

Abstract
Objective
The prominent histopathological feature of the amyotrophic lateral sclerosis (ALS) is the presence of intracellular inclusions in degenerating neurons and their axons. The appearance and localization of these pathological structures depend on an aggregated protein that forms their scaffold. We investigated if γ-synuclein, an aggregation-prone protein highly expressed in healthy motor neurons, and predominantly localized in their axons and synaptic terminals is involved in ALS pathology.
Methods
Immunostaining of histological sections and sequential protein extraction from postmortem neural samples followed by immunoblotting.
Results
Immunohistochemical screening revealed a subset of sporadic (9 of 31) and familial (8 of 23) ALS cases with a novel type of pathology characterized by the accumulation of γ-synuclein in distinct profiles within the dorsolateral column. Sequential fractionation of proteins from the spinal cord tissues revealed detergent-insoluble γ-synuclein species specifically in the dorsolateral corticospinal tracts of a ALS patient with γ-synuclein-positive profiles in this region. These profiles are negative for protein markers commonly found in pathological inclusions in the spinal cord of ALS patients and most probably represent degenerated axons of upper motor neurons that have lost their neurofilaments. A subset of these profiles was found in association with phagocytic cells positive for Mac-2/Galectin-3. A smaller subset of studied ALS cases (4 of 54) contained large cytoplasmic inclusions in the cell body of remaining spinal motor neurons.
Interpretation
Our observations suggest that pathological aggregation of γ-synuclein might contribute to the pathogenesis of ALS.
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating motor neuron disease characterized by the selective loss of discrete populations of upper and lower motor neurons. A histopathological hallmark of both familial (fALS) and the more common sporadic (sALS) forms of the disease is the presence of distinctive intracellular inclusions in degenerating neurons and their axons. Formation of these pathological structures has been linked with the accumulation and aggregation of several proteins, including those mutated in fALS, namely SOD1,1 TDP-43,2 FUS/TLS,3,4 neurofilament subunits,5 optineurin,6,7 ubiquilin-28 and even dipeptide products of unconventional repeat-associated non-ATG initiated translation of GGGGCC repeats in C9ORF72 locus.9,10 However, some proteins that are constituents of pathological inclusions have never been genetically associated with fALS or predisposition to sALS, examples include p38MAPK,11 galectin-1,12 Smad2/3,13 RBM4514 and p54nrb/NONO.15 It is likely that other, yet to be identified proteins may be involved in ALS pathology.
γ-Synuclein presents a feasible candidate for such a protein due to an auspicious combination of properties. Structurally it is closely related to α-synuclein and shares its ability to aggregate in vitro16,17 and in vivo,18 which is believed to be a crucial trigger of neurodegeneration in α-synucleinopathies. Motor neurons express high levels of γ-synuclein, with the protein highly abundant in their axons, where it might affect organization of axonal cytoskeleton.18–21 Axonal or periaxonal accumulation of γ-synuclein has been found to accompany major histopathological changes in several neurodegenerative diseases.22–24 Finally, genetically unaltered γ-synuclein aggregates, fibrillates and forms axonal and perikaryal inclusions when overexpressed in neurons of transgenic mice, which is coupled with progressive and selective loss of motor neurons and the development of motor deficiency.18,25 We therefore undertook a search for any evidence of γ-synuclein pathology in patients with sporadic and familial ALS, a disease where such pathology has not yet been assessed, using a set of previously histopathologically characterized spinal cord samples.
