

Abstract
Linkage disequilibrium (LD) between densely spaced, polymorphic genetic markers in humans and other species contains information about historical population size. Inferring past population size is of interest both from an evolutionary perspective (e.g., testing the “out of Africa” hypothesis of human evolution) and to improve models for mapping of disease and quantitative trait genes. We propose a novel multilocus measure of LD, the chromosome segment homozygosity (CSH). CSH is defined for a specific chromosome segment, up to the full length of the chromosome. In computer simulations CSH was generally less variable than the r2 measure of LD, and variability of CSH decreased as the number of markers in the chromosome segment was increased. The essence and utility of our novel measure is that CSH over long distances reflects recent effective population size (N), whereas CSH over small distances reflects the effective size in the more distant past. We illustrate the utility of CSH by calculating CSH from human and dairy cattle SNP and microsatellite marker data, and predicting N at various times in the past for each species. Results indicated an exponentially increasing N in humans and a declining N in dairy cattle. CSH is a valuable statistic for inferring population histories from haplotype data, and has implications for mapping of disease loci.
The large number of densely spaced, polymorphic genetic markers generated by modern genomics is a powerful tool for answering genetic questions. For instance, they are being used to fine-scale-map trait genes (Pritchard and Przeworski et al. 2001) and to infer the history of the human population (Reich et al. 2001). Inferring past population size is of interest both from an evolutionary perspective (e.g., testing the “out of Africa” hypothesis of human evolution) and to improve models for the mapping of disease and quantitative trait genes.
Under a neutral model with constant effective population size (N), the homozygosity of a marker, the probability of sampling two identical alleles from the population, can be used to estimate N, provided the mutation rate is known (e.g., Kuhner et al. 1998; Slatkin and Bertorelle 2001). If N has changed in the past, the homozygosity will estimate a form of average N. The higher the mutation rate, the less events from the distant past remain relevant and thus the average N estimated will reflect N in the more recent past. Similarly, linkage disequilibrium (LD) can be used to estimate N if the recombination rate is known. As will be shown in this paper, LD over large recombination distances estimates N in the more recent past than LD over short recombination distances. Use of LD rather than individual marker homozygosity has the advantage that the recombination rate is more controllable than the mutation rate (by selecting the length of chromosome segments), and more recent N can be estimated because recombination rates can be much higher than mutation rates. Therefore, although estimates of average past population size from n unlinked loci can be more accurate than from n linked loci (Kuhner et al. 1998), using LD between n linked loci can provide additional information on historical changes in population size.
Most measures of LD, such as r2 and related measures (Devlin and Risch 1995; Weir 1996), quantify the association between a pair of loci. Higher-order association coefficients analogous to r2 can be defined for groups of 3, 4, or more loci, but they have not been found to be a practical value (Hill 1981). Such higher-order LD measures also ignore the essentially linear nature of chromosomes and of recombination. An ideal multilocus measure of LD would take account of this linearity and capture as much as possible of the information content of the data (no single statistic could contain all the information). In addition, it would be desirable if the measure of LD used had a simple expectation, at least under standard models such as the neutral model.
Definition of Chromosome Segment Homozygosity (CSH)
We propose a novel multilocus measure of LD, the chromosome segment homozygosity (CSH). CSH is the probability that two chromosome segments of the same size and location drawn at random from the population are from a common ancestor, without intervening recombination. CSH is defined for a specific chromosome segment, up to the full length of the chromosome. The CSH cannot be directly observed from marker data but has to be inferred from marker haplotypes for segments of the chromosome. Consider a segment of chromosome with marker locus A at the left-hand end of the segment and marker locus B at the other end of the segment. The alleles at A and B define a haplotype. Two such segments are chosen at random from the population. The probability that the two haplotypes are identical by state (IBS) is the haplotype homozygosity (HH). The two haplotypes can be IBS in two ways:
(1) The two segments are descended from a common ancestor without intervening recombination, so are identical by descent (IBD); or
(2) The two haplotypes are identical by state but not IBD.
The probability of (1) is CSH. Now let x = the probability that A is homozygous when the chromosome segment is not IBD, and let y = the probability that B is homozygous when the chromosome segment is not IBD. Assuming the two loci behave independently in this case, the probability of (2) is
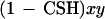 |
Then the probability of observing homozygosity at A is
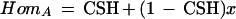 |
Solving for x,
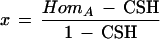 |
And similarly,
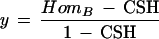 |
Substituting the last two equations into the first, and summing over (1) and (2) to get the probability of haplotype homozygosity, we get
 |
This equation can be solved for CSH when the haplotype homozygosities and individual marker homozygosities are observed from the data. For more than two markers, the predicted haplotype homozygosity can be calculated in an analogous but more complex manner (see Methods).
