/pcp/pcs001. [DOI] [PubMed] [Google Scholar]
- 87.Yang C, Xu Z, Song J, Conner K, Vizcay Barrena G, Wilson ZA. Arabidopsis MYB26/MALE STERILE35 regulates secondary thickening in the endothecium and is essential for anther dehiscence. Plant Cell. 2007;19:534–548. doi: 10.1105/tpc.106.046391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Riechmann JL, Meyerowitz EM. The AP2/EREBP family of plant transcription factors. Biol Chem. 1998;379:633–646. doi: 10.1515/bchm.1998.379.6.633. [DOI] [PubMed] [Google Scholar]
- 89.Elliott RC, Betzner AS, Huttner E, Oakes MP, Tucker WQ, Gerentes D, Perez P, Smyth DR. AINTEGUMENTA, an APETALA2-like gene of Arabidopsis with pleiotropic roles in ovule development and floral organ growth. Plant Cell. 1996;8:155–168. doi: 10.1105/tpc.8.2.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lin WC, Shuai B, Springer PS. The Arabidopsis LATERAL ORGAN BOUNDARIES-domain gene ASYMMETRIC LEAVES2 functions in the repression of KNOX gene expression and in adaxial-abaxial patterning. Plant Cell. 2003;15:2241–2252. doi: 10.1105/tpc.014969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Evans MMS. The indeterminate gametophyte1 gene of maize encodes a LOB domain protein required for embryo sac and leaf development. Plant Cell. 2007;19:46–62. doi: 10.1105/tpc.106.047506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang H, Tang W, Zhu C, Perry SE. A chromatin immunoprecipitation (ChIP) approach to isolate genes regulated by AGL15, a MADS domain protein that preferentially accumulates in embryos. Plant J. 2002;32:831–843. doi: 10.1046/j.1365-313X.2002.01455.x. [DOI] [PubMed] [Google Scholar]
- 93.Rowland O, Ludwig AA, Merrick CJ, Baillieul F, Tracy FE, Durrant WE, Fritz-Laylin L, Nekrasov V, Sjölander K, Yoshioka H, Jones JD. Functional analysis of Avr9/Cf-9 rapidly elicited genes identifies a protein kinase, ACIK1, that is essential for full Cf-9-dependent disease resistance in tomato. Plant Cell. 2005;17:295–310. doi: 10.1105/tpc.104.026013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Camehl I, Oelmüller R. Do ethylene response factors-9 and −14 repress PR gene expression in the interaction between Piriformospora indica and Arabidopsis? Plant Signal Behav. 2010;5:932–936. doi: 10.4161/psb.5.8.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Libault M, Wan J, Czechowski T, Udvardi M, Stacey G. Identification of 118 Arabidopsis transcription factor and 30 ubiquitin-ligase genes responding to chitin, a plant-defense elicitor. Mol Plant Microbe Interact. 2007;20:900–911. doi: 10.1094/MPMI-20-8-0900. [DOI] [PubMed] [Google Scholar]
- 96.Jensen PJ, Fazio G, Altman N, Praul C, McNellis TW. Mapping in an apple (Malus x domestica) F1 segregating population based on physical clustering of differentially expressed genes. BMC Genomics. 2014;15:261. doi: 10.1186/1471-2164-15-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1: Sample collection. Three key time points along grape berry development corresponding to stages E-L 15 (single flowers in compact groups), E-L 27 (young berries enlarging) and E-L 38 (berries harvest-ripe) of the modified E-L system 36 were matched to the number of days from bloom (DFB) and could be assigned to two main categories: “before” (E-L 15) and “after” (E-L 27 and 38) fertilization. (A) diagram showing the match of sampling dates (expressed as E-L stages) to days from bloom in reference 4. (B) picture of the materials collected from the two lines at each sampling date. (DOCX 233 KB)
Additional file 2: Table S1: Genotypic characterization of the wild-type and the mutant. Fifty-eight SSR (simple sequence repeat) markers, spread across the nineteen chromosomes of grapevine genome, were used to genotype the wild-type and the mutant. Marker details and PCR conditions are described in Methods. Symbols: * SSR markers commonly used to discriminate grapevine varieties, - indicates homozygous or null allele. (XLSX 35 KB)
Additional file 3: Text S1: Description of the procedure adopted to rank the transcripts by order of magnitude. P-values were computed to reflect the significance of the difference between 2 counts (n1 and n2 corresponding to any two library combination out of the six libraries, independently of the genotype) using a binominal model. (DOCX 117 KB)
Additional file 4: Table S2: List of the genes analyzed in real-time PCR and primers used for their amplification. The table reports the gene IDs, gene annotations and sequences of the primer sets used to analyze the transcriptional profile of 14 genes in addition to the two constitutive genes used as reference for normalization. (XLSX 23 KB)
Additional file 5: Table S3: Summary of read mapping to v1_mRNA version of 12x grapevine genome draft annotation. Short-read alignment and mapping of all the reads were carried on the 12x v1 annotation of the grapevine genome PN40024 38 using BWA (Burrows Wheeler Aligner) software 39. The mapping results were processed with SAMtools 40 to extract for each transcript the number of mapped reads and determine, whether their mapping position is unique. Reads mapping to several positions on the reference sequence with the same “mapping quality” (i.e. number of mismatches and quality of the bases generating the mismatches) were attributed at random to one of them with a “0” mapping quality. (XLSX 11 KB)