Materials and Methods
Patient data and analysis of tissue samples
Human tissue samples were obtained from the MRC London Neurodegenerative Diseases Brain Bank and Sheffield Brain Tissue Bank. Consent was obtained from all subjects for autopsy, histopathological assessment and research in accordance with local and national Ethics Committee approved donation. For each case a detailed post-mortem histopathological report produced by a qualified pathologist was obtained after completion of the primary assessment of γ-synuclein staining that was carried out blindly. The spinal cord tissues were histologically assessed from 31 cases of sALS (21 males, 10 females; mean age±SEM 65.2 ± 1.9 years, mean post-mortem delay ± SEM 34.7 ± 4.1 h), 17 cases of fALS with C9ORF72 expansion (nine males, eight females; mean age ± SEM 62.2 ± 2.2 years, mean post-mortem delay ± SEM 37.6 ± 7.6 h); three cases of SOD1-fALS (one male, two females; mean age ± SEM 53.7 ± 7.7 years, mean post-mortem delay ± SEM 27.7 ± 18.4 h), three cases of FUS-fALS (one male, two females; mean age ± SEM 34.3 ± 0.7 years, mean post-mortem delay ± SEM 38.3 ± 16.4 h), six Alzheimer's disease cases (three males, three females; mean age ± SEM 76.7 ± 4.4 years, mean post-mortem delay ± SEM 26.2 ± 8.5 h), one Lewy body dementia case (male; age 88 years, post-mortem delay 18 h), one Parkinson's disease case (male, age 74, post-mortem delay 10 h) and 13 control cases with no clinical history or histological evidence of neurodegeneration (eight males, five females; mean age ± SEM 68.7 ± 3.4 years, mean post-mortem delay ± SEM 46.5 ± 7.3 h). For details of ALS and control cases see Table1.
Table 1.
Summary of sALS, fALS, and control cases assessed in this study
| Case | Sex | Age | PMD | Corticospinal tract γ-synuclein-positive profiles | γ-synuclein-positive inclusions in motor neuron cell bodies | γ-synuclein amino acids encoded by allelic variants (SNP in the exon IV) |
|---|---|---|---|---|---|---|
| Amyotrophic lateral sclerosis with SOD1 mutation | ||||||
| 01 | F | 46 | 5 | − | − | Glu/Glu |
| 02 | F | 69 | 64 | + | − | Glu/Glu |
| 03 | M | 46 | 14 | + | − | Glu/Glu |
| Amyotrophic lateral sclerosis with FUS mutation | ||||||
| 04 | F | 35 | 20 | − | − | Glu/Glu |
| 05 | M | 33 | 71 | − | − | Glu/Val |
| 06 | F | 35 | 24 | − | − | Glu/Glu |
| Amyotrophic lateral sclerosis with hexanucleotide expansion in C9ORF72 locus | ||||||
| 59 | M | 72 | 96 | + | − | Val/Val |
| 60 | M | 46 | 4 | − | + | Val/Val |
| 61 | M | 59 | 60 | + | − | Glu/Val |
| 62 | F | 58 | 2 | − | − | Glu/Glu |
| 63 | M | 46 | 15 | − | + | Val/Val |
| 64 | F | 76 | 10 | − | − | Glu/Val |
| 65 | F | 64 | N/A | − | − | Glu/Glu |
| 66 | F | 62 | 63 | + | − | Glu/Val |
| 67 | F | 64 | 7 | + | − | Glu/Glu |
| 68 | M | 72 | 96 | − | − | Val/Val |
| 69 | F | 69 | 55 | + | − | Val/Val |
| 70 | M | 48 | 23 | − | − | Glu/Glu |
| 71 | F | 67 | 24 | − | − | Glu/Glu |
| 72 | F | 64 | 7 | − | − | Glu/Glu |
| 73 | M | 68 | 31 | + | + | Glu/Glu |
| 74 | M | 64 | 48 | − | − | Glu/Val |
| 75 | M | 59 | 60 | − | − | Glu/Val |
| Sporadic amyotrophic lateral sclerosis | ||||||
| 07 | M | 61 | 44 | − | − | Glu/Val |
| 08 | M | 71 | 58 | − | − | Glu/Val |
| 09 | M | 82 | 13 | − | − | Val/Val |
| 10 | F | 57 | 48 | − | − | Glu/Glu |
