

Abstract
The primary function of the blood-brain barrier (BBB) /neurovascular unit is to protect the CNS from potentially harmful xenobiotic substances and maintain CNS homeostasis. Restricted access to the CNS is maintained via a combination of tight junction proteins as well as a variety of efflux and influx transporters that limits the transcellular and paracellular movement of solutes. Of the transporters identified at the BBB, P-glycoprotein (P-gp) has emerged as the transporter that is the greatest obstacle to effective CNS drug delivery. In this chapter we provide data to support intracellular protein trafficking of P-gp within cerebral capillary microvessels as a potential target for improved drug delivery. We show that pain induced changes in P-gp trafficking are associated with changes in P-gp’s association with caveolin-1, a key scaffolding/trafficking protein that co-localizes with P-gp at the luminal membrane of brain microvessels. Changes in co-localization with the phosphorylated and non-phosphorylated forms of caveolin-1, by pain, are accompanied by dynamic changes in the distribution, relocalization and activation of P-gp “pools” between microvascular endothelial cell subcellular compartments. Since redox sensitive processes may be involved in signaling disassembly of higher order structures of P-gp, we feel that manipulating redox signaling, via specific protein targeting at the BBB, may protect disulfide bond integrity of P-gp reservoirs and control trafficking to the membrane surface providing improved CNS drug delivery. The advantage of therapeutic drug “relocalization” of a protein is that the physiological impact can be modified, temporarily or long term, despite pathology-induced changes in gene transcription.
Keywords: P-glycoprotein, protein trafficking, peripheral inflammatory pain, ATP-binding cassette transporter, blood-brain barrier, drug delivery, neurovascular unit, redox sensitive protein pools, reactive oxygen species
Introduction
The blood-brain barrier (BBB) is a formidable physical and biochemical barrier to effective drug delivery to the brain, thus limiting the ability to effectively treat central nervous system (CNS) disorders. Within the past decade, intense research efforts have focused on directly targeting the BBB for optimization of drug delivery (Ronaldson & Davis, 2013). Such BBB targets include influx organic anion transporting peptides (Oatp 1a4) and efflux P-glycoprotein (P-gp) transporters that are expressed at the level of the brain microvascular endothelium. Instead of physically circumventing the BBB by using various tissue disruption/damaging, mechanical techniques such as osmotic shock, microdialysis or intracerebroventricular (icv) injection, targeting transporters enable development of novel chemical approaches to utilize endogenous barrier components to deliver drugs to the brain thereby providing a unique opportunity to improve efficacy of existing therapies while promoting development of new ones (Ronaldson & Davis, 2013). This chapter provides an overview of BBB biology and focuses on the “ 800 pound gorilla”, known as the P-gp transporter, which is responsible for many failures of CNS developmental therapeutics. Furthermore, we highlight various techniques that have been developed to circumvent the BBB for CNS drug delivery, with a particular emphasis on opportunities provided by targeting endogenous BBB transporter systems, such as P-gp and Oatp 1a4.
The Blood-Brain Barrier/Neurovascular Unit
The neurovascular unit (NVU) is comprised of cellular constituents (i.e., endothelial cells, astrocytes, microglia, pericytes, neurons) and the extracellular matrix (Ronaldson & Davis, 2013) (Figure 1). The concept of the NVU emphasizes that brain function and dysfunction requires coordinated interaction between the various NVU components. Disruption of any NVU component, either as a result of a physiological, pathological or pharmacological stressor, can alter BBB integrity, subsequently modifying brain microvascular permeability and drug delivery (Ronaldson & Davis, 2013; Rolfe & Brown, 1997; Hawkins & Davis, 2005).
Figure 1. Transporters expressed in cells comprising the neurovascular unit (NVU).

A large number of transporters are expressed on capillary endothelial cells, astrocytes, microglia, and neurons. Transporter systems aid in transport of nutrients, peptides, drugs and ions into the brain parenchyma and as well as efflux of waste and potentially neurotoxic substance out of the brain. Arrows indicate the proposed direction of substrate transport. (Adapted from (Sanchez-Covarrubias et al., 2014)
Endothelial Cells and the Blood-Brain Barrier
The CNS is the most sensitive and critical organ system in the human body. Therefore, proper function requires precise regulation of the brain extracellular milieu. Additionally, CNS metabolic demands are considerable, with the CNS accounting for approximately 20% of overall oxygen consumption in humans (Oldendorf et al., 1977). The interface between the brain and the systemic circulation must possess highly selective and efficient mechanisms that are capable of facilitating nutrient transport, regulating ion balance, and providing a barrier to potentially toxic substances. Specifically, brain entry of some substances must be permitted while permeation of others must be limited. This homeostatic function of the cerebral microvasculature occurs primarily at the level of brain microvascular endothelial cells, the principal cell type of the BBB/NVU.
Compared to peripheral vasculature, BBB endothelial cells are characterized by increased mitochondrial content, high transendothelial electrical resistance (TEER), minimal pinocytosis activity, and lack of fenestrations (Hawkins & Davis, 2005; Oldendorf et al., 1977). Increased mitochondrial content is essential for these cells to maintain various active transport mechanisms such as those utilized to transport ions, nutrients, and waste products into and out of the brain parenchyma, thus contributing to the precise regulation of the CNS microenvironment and ensures proper neuronal function. Cell polarity of endothelial cells is ascribed to differing functional expression of transporter proteins and metabolic enzymes that are differentially expressed on the luminal and abluminal membranes. Such different biochemical characteristics of the luminal and abluminal plasma membranes further contribute to the high selectivity of the BBB (Betz et al., 1980; Sanchez del Pino et al., 1995; Vorbrodt & Dobrogowska, 2003).