In this paper we show that CSH is generally a less variable statistic than r2 when effective population size is constant. We then derive the expectation of CSH under a neutral model with changing N. Simulated data are used to validate the accuracy of the expectation. Finally, we use CSH to estimate the N at various times in the past in a human population (where we expect N has been increasing) and in a cattle population (where we expect N has been decreasing).
RESULTS AND DISCUSSION
In a population of constant effective size N, the approximate expectation of CSH is 1/(4Nc + 1), which is the same as the approximate expectation for r2, where c is the length of the chromosome segment in morgans (Sved 1971). To test the agreement between expectation and observed results, a chromosome segment of 10 cM containing 11 markers was simulated with a mutation-drift model, with a constant N of 1000. The average heterozygosity of markers was 0.65, and the number of alleles segregating was ∼5–10 per marker. The simulation gave a total of 55 haplotype configurations: 10 different haplotype regions of 1 cM with two markers, 9 different haplotype regions of 2 cM with three markers, and up to a single haplotype region of length 10 cM with 10 markers. A total of 200 replicate populations were simulated. The results (Fig. 1) indicate CSH and r2 have a similar mean, but different variance. The means of both statistics were close to 1/(4Nc + 1), except for the 1-cM haplotype, in which r2 was less than the expectation. In our simulations, marker allele frequencies were the result of drift and mutation, and follow a U-shaped distribution (e.g., Kimura and Crow 1964), often <0.05 or >0.95. Hudson (1985) showed in simulations that r2 was lower than expected when allele frequencies were <0.05 or >0.95. This may explain the lower-than-expected r2 value we observed for the 1-cM chromosome segments. The CSH does not appear to be sensitive to allele frequency.
Figure 1.



(A) CSH and r2 from the simulated data set. Results are averaged over all haplotype regions of a given length, and over 200 replicates. (B) Coefficient of variation of r2 and CSH over all haplotype regions of the same length, across 200 replicates. (C) CSH from the data simulated with either an infinite alleles model or a stepwise mutation model. Results are averaged over all haplotype regions of a given length, and over 200 replicates.
The CSH had a lower coefficient of variation (CV) than r2, provided there were more than three markers in the haplotype (Fig. 1B), indicating that it is a less variable statistic to estimate LD than pairwise measures.
Additional simulation indicated the decreasing variation of CSH was a result of an increasing number of markers in the chromosome segment rather than increasing haplotype length (data not shown). This is a major advantage of using CSH to measure LD rather than two locus measures such as r2 (for such measures variability of LD for a given chromosome segment cannot be reduced using additional markers).
With the infinite alleles model, all identical by state alleles are also IBD. Although this is not one of our assumptions in the derivation of CSH, we investigated estimates of CSH under an alternate mutation model. With microsatellite markers, multiple mutations can occur in the same marker, and two or more mutations can recover the initial allelic state. A stepwise mutation model assumes an equal probability of increasing the size of the allele by 1 and decreasing the size of the allele by 1, and is suitable for modeling microsatellite markers (e.g., Shriver et al. 1993). We simulated a population similar to that used to produce the results in Figure 1A, but with a stepwise mutation model. The estimated CSH from these data was almost identical to estimates of CSH using data from the infinite alleles model (Fig. 1C).
In Figure 2 CSH was recorded directly from simulated data, rather than estimated from marker haplotypes as in Figure 1. Again, where population size was constant over all generations (CONS), CSH was very close to the values predicted by 1/(4Nc + 1).
Figure 2.


Chromosomal homozygosity for different lengths of chromosome (given the recombination rate) for populations: (A) CONS (constant population size), (B) LINI (linearly increasing population size), (C) LIND (linearly decreasing population size), and (D) EXPI (exponentially increasing population size). The expected value of chromosomal homozygosity, 1/(4Nc + 1), is given on each graph for the maximum and minimum population sizes of each population. Standard error bars indicate variation among the 50 replicates.
