

Abstract
Background
Mortierella alpina is an oleaginous fungus used in the industrial scale production of arachidonic acid (ARA). In order to investigate the metabolic characteristics at a systems level and to explore potential strategies for enhanced lipid production, a genome-scale metabolic model of M. alpina was reconstructed.
Results
This model included 1106 genes, 1854 reactions and 1732 metabolites. On minimal growth medium, 86 genes were identified as essential, whereas 49 essential genes were identified on yeast extract medium. A series of sequential desaturase and elongase catalysed steps are involved in the synthesis of polyunsaturated fatty acids (PUFAs) from acetyl-CoA precursors, with concomitant NADPH consumption, and these steps were investigated in this study. Oxygen is known to affect the degree of unsaturation of PUFAs, and robustness analysis determined that an oxygen uptake rate of 2.0 mmol gDW−1 h−1 was optimal for ARA accumulation. The flux of 53 reactions involving NADPH was significantly altered at different ARA levels. Of these, malic enzyme (ME) was confirmed as a key component in ARA production and NADPH generation. When using minimization of metabolic adjustment, a knock-out of ME led to a 38.28% decrease in ARA production.
Conclusions
The simulation results confirmed the model as a useful tool for future research on the metabolism of PUFAs.
Electronic supplementary material
The online version of this article (doi:10.1186/s12918-014-0137-8) contains supplementary material, which is available to authorized users.
Keywords: Mortierella alpina, Arachidonic acid, Genome-scale metabolic model, Polyunsaturated fatty acids, Malic enzyme
Background
Mortierella alpina is an oleaginous zygomycete, that can accumulate lipids to 50% of its dry weight in the form of triacylglycerols [1]. The important ω-6 polyunsaturated fatty acid (PUFA) arachidonic acid (ARA) can account for over 50% of the lipid content [2]. M. alpina is nonpathogenic and nonallergenic, including the spores produced during the industrial production of ARA [3] which is widely used in food ingredients [4]. ARA has been produced at levels up to 19.8 g/L in 5 L cultures grown over 7 days [5]. Various methods have been attempted in order to improve ARA production including screening potentially higher yielding mutant strains following treatment with N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) [6]. This work led to the generation of M. alpina strain Y11 which possessed lowered ω-3 desaturation activity, and ARA production was 2.1 fold (2.21 g/L) higher than strain 1S-4. Optimizing the culture medium and fixing the ratio of defatted soybean meal to potassium nitrate at 2:1 gave a fourfold increase (6.0 g/L) in ARA production [7]. In another study the fermentation process was optimized, and a two-stage temperature-shift strategy increased ARA production by 26.1% (9.2 g/L) [8]. Additionally, overexpressing GLELO gene using a genetic manipulation approach increased ARA production by 101.2% (5.05 g/L) [9].
Genetic engineering of M. alpina for enhanced ARA production remains an attractive proposition and for further research. Early work identified desaturases as key enzymes in PUFA synthesis. Specifically, Δ5 desaturase [10], which catalyzes dehydrogenation of dihomo-γ-linolenic acid (DGLA) to form ARA, was isolated and functionally characterized. The rate-limiting step for ARA biosynthesis is catalyzed by elongase which converts γ-linolenic acid (GLA) to DGLA [11,12]. NADH-cytochrome b5 reductase (Cb5R), an electron carrier and a major component of the cytochrome b5-dependent electron transport system, is also crucial. Cb5R catalyzes several different reduction reactions, including the desaturation and elongation of acyl chains built from acetyl-CoA during PUFA synthesis [13]. Despite various studies that have identified the importance of these enzymes in PUFA synthesis and metabolism [2,14], their exact roles are not completely understood, and neither are the pathways through which glucose relates to PUFA biosynthesis and metabolism.
A genome-scale metabolic model (GSMM) is an indispensable tool for the study of metabolism that adopts a systems biology approach to integrate data from genomics, transcriptomics, proteomics and metabolomics. It has been widely used in the analysis of the network properties of metabolism [15], prediction and analysis of organism growth phenotypes [16], model-based interpretation of experimental data [17], and metabolic engineering [18]. Oleaginous organisms such as M. alpina can accumulate large quantities of lipids, but maximizing lipid production is complicated by the complexity of the regulatory mechanisms associated with lipid metabolism. It is generally difficult to identify key metabolic modules contributing to lipid physiology. Using reconstruction GSMM, we can systematically analyze the function of each gene and metabolic reaction and model the effects using flux balance analysis (FBA). Specific pathways can be understood based on the model of the whole metabolic network, and strain design strategies can also be used to guide metabolic engineering experiments. Two GSMM studies on Yarrowia lipolytica (iNL895 [19] and iYL619_PCP [20]) have been published along with recent modeled networks of Mucor circinelloides and M. alpina [21]. GSMM studies therefore provide a new approach to investigating the complex lipid metabolism in M. alpina. Vongsangnak et al. (2013) [21] previously published a M. alpina network model, however, this was a refined network that could only be used to investigate genome annotation and metabolic routes, and not flux distribution or phenotypic behaviors [21]. To systematically study flux distribution and the mechanism of lipid accumulation, we reconstructed a new M. alpina GSMM and used the COBRA Toolbox [22] for subsequent research.
In this study, a genome-scale metabolic model (iCY1106) of M. alpina ATCC 32222 was reconstructed based on sequencing results [2]. Using this model, we first identified essential genes and reactions in fermentation medium containing sodium nitrate as a nitrogen source [23]. The de novo synthesis pathways of PUFAs such as ARA and eicosapentaenoic acid (EPA) were subsequently resolved. The roles of acetyl-CoA and NADPH in the regulation of PUFA biosynthesis were probed, and important genes and reactions were systematically analyzed using FBA, flux variability analysis (FVA), and minimization of metabolic adjustment (MOMA).
Results and discussions
Characterization of the M. alpina GSMM iCY1106
The genome-scale metabolic model iCY1106 reconstructed in this study included 1106 genes representing 9.51% of the 11631 protein-coding genes in the genome of M. alpina ATCC 32222. The model consisted of 1854 reactions and 1732 metabolites (Table 1). Compared with the previous model constructed by Vongsangnak [21], iCY1106 is more detailed and contains a higher degree of functional information that can be used in in silico simulations. During the construction of iCY1106, some missing gene were added from the Vongsangnak’s model such as MA-090-247, which encodes an asparaginase (EC: 3.5.1.1), and catalyzes the transformation of L-asparagine into L-aspartate. When compared with the existing model, many improvements in iCY1106 are apparent. (1) A total of 139 transport proteins were annotated based on TCDB and TransportDB. (2) A total of 216 exchange reactions were added into model iCY1106. (3) Biomass composition of M. alpina was calculated based on literature mining. (4) Additional chemical reactions were also included. (5) The reversibility of some reactions was redefined according to MetaCyc and BioPath databases. For example, in the published model, the reaction catalyzed by aldehyde dehydrogenase (EC: 1.2.1.3) was reversible, whereas this reaction is irreversible according to BioPath. The flux distribution of iCY1106 was normal following this change.
Table 1.
General features of model i CY1106
| Features | i CY1106 | Existed model a |
|---|---|---|
| Genome feature | ||
| Genome size (Mb) | 38.38 | 38.38 |
| Total open reading frames (ORFs) | 11631 | 11631 |
| Metabolic model | ||
| Total reactions | 1854 | 1183 |
| Biochemical reactions | 1391 | 1183 |
| Transport reactions | 247 | none |
| Exchange reactions | 216 | none |
| Metabolites | 1732 | 1660 |
| ORFs associated in model | 1106 | 1042 |
| ORF coverageb (%) | 9.51 | 8.95 |
aThe existed model was constructed by Vongsangnak et al. [21]; bThe number of ORFs incorporated in model iCY1106 divided by the total number of ORFs.