| 11 | M | 55 | 54 | − | − | Glu/Val |
| 12 | F | 78 | 69 | − | − | Glu/Glu |
| 13 | M | 72 | 19 | − | − | Glu/Val |
| 14 | F | 80 | 37 | − | − | Glu/Val |
| 15 | M | 68 | 8 | + | − | Glu/Val |
| 16 | M | 60 | 53 | + | − | Glu/Val |
| 17 | F | 68 | 49 | + | − | Glu/Glu |
| 18 | M | 72 | 26 | + | − | Glu/Glu |
| 19 | M | 78 | 2 | + | − | Glu/Val |
| 20 | M | 59 | 77 | + | − | Glu/Glu |
| 21 | M | 67 | 70 | + | − | Glu/Val |
| 22 | M | 73 | 42 | + | − | Glu/Glu |
| 44 | F | 59 | 30 | − | + | Glu/Glu |
| 45 | M | 70 | 86 | − | − | Glu/Glu |
| 46 | M | 68 | N/A | − | − | Glu/Val |
| 47 | F | 69 | 12 | − | − | Glu/Glu |
| 48 | M | 66 | 8 | − | − | Glu/Glu |
| 49 | M | 65 | 9 | − | − | Glu/Glu |
| 50 | F | 41 | 24 | − | − | Glu/Val |
| 51 | F | 75 | 24 | − | − | Glu/Glu |
| 52 | M | 79 | 24 | − | − | Val/Val |
| 53 | M | 51 | N/A | − | − | Glu/Val |
| 54 | M | 51 | N/A | − | − | Glu/Val |
| 55 | M | 53 | 16 | − | − | Glu/Glu |
| 56 | F | 70 | 24 | + | − | Glu/Glu |
| 57 | M | 51 | 22 | − | − | Glu/Glu |
| 58 | F | 51 | 24 | − | − | Glu/Glu |
| Normal control (no evidence of neurological pathology) | ||||||
| 23 | F | 90 | 50 | − | − | Glu/Glu |
| 24 | M | 62 | 80 | − | − | N/A |
| 25 | F | 79 | 73 | − | − | N/A |
| 26 | M | 64 | 71 | − | − | N/A |
| 27 | M | 80 | 48 | − | − | Glu/Glu |
| 28 | F | 55 | 95 | − | − | N/A |
| 29 | M | 54 | 31 | − | − | Glu/Val |
| 30 | M | 79 | 24 | − | − | N/A |
| 39 | M | 64 | 12 | − | − | Glu/Glu |
| 40 | F | 62 | 36 | − | − | Glu/Val |
| 41 | M | 78 | 43 | − | − | Glu/Glu |
| 42 | M | 51 | 25 | − | − | Glu/Glu |
| 43 | F | 75 | 16 | − | − | Glu/Val |
| Non-ALS neurodegenerative diseases | ||||||
| 31 | M | 75 | 6 | − | − | N/A |
| 32 | M | 62 | 44 | − | − | N/A |
| 33 | F | 93 | 6 | − | − | N/A |
| 34 | M | 75 | 55 | − | − | N/A |
| 35 | F | 71 | 33 | − | − | N/A |
| 36 | F | 84 | 13 | − | − | N/A |
| 37 | M | 88 | 18 | − | − | N/A |
| 38 | M | 74 | 10 | − | − | N/A |
N/A, data unavailable; −, negative; +, positive; fALS, familial amyotrophic lateral sclerosis; sALS, sporadic amyotrophic lateral sclerosis.
Primary antibodies
Anti-human γ-synuclein rabbit-polyclonal SK109 antibody21 was used at 1:250 dilution for immunohistochemistry, 1:100 dilution for immunofluorescence and 1:500 dilution for Western blotting. Anti-human γ-synuclein goat-polyclonal E20 antibody (Santa Cruz Biotechnology Inc., Dallas, TX) was used at 1:100 dilution for immunohistochemistry and 1:250 dilution for Western blotting. To prove the specificity of γ-synuclein staining some sections were immunostained with SK109 or E20 antibody preincubated for 1-h with purified recombinant human γ-synuclein at 0.1 mg/mL concentration. Anti-ubiquitin mouse monoclonal Ubi-1 antibody (Zymed Laboratories Inc., South San Francisco, CA) was used at 1:100 dilution for immunohistochemistry and immunofluorescence. Anti-phospho(S409/410-2)TDP-43 rabbit-polyclonal antibody (Cosmo Bio Co. Ltd., Tokyo, Japan) was used at 1:3000 dilution for immunohistochemistry. For immunofluorescence, anti-neurofilaments mouse monoclonal SMI312 antibody (Covance, Princeton, NJ) was used at 1:100 dilution, anti-Mac-2/Galectin-3 mouse monoclonal B2C10 antibody (BD Pharmigen, Jose, CA) – at 1:100 dilution and anti-p62 mouse monoclonal clone 3 antibody (BD Pharmigen) – at 1:100 dilution.