A second set of data was simulated to illustrate the effect of past N on CSH at different lengths of chromosome. In these data, marker alleles were not simulated because the identity of chromosome segments was tracked directly. We simulated four populations, a population of constant N (CONST), a population with linearly increasing N (LINI), a population with linearly decreasing N (LIND), and a population with exponentially increasing N (EXPI). When the population size was either linearly (LINI) or exponentially (EXPI) increasing, or linearly decreasing (LIND), CSH at small recombination rates agreed with the expected CSH based on population size many generations ago, whereas CSH at large recombination rates agreed with expected CSH based on more recent population size (Fig. 2). These results concur with those of Hill (1981), who found that estimates of N from LD for very tightly linked genes were more dependent on long-term than on recent population history.
When population size is changing linearly, the expectation of CSH is ∼1/(4Ntc + 1), where Nt was the population size 1/(2c) generations ago. Effective population size 1/(2c) generations into the past was predicted from CSH (Fig. 3). Our method for predicting N assumes constant linear population growth from generation 1. Although this population growth model does not hold for any of the populations we have simulated, the estimates of N were in approximate agreement with the actual N for LINI and LIND. For EXPI, the later estimates of N agree reasonably well with actual N; however, N for 500, 200, and 100 generations ago was somewhat overestimated. The widths of the 95% confidence intervals, averaged over time, on the estimates of N for each population were 183, 260, 141, and 219 for CONST, LINI, LIND, and EXPI, respectively. For example, 20 generations ago the estimate of effective population size for CONST was 1062, and the 95% confidence interval was 971–1154. Confidence intervals were generally smaller with lower population sizes.
Figure 3.

Simulated and estimated effective population size over time for four populations; (CONST) constant population size from 0 to 6050 generations ago; (LINI) increase in population size in the last 50 generations from 1000 to 5000; (LIND) decrease in population size in the last 50 generations from 1000 to 100; (EXPI) increase in population size in the last 50 generations from 1000 to 11290. SIM and EST identify the simulated and estimated population sizes for each population.
CSH was calculated from a human haplotype data set including 24 SNPs and 2 microsatellites in a 1-cM region (Moffat et al. 2000). To validate that CSH was accurately estimated with the marker densities and heterozygosities in this data set, we simulated a population of constant N = 5000, using the mutation-drift model, with similar marker density, marker heterozygosities, and haplotype lengths to those observed by Moffat et al. (2000) in their data set. The value of CSH observed from the simulated data sets was similar to the expectation of CSH with N = 5000, and predictions of Nt were reasonably accurate, although Nt was somewhat overestimated in more recent generations (Table 1). The coefficient of variation for CSH was higher from this simulation compared with simulations with more heterozygous markers. Initial investigation showed that the values of CSH from the real data set were extremely variable for similar lengths of haplotype. To clarify the extent and variability of CSH, and Nt at different t from CSH, we first averaged CSH values in 0.05-cM bins. The first bin contained CSH for haplotypes 0–0.05 cM, and so on. The c value used to calculate t was the midpoint of these bins, and Nt was inferred from the average of CSH within a bin. The CSHs in the human data set at large lengths of haplotype were consistent with N = 15,000 (Fig. 4A). At short lengths of haplotype, CSH was closer to that expected when N = 5000. The Nt values indicated exponential growth in the human effective population size (Fig. 4B). Chromosomal homozygosity at very closely linked markers is needed to estimate effective population size many generations into the past. The marker spacing in our data set allowed us to calculate Nt up to 2000 generations into the past, although the next oldest prediction of Nt is many generations later. The situation improves in more recent history, with less time between predicted values of Nt. An attempt was made to assess the variability in estimates of Nt at the different times in the past, by calculating the 95% confidence of CSH within a bin, and calculating Nt for the upper and lower confidence interval. This 95% confidence interval captures the variation in CSH due to the process of gametic sampling of similar lengths of haplotype at different chromosomal locations. The range of Nt values for times in the recent past was extremely variable, with Nt for times in the distant past less so. For example, at 2000 generations ago, the lower limit of Nt was 3749, and the upper limit was 8376, whereas for 182 generations ago, the lower limit was 5146, and the upper limit was 26,932.
Table 1.
