

Abstract
With the increasing amount of available genome sequences, novel tools are needed for comprehensive analysis of species-specific sequence characteristics for a wide variety of genomes. We used an unsupervised neural network algorithm, a self-organizing map (SOM), to analyze di-, tri-, and tetranucleotide frequencies in a wide variety of prokaryotic and eukaryotic genomes. The SOM, which can cluster complex data efficiently, was shown to be an excellent tool for analyzing global characteristics of genome sequences and for revealing key combinations of oligonucleotides representing individual genomes. From analysis of 1- and 10-kb genomic sequences derived from 65 bacteria (a total of 170 Mb) and from 6 eukaryotes (460 Mb), clear species-specific separations of major portions of the sequences were obtained with the di-, tri-, and tetranucleotide SOMs. The unsupervised algorithm could recognize, in most 10-kb sequences, the species-specific characteristics (key combinations of oligonucleotide frequencies) that are signature features of each genome. We were able to classify DNA sequences within one and between many species into subgroups that corresponded generally to biological categories. Because the classification power is very high, the SOM is an efficient and fundamental bioinformatic strategy for extracting a wide range of genomic information from a vast amount of sequences.
[Supplemental material is available online at www.genome.org.]
In addition to protein-coding information, genome sequences contain a wealth of information of interest in many fields of biology, from molecular evolution to genome engineering. G+C% has been used as a fundamental characteristic of individual genomes, but the G+C% is apparently too simple a parameter to differentiate a wide variety of genomes of known sequences. Oligonucleotide frequency can be used to distinguish genomes, because oligonucleotide frequencies vary significantly among genomes; dinucleotide frequencies, for example, are shown to be genome signatures for both prokaryotes and eukaryotes (Nussinov 1984; Karlin et al. 1997; Karlin 1998; Gentles and Karlin 2001). Comprehensive analyses of oligonucleotide frequencies in a wide variety of genomes are thought to provide fundamental knowledge of individual genomes, namely, key combinations of oligonucleotides responsible for the biological properties of the different genomes and genome portions. We applied Kohonen's self-organizing map (SOM) to create graphical representations of oligonucleotide frequencies from which we could extract a wide range of genomic information. The unsupervised neural network algorithm is an effective tool for clustering and visualizing high-dimensional data; it converts complex nonlinear relations among high-dimensional data into simple geometric relations that can be viewed in two dimensions (Kohonen 1982, 1990; Kohonen et al. 1996).
We and others have used SOMs to characterize codon usage patterns of a wide variety of bacteria (Kanaya et al. 1998; Wang et al. 2001). We introduced a new feature to the SOM for studies of genomic sequences that makes the learning process independent of the order of data input (Abe et al. 1999), and we characterized codon usage in 60,000 genes from 29 bacterial species (Kanaya et al. 2001). SOMs were particularly useful, not only in searching for horizontally transferred genes, but also in predicting the donor genomes of the transferred genes. In the present study, SOMs were constructed with di-, tri-, and tetranucleotide frequencies for a total of 17,000 10-kb and 170,000 1-kb genomic sequences of 65 prokaryote genomes and a total of 46,000 10-kb and 460,000 1-kb segments of 6 eukaryote genomes. The resulting SOMs for the 16-, 64-, and 256-dimensional spaces (for di-, tri-, and tetranucleotide frequencies, respectively) revealed clear separations between inter- and intraspecies sequences that generally corresponded to biological categories. Comparative analysis of interspecies oligonucleotide frequencies could provide insight into hidden signatures in genome sequences established during evolution. For example, characteristic under-representation of palindromic tetranucleotides was observed in genomes of bacteria that have genes encoding restriction enzymes that cleave at these tetranucleotides. We also discuss the usefulness of SOMs for comprehensive searches for signal and motif sequences in the genomes.