In model iCY1106, four compartments (extracellular, cytoplasmic, peroxisome and mitochondrial compartments) were linked by 141 trans-plasma membrane transport reactions, 98 cytoplasmic-mitochondrial transport reactions and eight cytoplasmic-peroxisome transport reactions (Additional file 1). The extracellular compartment contained 267 reactions, including 216 exchange reactions and 51 metabolic reactions. These metabolic reactions catalyzed by extracellular enzymes may enable M. alpina to adapt to different environmental conditions. For example, M. alpina grows on different carbohydrates such as glycerol [24], sucrose [24], d-mannose [25], and raffinose [25] using various extracellular galactosidases. The peroxisome compartment contained 34 reactions mostly related to glyoxylate metabolism.
Metabolic reactions in the model iCY1106 were divided into ten metabolic subsystems (Figure 1). The largest subsystem was lipid metabolism (fatty acid biosynthesis, fatty acid elongation, steroid, glycerolipid, glycerophospholipid and sphingolipid metabolism) that accounted for 23.53% of the total reactions. Reactions involving the synthesis of PUFAs such as DGLA, ARA, and EPA are listed in model iCY1106. For most subsystems, most categories were more abundant in iCY1106 than in the Vongsangnak’s model, with the exception of Amino Acid Metabolism and Nucleotide Metabolism (Figure 1a). Since there were six compartments (cytosol, mitochondria, peroxisome, extracellular, plasma membrane and unclassified compartment) in Vongsangnak’s model, some reactions occur in multiple compartments at the same time. When ignoring compartments, not including transport reactions and exchange reactions, these two models shared 566 reactions (Additional file 2). Vongsangnak’s model and iCY1106 included 179 and 621 unique reactions, respectively (Figure 1b). Compared with model iYL619_PCP of the oleaginous yeast Y. lipolytica [20], lipid metabolism accounted for a higher proportion (16.7%, 191 of total 1142 reactions) of reactions in iCY1106. Amino acid and carbohydrate metabolism subsystems were 13.17% and 11.66%, respectively, in iCY1106.
Figure 1.
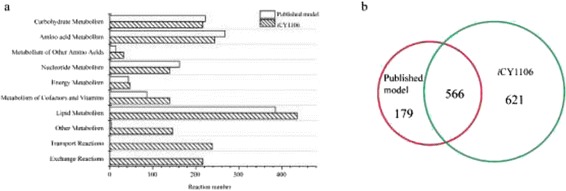
The comparison between Vongsangnak’s model and i CY1106. a: reaction distribution in subsystems between these two models; b: shared reactions and unique reactions between models.
Identification of essential genes and reactions in iCY1106
When cultured on minimal growth medium (MG) containing glucose, KH2PO4, MgSO4 and NaNO3 [23], 86 M. alpina genes (7.78% of the total) were identified as essential for growth using the FBA method (Additional file 3). In contrast, on yeast extract (YE) medium (based on MG but including all 20 regular amino acids) only 49 genes were identified as essential. On MG medium, over half of the essential genes were involved in amino acid (36.05%) and nucleotide metabolism (23.26%; Figure 2). However on YE medium, most of the essential genes were associated with nucleotide metabolism (38.78%).
Figure 2.
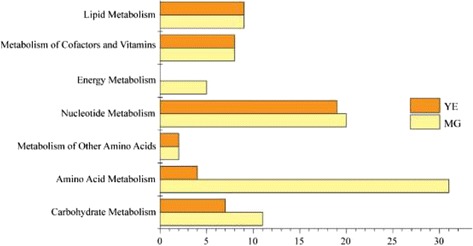
Essential genes identified by MG and YE medium in model i CY1106.
The identified genes were compared with the database of essential genes (DEG), and 40 genes were found to share high homology with genes identified by DEG (identity ≥ 40%, E-value ≤ 1.0E-30; Table 2). One gene, UTP-glucose-1-phosphate uridylyltransferase (MA-090-485), is involved in the synthesis of the cell wall component 1,3-beta-D-glucan (Table 2). MA-090-485 is highly homologous with the Saccharomyces cerevisiae enzyme (sequence identity = 62.58%, e-value = 0) identified in the DEG search. Similarly, nucleoside diphosphate kinase (MA-120-261), which is involved in the synthesis of CTP and GTP, also shared high homology with one of the essential genes identified by DEG (identity = 66.89%, e-value = 2.0E-68). Besides, in MG, there were 46 genes that did not match with DEG (Additional file 3). Of these, when nitrate was used as nitrogen source, nitrate reductase (MA-291-2) and nitrite reductase (MA-291-3), which together convert nitrate into NH3 for cell growth, were essential under this condition. Another two essential genes, delta 5 desaturase (MA-326-160) and delta 12 desaturase (MA-334-414) are vital for the synthesis of ARA in M. alpina. In addition, most of these essential genes were distributed in the Amino Acid Metabolism category (28.3%). This was understandable since essential gene identified by DEG were in a rich medium in which genes involving amino acid synthesis may not be essential.
Table 2.
Essential genes identified on different mediums which are high homology with DEG
| Gene | Enzyme | Essentiality | Subsystem | |
|---|---|---|---|---|
| MG | YE | |||
| MA-133-4 | Ribose-5-phosphate isomerase | E | E | Carbohydrate metabolism |
| MA-090-485 | UTP-glucose-1-phosphate uridylyltransferase | E | E | |
| MA-101-200 | Mannose-6-phosphate isomerase | E | E | |
| MA-120-141 | Phosphoacetylglucosamine mutase | E | E | |
| MA-184-380 | Acetyl-CoA carboxylase | E | E | |
| MA-072-196 | Argininosuccinate lyase | E | NE | Amino acid metabolism |
| MA-213-50 | Adenylosuccinate synthase | E | E | |
| MA-213-539 | Adenylosuccinate lyase | E | E | |
| MA-326-6 | Aspartate carbamoyltransferase | E | E | |
| MA-334-434 | Phosphoribosylanthranilate isomerase | E | NE | |
| MA-120-148 | 2-Acetolactate methylmutase | E | NE | |
| MA-090-434 | Dihydroxy-acid dehydratase | E | NE | |
| MA-139-331 | Homoaconitate hydratase | E | NE | |
| MA-323-77 | Saccharopine dehydrogenase | E | NE | |
| MA-326-106 | Ornithine carbamoyltransferase | E | NE | |
| MA-101-393 | Imidazole-4-carboxamide isomerase | E | NE | |
| MA-184-558 | Imidazoleglycerol-phosphate dehydratase | E | NE | |
| MA-090-452 | Glutamine amidotransferase:cyclase | E | NE | |
| MA-320-159 | 3-dehydroquinate synthase | E | NE | |
| MA-139-157 | Chorismate synthase | E | NE | |
| MA-073-62 | 3-deoxy-7-phosphoheptulonate synthase | E | NE | |
| MA-213-547 | Tryptophan synthase | E | NE | |
| MA-297-40 | Chorismate mutase | E | NE | |
| MA-120-157 | Thioredoxin reductase | E | E | |
| MA-173-30 | Purine-nucleoside phosphorylase | E | E | Nucleotide metabolism |
| MA-213-65 | IMP dehydrogenase | E | E | |
| MA-120-261 | Nucleoside diphosphate kinase | E | E | |
| MA-139-347 | Guanylate kinase | E | E | |
| MA-120-138 | Ribonucleoside-diphosphate reductase | E | E | |
| MA-055-211 | Ribonucleoside-diphosphate reductase | E | E | |
| MA-323-58 | Ribonucleoside-diphosphate reductase | E | E | |
| MA-334-356 | Thymidylate synthase | E | E | |
| MA-153-455 | 3(2),5-bisphosphate nucleotidase | E | NE | |
| MA-111-23 | phosphoadenylyl-sulfate reductase | E | NE | Energy metabolism |
| MA-182-360 | 6,7-dimethyl-8-ribityllumazine synthase | E | E | Cofactors and vitamins |
| MA-184-368 | Riboflavin synthase | E | E | |
| MA-210-311 | Aspartate dehydrogenase | E | E | |
| MA-055-340 | Fatty-acyl-CoA synthase | E | E | Lipid metabolism |
| MA-162-131 | Palmitoyl-protein thioesterase | E | E | |
| MA-334-239 | Very-long-chain enoyl-CoA reductase | E | E | |
E: essential gene; NE: non-essential gene. MG: minimal medium; YE: yeast extract medium.