Results From Simulation of a 0.5-cM Chromosome Segment Containing 10 Markers, With N = 5000
| Length of segment (cM) | Markers in segment | Expected CSHa | Observed CSHb | CVc (%) | t (generations ago) | Nt | 95% confidence intervald |
| 0.06 | 2 | 0.083 | 0.090 | 121 | 900 | 4540 | 3581–6098 |
| 0.11 | 3 | 0.043 | 0.046 | 147 | 450 | 4671 | 3363–7477 |
| 0.17 | 4 | 0.029 | 0.024 | 98 | 300 | 6028 | 4759–8184 |
| 0.22 | 5 | 0.022 | 0.023 | 80 | 225 | 4875 | 3949–6347 |
| 0.28 | 6 | 0.018 | 0.013 | 71 | 180 | 6679 | 5449–8610 |
| 0.33 | 7 | 0.015 | 0.011 | 72 | 150 | 6784 | 5411–9074 |
| 0.39 | 8 | 0.013 | 0.008 | 79 | 129 | 8028 | 6072–11,815 |
| 0.44 | 9 | 0.011 | 0.006 | 68 | 113 | 8806 | 6578–13,283 |
| 0.50 | 10 | 0.010 | 0.007 | 63 | 100 | 7377 | 5110–13,194 |
Results are from analysis of haplotypes after 30,000 simulated generations of breeding under the mutation drift model. The average heterozygosity of the markers was 0.39. The markers were equally spaced, with 0.06 cM between markers.
Calculated as 1/(4Nc + 1), where N = 5000 and c is the length of the segment in morgans.
The average results from 10 replicates. In all replicates, the minimum heterozygosity of the markers was 0.05, as this was the minimum heterozygosity of the markers in the Moffat et al. (2001) data set.
From pooled results over segments of the same length within a replicate and across replicates.
To calculate the 95% confidence intervals for Nt, a 95% confidence interval for the observed CSH was calculated as average CSH ± 2SE. Then the upper and lower bounds of CSH were used to calculate the upper and lower bounds of the 95% confidence interval for Nt.
Figure 4.


(A) Chromosomal homozygosity for increasing lengths of haplotype from the data of Moffat et al. (2000). The upper (solid) line is the expected CSH when the effective population size is 5000. The lower (dashed) line is the expected CSH when the effective population size is 15,000. (B) Effective population size of the human population ancestral to the sample used, up to 2000 generations ago. (C) Chromosomal homozygosity from the dairy cattle data set. Also plotted are the expected values of CSH when N = 1000 and N = 250. (D) Effective population size of the dairy cattle population ancestral to the sample used, up to 167 generations ago.
We compared the results from our method for the human data set, in which Nt has been increasing, with a species in which Nt has been decreasing over time. Accordingly, we sampled marker haplotypes from the Holstein-Friesian dairy cattle population. The marker data were 16 microsatellites on Chromosome 20 covering a 65-cM chromosome segment, sampled from 264 Australian Holstein-Friesian cows. In the dairy cattle data set, CSH at large lengths was consistent with N = 250 (Fig. 4C). At short lengths of haplotype, the observed CSH was more consistent with N = 1000. The two large values of CSH at 7.9 cM and 11.2 cM are for long chromosome segments with only two markers, the situation in which CSH is most variable. When we calculated Nt and t from CSH at c morgans, the results indicated a recent decline in the effective population size of dairy cattle (Fig. 4D). Because of the wide spacing of markers in our sample, there is little information on effective population size of dairy cattle more than 100 generations (∼400 yr) in the past. The one data point 167 generations in the past certainly indicates that the historical effective population size was much larger than the present effective population size.
The simulation study confirmed that CSH could be used to predict approximate effective population size at various times in the past. The estimates of past N were qualitatively correct although not numerically precise. In theory, more information could be extracted from the data. For instance, the frequencies of each haplotype contain information—many rare haplotypes imply an increasing population size (Slatkin and Bertorelle 2001). However, in practice there may be no method that is highly precise because of the need to make numerous assumptions in any method. A strength of the CSH is its simplicity. It also makes clear the close analogy between mutation affecting homozygosity at individual loci and recombination affecting LD at multiple loci. LD provides information equivalent to that from a mutation rate that can be controlled (by choosing the length of chromosome segment) and that can take much higher values than mutation rates at individual loci.