RESULTS
Species-Specific Oligonucleotide Frequencies in Bacterial Genomes
SOMs were constructed with di-, tri-, and tetranucleotide frequencies for ∼17,000 genomic 10-kb sequences derived from the 65 bacterial genomes whose complete sequences are known. As the first step to obtaining the initial weight vectors, frequencies for the 17,000 non-overlapping segments were analyzed by principal component analysis (PCA). This is based on the knowledge that multivariate analyses including PCA successfully classified gene sequences into groups corresponding to known biological categories when the numbers of sequences and species were much smaller than that analyzed here (Grantham et al. 1980; Medigue et al. 1991; Sharp and Matassi 1994; Andersson and Sharp 1996). After 40 learning cycles, oligonucleotide frequencies of genome sequences were effectively reflected as the weight vectors in SOMs (Fig. 1A–C). Comparison of the sequence classification into lattice points of the final weight vectors with that of the initial vectors set by the first and second principal components of PCA (Fig. 1G) showed that sequences of a single species were much more tightly clustered in the final vectors. Lattices that include sequences from a single species are indicated in color, and those including sequences of more than one species are indicated in black. In the SOMs, sequences of most species were separated into species-specific nonoverlapping zones (Fig. 1A–C). In contrast, the resolving power of the conventional PCA method that was estimated with the initial vectors (Fig. 1G) was poor. The contiguous nonintermingling zones that contained sequences of a single species were very limited when compared with the contiguous nonoverlapping zones obtained with SOMs.
Figure 1.

SOMs for 10-kb sequences of 65 bacterial genomes. (A,B,C) Di-, tri-, and tetra-SOMs, respectively. Lattices that include sequences from more than one species are indicated in black, and those that include sequences from a single species are indicated in color as detailed in the figure above. (D,E,F) G+C% for each weight vector in di-, tri-, and tetra-SOMs, respectively. G+C% for each lattice vector was divided into five categories containing an equal number of lattices. The highest, second-highest, middle, second-lowest, and lowest G+C% categories are shown in dark red, light red, white, light blue, and dark blue, respectively. (G) Classification by the initial weight vectors set by PCA for the di-SOM. Lattices are colored as described in A–C.
Analysis of the weight vectors for individual lattices showed that strongly biased vectors were localized to the edge of the map, whereas those with weakly biased vectors were in the center. The G+C% for each weight vector in di-, tri-, and tetranucleotide SOMs (abbreviated as di-, tri-, and tetra-SOMs) was reflected mainly in the horizontal axis and increased from left to right; sequences of AT- and GC-rich bacteria were distributed on the left and right sides of the SOMs, respectively (Fig. 1D–F). Importantly, sequences with the same G+C% are separated by a complex combination of oligonucleotide frequencies resulting in species-specific separations. In other words, most of the 10-kb segments in each genome have a combination of oligonucleotides that reflect the respective genome like a signature, and SOMs can reveal the signature as representative weight vectors. The 170,000 nonoverlapping 1-kb sequences were also analyzed (Fig. 2). Species-specific separations were again observed, although the resolution was somewhat reduced. This shows that species-specific signatures are detectable even in a major population of the 1-kb sequences.
Figure 2.

SOMs for 1-kb sequences of 65 bacterial genomes. (A,B,C) Di-, tri-, and tetra-SOMs, respectively. Lattices are colored as described in Fig. 1,A–C. (D,E,F) G+C% for each weight vector is shown as described in Fig. 1, D–F. (G) Classification by the initial weight vectors for the di-SOM.
Intraspecies Separation for Bacterial Genomes
Detailed inspection of SOMs showed that each species was often split into two major zones that were composed of roughly equal numbers of data points. To illustrate this split more clearly, data points for each of seven representative species in the 10-kb tri-SOM (Fig. 1B) and in the 1-kb tri- and tetra-SOMs (Fig. 2B,C) were plotted with a single color (Fig. 3A–C). In the 1-kb SOMs, intraspecies separations were observed for all seven species, but in the 10-kb SOM, the separation was not observed for three species. To investigate the biological significance of the two zones, correlation with transcription polarities of protein coding sequences (CDSs) in the respective genomic segments was examined for Escherichia coli and Bacillus subtilis from CDSs data compiled in the DDBJ Genome Information Broker (http://gib.genes.nig.ac.jp/). Sequences belonging to each major zone in the 1-kb tetra-SOMs (Fig. 3C) are illustrated separately as red and blue bands below the diagrams that show transcription polarities of CDSs in the 200-kb segment (Fig. 3D,E). A red or blue band in each species showed clustering of contiguous 1-kb sequences belonging to one of the two zones in the 1-kb SOM. Each band coincided with the clustering of CDSs with one polarity, and borders between red and blue bands were usually located at positions corresponding to the switch positions for transcription polarity. In the cases of the three species for which the intraspecies separation was lost in the 10-kb SOM, switching of transcriptional polarities occurs within a 10-kb segment at higher probabilities than observed for the other four species (data not shown). These findings indicate that codon usage patterns contribute to the intraspecies separations and probably also to the interspecies separations.