Additional file 6: Table S4: Distribution of mapped reads among genomic features. A Python script was developed to determine the distribution of mapped reads among genomic features in the wild-type and the mutant. (XLSX 13 KB)
Additional file 7: Table S5: List of transcripts ranked by order of magnitude. P-values (or scores) were computed to reflect the significance of the difference between 2 counts (n1 and n2 corresponding to any two library combination out of the six libraries, independently of the genotype) using a binomial model. The model is described in Additional file 3. The p-values were log-transformed in order to allow for greater numerical stability in comparing extreme values. The sign of the p-value reflects the direction of the comparison (whether n1 is greater or lesser than n2). The smaller is the absolute p-value, the more significant is the difference between the counts. Next all the p-values and the ratios of expression between the counts were considered to compute a ranking value for each transcript. Afterwards the ranking values were used to sort the transcripts and show on top the biggest differences in expressions between two of the libraries. (XLSX 9 MB)
Additional file 8: Table S6: Transcript abundance measurement at each developmental stage. A lower limit of detection for expression estimate was designated to be an RPM of 0.5 or, if the RPM value was less than 0.5, at least five uniquely mapped reads with identity > 98% over 100 bp. (XLSX 11 KB)
Additional file 9: Figure S2: Gene overlap between the wild-type and the mutant in the first two and last two developmental stages. (A) Venn diagram showing shared and unique expressed genes between the wild-type and the mutant during the first two developmental stages E-L 15 and E-L 27. (B) Venn diagram showing shared and unique expressed genes between the wild-type and the mutant during the last two developmental stages E-L 27 and E-L 38. Abbreviations: WT = wild-type, MT = mutant. (PDF 146 KB)
Additional file 10: Table S7: List of all differentially expressed genes in the pairwise comparison of developmental stages in the wild-type and the mutant. Differential expression (DE) analysis was carried out on the raw uniquely mapped reads using the software DESeq with the following parameters: false discovery rate (FDR) ≤ 0.05, log2-fold change (FC) > 1. An in-house R script was written to group DE genes with similar expression pattern based on the adjusted p-values, such that transcripts displaying significant negative differences, significant positive differences and insignificant differences between time points were fitted to -1, 1 and 0, respectively. The first position in each triplet corresponds to the shift from stage E-L 15 to E-L 27, the second position to the shift from stage E-L 27 to E-L 38 and the third position to the shift from stage E-L 15 to E-L 38. The different sheets report the genes differentially expressed in both the wild-type and the mutant, specific to the wild-type and the mutant, respectively. Abbreviations: nd = not detected in a pairwise comparison; imp (impossible to divide) = when the basemean of one stage in a pairwise comparison is the denominator with value 0; hyphen (-) = when the basemean of one stage in a pairwise comparison is the numerator with value 0 (the corresponding logarithm doesn’t exist). (XLS 524 KB)
Additional file 11: Table S8: Functional enrichment analysis in the whole set of differentially expressed genes in the wild-type and the mutant. Using a hypergeometric test in the AgriGO toolkit, a gene ontology (GO) term was considered significantly enriched, if the false discovery rate (FDR) was < 0.05 and p-value < 0.01 when compared to all gene transcripts annotated in the reference genome (supported in AgriGO). The two sheets report the results of functional enrichment analysis for the differentially expressed genes in the wild-type and the mutant, respectively. Abbreviations: P = biological process, F = molecular function, C = cellular component, BG = background, Ref = reference. Asterisks indicate terms commonly enriched in the wild-type and the mutant. (XLSX 61 KB)
Additional file 12: Table S9: Functional enrichment analysis within groups of differentially expressed genes in the wild-type and the mutant. Using a hypergeometric test in the AgriGO toolkit, a gene ontology (GO) term was considered significantly enriched, if the false discovery rate (FDR) was < 0.05 and p-value < 0.01 when compared to all gene transcripts annotated in the reference genome (supported in AgriGO). The different sheets report the results of functional enrichment analysis within specific clusters of differentially expressed genes in the wild-type and the mutant. Abbreviations: P = biological process, F = molecular function, C = cellular component, BG = background, Ref = reference. Asterisks indicate terms commonly enriched in the wild-type and the mutant clusters 11 and 16. (XLSX 182 KB)
Additional file 13: Table S10: Proportion of RNA-Seq-derived candidate genes in the physical proximity of seed-related QTLs. Genes that were specifically expressed in either clone (with or without significant modulation between developmental stages) were mapped to each of eight chromosomes carrying seed-related QTLs, identified previously by [23, 46]. The proportion of genes falling within a 10-Mb region centered on each QTL peak was calculated, as described in [96]. Abbreviations: chr = chromosome. (XLSX 9 KB)