Simulation and verification of model iCY1106
FBA with constraints was used to investigate the metabolic properties of model iCY1106. YE medium was used to simulate the growth rate and ARA production, the maximum uptake rate of all 20 amino acids were set at 0.01 mmol gDW−1 h-1 [26], and the glucose consumption rate was set at 0.8 mmol gDW−1 h-1 [8], and 2.0 mmol gDW−1 h-1 [1] respectively, and the in silico growth rate were closed to experimental values (differences were only 0.8% and 9.48%; Table 3). To investigate ARA production in silico, the growth rate was fixed at 0.03 h-1 [8], and the ARA exchange reaction was set as the objective function. Under these conditions, the ARA rate was 0.128 mmol gDW−1 h−1 by FBA. This in silico value was lower than the experimentally determined value of 0.149 mmol gDW−1 h−1 (14.09%) [8], and this may due to the use of two different M. alpina strains (ATCC 32222 and ME-1) for modeling and experimental components, with ME-1 being the higher ARA producing strain [27]. Furthermore, the difficulties associated with accurately modeling the yeast extract may have resulted in a nutritional deficiency in the model.
Table 3.
Comparison of in silico and in vivo growth rates of M . alpina
| Media condition (mmol gDW −1 h −1 ) | Growth rate (h −1 ) | Ref. | |
|---|---|---|---|
| in vivo | in silico | ||
| Glc (v = 0.8) | 0.0696 | 0.0690 | [8] |
| Glc (v = 2.0) | 0.1708 | 0.1546 | [1] |
in vivo: experimental results; in silico: simulation results.
The capability of utilizing 14 different carbon sources for cell growth was qualitatively predicted by the model iCY1106 using FBA (Additional file 4). Each substance was the sole carbon source and the rate of uptake was 1.0 mmol gDW−1 h−1. Initially, there were four types of carbon source (ethanol, xylose, maltose, and rhamnose), and the model could not achieve growth with any individual carbon source. For ethanol, the reaction catalyzed by alcohol dehydrogenase (EC: 1.1.1.1) was irreversible, but should be reversible according to the MetaCyc database. For rhamnose, the lack of L-rhamnulose-1-phosphate lactaldehyde-lyase (EC: 4.1.2.19) meant that rhamnose could not generate glycerone phosphate, which could otherwise be used by the glycolysis pathway. After elimination of initial discrepancies using the continuous gap filling process, a 100% match was acquired. This indicated that model iCY1106 could predict catabolic pathways of various carbon sources including common sugars and alcohols. A similar FBA simulation of nitrogen sources (ammonium, nitrate, urea, and amino acids) also generated results that were consistent with experimental fermentation data. Both glutamate and glycine supported M. alpina growth, confirming that the model performed well for predicting the utilization of different carbon and nitrogen sources.
PUFA biosynthetic pathways based on GSMM
To further investigate the pathways associated with PUFA biosynthesis and metabolism, genome annotation, literature mining and KEGG database analysis were performed. ARA is a ω-6 PUFA synthesized by M. alpina (Figure 3). ARA (c204(6)) is synthesized from acetyl-CoA in 38 enzymatic steps, including the de novo synthesis of palmitic acid (c160) and the synthesis of very long chain fatty acids. During PUFA biosynthesis, all fatty acids should be in the form of acyl-CoA [2]. Desaturase and elongase were necessary for the conversion of octadecanoyl-CoA (c180coa) to ARA. Δ9 desaturase [28] (MA-055-51, MA-101-533, MA-184-235), Δ12 desaturase [29] (MA-334-414) and Δ6 desaturase [30,31] (MA-268-54, MA-101-36) catalyzed the conversion of c180coa into γ-linolenoyl-CoA (c183(6)coa) which was subsequently lengthened to 8,11,14-eicosatrienoyl-CoA (c203(6)coa) by an elongase such as ELO/GLELO [30] (MA-189-257), MAELO [32] (MA-184-206) or MALCE1 [33] (MA-073-327, MA-320-221). Δ5 desaturase [10] (MA-326-160) catalyzed c203(6)coa to arachidonyl-CoA (c204(6)coa), which was finally hydrolyzed to c204(6) by choloyl-CoA hydrolase (MA-049-4, MA-139-47).
Figure 3.
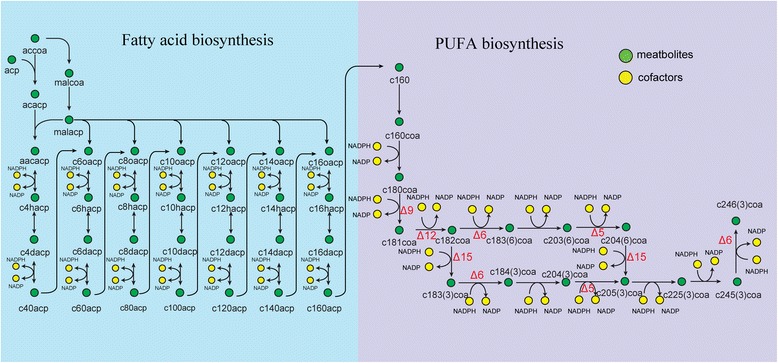
Synthesis pathway of PUFAs in M . alpina . (accoa: acetyl-CoA, acp: acyl-carrier protein, acacp: Acetyl-[acyl-carrier protein], malcoa: malonyl-CoA, malacp: malonyl-[acp], aacacp: acetoacetyl [acp], c4hacp: (3R)-3-Hydroxybutanoyl-[acp], c4dacp: but-2-enoyl-[acp], c40acp: butanoyl-[acp], c6oacp: 3-oxohexanoyl-[acp], c6hacp: 3-hydroxyhexanoyl-[acp], c6dacp: hex-2-enoyl-[acp], c60acp: hexanoyl-[acp], c8oacp: 3-oxooctanoyl-[acp], c8hacp: 3-hydroxyoctanoyl-[acp], c8dacp: oct-2-enoyl-[acp], c80acp: octanoyl-[acp], c10oacp: 3-oxodecanoyl-[acp], c10hacp: 3-hydroxydecanoyl-[acp], c10dacp: dec-2-enoyl-[acp], c100acp: decanoyl-[acp], c12oacp: 3-oxododecanoyl-[acp], c12hacp: 3-hydroxy-dodecanoyl-[acp], c12dacp: dodec-2-enoyl-[acp], c120acp: dodecanoyl-[acp], c14oacp: 3-oxomyristoyl-[acp], c14dacp: tetradec-2-enoyl-[acp], c14hacp: 3-hydroxymyristoyl-[acp], c140acp: myristoyl-[acp], c16oacp: 3-oxohexadecanoyl-[acp], c16hacp: (3R)-3-Hydroxypalmitoyl-[acp], c16dacp: hexadec-2-enoyl-[acp], c160acp: hexadecanoyl-[acp], c160: palmitic acid, c160coa: hexadecanoyl-CoA, c180coa: octadecanoyl-CoA, c181coa: octadecenoyl-CoA, c182coa: linoleoyl-CoA, c183(6)coa: gamma-Linolenoyl-CoA, c203(6)coa: 8,11,14-Eicosatrienoyl-CoA, c204(6)coa: Arachidonyl-CoA, c204(6): ARA, c183(3)coa: alpha-Linolenoyl-CoA, c184(3)coa: Stearidonoyl-CoA, c204(3)coa: (11Z,14Z,17Z)-Icosatrienoyl-CoA, c205(3)coa: eicosapentaenoyl-CoA, c225(3)coa: (7Z,10Z,13Z,16Z,19Z)-Docosapentaenoyl-CoA, c245(3)coa: (9Z,12Z,15Z,18Z,21Z)-Tetracosaheptaenoyl-CoA, c246(3)coa: (6Z,9Z,12Z,15Z,18Z,21Z)-Tetracosahexaenoyl-CoA, c226(3)coa: (4Z,7Z,10Z,13Z,16Z,19Z)-Docosahexaenoyl-CoA).