Histological techniques
Eight-μm thick sections of 10% formalin-fixed paraffin-embedded cervical and thoracic spinal cord and primary motor cortex were processed and immunostained as described previously.18,21 Immunohistochemical detection was carried out using Elite plus kits (Vector Laboratories, Burlingame, CA) and 3,30-diaminobenzidine as a substrate (Sigma-Aldrich, St. Louis, MO). For immunofluorescent detection anti-rabbit and anti-mouse secondary antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 546 or Alexa Fluor 633 (Invitrogen, Carlsbad, CA) were used at 1:1000 dilution and images were collected sequentially on a Leica TCS SP2 laser scanning confocal microscope (Leica Microsystems, CMS, GmbH, Mannheim, Germany).
Sequential protein extraction
A previously described protocol18 was used with minor modifications. Briefly, frozen neuronal tissues were homogenized in five volumes of high salt buffer (50 mmol/L Tris–HCl, pH 7.5; 750 mmol/L NaCl; 5 mmol/L EDTA) with protease inhibitors (Complete Mini from Roche Diagnostics Limited, Burgess Hill, UK) and centrifuged at 12,000g for 15 min at 4°C. The supernatant (PMS fraction) was centrifuged at 100,000g for 20 min at 4°C. The high-speed supernatant represented high salt soluble (HS) fraction. The pellet was washed in the same high salt buffer and re-extracted with high salt buffer containing 1% Triton-X100. The high-speed centrifugation produced the supernatant (HS/T fraction) and the pellet that was subsequently washed in HS/T buffer and re-extracted with RIPA buffer (50 mmol/L Tris-HCl, pH 8.0; 150 mmol/L NaCl; 5 mmol/L EDTA; 1% NP40; 0.5% SDS) as above to obtain RIPA soluble (RIPA) fraction. The final pellet was washed in RIPA buffer, resuspended in a gel loading buffer with 2% SDS and incubated in a boiling water bath for 10 min (SDS fraction). Aliquots of other fractions were mixed with an equal volume of 2× gel loading buffer and boiled for 5 min before loading on a 16% SDS-PAGE. The level of γ-synuclein in fractions was assessed by Western blotting using SK109 rabbit-polyclonal antibody as described previously.26
Results
Immunohistochemical assessment of transverse spinal cord sections representing 31 sporadic and 23 familial cases revealed a group of nine sALS and eight fALS patients with unusual structures, which were intensely stained by two antibodies that recognize human γ-synuclein (Fig.1A–D). High specificity of SK109 antibody has been demonstrated in our previous studies26,27 and preincubation of antibodies with recombinant human γ-synuclein (Fig.1D) but not other recombinant synucleins (data not shown) abolished this staining. The occurrence of these γ-synuclein-positive profiles was restricted to the corticospinal tract in the dorsolateral column of the spinal cord (Fig.1A). No such profiles were observed in any of studied 21 control spinal cord tissues from healthy individuals or patients with unrelated neurodegenerative diseases (see and Table1 for details). Occasional Corpora amylacea were detected within the corticospinal tracts, however, the morphology and distribution of these bodies did not match that of the γ-synuclein-positive profiles. Generally round or slightly elliptical shape of profiles detected on transverse sections may suggest that they represent either small spheroid-shaped structures or transsections of rod-like structures orientated along the spinal cord rostrocaudal axis. Immunostaining of longitudinal sections along the rostrocaudal axis of spinal cords prepared from re-orientated paraffin blocks of three patients with γ-synuclein-positive profiles revealed elongated or rod-like γ-synuclein-positive structures, which thickness (∼10 μm) accurately correlated with the diameter of profiles seen on transverse sections (Fig.1E). Taking into consideration high abundance of γ-synuclein in axons of many types of neurons18–21 it was feasible to suggest that observed structures represented damaged axons. However, double immunofluorescent staining demonstrated that these structures are negative for a pan-axonal neurofilament marker (Fig.2A and B), indicating that they are not simply damaged axons but products of a more specific accumulation/aggregation process. Further double immunofluorescent or immunohistochemical staining of parallel sections with antibodies against γ-synuclein and proteins that often aggregate in the nervous system of ALS patients demonstrated that γ-synuclein-positive profiles detected within the dorsolateral column are negative for TDP-43, p62 and ubiquitin, although in a number of cases typical inclusions stained for these proteins were frequent in the anterior horn motor neurons in the same sections (Figs.1F, G and 2C). Retrospective analysis of the available clinical and histopathological data showed that in sALS these profiles were present not only in the more common cases with profound loss of upper motor neurons (observed in 4 of 11 analyzed cases), but also in cases with insignificant loss of these cells (observed in 4 of 5 analyzed cases). Furthermore, γ-synuclein-positive profiles were detected in sALS cases with either mild or severe myelin pallor, suggesting that there is no correlation between their presence and the severity of damage to descending motor fibers.