Figure 3.

Intraspecies separations and tetranucleotide distributions in SOMs for bacterial genomes. (A,B,C) The 10-kb tri-, 1-kb tri-, and 1-kb tetra-SOMs. Seven representative species with two major zones are indicated in color as detailed in the figure above. In C, the two major zones of B. subtilis or E. coli are noted with red or blue arrows with the letter B or E, respectively. (D) Transcriptional polarity and SOM separation for B. subtilis sequences. Two transcriptional polarities of CDSs in the 200-kb B. subtilis segment with a replication origin are presented separately in the top two panels; this was obtained from the DDBJ Web site (http://gib.genes.nig.ac.jp/). Below the two panels, contiguous 1-kb sequences within the 200-kb segment and belonging to the two major zones marked with red and blue arrows in C are shown separately with the red and blue bands, respectively. (E) Transcriptional polarity and SOM separation for E. coli sequences. A 200-kb E. coli segment lacking a replication origin was analyzed as described in D. (F) Tetranucleotide distribution in the 1-kb tetra-SOM for bacteria. Levels of each tetranucleotide for all lattice vectors in the tetra-SOM of Fig. 2C were divided into five categories containing an equal number of lattices, and the highest, second-highest, middle, second-lowest, and lowest categories are shown with different levels of red and blue as described in Fig. 1,D–F. Zones for bacteria that have genes encoding a restriction enzyme that recognizes the respective tetranucleotide are noted by light blue lines with the following numbers to show the species name: (1) H. pylori; (2) M. jannaschii; (3) S. aureus; (4) S. pneumoniae; (5) P. abyssi; (6) P. horikoshii; (7) A. fulgidus; (8) A. pernix; and (9) D. radiodurans. For other palindromic tetranucleotides, see Supplementary Data 2. Of 17 restriction enzymes from 11 bacteria, the respective tetranucleotides were under-represented in 15 instances.
Genome segments introduced through horizontal transfer from distantly related organisms are known to retain the sequence characteristics of the donor genome and can be distinguished from those of the acceptor genome (Lawrence and Ochman 1997, 1998). For example, genes transferred from other genomes often have codon-usage patterns distinct from those of their intrinsic genomes (Grantham et al. 1980; Ikemura 1985; Medigue et al. 1991). We showed previously that SOMs are useful for identifying horizontally transferred genes and, importantly, for predicting the donor genomes of the transferred genes (Kanaya et al. 2001). There are characteristic data points in Figure 3A that are located away from the major zones of individual species. Those sequences that have oligonucleotide frequencies clearly distinct from those of major zones should correspond, at least in part, to genome portions that have been transferred horizontally from other genomes. To test this possibility, we examined 10-kb sequences from E. coli that were located outside of the territories of both E. coli and a closely related bacterium Salmonella typhimurium. When the sequences in the S. typhimurium territory were excluded, the next highest number of E. coli sequences was found in the Yersinia pestis territory. We then focused the five sequences in the Y. pestis territory commonly found in the di-, tri-, and tetra-SOMs. Within these sequences, there were 37 known genes, 23 of which had significant homology with Y. pestis genes. For example, at the amino acid level, 6 of 23 proteins had identity levels greater than 60%, and the highest was 80%, which was significantly higher than the 40% calculated for the average identity for the orthogonal pairs of E. coli and Y. pestis proteins (Deng et al. 2002). Furthermore, three genes were homologous with the phage-encoded genes, and one was homologous with a transposon gene. These findings support the prediction that these genes may have been transferred horizontally into the E. coli genome from other organisms.