M. alpina is capable of producing ARA and EPA, and EPA can be produced via the ω-3 or the ω-6 PUFA biosynthetic pathways through ARA desaturation [34]. During EPA biosynthesis, Δ15 (ω3) desaturase (MA-072-10) is the key enzyme and was the first fungal desaturase known that uses both 18-carbon and 20-carbon ω-6 PUFAs as substrates [35]. In the ω-3 pathway, (11Z,14Z,17Z)-icosatrienoyl-CoA (c204(3)coa) was used as substrate, while the ω-6 pathway utilized c204(6)coa.
Both the maximum and minimum flux of oxygen absorption of DGLA, ARA and EPA production in silico were found to be 1.552, 1.674 and 1.891 mmol gDW−1 h−1 respectively, using FVA. This indicated that the degree of PUFA unsaturation was dependent on oxygen content. Robustness analysis was used to elucidate the effects of oxygen uptake rate on PUFA biosynthesis. The oxygen uptake rate was perturbed by constraints-based flux analysis between 0 and 20 mmol gDW−1 h−1. The minimum growth rate was set at 0.03 h−1, with ARA exchange reaction as the objective function. When oxygen absorption was lower than 2.0 mmol gDW−1 h−1, ARA production increased with oxygen absorption rate (Additional file 5a). However, beyond this range, ARA production gradually declined with increasing oxygen uptake rate. As reported by Higashiyama et al., when the DO concentration range was 10-15 ppm, the ARA yield was enhanced 1.6-fold over a DO of 7 ppm, and a DO between 20 and 50 ppm drastically decreased ARA production [36]. This indicated that excessive oxygen exposure during fermentation could impair ARA overproduction. During ARA biosynthesis, high oxygen concentrations may affect metabolism and cell growth in the filamentous mycelia, and beta-oxidation of fatty acids may be needed to obtain the extra energy required for adaptation to the high oxygen conditions.
Regulation of PUFA biosynthesis and metabolism based on GSMM
Acetyl-CoA is the essential two-carbon donor molecule for fatty acid synthesis. In model iCY1106, there were ten pathways that could produce acetyl-CoA, including amino acid degradation (from aspartate, isoleucine, leucine, and lysine), fatty acid beta-oxidation, and other acetyl-CoA generation processes (phosphoenolpyruvate [PEP], malate, L-lactate, acetate, and citrate) (Figure 4). However, only the pathway from PEP to pyruvate could generate sufficient flux for synthesis of PUFAs on the fermentation medium. During the growth stage, the flux of acetyl-CoA generated by pyruvate was 1.22 mmol gDW−1 h−1, while this value was 1.56 mmol gDW−1 h−1 during the product synthesis stage. This corresponds to a 27.87% increase, which would ensure acetyl-CoA was available for ARA production due to the enhanced activity of the pyruvate dehydrogenase complex (EC: 1.2.4.1, 2.3.1.12, 1.8.1.4) [37]. Although aspartate, isoleucine, leucine, and lysine could all be used to generate acetyl-CoA for ARA production. Asparate has four carbon, while isoleucine, leucine, and lysine all contains six carbon. When the maximum uptake rate of asparate and the other amino acids were set at 0.15 mmol gDW−1 h−1 and 0.1 mmol gDW−1 h−1, respectively. FBA showed only that leucine and lysine could increase ARA production by 14.06% and 13.28%.
Figure 4.
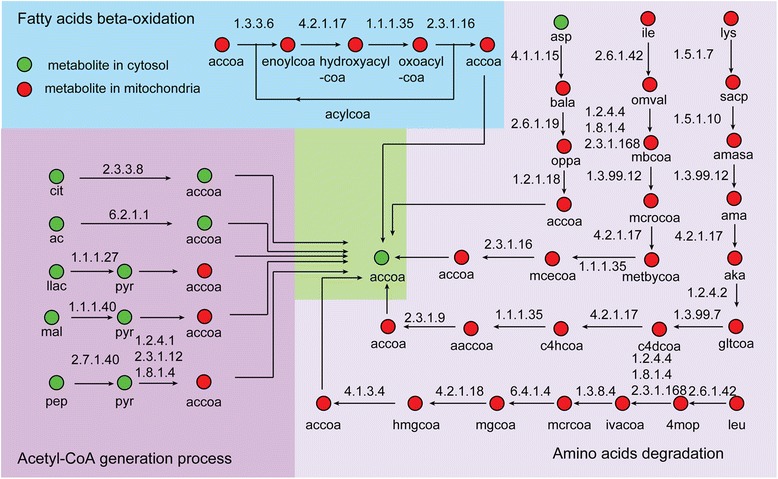
The sources of acetyl-CoA in M . alpina . (cit: citrate, ac: acetate, llac: l-lactate, mal: malate, pep: phosphoenolpyruvate, pyr: pyruvate, accoa: acrtyl-CoA, asp: l-aspartate, bala: beta-alanine, oppa: 3-oxopropanoate, ile: l-isoleucine, omval: (s)-3-nethyl-2-oxopentanoate, mbcoa: 2-methylbutanoyl-CoA, mcrocoa: 2-methylcrotonoyl-CoA, metbycoa: (2S,3S)-3-Hydroxy-2-methylbutanoyl-CoA, mcecoa: 2-Methylacetoacetyl-CoA, lys: l-lysine, sacp: l-saccharopine, amasa: l-2-aminoadipate 6-semialdehyde, ama: l-2-aminoadipate, aka: 2-oxoadipate, gltcoa: glutaryl-CoA, c4dcoa: crotonoyl-coa, c4hcoa: (S)-3-Hydroxybutanoyl-CoA, aaccoa: acetoacetyl-CoA, leu: l-leucine, 4mop: 4-methyl-2-oxopentanoate, ivacoa: 3-isovaleryl-coa, mcrcoa: 3-nethylcrotonyl-CoA, mgcoa: trans-3-methylglutaconyl-CoA, hmgcoa: (s)-3-hydroxy-3-methylglutaryl-coa).