Of the many transporters expressed at the BBB endothelium, several have been implicated in influx and/or efflux of drugs into the CNS. Examples of efflux transporters include P-gp (Bendayan et al., 2002), Breast cancer resistance protein (BCRP in humans; Bcrp in rodents) and Multidrug resistance proteins (MRPs in humans; Mrps in rodents). Additionally, transporters that facilitate drug entry into the brain are also expressed at the BBB. For example, OATPs (humans)/Oatps (rodents), whose transport are bidirectional and have been shown to mediate drug transport into the brain, are also expressed on capillary endothelial cells. Other examples of uptake transporters that are endogenously expressed at the BBB include organic anion transporters (OATs in humans; Oats in rodents), organic cation transporters (OCTs in humans; Octs in rodents), nucleoside transporters, monocarboxylate transporters (MCTs in humans; Mcts in rodents), and mechanisms for peptide transport. Shown in Figure 1 is a representation of the location of several of these transporters on NVU cell types.
Transport Across the Brain Barriers
Several disorders of the CNS remain difficult to treat pharmacologically due to an inability of many drugs to attain efficacious concentrations in the brain. In part, this is due to active efflux transport processes that restrict blood-to-brain drug uptake. However, drugs may still cross brain barriers (i.e. BBB, brain cerebral spinal fluid (BCSF) barrier) and accumulate in the CNS by various mechanisms that favor uptake including passive diffusion, transcytosis, carrier-mediated transport, and endocytosis. A brief description of each process is provided in Figure 2.
Figure 2. Methods of drug transport across the blood - brain barrier.
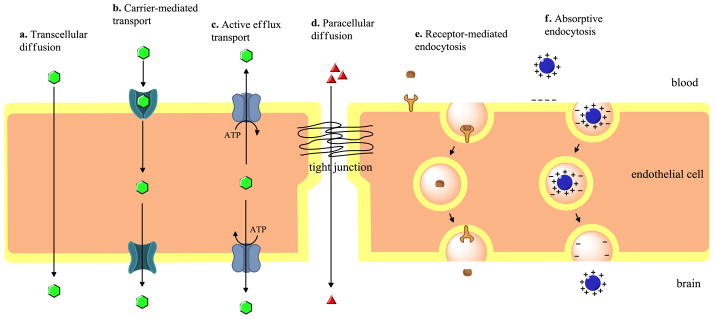
This figure describes the various routes of delivery of xenobiotics across the BBB from transcytosis to paracellular delivery to various types of transporter-based delivery. (Adapted from (Sanchez-Covarrubias et al., 2014)
P-glycoprotein
P-gp is a 170-kDa efflux transporter encoded by the MDR gene (Gottesman et al., 1995). Two MDR isoforms have been identified in human tissues, MDR-1 and MDR-2 (Chen et al., 1986; Roninson et al., 1986); however, P-gp expression in rodent tissues is encoded by three distinct mdr isoforms (mdr-1a, mdr-1b, and mdr-2 in rats and mdr1, mdr2 and mdr3 in mice). While over expression of MDR-1/mdr-1a/mdr1b confers the MDR phenotype (Gottesman et al., 1995; Ueda et al., 1987), MDR-2/mdr-2 is primarily expressed in the liver and is involved in transport of phosphatidylcholine into bile (Gottesman et al., 1995; Smit et al., 1993). In humans, the MDR1 gene product is 1280 amino acids in length and consists of two homologous halves, each containing six transmembrane domains. Each homologous half also contains one ATP-binding site. Two to four glycosylation sites have been located on the first extracellular loop. Although glycosylation is necessary for localization to the plasma membrane, it is not necessary for transport function. Studies using glycosylation-deficient P-gp found lower levels of this transporter at the cell surface but transport function remained unaffected (Gribar et al., 2000). Mature P-gp is phosphorylated on the linker region between the two homologous halves (TM6-TM7) (Gottesman et al., 1995). Phosphorylation may protect non-glycosylated P-gp from being degraded by endoplasmic reticulum proteases or from undergoing proteasomal degradation prior to glycosylation and trafficking to the plasma membrane. For example, in vitro studies have demonstrated that activation of Pim-1 kinase, a serine/threonine kinase, decreased P-gp degradation and increased cell surface expression (Xie et al., 2010), which suggests that phosphorylation may be a critical step in processing of a mature and functional P-gp transporter and a potential point to target for improved CNS drug delivery (Ronaldson & Davis, 2013). Studies using rat brain endothelial cells in vitro have also demonstrated that the physical interaction between P-glycoprotein with caveolin-1 is enhanced by tyrosine-14-phosphorylation of caveolin-1 (Barakat et al., 2007).
Since its initial discovery in Chinese hamster ovary cells (Ling & Thompson, 1974), P-gp expression has been observed in multiple barrier and non-barrier cell types, including kidney, liver, gastrointestinal tract, placenta, and testes (Juliano & Ling, 1976). In the brain, P-gp is localized to both the luminal and abluminal membranes of the BBB endothelium (Bendayan et al., 2006) and to the apical plasma membrane of choroid plexus epithelial cells (Rao et al., 1999). Expression of P-gp at the BBB likely evolved to protect the CNS from exposure to potentially neurotoxic xenobiotics and to maintain the precise homeostatic environment required for proper neuronal function (Sharom, 2007). Evolution favors adaptation and the maintenance of homeostasis and this is why P-gp has evolved as central to brain function during times of stress. The importance of P-gp’s role in CNS protection is highlighted by studies using mdr1a/mdr1b knockout mice. Mdr-1a/mdr1b null mice showed a 100-fold increase in brain uptake of ivermectin, a neurotoxic pesticide, when compared to their wild-type counterparts. Furthermore, mdr1a/mdr1b null mice displayed multiple symptoms of ivermectin toxicity (i.e., tremors, paralysis, coma, and death) that are directly attributed to increased brain penetration (Schinkel et al., 1994). Similar observations were reported in collies where increased sensitivity to ivermectin was directly correlated to a complete absence of the mdr1 gene (Doran et al., 2005). Additionally, P-gp expression has been detected in brain parenchyma cellular compartments such as astrocytes, microglia, and neurons (Golden & Pardridge, 1999; Schlachetzki & Pardridge, 2003; Ronaldson et al., 2004; Lee et al., 2001; Volk et al., 2004). Each of these observations point to the real possibility that P-gp has developed and evolved as the primary “gate keeper” that is critical in maintaining a safe, non-toxic environment in the brain and CNS that limits entry of many potentially toxic drugs such as morphine and other opioids.