SOMs for Eukaryotic Genomes
The protein-coding portion of each eukaryotic genome, especially in higher eukaryotes, is reduced appreciably in comparison with that of bacterial genomes. Therefore, genome signatures derived from species-specific codon usage should be less prevalent than those observed for prokaryotes. We examined di-, tri-, and tetranucleotide frequencies for six eukaryote genomes (a total of 460 Mb) including four genomes (Saccharomyces cerevisiae, C. elegans, Arabidopsis thaliana, Drosophila melanogaster) that have been sequenced completely, Plasmodium falciparum chromosomes 2 and 3, and human chromosomes 20, 21, and 22. The 46,000 nonoverlapping 10-kb segments from these 6 eukaryote genomes were analyzed (Fig. 4A–C). Most of the 10-kb segments were separated according to species. For example, more than 95% of the human sequences were located in the human territories, which are marked in red in Figure 4, A–C. This shows that SOM separations, which were obtained without any species information, closely fit separations among species, and thus, the unsupervised algorithm can recognize in most 10-kb sequences, the species-specific characteristic (a key combination of oligonucleotide frequencies) that is the representative signature of each genome.
Figure 4.

SOM for 10-kb sequences of six eukaryotes. (A,B,C) Di-, tri-, and tetra-SOMs, respectively. Lattices that include sequences from more than one species are indicated in black, and those that include sequences from a single species are indicated in color as detailed in the figure above. (D,E,F) G+C% for each weight vector in di-, tri-, and tetra-SOMs, respectively. G+C% for each lattice vector is shown as described in Fig. 1, D–F. (G) Classification by the initial weight vectors for the di-SOM.
The G+C% calculated for each weight vector in di-, tri-, and tetra-SOMs are shown in Figure 4, D–F. It is apparent that sequences with the same G+C% are separated by a complex combination of oligonucleotide frequencies resulting in species-specific separations. Underlying representation in SOMs enables us to retrieve characteristic sequence patterns for individual genomes and genome regions. The frequency of each dinucleotide in the weight vector for each lattice in the di-SOM is illustrated in red and blue (Fig. 5). Complementary pairs of dinucleotides (e.g., AA vs. TT) had similar distribution patterns. This indicates that when the sequence complementary to the sequence registered by the International DNA Databank is used for a certain genome, general patterns may not change appreciably. Lines in all panels in Figure 5 represent the species borders observed in the di-SOM. Species borders coincide with regions of transition between the red and blue levels for several dinucleotides, which correspond to the diagnostic dinucleotides for the species border formation. For example, the CG dinucleotide deficiency (dark blue zones in the CG panel) is a factor responsible for separation of human sequences (red in Fig. 4A) from Drosophila (pink) and Arabidopsis (green) sequences. Levels of CG, GC, AG, and GA contributed appreciably to separation of Drosophila sequences from others. It should be stressed that the SOM utilizes complex combinations of many more dinucleotides for the sequence separations in an area-dependent manner. This is because SOMs implement nonlinear projection from the multi-dimensional space of input data onto a two-dimensional array of weight vectors (Kohonen 1982, 1990; Kohonen et al. 1996).
Figure 5.

Dinucleotide distribution in 10-kb di-SOM for six eukaryotes. Levels of each dinucleotide for all lattice vectors in the di-SOM of Fig. 4A were divided into five categories containing an equal number of lattices and the categories are shown as described in Fig. 3F. Species borders in the di-SOM (Fig. 4A) are marked by lines. Major zones for four species were noted in the CG panel as follows: A. thaliana (A), C. elegans (C), D. melanogaster (D), and human (H).
In similar fashion, trinucleotide levels for each representative vector in the tri-SOM were analyzed. Again, species borders often coincided with regions of sharp transition between the red and blue levels for various diagnostic trinucleotides (Fig. 6). For humans, high levels of AGG, CAG, CCC, CCT, CTG, and GGG as well as low levels of ACG, CGA, CGT, and TCG were observed, and for Drosophila, high levels of GCA and TGC and low levels of AGA, GCC, TCA, TCT, and TGA were observed. The under-representation of CNG in a major portion of the Arabidopsis territory is thought to be related to cytosine methylation in CNG trinucleotides (Lindroth et al. 2001). The SOM utilizes complex combinations of many trinucleotides for species separation in an area-dependent manner; the 64 panels for all trinucleotides are presented as supplementary data 1.
Figure 6.

Trinucleotide distribution in 10-kb tri-SOM for six eukaryotes. Levels of each trinucleotide for all lattice vectors in the tri-SOM of Fig. 4B were divided into five categories and shown as described in Fig. 3F. Species borders are shown as described in Fig. 5. (A) Human. Six diagnostic trinucleotides with high frequencies and four with low frequencies. (B) D. melanogaster. Two diagnostic trinucleotides with high frequencies and four with low frequencies. (C) A. thaliana. Four diagnostic trinucleotides with high frequencies and four with low frequencies (CNG).