Figure 1.

Immunohistochemical staining of sections through cervical spinal cord of ALS patients demonstrated selective presence in the dorsolateral corticospinal tract region (A) of rounded, unevenly stained γ-synuclein-positive profiles (insert). These profiles could be revealed using either rabbit-polyclonal antibodies (SK109, B and E) or goat-polyclonal antibodies (E20, C) and preincubation of antibodies with recombinant human γ-synuclein abolished staining (D). Immunostaining of sections cut through the corticospinal tract region along the rostrocaudal axis revealed elongated or rod-shaped γ-synuclein-positive profiles (white arrowheads in E). Immunostaining for ubiquitin (F) or phosphorylated TDP-43 (G) failed to detect large ubiquitinated profiles (comparable to those containing γ-synuclein) in the lateral corticospinal tracts of patients, although in the same sections ubiquitinated skein-like inclusions (Fi) or cytoplasmic TDP-43-positive structures (Gi) were observed in the ventral horn motor neurons. (A) represents case 19, (B, C, D, F) represent case 18, (E) represents case 67 and (G) represents case 22. Scale bars = 1 mm for A-left, 100 μm for A-right, 10 μm for the inset in A and 50 μm for B–G. ALS, amyotrophic lateral sclerosis.
Figure 2.

Immunofluorescent staining of the dorsolateral corticospinal tract region of ALS cases for γ-synuclein (SK109 antibody) and various marker proteins. Double immunostaining with pan-axonal neurofilament marker (NF) showed the absence of γ-synuclein co-localization with axonal neurofilaments within the rod-like γ-synuclein-positive profiles (large arrowheads) on sections cut through the corticospinal tract region along the rostrocaudal axis (A, case 67) or on transverse sections (B, case 19). Note strong neurofilament staining of multiply healthy looking axons on these sections with different degree of co-staining for γ-synuclein (arrows show axons strongly positive for both NF and γ-synuclein, and small arrowheads show axons strongly positive for NF but weakly for γ-synuclein). γ-synuclein-positive profiles were also negative for p62 (C, case 18) although p62-positive skein-like inclusions were detected in the anterior horn motor neurons (inset, small arrows) on the same section. Some γ-synuclein-positive profiles were found engulfed by cells positive for phagocytic activity marker Mac-2/Galectin-3 (D – arrow, case 02). Secondary anti-mouse antibody conjugated with Alexa Fluor 546 (A) or Alexa Fluor 633 (B–D) were utilized to assess co-localization of different proteins with γ-synuclein detected using secondary anti-rabbit antibody conjugated with Alexa Fluor 488. Scale bars = 10 μm for B and 20 μm for A, C, D and the inset in C. ALS, amyotrophic lateral sclerosis.
Within the profiles the staining was observed to have a mesh-like appearance (Figs.1, 2) suggesting that they might be composed of aggregated γ-synuclein. To biochemically assess for the presence of aggregated γ-synuclein species we carried out sequential protein fractionation on tissue samples dissected from the frozen spinal cord of a sALS patient histologically proven to be positive for γ-synuclein-positive profiles in the dorsolateral column but not in the anterior horn (case 15). Western blot analysis of these protein fractions revealed the presence of γ-synuclein in the detergent-insoluble fraction of tissues dissected from the dorsolateral column, but not from the anterior horn areas (Fig.3). The large amounts of γ-synuclein seen in the soluble fractions should be attributed to high levels of this protein in multiple healthy or less damaged axons located in the dissected areas of the spinal cord. The presence of γ-synuclein species with slow mobility in the denaturing SDS–PAGE was not typical even for the detergent-insoluble fractions, probably reflecting an intrinsic feature of aggregates formed by γ-synuclein, as such high molecular mass species were previously found to be scarce even in the spinal cord samples of γ-synuclein transgenic mice with a high burden of pathological γ-synuclein aggregates.18 Similar analysis detected γ-synuclein in the detergent-insoluble fraction extracted from the dorsolateral column of another profiles-positive sALS case but not in a profiles-negative sALS or healthy control cases, representative Western blots are shown in Figure S1. We also detected detergent-insoluble γ-synuclein in the motor cortex of patients with FTLD-MND (Fig. S1).