P-glycoprotein also has an immense substrate and drug profile that renders it a formidable obstacle to any/all CNS drug delivery. In fact, the number of compounds known to be P-gp substrates is continuously expanding as more and more research is done. P-gp substrates are generally non-polar, weakly amphipathic compounds that vary considerably in molecular size. For example P-gp is known to transport small molecule drugs such as daunorubicin (563.99 Da) as well as larger molecules such as actinomycin D (1255.42 Da) (Sharom, 2007). The list of known substrate categories includes, but is not limited to, antibiotics, calcium channel blockers, cardiac glycosides, chemotherapeutics, immunosuppressants, anti-epileptics, anti-depressants, and HIV-1 protease inhibitors (Sun et al., 2004; Demeule et al., 2002). Additionally, recent studies have demonstrated that many HMG CoA reductase inhibitors (i.e., pitavastatin, pravastatin) are transported across biological membranes by P-gp (Shirasaka et al., 2011a; Shirasaka et al., 2011b). Studies have also shown that opioid analgesic drugs such as morphine and the opioid peptide DPDPE are directly extruded from brain tissue by P-gp (Figure 3) (Seelbach et al., 2007; Ronaldson et al., 2011; Chen & Pollack, 1998; Chen & Pollack, 1997). Endogenous substrates of P-glycoprotein may include cytokines, lipids, steroid hormones, and peptides (Sharom, 2007).
Figure 3. P-glycoprotein transporter – can it be targeted?

Although preclinical evidence suggests that P-glycoprotein transport activity can be modulated with small molecule inhibitors, clinical evidence indicates that this approach cannot work. Use of small molecule inhibitors to block P-glycoprotein in clinical settings has resulted in significant toxicity associated with increased deposition of drug in peripheral tissues or due to high concentrations of the inhibitor itself. Morphine is a good example of the perils of blocking P-glycoprotein to increase drug delivery to the brain. In the setting of functional P-glycoprotein, only 0.02% of systemic morphine is able to permeate the blood-brain barrier. Blockade of P-glycoprotein at the blood-brain barrier would significantly increase this amount and lead to clinically significant adverse drug reactions (i.e., seizures).
Additionally, several substrates of P-gp have been found to be competitive transport inhibitors. Examples of such drugs include calcium channel blockers (i.e., verapamil), antipsychotics (i.e., chlorpromazine), immunosuppressive agents (i.e., cyclosporine A) and the cyclosporine A analog PSC 833 (i.e., valspodar) (Sharom, 2007). HMG CoA reductase inhibitors have also been found to block P-gp transport function and several studies are exploring the possibility of using these drugs to reverse P-gp induced drug resistance in tumor cells (Goard et al., 2010).
Drug Delivery to the Central Nervous System: Strategies developed to circumvent brain barrier sites
The blood-brain barrier (BBB) is a formidable obstacle to drug delivery. Transcellular permeability of compounds across the BBB is complex and regulated by expression of various transporter proteins (Figure 2). In fact, the overall balance of these transporters is a critical determinant in CNS permeation of multiple therapeutic drugs. Restricted entry of therapeutic compounds into the CNS results in ineffectual treatment of CNS disorders such as epilepsy, brain cancer, HIV-associated neurocognitive disease, cerebral hypoxia, ischemic stroke, and peripheral inflammatory pain. Therefore, several therapeutic strategies have been developed to circumvent the BBB and improve CNS drug delivery. Among those developed, some efforts have involved invasive procedures such as forced, mechanical opening of the BBB that can cause undesirable side effects and extensive tissue pathology. Other efforts have focused on circumventing those efflux transporters (i.e., P-gp, MRPs/Mrps, BCRP/Bcrp) that severely limit entry of therapeutic compounds into the brain. While efflux transporter inhibition has achieved modest success in improving CNS drug permeability, their utility is greatly limited by adverse drug reactions that may occur due to increased drug concentrations in the brain and other peripheral tissues. Recently, there is a growing interest in exploiting other transport systems to improve drug delivery, including targeting endogenous influx transporters expressed at brain barrier sites such as Oatp1a4. The following section will provide a brief overview of several methods for drug delivery to the CNS that have been developed to date as well as suggest novel approaches based on recent findings.
Inhibition of Brain Barrier Efflux Transporters
In order to circumvent efflux transporters at brain barrier sites, particularly P-gp, pharmacological inhibitors have been developed with the intent of enabling greater penetration of drugs into the CNS. Such studies have shown mixed results with regards to efficacy and safety of these inhibitory compounds. The first generation of P-gp inhibitors were identified in the early 1980s. Despite their ability to inhibit P-gp transport activity and increase delivery cellular drug permeation, the doses required to be effective inhibitors were extremely high and resulted in both toxicity and unwanted pharmacokinetic interactions (Thomas & Coley, 2003). Second generation inhibitors, such as PSC833 (i.e., valspodar), are much more potent than their predecessors and do exhibit less toxicity. However, PSC833 demonstrated disappointing results in clinical studies, with only modest increases in CNS drug delivery (Thomas & Coley, 2003). Additionally, this generation of inhibitors significantly inhibited metabolism and excretion of cytotoxic agents. These unexpected effects necessitated reduction in chemotherapy doses to levels that were no longer efficacious (Thomas & Coley, 2003).