Intraspecies Separation Observed for Eukaryote Genomes
Human sequences had two and one satellite zones in the di- and tri-SOMs, respectively (red minor zones in Fig. 4A,B). Genomes of warm-blooded vertebrates, such as humans, are known to be composed of long-range segmental G+C% distributions isochores (Bernardi et al. 1985; Ikemura and Aota 1988; Bernardi 1989; Gautier 2000; Eyre-Walker and Hurst 2001). Correlation of the segmental G+C% distributions with SOM separations was observed. For example, ∼500 10-kb sequences belonging to the satellite red zone located at top at the left side in the di- and tri-SOMs were practically common between the two SOMs, and the G+C% was between 30% and 33%, which corresponds to very AT-rich sequences in the human genome. Four-fifths of the sequences were derived from very AT-rich gene-desert regions on chromosome 21 (Hattori et al. 2000), which correspond to L1 isochores (Saccone et al. 1999) and replicate very late during S phase (Watanabe et al. 2002). The SOM can unveil specific genome portions with distinct characteristics as intraspecies separations. Drosophila sequences were split into two major zones in the tetra-SOM (pink in Fig. 4C), and this split was associated with G+C%.
SOMs With 1-kb Eukaryote Sequences
To determine the usefulness of SOMs for analysis of genomes with respect to functional aspects, we investigated the effects of shortening the sequence length on SOM separations, because 10-kb segments appear to be too large for such studies. SOMs were constructed with the dinucleotide frequencies for a total of 460,000 nonoverlapping 1-kb sequences from six eukaryotes. Clear separations of species were observed, but territories of individual species were split into several zones (Fig. 7A). Mirror symmetric distributions were apparent for sequences of each genome. The G+C% in the SOM was reflected mainly on the horizontal axis, and the complementarity of oligonucleotides was reflected on the vertical axis. We examined the possible factors responsible for the separations in the 1-kb SOM by analyzing dinucleotide levels for each lattice. The best example was the CG dinucleotide level shown in Figure 7B. All Drosophila zones (pink in Fig. 7A) corresponded primarily to the CG-rich zones (red in Fig. 7B), and all human zones (red in Fig. 7A) corresponded primarily to the CG-poor zones (blue in Fig. 7B), except for one clear characteristic zone that is marked by an arrow. This CG-rich human zone is thought to have CpG-island sequences that are often present in the regulatory regions for transcription. The finding that use of shorter sequences can identify intraspecies separations, rather than intermingling different species sequences, demonstrates the usefulness of this method for discovery of local, functional sequence characteristics. The average number of sequences per lattice in the 1-kb SOM was 11. The actual number of sequences classified into each lattice that is composed of sequences from a single species is shown by the height of the colored rod. There were many apparently high rods. Systematic analyses of these characteristic rods might provide unique, biologically significant information.
Figure 7.

Di-SOM for 1-kb sequences of six eukaryotes. (A) Di-SOM. Lattices are colored as described in Fig. 4A. (B) CG dinucleotide levels for all weight vectors were calculated and shown as described in Fig. 5. The CG-rich zone in the human territories is noted with an arrow. (C) Three-dimensional presentation of the di-SOM. Number of sequences classified into each lattice that has sequences from a single species is presented with the height of the colored rod.
It should be noted that many of the 1-kb segments are free of species-specific ubiquitous repetitive elements, such as Alu or L1 elements in the human genome. The sequences with or without repetitive elements were found to be colocalized in the major zones of individual species. Detailed inspection showed that 10-kb human sequences with or without Alu or L1 elements were also colocalized in the human major zones on the 10-kb SOMs. Therefore, the major factors responsible for the species-specific separations of eukaryote sequences do not appear to be ubiquitous repetitive elements. Factors responsible for the separations could be characteristics that are more extensively embedded than repetitive elements.