In model iCY1106, acetyl-CoA can be consumed by amino acid synthesis, amino sugar metabolism and fatty acid synthesis as well as ARA production (Additional file 5b). During the synthesis of six amino acids (threonine, methionine, lysine, cysteine, ornithine, and leucine), acetyl-CoA was required. FBA results showed that during the growth stage, 96.5% of the acetyl-CoA flux was used to synthesize fatty acids and 2.58% was used for amino acid synthesis. In contrast, during the product synthesis stage, the flux of acetyl-CoA used for fatty acid synthesis accounted for 99.52%, which indicates that amino acid synthesis was inhibited during ARA production. Limiting the nitrogen source can be an effective strategy to control amino acids biosynthesis [38], in which the lipid yield may be increased despite decreases in mycelia concentration.
During the growth stages, the flux of acetyl-CoA used to synthesize malonyl-CoA, catalyzed by acetyl-CoA carboxylase (ACC, EC 6.4.1.2), was 0.79 mmol gDW−1 h−1. In contrast during the ARA production stage, the corresponding flux was 1.43 mmol gDW−1 h−1 (81.0%). ACC catalyzes the first committal step of the fatty acid biosynthetic pathway, and should be overexpressed to maximize ARA production [39]. Glutamate has been shown to activate ACC, and adding glutamate to the culture medium led to an increase in total lipid (21.81%) and ARA yield (66.07%) [40]. As the M. alpina growth rate in batch culture ranged from 0.06 h−1 to 0.19 h-1 [1,7,8,41,42], the glutamate uptake rates used for the in silico analysis should be set between 0 and 1.8 mmol gDW−1 h−1. When the glutamate uptake rate was increased, ARA production increased from 0.128 mmol gDW−1 h−1 to 0.355 mmol gDW−1 h−1 (Additional file 5c).
During PUFA biosynthesis, NADPH is a necessary cofactor for de novo fatty acid synthesis involving desaturation and elongation. In model iCY1106, there were 172 reactions involving NADPH (Additional file 6). With ARA production fixed at 0.059 mmol gDW−1 h−1 (μ = 0.06 h−1) and 0.128 mmol gDW−1 h−1 (μ = 0.03 h−1), the flux of NADPH reactions were investigated. In total, 53 NADPH reactions exhibited flux changes of > 10−6 mmol gDW−1 h−1 (Figure 5).
Figure 5.
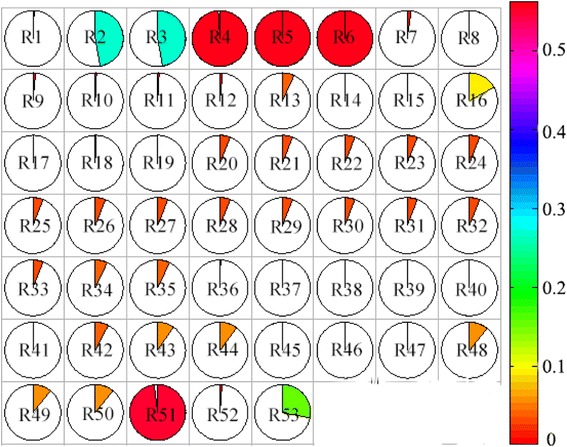
Changes of reactions involving NADPH at different ARA levels. (Different colors represent different changes on NADPH flux. Flux obviously changed reactions are listed. R2: D-glucose 6-phosphate + NADP ⬄ 6-phospho-D-glucono-1,5-lactone + NADPH + H, R3: 6-phospho-D-gluconate + NADP - > D-ribulose 5-phosphate + CO2 + NADPH + H, R4: (S)-malate + NADP - > pyruvate + CO2 + NADPH, R5: L-glutamate 5-semialdehyde + NAD P+ H2O - > L-glutamate + NADPH + H, R6: 1-pyrroline-5-carboxylate + NADPH + H ⬄ L-proline + NADP, R16: Nitrite + 3 NADPH + 5 H - > Ammonia + 3 NADP + 2 H2O, R51: N-acetyl-L-glutamyl 5-phosphate + NADPH + H ⬄ N-acetyl-L-glutamate 5-semialdehyde + NADP + Phosphate, R53: (S)-3-hydroxy-3-methylglutaryl-CoA + 2 NADPH + 2 H ⬄ (R)-mevalonate + CoA + 2 NADP).
Further research showed that only five reactions (R2, R3, R4, R5, and R6) involving accumulation of NADPH were significantly altered. When ARA production was increased, three reactions (R4, R5, and R6) underwent flux increases, which corresponded to enhanced enzyme activities due to up-regulation of the genes encoding the enzymes. Of these, malic enzyme (ME, EC 1.1.1.40), which catalyzes the conversion of malate to pyruvate (R4), is considered a key enzyme in lipogenesis in M. alpina. ME expression and enzyme activity are enhanced during ARA production [43], and the minimization of metabolic adjustment (MOMA) method was used to investigate the role of this enzyme. With the biomass and ARA flux maximized by ME deletion using MOMA, 410 (22.11%) reactions showed flux changes (Additional file 6) compared with the wild type model. The ARA exchange reaction flux was lowered from 0.128 mmol gDW−1 h−1 to 0.079 mmol gDW−1 h−1, a decrease of 38.28%. Additionally, all reactions involving NADPH consumption reactions other than R13, R16, R51, and R53 were associated with lipid metabolism. These results indicated that the increase in ARA production was directly correlated with the NADPH consumption rate.
Conclusions
In this study, a genome-scale metabolic model (iCY1106) for Mortierella alpina ATCC 32222 was successfully reconstructed using knowledge from the scientific literature and publicly accessible databases. The model comprised 1106 genes, 1854 reactions and 1732 metabolites, and included 247 transport reactions and 216 exchange reactions. Following a series of simulations and verification by measurement of growth rates and substrate usage, the model was found to agree with published literature. The model was used to investigate the effects of the important precursors acetyl-CoA and NADPH on the biosynthesis of PUFAs such as ARA and EPA. FBA results showed that enhancing the pyruvate dehydrogenase complex increased acetyl-CoA availability for increasing ARA production. Limiting the nitrogen source was an effective method for reducing acetyl-CoA consumption, and malic enzyme was found to be a key node in the regulation of NADPH in ARA biosynthesis. Model iCY1106 could serve as a useful predictive tool for future systems biology studies to guide the genetic engineering of M. alpina to improve the production of industrially important metabolites.
Methods
Construction of the M. alpina GSMM
The draft model was constructed by amassing reactions from the genome-scale metabolic models of genetically related organisms Aspergillus terreus [44], Pichia pastoris [45] and Yarrowia lipolytica [20]. Reactions were chosen based on orthologs shared between M. alpina and the three reference organisms identified by protein sequence similarity searches using BLAST. Protein sequences from A. terreus NIH 2624, P. pastoris GS 115 and Y. lipolytica CLIB 122 were downloaded from UniProt [46]. Open reading frame (ORF) information for M. alpina ATCC 32222 was provided by Yong Q. Chen based on sequencing results. The iJL1454 GSMM was used as a reference. Additionally, iLC915 [45] and iYL619_PCP [20] were also used for comparison since both models were concerned with lipid metabolism and overproduction. To ensure accuracy, only sufficiently similar orthologs with e-values ≤10−30 and sequence identities ≥40% were included [47]. To expand and update the draft model, the genome of M. alpina was re-annotated by submitting ORFs to the KAAS online annotation server [48]. Metabolic reactions absent from the draft model were added from the KEGG database [49] based on KAAS annotation results. MetaCyc [50] and BioPath [51] databases were used to judge reaction reversibility. Compartmentalization information assigned to reactions was determined by subcellular localization prediction tools CELLO [52] and WoLF PSORT. [53] BaCelLo [54] was also used for proteins that were difficult to determine with the other tools. Transport information was obtained by cross-referencing BLATSp searches and the Transporter Classification Database TCDB [55].