The variation in LD arises from two sampling processes (Weir and Hill 1980). The first sampling process reflects the sampling of gametes to form successive generations, and is dependent on finite population size. The second sampling process is the sampling of individuals to be genotyped from the population, and is dependent on the sample size, n. Unless n is sufficiently large, the effect of Nt on CSH is likely to be swamped by sampling effects resulting from choosing only a fraction of individuals to be genotyped from the population in the present generation (e.g., Weir and Hill 1980). Hill (1981) discussed the sample size necessary to obtain precise estimates of population size (from r2 in his case), and showed CV(N) is approximately
 |
where n is the number of haplotypes sampled and k is the number of pairs of loci used in the estimate. Sample size must therefore be large relative to 4Nc to precisely estimate N. This conclusion is also likely to be true for estimates of Nt from CSH (even though the variability of CSH is reduced relative to r2 as the number of markers on the chromosome segment increases). For a given sample size, Hill's conclusion indicates that because the variability of the estimate of Nt will increase as the length of the chromosome segment used to estimate CSH (and then Nt) increases, the recent population size will be estimated less accurately than the population size many generations in the past. This concurs with our results, in which the 95% confidence interval of Nt in the recent past was much larger than for Nt in the more distant past. For the human data, the accuracy of estimates of recent population size is further eroded by the rapidly increasing value of Nt. Unrealistically large sample sizes would be necessary to obtain accurate estimates of Nt in the very recent past.
As all chromosome segments within the genome are subject to the same Nt, the variability of estimates of Nt from LD caused by finite population size (the history of sampling gametes) could be reduced by averaging LD over chromosome segments of equal recombination length. An ideal data set for estimating Nt from CSH would contain many equally spaced markers across a number of chromosomes, so results for haplotypes of the same recombination length in different parts of the genome could be averaged to obtain more accurate estimates of Nt. This is analogous to the result from Kuhner et al. (1998) that a gain in precision is obtained by sampling multiple unlinked loci.
Our estimates of past N for both cattle and humans agree with what is historically known about these populations. Using a dense genome-wide SNP map in humans, Reich et al. (2001) calculated D‘, and then used simulation to infer what pattern of change in N could give the observed results. They concluded that the European population passed through a bottleneck 27,000–53,000 years ago, and proposed the bottleneck led to an inbreeding coefficient of 0.2. This level of inbreeding could be caused by a bottleneck of 50 individuals for 20 generations, 1000 individuals for 400 generations, or any other combination with the same ratio. We estimated that N for the population ancestral to our sample 2000 generations ago (∼30,000 yr ago) was ∼5000 and that this size lasted for thousands of years.
Shifman and Darvasi (2001) compared the amount of linkage disequilibrium at various distances in isolated populations (e.g., Finnish, Ashkenazi, and Sardinian) to that in an outbred population (CEPH). They found that at short distances (<200 kb), there was a similar amount of LD in isolated and outbred populations, whereas at long distances (>200 kb), there was up to six times more LD in the isolated populations. They concluded that LD was similar for all populations at short distances because processes other than recombination, such as mutation, determined the amount of LD at <200 kb. At >200 kb, recombination was the main determinant of LD and so LD differed greatly between the different populations as a result of their different N. Given our result of t = 1/2c, LD at 200 kb would reflect the population size 213 generations or ∼5000 yr ago. Hence, another interpretation of Shifman and Darvasi’s (2001) results is that LD at <200 kb reflects N of the common ancestral population to both the isolated and outbred populations, and is therefore similar regardless of the present population size.
Sabatti and Risch (2002) recently investigated the relationship between two-locus haplotype homozygosity and linkage disequilibrium, and illustrated how haplotype homozygosity can be used to measure and test for multilocus LD. Their new measure is based on the population or sample frequencies of haplotypes (like the HH in this study) but, unlike our definition of CSH, does not take account of the linear nature of chromosomes and recombination and does not model homozygosity by descent.
Domestication, breed formation, and artificial breeding technologies have all served to reduce the effective population size of the world dairy cattle population. Because of the wide spacing of the markers in our data set, we can only infer population sizes up to 167 generations (∼700 yr) ago. This is prior to the emergence of Holstein-Friesians as a separate breed, which is estimated to have occurred ∼200 yr ago (Bradley and Cunningham 1998). Our results certainly indicate that the effective population size of the ancestral dairy population has declined sharply between 200 yr ago and the present. The data also contain some long (0.25-M) haplotypes, the LD at which can be used to infer recent effective population size. Our data indicate the recent effective population size (in the last 5–6 generations) to be ∼150, although there is variation around this value. An N of 150 is larger than, but similar in magnitude to, estimates of ∼100 based on the rate of inbreeding in Holstein populations (Young and Seykora 1996).