DISCUSSION
Biological Implications of SOM Separations and Genome Signatures
To investigate the biological significance of diagnostic oligonucleotides for SOM separations, we examined the correlation of levels of palindromic tetranucleotides with respective restriction enzyme systems by referring to the restriction enzyme database (REBASE; http://vent.neb.com/∼vincze/genomes/). Restriction site tetranucleotides were under-represented in 10 of the 11 bacteria that have genes encoding 4-base cutter enzymes (blue in Fig. 3F). This finding is consistent with that of Karlin et al. (1997) on compositional biases of bacterial genomes, again indicating that SOMs can effectively classify sequences according to biological categories. The 256 panels for all tetranucleotides in the prokaryote and eukaryote SOMs are presented as supplementary data 2 and 3, respectively. We then considered the biological significance of diagnostic tetranucleotides in eukaryotes. One possible explanation is a contributory effect of levels of the di- and trinucleotide components of tetranucleotides. For example, tetranucleotides containing the CG dinucleotide were clearly under-represented in the human territory (supplementary data 3). Transition zones of various other tetranucleotides (e.g., CTCA and CTGA) were also sharp and coincided exactly with species borders. Such sharp transitions and exact coincidences were not typical for the dinucleotide components (e.g., CT, TC, and CA in Fig. 5). As found in the restriction enzyme analysis, some tetranucleotides may have biological significance. Species-specific characteristics for DNA synthesis and repair enzymes, as well as sequence preferences of ubiquitous DNA-binding proteins, may be responsible for differences in oligonucleotide distribution between species. In the cases of signal and motif sequences, such as transcription factor-binding sites, they may be biased from the random occurrence statistically calculated from the genome base composition. This prediction is consistent with the finding that GAGA/TCTC, which is a transcription signal in Drosophila (Soeller et al. 1993), was under-represented in the Drosophila genome (supplementary data 3). SOMs for longer oligonucleotides such as penta- and hexanucleotides may reveal a wide range of signal and motif sequences, because these sequences are typically longer than tetranucleotides. A preliminary study of the correlation of diagnostic oligonucleotide with sequence motifs for transcription factors suggested that such sequences are often under-represented in a major portion of the respective genome, and, therefore, may contribute to genome signatures. Because species-specific separations in SOMs are very clear, SOMs may provide fundamental guidelines for identifying molecular mechanisms that established genome signatures of individual species during evolution. The present analysis is an example of comparative genomics, and the results obtained were affected by choice of the genomes used. As a strategy to reduce this effect, most (if not all) genomes that have been sequenced completely were analyzed.
METHODS
Genome Sequences
The DNA sequences of 65 bacterial genomes, itemized in the Figure 1 legend, were obtained from the DDBJ GIB Web site (http://www.ddbj.nig.ac.jp/), and those of the 6 eukaryotes itemized in the Figure 4 legend were obtained from the GenBank Web site (http://www.ncbi.nlm.nih.gov/Genbank/).
Self-Organizing Map
The SOM is an unsupervised neural network algorithm that implements a characteristic nonlinear projection from the high-dimensional space of input data onto a two-dimensional array of weight vectors (Kohonen 1982, 1990; Kohonen et al. 1996). It is thought of as a flexible net that is spread into the multi-dimensional data cloud. Because the net is a two-dimensional array, it can be visualized easily. The weight vectors (wij) are arranged in the two-dimensional lattice denoted by i (=0, 1,…, I-1) and j (= 0, 1,…, J-1). The learning process of the present SOM was designed to be independent of the order of input of vectors on the basis of batch-learning SOM as we reported previously (Abe et al. 1999; Kanaya et al. 2001). In the original method, the initial weights vectors wij are set by random values (Kohonen 1990; Kohonen et al. 1996), but in the present method, the vectors are initialized by PCA (Step 1). For mapping multidimensional space data onto a plane, PCA rotates the vector space with the eigenvectors (the principal components) of the covariance matrix as a new basis. The principal components are orthogonal, and the plane spanned by the two first components, PC1 and PC2, was usually used for a linear data projection. Weights in the first dimension (I) were arranged into 150 lattices for 10-kb sequences, or 350 lattices for 1-kb sequences, corresponding to a width of five times the standard deviation (5ς1) of the first principal component; and the second dimension (J) was defined by the nearest integer greater than ς2/ς1 × 150 (or 350). The weight vector on the ijth lattice was represented as follows:
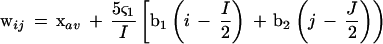 |
1 |
in which xav is the average vector for oligonucleotide frequencies, and b1 and b2 are eigenvectors for the first and second principal components. In Step 2, the Euclidean distances between the input vector xk and all weight vectors wij were calculated; then xk was associated with the weight vector (called wi'j') with minimal distance. After associating all input vectors with weight vectors, updating was done according to Step 3.