Figure 3.
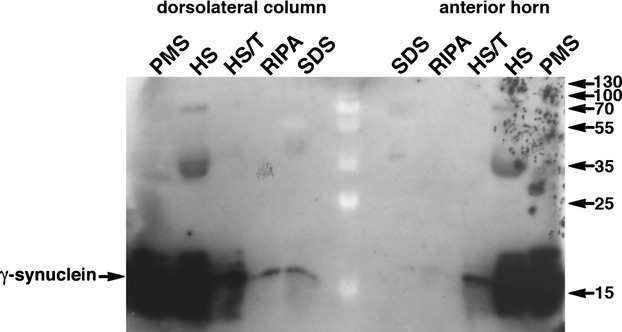
Detection of γ-synuclein in post-mitochondrial supernatant (PMS) and fractions obtained from it by sequential extraction with high salt (HS), high salt/Triton-X100 (HS/T), RIPA buffer (RIPA) and boiling in 2% SDS (SDS). A Western blot probed with SK109 antibody (A) shows the presence of detergent-insoluble γ-synuclein species in the area of corticospinal tract (dorsolateral column) but not anterior horn of the spinal cord of the ALS patient (case 15). Sizes of protein markers visible as white shades in the middle lane of the Western blot are shown in kDa on the right.
DAPI-positive nuclei were occasionally observed in close proximity to γ-synuclein-positive rod-like structures on longitudinal sections (Fig.2A). In transverse sections partial overlap of some nuclei with γ-synuclein-positive profiles was seen (Fig.2B and C), suggesting that certain phagocytic cells envelop or internalize fragments of these structures. Internalization of axon-derived γ-synuclein aggregates by specialized astrocytes in the optic nerve of a mouse model of glaucoma28 and it is feasible that a similar process might take place in the dorsolateral column of the spinal cord of ALS patients. A staining pattern compatible with this hypothesis was revealed by co-immunostaining for γ-synuclein and Mac-2/galectin-3 (Fig.2D), a marker of phagocytic cells, which has also been previously linked to ALS.29
A pale diffuse cytoplasmic γ-synuclein staining characteristic for spinal motor neuron cell bodies of healthy individuals is also typical for the majority of remaining spinal motor neurons in ALS patients (Fig.4A). However, in one sALS and three fALS cases with C9ORF72 repeat expansion (see Table1) we found motor neurons with large γ-synuclein-positive cytoplasmic inclusions (Fig.4A and B). Only in one of these cases γ-synuclein-positive profiles in the corticospinal tract were also present. No clinical characteristics that clearly distinct these four from other studied ALS cases were noted (Table S1). Retrospective analysis of available histopathological data revealed that in spinal motor neurons of these four patients, p62- and TDP-43-positive inclusions were more abundant than rarely observed γ-synuclein-positive inclusions, suggesting that these are different structures. We did not found cytoplasmic inclusions stained by γ-synuclein antibody in upper motor neurons of several studied sALS cases but in one of them (case 18) γ-synuclein-positive ballooned neuritis and amorphous extracellular deposits were observed in the motor cortex (Fig.4C). Finally, we also found no correlations between the presence of cytoplasmic inclusions or corticospinal tract profiles and the presence of human γ-synuclein allelic variants encoding either valine or glutamic acid in the position 110 of the protein amino acid sequence (Table1).
Figure 4.