We have shown that our novel multilocus measure of linkage disequilibrium can be used to estimate past effective population size. A similar approach can be used for LD mapping of genes for complex traits (Meuwissen and Goddard 2001). Chromosome segments that are IBD contain the same allele, except for mutation, at any gene within the segment. Therefore, the trait values of people that share IBD chromosome segments will be correlated if there is a gene affecting the trait located within the segment.
METHODS
Simulated Data Sets
Two types of simulated data were used. A diploid population, of N = 1000, was simulated for 6000 generations with either an infinite alleles or stepwise mutation model. Each individual in the population consisted of a pair of chromosomes, and was either male or female (probability 0.5). Each chromosome was 10 cM long, and had 11 marker loci. To create an offspring, a pair of parents of different sex was randomly chosen from the population. For each parent in a mating pair, a gamete was formed from its chromosome pairs by sampling the number of crossovers for each chromosome pair from a Poisson distribution, with mean of 0.1. Crossover points were randomly positioned along chromosome pairs. The haploid gametes were mutated at a rate of 5 × 10−4 per locus per gamete per generation. In the infinite alleles model, if a locus was mutated, a new allele was added. In the stepwise mutation model, the allele was either increased by 1 or decreased by 1, with probability 0.5 of each occurrence. The results presented are the average of 200 replicate populations. This simulation model was also used to evaluate CSH with other population sizes. The number of generations for which the population was simulated was always 6N. The heterozygosity of markers was decreased in some simulations by decreasing the mutation rate.
In the second simulated data set, marker alleles were not used because the identity of chromosome segments was tracked directly. To demonstrate the effect of past N on CSH, we simulated four populations, a population of constant N (CONST), a population with linearly increasing N (LINI), a population with linearly decreasing N (LIND), and a population with exponentially increasing N (EXPI). Each population consisted of N individuals, such that an individual comprises a pair of chromosome segments. To form a new individual, two individual parents were selected at random from the population. Each of these contributed a chromosome to the progeny. There was a probability c that the chromosome from a parent was a recombinant and hence a “new” chromosome segment. Each population was simulated with seven values of c, 0.001, 0.0025, 0.005, 0.01, 0.025, 0.05, and 0.1. All populations began with 6000 generations with N = 1000. Then 50 (CONS, LINI, LIND) or 100 generations (EXPI) of breeding with changing N followed. Table 2 describes the change in N over generations for each population. Fifty replicates of each population were simulated. Using the observed CSH, the formula CSH = 1/(4Nc + 1) was solved for N to estimate N at 500, 200, 100, 50, 20, and 5 generations into the past for each population, the times corresponding to 1/2c for the values of c we have used.
Table 2.
Simulated Change in N for Populations CONST, LINI, LIND, and EXPI
| Population | Change in population size after generation 6000 | Minimum population size (generation) | Maximum population size (generation) |
| CONS | Nt = Nt−1 | 1000 (1–6050) | 1000 (1–6050) |
| LINI | Nt = Nt−1 + 80 | 1000 (1–6000) | 5000 (6050) |
| LIND | Nt = Nt−1 − 18 | 100 (6050) | 1000 (1–6000) |
| EXPI | Nt = 1.025Nt−1 | 1000 (1–6000) | 11,290 (6100) |
Human Data Set
The data set of Moffat et al. (2000) was retrieved from the Web site http://www.well.ox.ac.uk/asthma/public/TCR/index.html. The data consisted of 24 SNPs and 2 microsatellites in an 850-kb section of the TCR locus on Chromosome 14q. To derive haplotypes, 159 nuclear and extended families were genotyped, and the LD between markers was investigated in 600 haplotypes from unrelated individuals (the parents). CSH were summarized into 0.05-cM bins. The first bin contained CSH for haplotypes 0–0.05 cM, and so on. The c value used to calculate t was the midpoint of these bins, and Nt was inferred from the average of CSH within a bin. The 95% confidence interval for estimates within a bin was calculated as the bin average CSH ± 2SE. The data set was too small to reliably calculate CSH at >0.3 cM, as the value of the CSH estimates became much smaller than the sampling variance caused by sampling only 600 chromosomes from the population. Therefore, we only considered haplotypes less than this length.