An ability to selectively modulate P-gp activity has the potential to impact treatment of numerous CNS pathologies and alter disease progression. Accumulated research suggests that P-gp affects CNS drug uptake in a plethora of diseases including inflammation, pain, epilepsy, HIV, brain cancer (Seelbach et al., 2007; McCaffrey et al., 2012; Miller et al., 2008; Zhang et al., 2012) and cerebral ischemia (Spudich et al., 2006; Miller et al., 2008 and Thompson & Ronaldson in the current volume). These data support a role for P-gp in treatment response. Although there is consensus that P-gp plays a role in the ability to treat disease at the level of the CNS, there are inconsistencies in the data particularly when P-gp inhibitors are used. Further, the failure of available P-gp inhibitors to improve clinical outcome has been discouraging. There is also some evidence that P-gp is involved in Alzheimer disease (Cirrito et al., 2005; Hartz et al., 2010); decreased P-gp function has been observed in Alzheimer patients compared to healthy control subjects (van Assema et al., 2012). These data suggest that P-gp can impact disease progression. However, there is a study showing that β-amyloid clearance in a rat Alzheimer disease model did not decrease after treatment with P-gp inhibitors (Ito et al., 2006). Each of the pathologies in which P-gp potentially plays a role is complex and the contribution of P-gp to treatment response or pathogenesis is still being defined. The complexity of this issue indicates that an understanding of signaling and trafficking pathways that lead to increased (or decreased) P-gp activity in each pathological condition is necessary to identify novel drug targets and regulate P-gp in a context-dependent way. A more nuanced approach to P-gp regulation may succeed where direct inhibition of P-gp has failed.
Recent work elucidating mechanisms that regulate changes in P-gp functional expression have suggested discrete signaling pathways that can be targeted to impair P-gp function and improve CNS drug delivery. Targeting such pathways is an attractive alternative to global inhibition of P-gp as it can lead more precise control of P-gp in specific target tissues and/or preservation of basal P-gp activity, which is critical for neuroprotection (Ronaldson & Davis, 2013). Recently, the role of sphingolipid signaling in regulating basal levels of P-gp activity was investigated. Using a confocal-based activity assay that used rat brain capillaries, investigators determined that basal activity levels were regulated via signaling through the sphingosine-1-phosphate receptor (S1PR) (Cannon et al., 2012). Exposure of brain capillaries to sphingosine-1-phosphate (S1P), a bioactive lipid metabolite and endogenous ligand for S1PR1, resulted in a reduction in P-gp-mediated drug efflux (Cannon et al., 2012). Removal of S1P from the capillary media restored P-gp efflux activity to levels seen in the control group, demonstrating that signaling via S1PR can allow for transient modulation of P-gp-mediated efflux activity (38). Changes in P-gp function observed in isolated brain capillaries were validated in vivo using the in situ perfusion technique. Animals treated with S1P or the S1P analog, fingolimod (FTY720), exhibited increased brain uptake of radiolabeled verapamil, loperamide, and paclitaxel, demonstrating reduced P-gp activity in vivo (Cannon et al., 2012). The use of an S1PR1-specific antagonist or inhibition of G-protein-coupled receptor signaling blocked this effect (Cannon et al., 2012). While targeting S1PR1 directly may prove to be a useful method for controlling efflux transport at the BBB, data from this study also suggest that additional targets for therapeutic development lie downstream in the S1PR signaling pathway. Characterization of the signaling events that result in S1P production ((Cannon et al., 2012) and references therein) and the recent finding that Mrp1 is critical for the sphingolipid signaling events that alter P-gp activity at the mouse BBB (Cartwright et al., 2013) indicate that there is also potential for therapeutic intervention upstream of S1PR signaling.
Our studies measuring the effects of peripheral inflammatory pain, induced by λ-carrageenan injection into the rat footpad, on CNS drug delivery indicate that the protein-protein interactions that govern P-gp trafficking and complex formation are also potential drug targets. Protein transport from one subcellular location to another results directly from specific protein-protein interactions that are governed by unique motifs encoded within a protein’s primary sequence. P-gp has a binding motif in its N-terminus for caveolin-1, a key scaffolding protein. Studies using rat brain endothelial cells in vitro have demonstrated that the physical interaction between P-glycoprotein with caveolin-1 is enhanced by tyrosine-14-phosphorylation of caveolin-1, and that the binding of P-glycoprotein to caveolin-1 negatively regulates P-glycoprotein function (Barakat et al., 2007). Our in vivo work showed that peripheral inflammatory pain causes a redistribution of P-gp, total caveolin-1 and tyrosine-14-phosphorylated caveolin-1 in rat brain microvessels suggesting movement of these proteins to different subcellular locations (McCaffrey et al., 2012).(Figure 5). Trafficking of these proteins is accompanied by increased P-gp activity (McCaffrey et al., 2012) and decreased accumulation of morphine in the brain (Seelbach et al., 2007). These data suggest that a further characterization of peripheral inflammatory pain-induced trafficking events will identify potential therapeutic targets. Additionally, demonstration of a vascular endothelial growth factor (VEGF)-induced rapid reduction of P-gp efflux function at the BBB through endocytosis further highlights the potential that therapeutic manipulation of a trafficking pathway may have in temporarily reducing P-glycoprotein efflux activity at the luminal membrane (Hawkins et al., 2010).
Figure 5. P-glycoprotein (P-gp) Trafficking within Caveolin-Enriched Domains modulated by Peripheral Inflammatory Pain.

These data show that the previously characterized increase, in vivo, of P-glycoprotein functional activity, at 3 h post inflammatory pain, was associated with a dynamic redistribution of P-glycoprotein between micro-vascular, endothelial cells, subcellular compartments. The top panel shows the typical OptiPrep density gradient profile of fractions from previously intact rat microvessels. P-gp and the key scaffolding protein caveolin were both shown to be associated with luminal membrane enriched fractions 15 and 16 (Bottom panel) and dynamically trafficked to higher density fractions after the pain stimulus. No trafficking change was noted for the brain efflux transporter Mrp4 after a peripheral pain stimulus to the rats. (Adapted from (McCaffrey et al., 2012)
Peripheral inflammatory pain causes disassembly of high molecular weight complexes containing P-gp (McCaffrey et al., 2012). The disassembly process includes a loss or rearrangement of disulfide bonds that are accessible to aqueous reducing agents (McCaffrey et al., 2012). These data suggest that redox processes are involved in the activation/trafficking of P-gp. Recent data indicate that drug-mediated ATPase activity in P-gp depends on formation of specific disulfide bonds after the binding of the ATP molecule (Loo et al., 2013). The redox dependent changes we see could be direct, i.e. rearrangements of disulfide bonds within P-gp complexes catalyzed by enzymatic processes, or occur via redox dependent signaling pathways. An ability to block disassembly of higher order P-gp complexes and maintain a major portion of the P-gp in an inactive form in reservoirs would provide a unique opportunity to titrate P-gp activity as described in Figure 6. This would allow maintenance of basal P-gp activity to protect against xenobiotic toxicity while preventing the increased P-gp activity that inhibits effective CNS drug delivery. With the increasing availability of redox-based therapeutics, characterizing the redox reactions and signaling that occur during P-gp trafficking/activation after a peripheral inflammatory pain stimulus could suggest new applications of redox-based drugs to improve CNS drug delivery (Figure 6).