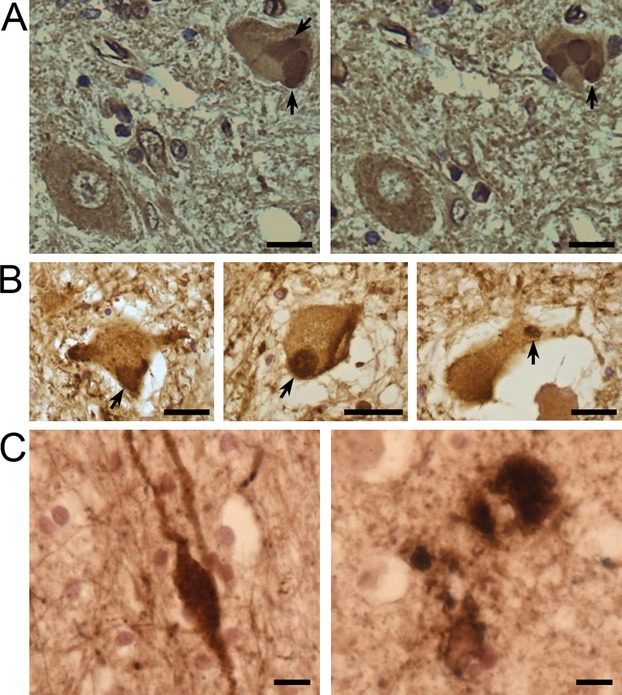
Immunohistochemical staining of anterior horn motor neurons and motor cortex of ALS patients with antibodies against γ-synuclein. On two adjacent sections through the spinal cord of a sALS patient (case 60) E20 goat-polyclonal antibodies detect diffuse cytoplasmic staining in the cell body of a motor neuron in the left bottom corner and large inclusions (arrows) in the cell body of a motor neuron in the right top corner (A). Similar cytoplasmic inclusions were revealed in motor neurons of case 63 (B, left panel) and case 73 (B, middle and right panels) fALS patients with C9ORF72 repeat expansion using SK109 rabbit-polyclonal antibodies. In the motor cortex of a sALS patient (case 18), γ-synuclein-positive ballooned neurites (left panel) and amorphous deposits (right panel) were detected using SK109 antibody. Scale bars = 50 μm for A, B and 10 μm for C. fALS, familial amyotrophic lateral sclerosis; sALS, sporadic amyotrophic lateral sclerosis.
Discussion
We identified a novel histopathological feature of ALS, γ-synuclein-positive structures present in the spinal cord of approximately 30 percent of ALS patients we assessed. The most abundant (found in 17 of 58 cases included in this study) were pathological profiles that present exclusively in the dorsolateral column where the descending axons of upper motor neurons are localized. They have a distinct morphology and could be revealed by immunostaining with antibodies specific to γ-synuclein, but not with antibodies to other proteins typically associated with ALS pathology. Large γ-synuclein-positive inclusions in the cell bodies of some spinal motor neurons were observed in fewer ALS cases (4 of 58) and only in one case were both types of structures found.
Although γ-synuclein is not a constituent of histopathological hallmarks in any neurodegenerative diseases, abnormal structures positive for this protein have been observed in the brain of some patients with Parkinson's disease, Lewy body dementia and Hallervorden-Spatz syndrome.22,23 However, the profiles we describe in the spinal cord of ALS patients are clearly different from previously described γ-synuclein-positive structures in these diseases. The axonal spheroids seen in the stratum moleculare of the dentate gyrus of Parkinson's disease and Lewy body dementia cases were several times smaller and pretreatment of sections with formic acid was necessary for their detection.23 Spheroids detected using an antibody against γ-synuclein in the cingulate and midfrontal cortices of patients with Hallervorden-Spatz syndrome were also detected with antibodies to NF proteins and ubiquitin,22 whereas γ-synuclein-positive profiles in the dorsolateral column of ALS patients were negative for these proteins. Thus, these newly identified profiles represent a truly novel histopathological feature of neurodegenerative diseases. Because these profiles were found in both sALS, SOD1-fALS and C9ORF72-fALS cases it might be concluded that their formation is a common downstream event in pathogenesis of both sporadic and at least certain familial forms of ALS. Further, we suggest that γ-synuclein-positive profiles could be formed at early stages or in particular types of upper motor neuron atrophy due to their presence not only in cases with profound loss of upper motor neurons and severe damage to descending motor fibers, but also in cases with mild changes to these neurons and their axons.
The ability of wild type γ-synuclein to form aggregates and fibrils in vivo has been previously demonstrated in mouse models.18,28 The presence of detergent-insoluble γ-synuclein species in the extracts from the profile-positive dorsolateral column but not from the profile-negative areas suggests the profiles observed in the spinal cord of ALS patients are also composed of aggregated γ-synuclein.