Cattle Data Set
The resource population for the dairy cattle data set consisted of four Holstein-Friesian sire families. Sire A had 22 daughters, Sire B 38 daughters, Sire C 74 daughters, and Sire D 130 daughters. Each daughter had a unique dam. Daughters were genotyped for 15 microsatellite markers on Chromosome 20. The markers were BM1225, RM310, ILSTS068, BMS2361, AGLA29, BM4107, ILSTS072, BMS703, BM5004, BMS3517, HEL12, RM106, BMS1282, TGLA304, and BMS1719(see http://www.thearkdb.org/browser?species=cow for details). The markers bracketed a length of 65 cM, with various spacings between the markers. As the daughters were from four sire families, the paternal and maternal marker haplotypes could be determined. We estimated CSH from the maternal haplotypes only, as CSH from the paternal haplotypes would reflect CSH in each of the four sires, rather than in the wider population. The data set was too small to reliably calculate CSH at >25 cM, as the value of the CSH estimates became much smaller than the sampling variance caused by sampling only 264 chromosomes from the population. Therefore, we only considered haplotypes <25 cM.
Calculation of CSH and r2
We cannot observe CSH directly from the marker haplotypes. Instead, we estimate CSH from the observed homozygosity of haplotypes (HH). HH is defined as the probability that two chromosome segments drawn at random from the population have identical marker haplotypes. The value of HH in the population is estimated from the sample as
 |
where there were n haplotypes in the population, and pi was the frequency of the i-th haplotype. This formula corrects HH for sampling effects (following Hill 1981).
The algorithm to calculate CSH with multiple markers proceeds as follows. The number of markers in the chromosome segment is m, and an array, CSH, stores CSH for the chromosome segment between markers i and j. The algorithm calculates values of CSHij for all possible combinations of j > i.Step 1. For i = 1 to m − 1, and j = i + 1 (the case of two adjacent markers), calculate CSHij using the definition given above.Step 2. For i = 1 to m − 2, and j = i + 2, k = j − i = 2 (three adjacent markers), generate the 2k − 1 possible recombination configurations for the segments between the markers (k is the number of adjacent markers). Representing 0 as no recombination, and 1 as a recombination, the four possible recombination configurations are 00, 10, 01, 11. The recombination configurations can be quickly found by writing 0 to 2k − 1 − 1 as binary numbers. The probability of 00 is CSHij. To calculate the probability of the other recombinations (Probl for l = 1–3), the rules of Meuwissen and Goddard (2001) are used, except the values of f(c…) are replaced by the appropriate CSH for two markers calculated in Step 1. As some Probl values also contain CSHij, a search must be performed for the value of CSHij that minimizes
 |
This value was taken as the value of CSHij.Step 3. For increasing k (4, 5, …), repeat Step 3 until j − i = m − 1.
The value of r2 was calculated as described by Hudson (1985), with a correction for sampling described by Hill (1981).
Estimation of Past N From CSH
We wish to determine the effective population size, Nt, t generations ago from CSH. Let Pt be the probability that two chromosome segments of length c coalesce by generation t. Pt is a cumulative probability, and time is measured from the present t = 0 to generation t in the past. Then the probability that coalescence occurs exactly in generation t is pt = Pt − Pt − 1. Then, in the standard coalescence model, pt = (1 − Pt − 1)/2Nt, where (1 − Pt − 1) is the probability that coalescence hasn't already happened and 1/2Nt is the probability that the two random chromosomes have a common ancestor in the previous generation (Kingman 1982). This can be expressed in a continuous rather than discrete form as pt = dPt/dt = (1 − Pt)/(2Nt).
The probability that there has been no recombination in either chromosome over the t generations is (1 − c)2t, which, for small c, is approximately equal to e(−2ct). Therefore, the probability that coalescence happens at generation t and there has been no recombination is
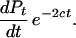 |
The total probability that coalescence happens before recombination (the CSH) is the sum of this expression over all t values from 0 to infinity,
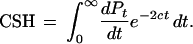 |
If we make the assumption that N is linear with time, such that 2Nt = α + βt, then
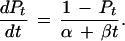 |
Taking the logarithm of both sides of this equation gives
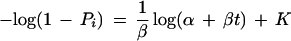 |
where K is a constant. Rearranging this formula gives
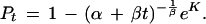 |
At t = 0, and Pt = 0, eK = α1/β. Substituting α1/β for eK and rearranging gives
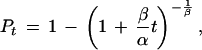 |
which is differentiated to give
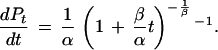 |
This expression is approximately equal to
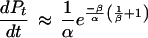 |
Substituting
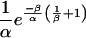 |
for dPt/dt in the second equation gives
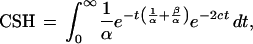 |
and after integration,
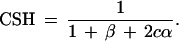 |
Now
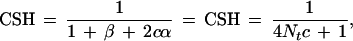 |
where 2Nt = α + β/2c. That is, Nt is the effective population size at t = 1/2c generations in the past.