Figure 6.
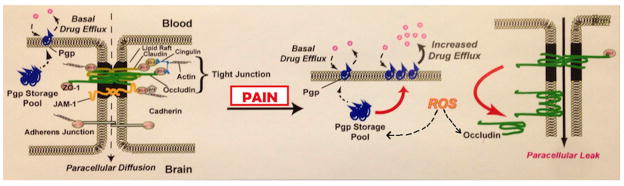
Model Indicating Proposed Molecular Targets of Peripheral Inflammatory Pain-Induced Reactive Oxygen Species (ROS) that Account for the Compromised Blood-Brain Barrier Observed During Peripheral Inflammatory Pain.
Our studies measuring the effects of peripheral inflammatory pain on CNS drug delivery indicate that transforming growth factor-β1 (TGF-β1) signaling could also be manipulated to improve drug uptake into the brain. TGF-β1 is a critical regulator of brain microvascular homeostasis (Lebrin et al., 2005). During peripheral inflammatory pain, serum TGF-β1 decreased concomitant with activin receptor-like kinase 1 (ALK1)/ activin receptor-like kinase 5 (ALK5) signaling (Ronaldson et al., 2009). Administration of diclofenac, a commonly prescribed non-steroidal anti-inflammatory (NSAID), prevented decreases in serum TGF-β as well as reduced microvascular expression of ALK1/ALK5, suggesting that inflammatory pain in the periphery is directly involved in overall regulation of the TGF-β signaling pathway (Ronaldson et al., 2011). Furthermore, pharmacological inhibition of TGF-β/ALK5 signaling using SB431542 increased the Oatp1a4 drug uptake transporter functional expression both in animals subjected to peripheral inflammatory pain and in corresponding saline controls (Ronaldson et al., 2011). Although studies in immortalized mouse brain endothelial cells (MBE4) have shown involvement of ALK5-mediated signaling in P-gp transporter regulation (Dohgu et al., 2004), we are the first to report TGF-β/ALK5 signaling regulation of any endogenous BBB drug uptake transporter. Our work on TGF-β/ALK5 signaling demonstrated that this pathway could regulate permeability at the BBB both by altering the structure of tight junction protein complexes and by increasing functional expression of an influx transporter. Furthermore, these studies highlight the potential of the TGF-β/ALK5 pathway as a pharmacological target that can be utilized to precisely control functional expression of a BBB influx transporter for optimization of CNS drug delivery.
Conclusion
The field of BBB biology and particularly the study of endogenous xenobiotic transport systems have rapidly advanced over the past 2 decades. For example, it is now well-established that tight junctions between the capillary endothelial cells effectively limits paracellular drug diffusion while expression of various efflux transporters (i.e. P-gp, OATPs/Oatps, MRPs/Mrps, BCRP/Bcrp) interact with a multitude of therapeutic compounds, further restricting their influx or efflux at the brain parenchyma. Additionally, many previous studies reported on the controversial ability of drug transporters (i.e., Oatp1a4 and P-gp) to act as facilitators of brain drug uptake. Now, it is beginning to be appreciated that endogenous BBB transporters can facilitate uptake of xenobiotics from blood to the brain, thereby rendering these transport proteins potential targets for optimizing CNS drug delivery. Furthermore, molecular machinery involved in regulating endogenous BBB transport systems (i.e., TGF-β/ALK5 signaling, nuclear receptor systems, protein – protein signaling) and mechanisms governing intracellular trafficking of BBB transporters are just now being identified and characterized. These critical discoveries have identified multiple molecular targets that can be exploited for optimization of CNS delivery of therapeutic agents. Such studies are particularly critical for newly developed therapeutics such as opioid analgesic peptides. In fact, many novel opioid peptides have been recently produced and have shown analgesic efficacy (Largent-Milnes et al., 2010; Yamamoto et al., 2009); however, molecular and trafficking mechanisms involved in their CNS delivery have yet to be identified. Discovery of mechanisms that determine brain permeation of these peptides will undoubtedly enable more efficient analgesia and an improved utility of these compounds as potential therapeutics. Perhaps targeting of novel opioid peptides to influx transporters such as Oatp1a4, or efflux transporters such as P-gp, which are already known to be involved in opioid peptide transport at the BBB (Ronaldson et al., 2011), will lead to significant advancements in the field of opioid pharmacology and pain management. Additionally, identification and characterization of intracellular signaling pathways such as reactive oxygen species sensitive pathways (Lochhead et al., 2010; Lochhead et al., 2012), and protein trafficking mechanisms (Ronaldson & Davis, 2013; Hawkins & Davis, 2005; McCaffrey et al., 2012; Lochhead et al., 2010) that can regulate functional expression/activity of uptake or efflux transporters provides an additional approach for pharmacological modulation/control of drug transporter systems in an effort to deliver therapeutics to the CNS. Future work will continue to provide more insight on the interplay of tight junction protein complexes, transporters, and intracellular protein-protein signaling pathways at the BBB and how these systems can be effectively targeted. Therapeutic manipulation of protein trafficking is only recently emerging as a novel means of modulating protein function. The advantage of therapeutic drug development focused on “relocalization” of a protein, such as P-gp or Oatp1a4, is that its physiological impact can be modified, temporarily or long term, despite pathology-induced changes in gene transcription. By targeting the trafficking of P-gp or Oatp 1a4 as a novel, reversible means of optimizing CNS drug delivery, data derived from studies described in this chapter, and ongoing work in several laboratories to understand the composition of the storage pools of transporters and how they are released in response to stress will enable achievement of more precise drug concentrations within the CNS and improved treatment for pathological conditions.