To refine the draft model, the gapFind [56] program in the COBRA Toolbox [22] was used to identify metabolic gaps in draft model and literature data were used to fill these gaps. The metabolites in each reaction were characterized based on their chemical formulae and neutral charges, which were obtained using CHEBI [57] and PubChem [58].
Biomass composition
The biomass equation of M. alpina was assumed to have six components: proteins, DNA, RNA, lipids, the cell wall and the small molecule pool [59,60]. Since no detailed information on M. alpina DNA and RNA was available, the ratio was assumed to be the same as in the related Aspergillus niger [61]. The nucleotide and amino acid composition were calculated based on the M. alpina ATCC 32222 genome [2], as no specific experimental data were available. Similarly, the cell wall composition was calculated based on the typical fungal cell wall structure [62]. The lipid composition was calculated based on the current literature [63]. For calculation of energetic parameters, the growth and non-growth associated ATP maintenance values (GAM and NGAM, respectively) were assumed to be the same as those in the central carbon metabolic model of A. niger [64]. Detailed biomass composition information can be found in Additional file 7.
Simulation and analysis
The reconstructed metabolic network was converted into stoichiometric matrix (S = M * N) using the Matlab program, where M represents metabolites and N represents reactions [65]. The basic tools used for model analysis were flux balance analysis (FBA) and flux variability analysis (FVA). GLPK was used for linear programming [22], and Gurobi was used for quadratic programming [66]. In silico analysis included growth simulation, gene and reaction essentiality analysis, robustness analysis, and minimization of metabolic adjustment. Analyses were performed according to the instructions for the COBRA Toolbox [22]. Constraints used in each analysis are mentioned in the results section. To analyze model parameters relevant to cell growth, the biomass equation was selected as the objective function. For analysis of ARA production, the exchange reaction of ARA was the objective function.
Acknowledgements
The author would like to thank Dr Jens Nielsen, Professor at Chalmers University of Technology, for technological help on the model reconstruction. This work was supported by the Program for Innovative Research Team in University (IRT1249), the Provincial Outstanding Youth Foundation of Jiangsu Province (BK2012002), the National Natural Science Foundation of China (21422602), the Program for Young Talents in China, the National High-Tech R & D Program of China (No. 2014AA021701). The Program for New Century Excellent Talents (NCET-13-0831), the National Natural Science Foundation of China (No. 31125021, No. 21276108), and the Fundamental Research Funds for the Central Universities (No. JUSRP51320B).
Abbreviations
- ARA
Arachidonic acid
- PUFAs
Polyunsaturated fatty acids
- ME
Malic enzyme
- GSMM
Genome-scale metabolic model
- MNNG
N-methyl-N’-nitro-N-nitrosoguanidine
- DGLA
Dihomo-γ-linolenic acid
- GLA
γ-linolenic acid
- Cb5R
NADH-cytochrome b5 reductase
- FBA
Flux balance analysis
- EPA
Eicosapentaenoic acid
- FVA
Flux variability analysis
- MOMA
Minimization of metabolic adjustment
- MG
Minimal growth
- YE
Yeast extract
- DO
Dissolved oxygen
- ACC
Acetyl-CoA carboxylase
- GAM
Growth associated ATP maintenance
- NGAM
Non-growth associated ATP maintenance
Additional files
Detailed information about model i CY1106.
Shared reactions between Vongsangnak’s model and i CY1106 (not including transport and exchange reactions, no compartments information).
Result of essential genes identified by FBA using minimum growth medium and yeast extract medium.
Simulation result for the capability of usage different carbon and nitrogen sources for i CY1106.
a Figure for the robustness analysis about oxygen and ARA. b Figure about the acetyl-CoA consumption pathways in iCY1106. c Figure for the robustness analysis about glutamate and ARA.
FBA simulation result for the reactions involving NADPH at different ARA levels.
Biomass composition information for M. alpina.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CY carried out the construction of iCY1106, performed the statistical analysis, and drafted the manuscript. NX carried out literature mining for M. alpina, provided help for the construct of model. HC, YQC and WC provided the refined genome sequence of M. alpina. LL participated in the design of the study and helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Chao Ye, Email: ziye0911@gmail.com.
Nan Xu, Email: nan1989111@gmail.com.
Haiqin Chen, Email: haiqinchen@jiangnan.edu.cn.
Yong Q Chen, Email: yqchen@jiangnan.edu.cn.
Wei Chen, Email: weichen@jiangnan.edu.cn.
Liming Liu, Email: mingll@jiangnan.edu.cn.
References
- 1.Ho S-Y, Jiang Y, Chen F. Polyunsaturated fatty acids (PUFAs) content of the fungus Mortierella alpina isolated from soil. J Agric Food Chem. 2007;55:3960–6. doi: 10.1021/jf0700071. [DOI] [PubMed] [Google Scholar]
- 2.Wang L, Chen W, Feng Y, Ren Y, Gu Z, Chen H, et al. Genome characterization of the oleaginous fungus Mortierella alpina. PLoS One. 2011;6:e28319. doi: 10.1371/journal.pone.0028319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shimizu S, Ogawa J, Kataoka M, Kobayashi M. Screening of novel microbial enzymes for the production of biologically and chemically useful compounds. Adv Biochem Eng Biotechnol. 1997;58:45–87. doi: 10.1007/BFb0103302. [DOI] [PubMed] [Google Scholar]
- 4.Streekstra H. On the safety of Mortierella alpina for the production of food ingredients, such as arachidonic acid. J Biotechnol. 1997;56:153–65. doi: 10.1016/S0168-1656(97)00109-0. [DOI] [PubMed] [Google Scholar]
- 5.Jin MJ, Huang H, Xiao AH, Zhang K, Liu X, Li S, et al. A novel two-step fermentation process for improved arachidonic acid production by Mortierella alpina. Biotechnol Lett. 2008;30:1087–91. doi: 10.1007/s10529-008-9661-1. [DOI] [PubMed] [Google Scholar]
- 6.Sakuradani E, Hirano Y, Kamada N, Nojiri M, Ogawa J, Shimizu S. Improvement of arachidonic acid production by mutants with lower n-3 desaturation activity derived from Mortierella alpina 1S-4. Appl Microbiol Biotechnol. 2004;66:243–8. doi: 10.1007/s00253-004-1682-7. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, Zhao JX, Chen HQ, Chen W, Zhang H. The preliminary study of fermentation conditions for polyunsaturated fatty acids production by Mortierella aplina. Sci Technol Food Int. 2011;11:216–9. [Google Scholar]
- 8.Peng C, Huang H, Ji X, Liu X, You J, Lu J, et al. A temperature-shift strategy for efficient arachidonic acid fermentation by Mortierella alpina in batch culture. Biochem Eng J. 2010;53:92–6. doi: 10.1016/j.bej.2010.09.014. [DOI] [Google Scholar]