In Step 3, the ijth weight vector was updated by
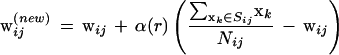 |
2 |
in which components of set Sij are input vectors associated with wi'j' satisfying i − β(r) ≤ i‘ ≤ i + β(r) and j − β(r) ≤ j′ ≤ j + β(r). The two parameters α(r) and β(r) are learning coefficients for the rth cycle, and Nij is the number of components of Sij. In the present study, α(r) and β(r) are set by
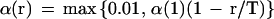 |
3 |
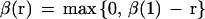 |
4 |
in which α(1) and β(1) are the initial values for the T-cycle of the learning process. In the present study, we selected 40 for T, 0.6 for α(1), and 20 for β(1). The learning process is monitored by the total distance between xk, and the nearest weight vector wI‘j‘, represented as
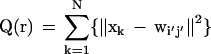 |
5 |
in which N is the total number of sequences analyzed.
The SOM program used for the sequence analyses “XanaMine” was obtained from Xanagen Inc. (URL; http://www.xanagen.com/index-e.html, E-mail; info@xanagen.com).
WEB SITE REFERENCES
http://gib.genes.nig.ac.jp/; DDBJ Genome Information Broker Web site.
http://vent.neb.com/∼vincze/genomes/REBASE; Restriction enzyme database.
http://www.ddbj.nig.ac.jp/; DDBJ Web site.
http://www.ncbi.nlm.nih.gov/Genbank/; GenBank Web site.
http://www.xanagen.com/index-e.html; SOM programs for genome analysis.
Acknowledgments
This work was supported by ACT—Japan Science and Technology Corporation and by the Advanced and Innovational Research Program in Life Sciences and a Grant-in-Aid for Scientific Research on Priority Areas (C) “Genome Science” from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We thank Ms. Nanayo Ishihara and Yoko Kosaka for technical assistance.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
E-MAIL tikemura@lab.nig.ac.jp; FAX 81-55-981-6794.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.634603.
REFERENCES
- 1.Abe T., Kanaya, S., Kinouchi, M., Kudo, Y., Mori, H., Matsuda, H., Carlos, D.C., and Ikemura, T. 1999. Gene classification method based on batch-learning SOM. Genome Inform. Ser. 10: 314-315. [Google Scholar]
- 2.Andersson S.G. and Sharp, P.M. 1996. Codon usage in the Mycobacterium tuberculosis complex. Microbiology 142: 915-925. [DOI] [PubMed] [Google Scholar]
- 3.Bernardi G. 1989. The isochore organization of the human genome. Annu. Rev. Genet. 23: 637-661. [DOI] [PubMed] [Google Scholar]
- 4.Bernardi G., Olofsson, B., Filipski, J., Zerial, M., Salinas, J., Cuny, G., Meunier-Rotival, M., and Rodier, F. 1985. The mosaic genome of warm-blooded vertebrates. Science 228: 953-958. [DOI] [PubMed] [Google Scholar]
- 5.Deng W., Burland, V., Plunkett, G., III, Boutin, A., Mayhew, G.F., Liss, P., Perna, N.T., Rose, D.J., Mau, B., Zhous, S., et al. 2002. Genome sequence of Yersinia pestis KIM. J. Bacteriol. 184: 4601-4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eyre-Walker and Hurst, L.D. 2001. The evolution of isochores. Nat. Rev. 2: 549-555. [DOI] [PubMed] [Google Scholar]
- 7.Gautier C. 2000. Compositional bias in DNA. Curr. Opin. Genet. Dev. 10: 656-661. [DOI] [PubMed] [Google Scholar]
- 8.Gentles A.J. and Karlin, S. 2001. Genome-scale compositional comparisons in eukaryotes. Genome Res. 11: 540-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grantham R., Gautier, C., Gouy, M., Mercier, R., and Pave, A. 1980. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 8: r49-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hattori M., Fujiyama, A., Taylor, T.D., Watanabe, H., Yada, T., Park, H.S., Toyoda, A., Ishii, K., Totoki, Y., Choi, D.K., et al. 2000. The DNA sequence of human chromosome 21. Nature 405: 311-319. [DOI] [PubMed] [Google Scholar]