Figure 4. P-glycoprotein (P-gp) at the Blood-Brain Barrier: the Greatest Molecular Challenge to CNS Drug Delivery.
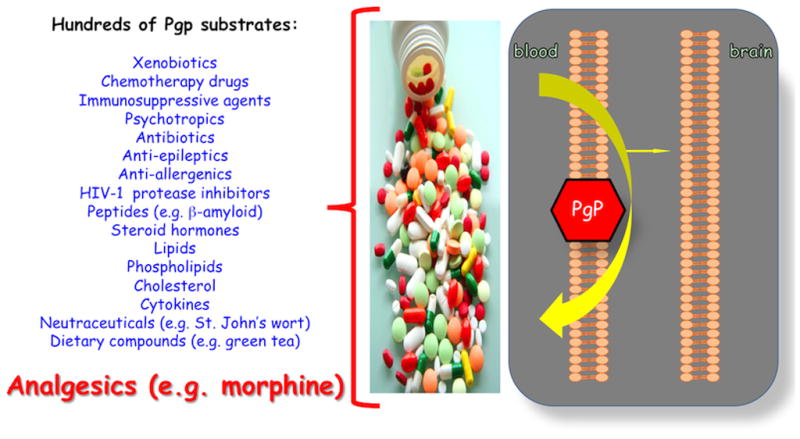
Note the significant cross section of drugs and xenobiotics that are all substrates for P-gp.
Acknowledgments
The authors acknowledge the financial support by the NINDS and NIDA of the NIH. RO1 Grants NS 42652-15 and DA 11271-16 to TPD funded this work.
Abbreviations
- ALK-1
activin-like receptor kinase-1
- ALK-5
activin-like receptor kinase-5
- BBB
blood brain barrier
- BCRP/Bcrp
breast cancer resistance protein
- BCSF
brain cerebral spinal fluid
- CNS
central nervous system
- MBE4
mouse brain endothelial cells
- MCTs/Mcts
monocarboxylate transporters
- MRPs/Mrps
multidrug resistance proteins
- NSAID
non-steroidal anti-inflammatory
- NVU
neurovascular unit
- OATP/Oatp
organic anion transporting peptides
- OATs/Oats
organic anion transporters
- OCTs/Octs
organic cation transporters
- P-gp
P-glycoprotein
- S1P
sphingosine-1-phosphate
- S1PR
sphingosine-1-phosphate receptor
- TEER
transendothelial resistance
- TGF-β
transforming growth factor beta
- VEGF
vascular endothelial growth factor
Footnotes
Conflict of Interest
The authors have no conflicts of interest to report.
References
- Barakat S, Demeule M, Pilorget A, Regina A, Gingras D, Baggetto LG, et al. Modulation of p-glycoprotein function by caveolin-1 phosphorylation. J Neurochem. 2007;101:1–8. doi: 10.1111/j.1471-4159.2006.04410.x. [DOI] [PubMed] [Google Scholar]
- Bendayan R, Lee G, Bendayan M. Functional expression and localization of P-glycoprotein at the blood brain barrier. Microsc Res Tech. 2002;57:365–380. doi: 10.1002/jemt.10090. [DOI] [PubMed] [Google Scholar]
- Bendayan R, Ronaldson PT, Gingras D, Bendayan M. In situ localization of P-glycoprotein (ABCB1) in human and rat brain. J Histochem Cytochem. 2006;54:1159–1167. doi: 10.1369/jhc.5A6870.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz AL, Firth JA, Goldstein GW. Polarity of the blood-brain barrier: distribution of enzymes between the luminal and antiluminal membranes of brain capillary endothelial cells. Brain Research. 1980;192:17–28. doi: 10.1016/0006-8993(80)91004-5. [DOI] [PubMed] [Google Scholar]
- Cannon RE, Peart JC, Hawkins BT, Campos CR, Miller DS. Targeting blood-brain barrier sphingolipid signaling reduces basal P-glycoprotein activity and improves drug delivery to the brain. Proc Natl Acad Sci USA. 2012;109:15930–15935. doi: 10.1073/pnas.1203534109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright TA, Campos CR, Cannon RE, Miller DS. Mrp1 is essential for sphingolipid signaling to p-glycoprotein in mouse blood-brain and blood-spinal cord barriers. J Cereb Blood Flow Metab. 2013;33:381–388. doi: 10.1038/jcbfm.2012.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Pollack GM. Extensive biliary excretion of the model opioid peptide [D-PEN2,5] enkephalin in rats. Pharm Res. 1997;14:345–350. doi: 10.1023/a:1012054222845. [DOI] [PubMed] [Google Scholar]
- Chen C, Pollack GM. Altered disposition and antinociception of [D-penicillamine(2,5)] enkephalin in mdr1a-gene-deficient mice. J Pharmacol Exp Ther. 1998;287:545–552. [PubMed] [Google Scholar]
- Chen CJ, Chin JE, Ueda K, Clark DP, Pastan I, Gottesman MM, et al. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell. 1986;47:381–389. doi: 10.1016/0092-8674(86)90595-7. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest. 2005;115:3285–3290. doi: 10.1172/JCI25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeule M, Regina A, Jodoin J, Laplante A, Dagenais C, Berthelet F, et al. Drug transport to the brain: key roles for the efflux pump P-glycoprotein in the blood-brain barrier. Vascul Pharmacol. 2002;38:339–348. doi: 10.1016/s1537-1891(02)00201-x. [DOI] [PubMed] [Google Scholar]
- Dohgu S, Yamauchi A, Takata F, Naito M, Tsuruo T, Higuchi S, et al. Transforming growth factor-beta1 upregulates the tight junction and P-glycoprotein of brain microvascular endothelial cells. Cell Mol Neurobiol. 2004;24:491–497. doi: 10.1023/B:CEMN.0000022776.47302.ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doran A, Obach RS, Smith BJ, Hosea NA, Becker S, Callegari E, et al. The impact of P-glycoprotein on the disposition of drugs targeted for indications of the central nervous system: evaluation using the MDR1A/1B knockout mouse model. Drug Metab Dispos. 2005;33:165–174. doi: 10.1124/dmd.104.001230. [DOI] [PubMed] [Google Scholar]
- Goard CA, Mather RG, Vinepal B, Clendening JW, Martirosyan A, Boutros PC, et al. Differential interactions between statins and P-glycoprotein: implications for exploiting statins as anticancer agents. Int J Cancer. 2010;127:2936–2948. doi: 10.1002/ijc.25295. [DOI] [PubMed] [Google Scholar]
- Golden PL, Pardridge WM. P-Glycoprotein on astrocyte foot processes of unfixed isolated human brain capillaries. Brain Research. 1999;819:143–146. doi: 10.1016/s0006-8993(98)01305-5. [DOI] [PubMed] [Google Scholar]