- 9.Takeno S, Sakuradani E, Tomi A, Inohara-Ochiai M, Kawashima H, Shimizu S. Transformation of oil-producing fungus, Mortierella alpina 1S-4, using zeocin, and application to arachidonic acid production. J Biosci Bioeng. 2005;100:617–22. doi: 10.1263/jbb.100.617. [DOI] [PubMed] [Google Scholar]
- 10.Abe T, Sakuradani E, Ueda T, Shimizu S. Identification of mutation sites on delta5 desaturase genes from Mortierella alpina 1S-4 mutants. J Biosci Bioeng. 2005;99:296–9. doi: 10.1263/jbb.99.296. [DOI] [PubMed] [Google Scholar]
- 11.Wynn JP, Ratledge C. Evidence that the rate-limiting step for the biosynthesis of arachidonic acid in Mortierella alpina is at the level of the 18:3 to 20:3 elongase. Microbiology. 2000;146:2325–31. doi: 10.1099/00221287-146-9-2325. [DOI] [PubMed] [Google Scholar]
- 12.Takeno S, Sakuradani E, Murata S, Inohara-Ochiai M, Kawashima H, Ashikari T, et al. Molecular evidence that the rate-limiting step for the biosynthesis of arachidonic acid in Mortierella alpina is at the level of an elongase. Lipids. 2005;40:25–30. doi: 10.1007/s11745-005-1356-6. [DOI] [PubMed] [Google Scholar]
- 13.Certik M, Sakuradani E, Kobayashi M, Shimizu S. Characterization of the second form of NADH-cytochrome b(5) reductase gene from arachidonic acid-producing fungus Mortierella alpina 1S-4. J Biosci Bioeng. 1999;88:667–71. doi: 10.1016/S1389-1723(00)87098-X. [DOI] [PubMed] [Google Scholar]
- 14.Sakuradani E, Ando A, Ogawa J, Shimizu S. Improved production of various polyunsaturated fatty acids through filamentous fungus Mortierella alpina breeding. Appl Microbiol Biotechnol. 2009;84:1–10. doi: 10.1007/s00253-009-2076-7. [DOI] [PubMed] [Google Scholar]
- 15.Mahadevan R, Schilling CH. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab Eng. 2003;5:264–76. doi: 10.1016/j.ymben.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 16.Varma A, Palsson BO. Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110. Appl Environ Microbiol. 1994;60:3724–31. doi: 10.1128/aem.60.10.3724-3731.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrett CL, Herring CD, Reed JL, Palsson BO. The global transcriptional regulatory network for metabolism in Escherichia coli exhibits few dominant functional states. Proc Natl Acad Sci U S A. 2005;102:19103–8. doi: 10.1073/pnas.0505231102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pharkya P, Burgard AP, Maranas CD. OptStrain: A computational framework for redesign of microbial production systems. Genome Res. 2004;14:2367–76. doi: 10.1101/gr.2872004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loira N, Dulermo T, Nicaud J-M, Sherman DJ. A genome-scale metabolic model of the lipid-accumulating yeast Yarrowia lipolytica. BMC Syst Biol. 2012;6:35–43. doi: 10.1186/1752-0509-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan P, Hua Q. Reconstruction and in silico analysis of metabolic network for an oleaginous yeast, Yarrowia lipolytica. PLoS One. 2012;7:e51535. doi: 10.1371/journal.pone.0051535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vongsangnak W, Ruenwai R, Xin T, Xinjie H, Hao Z, Shen B, et al. Genome-scale analysis of the metabolic networks of oleaginous Zygomycete fungi. Gene. 2013;521:180–90. doi: 10.1016/j.gene.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 22.Schellenberger J, Que R, Fleming RM, Thiele I, Orth JD, Feist AM, et al. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat Protoc. 2011;6:1290–307. doi: 10.1038/nprot.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu J, Peng C, Ji XJ, You J, Cong L, Ouyang P, et al. Fermentation characteristics of Mortierella alpina in response to different nitrogen sources. Appl Biochem Biotechnol. 2011;164:979–90. doi: 10.1007/s12010-011-9189-z. [DOI] [PubMed] [Google Scholar]
- 24.Yu A-Q, Zhu J-C, Zhang B, Xing L-J, Li M. Effects of different carbon sources on the growth, fatty acids production, and expression of three desaturase genes of mortierella alpina ATCC 16266. Curr Microbiol. 2011;62:1617–22. doi: 10.1007/s00284-011-9902-8. [DOI] [PubMed] [Google Scholar]
- 25.Nisha A, Venkateswaran G. Effect of culture variables on mycelial arachidonic acid production by Mortierella alpina. Food Bioprocess Tech. 2011;4:232–40. doi: 10.1007/s11947-008-0146-y. [DOI] [Google Scholar]
- 26.Boghigian BA, Armando J, Salas D, Pfeifer BA. Computational identification of gene over-expression targets for metabolic engineering of taxadiene production. Appl Microbiol Biotechnol. 2012;93:2063–73. doi: 10.1007/s00253-011-3725-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cong L, Peng C, Ji X, Li Z, You J, Lu J, et al. Progress in production of arachidonic acid by Mortierella alpina and genetic modification. Chin J Biotechnol. 2010;26:1232–8. [PubMed] [Google Scholar]
- 28.Abe T, Sakuradani E, Asano T, Kanamaru H, Shimizu S. Functional characterization of Δ9 and ω9 desaturase genes in Mortierella alpina 1S-4 and its derivative mutants. Appl Microbiol Biotechnol. 2006;70:711–9. doi: 10.1007/s00253-005-0115-6. [DOI] [PubMed] [Google Scholar]
- 29.Sakuradani E, Abe T, Matsumura K, Tomi A, Shimizu S. Identification of mutation sites on delta 12 desaturase genes from Mortierella alpina 1S-4 mutants. J Biosci Bioeng. 2009;107:99–101. doi: 10.1016/j.jbiosc.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 30.Chen R, Matsui K, Ogawa M, Oe M, Ochiai M, Kawashima H, et al. Expression of delta 6, delta 5 desaturase and GLELO elongase genes from Mortierella alpina for production of arachidonic acid in soybean glycine [max (L.) merrill] seeds. Plant Sci. 2006;170:399–406. doi: 10.1016/j.plantsci.2005.09.006. [DOI] [Google Scholar]
- 31.Sakuradani E, Shimizu S. Gene cloning and functional analysis of a second delta 6-fatty acid desaturase from an arachidonic acid-producing Mortierella fungus. Biosci Biotechnol Biochem. 2003;67:704–11. doi: 10.1271/bbb.67.704. [DOI] [PubMed] [Google Scholar]
- 32.Sakuradani E, Murata S, Kanamaru H, Shimizu S. Functional analysis of a fatty acid elongase from arachidonic acid-producing Mortierella alpina 1S-4. Appl Microbiol Biotechnol. 2008;81:497–503. doi: 10.1007/s00253-008-1675-z. [DOI] [PubMed] [Google Scholar]
- 33.Sakuradani E, Nojiri M, Suzuki H, Shimizu S. Identification of a novel fatty acid elongase with a wide substrate specificity from arachidonic acid-producing fungus Mortierella alpina 1S-4. Appl Microbiol Biotechnol. 2009;84:709–16. doi: 10.1007/s00253-009-1999-3. [DOI] [PubMed] [Google Scholar]
- 34.Bajpai P, Bajpai PK, Ward OP. Eicosapentaenoic acid (EPA) Production by Mortierella alpina ATCC 32222. Appl Biochem Biotechnol. 1991;31:267–72. doi: 10.1007/BF02921753. [DOI] [PubMed] [Google Scholar]