- 11.Ikemura T. 1985. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 2: 13-34. [DOI] [PubMed] [Google Scholar]
- 12.Ikemura T. and Aota, S. 1988. Global variation in G+C content along vertebrate genome DNA: Possible correlation with chromosome band structures. J. Mol. Biol. 203: 1-13. [DOI] [PubMed] [Google Scholar]
- 13.Kanaya S., Kudo, Y., Abe, T., Okazaki, T., Carlos, D.C., and Ikemura, T. 1998. Gene classification by self-organization mapping of codon usage in bacteria with completely sequenced genome. Genome Inform. Ser. 9: 369-371. [Google Scholar]
- 14.Kanaya S., Kinouchi, M., Abe, T., Kudo, Y., Yamada, Y., Nishi, T., Mori, H., and Ikemura, T. 2001. Analysis of codon usage diversity of bacterial genes with a self-organizing map (SOM): Characterization of horizontally transferred genes with emphasis on the E. coli O157 genome. Gene 276: 89-99. [DOI] [PubMed] [Google Scholar]
- 15.Karlin S. 1998. Global dinucleotide signatures and analysis of genomic heterogeneity. Curr. Opin. Microbiol. 1: 598-610. [DOI] [PubMed] [Google Scholar]
- 16.Karlin S., Mrazek, J., and Campbell, A. 1997. Compositional biases of bacterial genomes and evolutionary implications. J. of Bacteriol. 179: 3899-3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kohonen T. 1982. Self-organized formation of topologically correct feature maps. Biol. Cybern. 43: 59-69. [Google Scholar]
- 18.___, 1990. The self-organizing map. Proc. IEEE 78: 1464-1480. [Google Scholar]
- 19.Kohonen T., Oja, E., Simula, O., Visa, A., and Kangas, J. 1996. Engineering applications of the self-organizing map. Proc. IEEE 84: 1358-1384. [Google Scholar]
- 20.Lawrence J.G. and Ochman, H. 1997. Amelioration of bacterial genomes: Rates of change and exchange. J. Mol. Evol. 44: 383-397. [DOI] [PubMed] [Google Scholar]
- 21.___, 1998. Molecular archaeology of the Escherichia coli genome. Proc. Natl. Acad. Sci. 95: 9413-9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindroth M.A., Cao, X., Jackson, P.J., Zilberman, D., McCallum, M.C., Henikoff, S., and Jacobsen, E.S. 2001. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 292: 2077-2080. [DOI] [PubMed] [Google Scholar]
- 23.Medigue C., Rouxel, T., Vigier, P., Henaut, A., and Danchin, A. 1991. Evidence for horizontal gene transfer in Escherichia coli speciation. J. Mol. Biol. 222: 851-856. [DOI] [PubMed] [Google Scholar]
- 24.Nussinov R. 1984. Doublet frequencies in evolutionary distinct groups. Nucleic Acid Res. 10: 1749-1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saccone S., Federico, C., Solovei, I., Croquette, M.F., Valle, G.D., and Bernardi, G. 1999. Identification of the gene-richest bands in human prometaphase chromosomes. Chromosome Res. 7: 379-386. [DOI] [PubMed] [Google Scholar]
- 26.Sharp P.M. and Matassi, G. 1994. Codon usage and genome evolution. Curr. Opin. Gen. Dev. 4: 851-860. [DOI] [PubMed] [Google Scholar]
- 27.Soeller W.C., Oh, C.E., and Kornberg, T.B. 1993. Isolation of cDNAs encoding the Drosophila GAGA transcription factor. Mol. Cell Biol. 13: 7961-7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H.C., Badger, J., Kearney, P., and Li, M. 2001. Analysis of codon usage patterns of bacterial genomes using the self-organizing map. Mol. Biol. Evol. 18: 792-800. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe Y., Fujiyama, A., Ichiba, Y., Hattori, M., Yada, T., Sakaki, Y., and Ikemura, T. 2002. Chromosome-wide assessment of replication timing for human chromosomes 11q and 21q: Disease-related genes in timing-switch regions. Hum. Mol. Genet. 11: 13-21. [DOI] [PubMed] [Google Scholar]