- Gottesman MM, Hrycyna CA, Schoenlein PV, Germann UA, Pastan I. Genetic analysis of the multidrug transporter. Annu Rev Genet. 1995;29:607–649. doi: 10.1146/annurev.ge.29.120195.003135. [DOI] [PubMed] [Google Scholar]
- Gribar JJ, Ramachandra M, Hrycyna CA, Dey S, Ambudkar SV. Functional characterization of glycosylation-deficient human P-glycoprotein using a vaccinia virus expression system. J Membr Biol. 2000;173:203–214. doi: 10.1007/s002320001020. [DOI] [PubMed] [Google Scholar]
- Hartz AM, Miller DS, Bauer B. Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer’s disease. Mol Pharmacol. 2010;77:715–723. doi: 10.1124/mol.109.061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Rigor RR, Miller DS. Rapid loss of blood-brain barrier P-glycoprotein activity through transporter internalization demonstrated using a novel in situ proteolysis protection assay. J Cereb Blood Flow Metab. 2010;30:1593–1597. doi: 10.1038/jcbfm.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Ohtsuki S, Terasaki T. Functional characterization of the brain-to-blood efflux clearance of human amyloid-beta peptide (1–40) across the rat blood-brain barrier. Neurosci Res. 2006;56:246–252. doi: 10.1016/j.neures.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochimica et Biophysica Acta. 1976;455:152–162. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- Largent-Milnes TM, Yamamoto T, Nair P, Moulton JW, Hruby VJ, Lai J, et al. Spinal or systemic TY005, a peptidic opioid agonist/neurokinin 1 antagonist, attenuates pain with reduced tolerance. Br J Pharmacol. 2010;161:986–1001. doi: 10.1111/j.1476-5381.2010.00824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrin F, Deckers M, Bertolino P, ten Dijke P. TGF-beta receptor function in the endothelium. Cardiovasc Res. 2005;65:599–608. doi: 10.1016/j.cardiores.2004.10.036. [DOI] [PubMed] [Google Scholar]
- Lee G, Dallas S, Hong M, Bendayan R. Drug transporters in the central nervous system: brain barriers and brain parenchyma considerations. Pharmacol Rev. 2001;53:569–596. [PubMed] [Google Scholar]
- Ling V, Thompson LH. Reduced permeability in CHO cells as a mechanism of resistance to colchicine. J Cell Physiol. 1974;83:103–116. doi: 10.1002/jcp.1040830114. [DOI] [PubMed] [Google Scholar]
- Lochhead JJ, McCaffrey G, Quigley CE, Finch J, Demarco KM, Nametz N, et al. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J Cereb Blood Flow Metab. 2010;30:1625–1636. doi: 10.1038/jcbfm.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead JJ, McCaffrey G, Sanchez-Covarrubias L, Finch JD, Demarco KM, Quigley CE, et al. Tempol modulates changes in xenobiotic permeability and occludin oligomeric assemblies at the blood-brain barrier during inflammatory pain. Am J Physiol Heart Circ Physiol. 2012;302:H582–H593. doi: 10.1152/ajpheart.00889.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo TW, Bartlett MC, Clarke DM. Human P-glycoprotein contains a greasy ball-and-socket joint at the second transmission interface. J Biol Chem. 2013;288:20326–20333. doi: 10.1074/jbc.M113.484550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey G, Staatz WD, Sanchez-Covarrubias L, Finch JD, Demarco K, Laracuente ML, et al. P-glycoprotein trafficking at the blood-brain barrier altered by peripheral inflammatory hyperalgesia. J Neurochem. 2012;122:962–975. doi: 10.1111/j.1471-4159.2012.07831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DS, Bauer B, Hartz AM. Modulation of P-glycoprotein at the blood-brain barrier: opportunities to improve central nervous system pharmacotherapy. Pharmacol Rev. 2008;60:196–209. doi: 10.1124/pr.107.07109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldendorf WH, Cornford ME, Brown WJ. The large apparent work capability of the blood-brain barrier: a study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann Neurol. 1977;1:409–417. doi: 10.1002/ana.410010502. [DOI] [PubMed] [Google Scholar]
- Rao VV, Dahlheimer JL, Bardgett ME, Snyder AZ, Finch RA, Sartorelli AC, et al. Choroid plexus epithelial expression of MDR1 P glycoprotein and multidrug resistance-associated protein contribute to the blood-cerebrospinal-fluid drug-permeability barrier. Proc Natl Acad Sci USA. 1999;96:3900–3905. doi: 10.1073/pnas.96.7.3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77:731–758. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- Ronaldson PT, Bendayan M, Gingras D, Piquette-Miller M, Bendayan R. Cellular localization and functional expression of P-glycoprotein in rat astrocyte cultures. J Neurochem. 2004;89:788–800. doi: 10.1111/j.1471-4159.2004.02417.x. [DOI] [PubMed] [Google Scholar]