- 35.Sakuradani E, Abe T, Iguchi K, Shimizu S. A novel fungal omega 3-desaturase with wide substrate specificity from arachidonic acid-producing Mortierella alpina 1S-4. Appl Microbiol Biotechnol. 2005;66:648–54. doi: 10.1007/s00253-004-1760-x. [DOI] [PubMed] [Google Scholar]
- 36.Higashiyama K, Murakami K, Tsujimura H, Matsumoto N, Fujikawa S. Effects of dissolved oxygen on the morphology of an arachidonic acid production by Mortierella alpina 1S-4. Biotechnol Bioeng. 1999;63:442–8. doi: 10.1002/(SICI)1097-0290(19990520)63:4<442::AID-BIT7>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 37.Pettit FH, Pelley JW, Reed LJ. Regulation of pyruvate dehydrogenase kinase and phosphatase by acetyl-CoA/CoA and NADH/NAD ratios. Biochem Biophys Res Commun. 1975;65:575–82. doi: 10.1016/S0006-291X(75)80185-9. [DOI] [PubMed] [Google Scholar]
- 38.Koike Y, Cai HJ, Higashiyama K, Fujikawa S, Park EY. Effect of consumed carbon to nitrogen ratio of mycelial morphology and arachidonic acid production in cultures of Mortierella alpina. J Biosci Bioeng. 2001;91:382–9. doi: 10.1016/S1389-1723(01)80156-0. [DOI] [PubMed] [Google Scholar]
- 39.Davis MS, Solbiati J, Cronan JE., Jr Overproduction of acetyl-CoA carboxylase activity increases the rate of fatty acid biosynthesis in Escherichia coli. J Biol Chem. 2000;275:28593–8. doi: 10.1074/jbc.M004756200. [DOI] [PubMed] [Google Scholar]
- 40.Yu L, Qin W, Lan W, Zhou P, Zhu M. Improved arachidonic acids production from the fungus Mortierella alpina by glutamate supplementation. Bioresour Technol. 2003;88:265–8. doi: 10.1016/S0960-8524(02)00312-7. [DOI] [PubMed] [Google Scholar]
- 41.Eroshin V, Satroutdinov A, Dedyukhina E, Chistyakova T. Arachidonic acid production by Mortierella alpina with growth-coupled lipid synthesis. Process Biochem. 2000;35:1171–5. doi: 10.1016/S0032-9592(00)00151-5. [DOI] [Google Scholar]
- 42.Zeng Y, Ji X, Chang S, Nie Z, Huang H. Improving arachidonic acid accumulation in Mortierella alpina through B-group vitamin addition. Bioprocess Biosystems Eng. 2012;35:683–8. doi: 10.1007/s00449-011-0648-2. [DOI] [PubMed] [Google Scholar]
- 43.Wynn JP, Bin Abdul Hamid A, Ratledge C. The role of malic enzyme in the regulation of lipid accumulation in filamentous fungi. Microbiology. 1999;145:1911–7. doi: 10.1099/13500872-145-8-1911. [DOI] [PubMed] [Google Scholar]
- 44.Liu J, Gao Q, Xu N, Liu L. Genome-scale reconstruction and in silico analysis of Aspergillus terreus metabolism. Mol Biosyst. 2013;9:1939–48. doi: 10.1039/c3mb70090a. [DOI] [PubMed] [Google Scholar]
- 45.Caspeta L, Shoaie S, Agren R, Nookaew I, Nielsen J. Genome-scale metabolic reconstructions of Pichia stipitis and Pichia pastoris and in silico evaluation of their potentials. BMC Syst Biol. 2012;6:24–37. doi: 10.1186/1752-0509-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, et al. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 2004;32:D115–9. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian W, Skolnick J. How well is enzyme function conserved as a function of pairwise sequence identity? J Mol Biol. 2003;333:863–82. doi: 10.1016/j.jmb.2003.08.057. [DOI] [PubMed] [Google Scholar]
- 48.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–5. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caspi R, Foerster H, Fulcher CA, Kaipa P, Krummenacker M, Latendresse M, et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2008;36:D623–31. doi: 10.1093/nar/gkm900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reitz M, Sacher O, Tarkhov A, Trumbach D, Gasteiger J. Enabling the exploration of biochemical pathways. Org Biomol Chem. 2004;2:3226–37. doi: 10.1039/b410949j. [DOI] [PubMed] [Google Scholar]
- 52.Yu CS, Lin CJ, Hwang JK. Predicting subcellular localization of proteins for gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 2004;13:1402–6. doi: 10.1110/ps.03479604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horton P, Park K-J, Obayashi T, Fujita N, Harada H, Adams-Collier C, et al. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35:W585–7. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pierleoni A, Martelli PL, Fariselli P, Casadio R. BaCelLo: a balanced subcellular localization predictor. Bioinformatics. 2006;22:e408–16. doi: 10.1093/bioinformatics/btl222. [DOI] [PubMed] [Google Scholar]
- 55.Saier MH, Jr, Tran CV, Barabote RD. TCDB: the Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 2006;34:D181–6. doi: 10.1093/nar/gkj001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumar VS, Dasika MS, Maranas CD. Optimization based automated curation of metabolic reconstructions. BMC Bioinformatics. 2007;8:212–27. doi: 10.1186/1471-2105-8-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Degtyarenko K, de Matos P, Ennis M, Hastings J, Zbinden M, McNaught A, et al. ChEBI: a database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2008;36:D344–50. doi: 10.1093/nar/gkm791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009;37:W623–33. doi: 10.1093/nar/gkp456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nisha A, Muthukumar SP, Venkateswaran G. Safety evaluation of arachidonic acid rich Mortierella alpina biomass in albino rats-A subchronic study. Regul Toxicol Pharmacol. 2009;53:186–94. doi: 10.1016/j.yrtph.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 60.Shinmen Y, Kawashima H, Shimizu S, Yamada H. Concentration of eicosapentaenoic acid and docosahexaenoic acid in an arachidonic acid-producing fungus, Mortierella alpina IS-4, grown with fish oil. Appl Microbiol Biotechnol. 1992;38:301–4. [Google Scholar]
- 61.Andersen MR, Nielsen ML, Nielsen J. Metabolic model integration of the bibliome, genome, metabolome and reactome of Aspergillus niger. Mol Syst Biol. 2008;4:178. doi: 10.1038/msb.2008.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bowman SM, Free SJ. The structure and synthesis of the fungal cell wall. Bioessays. 2006;28:799–808. doi: 10.1002/bies.20441. [DOI] [PubMed] [Google Scholar]
- 63.Jareonkitmongkol S, Sakuradani E, Shimizu S. Isolation and characterization of a Delta 9-desaturation-defective mutant of an arachidonic acid-producing fungus, Mortierella alpina 1S-4. J Am Oil Chem Soc. 2002;79:1021–6. doi: 10.1007/s11746-002-0596-1. [DOI] [Google Scholar]
- 64.David H, Akesson M, Nielsen J. Reconstruction of the central carbon metabolism of Aspergillus niger. Eur J Biochem. 2003;270:4243–53. doi: 10.1046/j.1432-1033.2003.03798.x. [DOI] [PubMed] [Google Scholar]
- 65.Thiele I, Palsson BO. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 2010;5:93–121. doi: 10.1038/nprot.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yin W: Gurobi Mex: a MATLAB interface for Gurobi. Online athttp://convexoptimization.com/wikimization/index.php/gurobi_mex 2009.