WEB SITE REFERENCES
http://www.thearkdb.org/browser?species=cow; cattle data set.
http://www.well.ox.ac.uk/asthma/public/TCR/index.html; Moffat data set from human Chromosome 14q.
Acknowledgments
The authors thank W.G. Hill, T.H.E. Meuwissen, and two referees for useful comments on an earlier version of this manuscript. P.M.V. acknowledges support from the UK Biotechnology and Biological Sciences Research Council.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
E-MAIL Ben.Hayes@nre.vic.gov.au; FAX 61 39217 4359.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.387103. Article published online before print in March 2003.
REFERENCES
- 1.Bradley D.G. and Cunningham, E.P. 1998. Genetic aspects of domestication. In The genetics of cattle (eds. R. Fries & A. Ruvinski), pp. 15–32. CAB International, Oxon, UK.
- 2.Devlin B. and Risch, N. 1995. A comparison of linkage disequilibrium measures for fine-scale mapping. Genomics 29: 311-322. [DOI] [PubMed] [Google Scholar]
- 3.Hill W.G. 1981. Estimation of effective population size from data on linkage disequilibrium. Genet. Res. 38: 209-216. [Google Scholar]
- 4.Hudson R.R. 1985. The sampling distribution of linkage disequilibrium under an infinite allele model without selection. Genetics 109: 611-631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kimura M. and Crow, J.F. 1964. The number of alleles that can be maintained in a finite population. Genetics 49: 725-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kingman J.F.C. 1982. On the genealogy of large populations. In Essays in statistical science: Papers in honour of P.A.P. Moran (eds. J. Gani & E.J. Hannan), pp. 27–43. Applied Probability Trust, Sheffield. J. Appl. Prob., special volume 19A.
- 7.Kuhner M.K., Yamato, J., and Felsenstein, J. 1998. Maximum likelihood estimation of population growth rates based on the coalescent. Genetics 149: 429-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meuwissen T.H.E. and Goddard, M.E. 2001. Prediction of identity-by-descent probabilities from marker haplotypes. Genet. Select. Evol. 33: 605-634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moffat M.F., Traherne, J.A., Abcasis, G.R., and Cookson, W.O.C.M. 2000. Single nucleotide polymorphism and linkage disequilibrium within the TCR α/δ locus. Hum. Mol. Genet. 9: 1011-1019. [DOI] [PubMed] [Google Scholar]
- 10.Pritchard J.K. and Przeworski, M. 2001. Linkage disequilibrium in humans: Models and data. Am. J. Hum. Genet. 69: 1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reich E.D., Cargill, M., Bolk, S., Ireland, J., Sabeti, P.C., Richter, D.J., Lavery, T., Kouyoumjian, R., Farhadian, S.F., Ward, R., et al. 2001. Linkage disequilibrium in the human genome. Nature 411: 199-204. [DOI] [PubMed] [Google Scholar]
- 12.Sabatti C. and Risch, N. 2002. Homozygosity and linkage disequilibrium. Genetics 160: 1707-1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shifman S. and Darvasi, A. 2001. The value of isolated populations. Nat. Genet. 28: 309-310. [DOI] [PubMed] [Google Scholar]
- 14.Shriver M.D.L., Jin, L., Chakraborty, R., and Boerwinkle, E. 1993. VNTR allele frequency distributions under the stepwise mutation model: A computer simulation approach. Genetics 134: 983-993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slatkin M. and Bertorelle, G. 2001. The use of intraallelic variability for testing neutrality and estimating population growth rate. Genetics 158: 865-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sved J.A. 1971. Linkage disequilibrium and homozygosity of chromosome segments in finite population. Theoret. Pop. Biol. 2: 125-141. [DOI] [PubMed] [Google Scholar]
- 17.Weir B.S., 1996. Genetic data analysis II. Sinauer Associates, Sunderland, MA.
- 18.Weir B.S. and Hill, W.G. 1980. Effect of mating structure on variation in linkage disequilibrium. Genetics 95: 477-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young C.W. and Seykora, A.J. 1996. Estimates of inbreeding and relationship among registered Holstein females in the Unites States. J. Dairy Sci. 79: 502-505. [DOI] [PubMed] [Google Scholar]