- Ronaldson PT, Davis TP. Targeted drug delivery to treat pain and cerebral hypoxia. Pharmacol Rev. 2013;65:291–314. doi: 10.1124/pr.112.005991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronaldson PT, Demarco KM, Sanchez-Covarrubias L, Solinsky CM, Davis TP. Transforming growth factor-beta signaling alters substrate permeability and tight junction protein expression at the blood-brain barrier during inflammatory pain. J Cereb Blood Flow Metab. 2009;29:1084–1098. doi: 10.1038/jcbfm.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronaldson PT, Finch JD, Demarco KM, Quigley CE, Davis TP. Inflammatory pain signals an increase in functional expression of organic anion transporting polypeptide 1a4 at the blood-brain barrier. J Pharmacol Exp Ther. 2011;336:827–839. doi: 10.1124/jpet.110.174151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roninson IB, Chin JE, Choi KG, Gros P, Housman DE, Fojo A, et al. Isolation of human mdr DNA sequences amplified in multidrug-resistant KB carcinoma cells. Proc Natl Acad Sci USA. 1986;83:4538–4542. doi: 10.1073/pnas.83.12.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez del Pino MM, Peterson DR, Hawkins RA. Neutral amino acid transport characterization of isolated luminal and abluminal membranes of the blood-brain barrier. J Biol Chem. 1995;270:14913–14918. doi: 10.1074/jbc.270.25.14913. [DOI] [PubMed] [Google Scholar]
- Sanchez-Covarrubias L, Slosky LM, Thompson BJ, Davis TP, Ronaldson PT. Transporters at CNS barrier sites: obstacles or opportunities for drug delivery? Curr Pharm Des. 2014;20:1422–1449. doi: 10.2174/13816128113199990463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- Schlachetzki F, Pardridge WM. P-glycoprotein and caveolin-1alpha in endothelium and astrocytes of primate brain. Neuroreport. 2003;14:2041–2046. doi: 10.1097/00001756-200311140-00007. [DOI] [PubMed] [Google Scholar]
- Seelbach MJ, Brooks TA, Egleton RD, Davis TP. Peripheral inflammatory hyperalgesia modulates morphine delivery to the brain: a role for P-glycoprotein. J Neurochem. 2007;102:1677–1690. doi: 10.1111/j.1471-4159.2007.04644.x. [DOI] [PubMed] [Google Scholar]
- Sharom FJ. Multidrug Resistance Protein: P-glycoprotein. In: You GFaMME., editor. Drug Transporters: Molecular Characterization and Role in Drug Disposition. Hoboken, NJ: John Wiley & Sons, Inc; 2007. pp. 263–318. [Google Scholar]
- Shirasaka Y, Suzuki K, Nakanishi T, Tamai I. Differential effect of grapefruit juice on intestinal absorption of statins due to inhibition of organic anion transporting polypeptide and/or P-glycoprotein. J Pharm Sci. 2011a;100:3843–3853. doi: 10.1002/jps.22586. [DOI] [PubMed] [Google Scholar]
- Shirasaka Y, Suzuki K, Shichiri M, Nakanishi T, Tamai I. Intestinal absorption of HMG-CoA reductase inhibitor pitavastatin mediated by organic anion transporting polypeptide and P-glycoprotein/multidrug resistance 1. Drug Metab Pharmacokinet. 2011b;26:171–179. doi: 10.2133/dmpk.dmpk-10-rg-073. [DOI] [PubMed] [Google Scholar]
- Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75:451–462. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- Spudich A, Kilic E, Xing H, Kilic U, Rentsch KM, Wunderli-Allenspach H, et al. Inhibition of multidrug resistance transporter-1 facilitates neuroprotective therapies after focal cerebral ischemia. Nat Neurosci. 2006;9:487–488. doi: 10.1038/nn1676. [DOI] [PubMed] [Google Scholar]
- Sun J, He ZG, Cheng G, Wang SJ, Hao XH, Zou MJ. Multidrug resistance P-glycoprotein: crucial significance in drug disposition and interaction. Med Sci Monit. 2004;10:RA5–14. [PubMed] [Google Scholar]
- Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control. 2003;10:159–165. doi: 10.1177/107327480301000207. [DOI] [PubMed] [Google Scholar]
- Ueda K, Cardarelli C, Gottesman MM, Pastan I. Expression of a full-length cDNA for the human “MDR1” gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc Natl Acad Sci USA. 1987;84:3004–3008. doi: 10.1073/pnas.84.9.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Assema DM, Lubberink M, Bauer M, van der Flier WM, Schuit RC, Windhorst AD, et al. Blood-brain barrier P-glycoprotein function in Alzheimer’s disease. Brain. 2012;135:181–189. doi: 10.1093/brain/awr298. [DOI] [PubMed] [Google Scholar]
- Volk HA, Burkhardt K, Potschka H, Chen J, Becker A, Loscher W. Neuronal expression of the drug efflux transporter P-glycoprotein in the rat hippocampus after limbic seizures. Neuroscience. 2004;123:751–759. doi: 10.1016/j.neuroscience.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Vorbrodt AW, Dobrogowska DH. Molecular anatomy of intercellular junctions in brain endothelial and epithelial barriers: electron microscopist’s view. Brain Res Brain Res Rev. 2003;42:221–242. doi: 10.1016/s0165-0173(03)00177-2. [DOI] [PubMed] [Google Scholar]
- Xie Y, Burcu M, Linn DE, Qiu Y, Baer MR. Pim-1 kinase protects P-glycoprotein from degradation and enables its glycosylation and cell surface expression. Mol Pharmacol. 2010;78:310–318. doi: 10.1124/mol.109.061713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Nair P, Jacobsen NE, Vagner J, Kulkarni V, Davis P, et al. Improving metabolic stability by glycosylation: bifunctional peptide derivatives that are opioid receptor agonists and neurokinin 1 receptor antagonists. J Med Chem. 2009;52:5164–5175. doi: 10.1021/jm900473p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Kwan P, Zuo Z, Baum L. The transport of antiepileptic drugs by P-glycoprotein. Adv Drug Deliv Rev. 2012;64:930–942. doi: 10.1016/j.addr.2011.12.003. [DOI] [PubMed] [Google Scholar]