Normally γ-synuclein is abundant in axons of upper and lower motor neurons18–21 and therefore it is feasible to link the formation of the profiles with aggregation of γ-synuclein in axons due to or followed by their structural damage. A rod-like appearance of these structures on longitudinal sections is consistent with this suggestion. However, absence of neurofilament proteins within profiles argues against a simple explanation that they are remnants of damaged axons. In the previous studies we have demonstrated the loss of neurofilament staining in axons of cultured neurons upon overexpression of γ-synuclein20 and decreased abundance of neurofilament proteins in the sciatic nerve of transgenic mice overexpressing this protein.18,25 Although a molecular mechanism linking abnormal levels of γ-synuclein with disturbances of neurofilament metabolism or integrity of the neurofilament network is not clear, it is tempting to suggest that similar processes may contribute to the development of axonal pathology in ALS. It is feasible that at some stage of the pathology progression the local concentration of γ-synuclein in axons of upper motor neurons might increase, which in turn might trigger the loss of neurofilaments and aggregation of accumulating γ-synuclein with both processes contributing to further axonal damage.
Transmission of aggregated, axon-derived γ-synuclein species to a specific sub-class of astrocytes expressing Mac-2/galectin-3, a protein also linked to ALS,29 has been recently demonstrated in the optic nerve of a mouse model of glaucoma,28 which may explain previously observed accumulation of γ-synuclein in the optic nerve astrocytes of human glaucoma patients.24 Detection of some of the corticospinal tract γ-synuclein-positive profiles within Mac-2/galactin-3-positive cells allowed us to hypothesize that a similar process involving phagocytic cells occurs in the spinal cord of some ALS patients.
The presence of γ-synuclein-positive inclusions in the cell bodies of motor neurons, although rare, demonstrates that aggregation of this protein in neurons of ALS patients is not restricted to the axonal compartment. Both axonal pathology and cytoplasmic inclusions were also observed in transgenic mice overexpressing γ-synuclein.18,25 Further studies should reveal relationships between the two types of pathological structures described here and their contribution to the pathogenesis of ALS.
Acknowledgments
The authors thank the donors and their families for donating tissues to the Medical Research Council London Neurodegenerative Diseases Brain Bank and Sheffield Brain Tissue Bank, without which this research would not have been possible. We are grateful to Istvan Bodi, Andrew King, and Safa Al-Sarraj for sharing with us histopathological assessment data. This project was funded by the Wellcome Trust Programme Grant 075615/Z/04/z and Motor Neurone Disease Association (Buchman/Apr13/6096) to VLB, by RFBR research project no. 13-04-01633 A and RFBR KOMFI 13-04-40379-H13 to NN.
Author Contributions
All listed authors conducted the experiments. O. P., T. A. S., J. R. H., N. N., T. H., and V. L. B. discussed, planned, and supervised experiments. O. P. and V. L. B. wrote the manuscript.
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Western blot analysis of γ-synuclein in protein fractions obtained by sequential extraction of the dorsolateral column area samples of sALS patients with (+ve) or without (−ve) γ-synuclein profiles and one of healthy controls as well as the motor cortex samples of a FTLD-MND and control patients. Fraction designations as in Figure 2. Note that for these Western blots PMS and HS samples were diluted 100 times comparing to those used for the blot shown in Figure 2.
Clinical characteristic ALS patients with γ-synuclein-positive inclusions detected in the cell bodies of spinal motor neurons
References
- Shibata N, Hirano A, Kobayashi M, et al. Intense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J Neuropathol Exp Neurol. 1996;55:481–490. doi: 10.1097/00005072-199604000-00011. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano A, Donnenfeld H, Sasaki S, Nakano I. Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1984;43:461–470. doi: 10.1097/00005072-198409000-00001. [DOI] [PubMed] [Google Scholar]
- Hortobagyi T, Troakes C, Nishimura AL, et al. Optineurin inclusions occur in a minority of TDP-43 positive ALS and FTLD-TDP cases and are rarely observed in other neurodegenerative disorders. Acta Neuropathol. 2011;121:519–527. doi: 10.1007/s00401-011-0813-3. [DOI] [PubMed] [Google Scholar]
- Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–226. doi: 10.1038/nature08971. [DOI] [PubMed] [Google Scholar]
- Deng HX, Chen W, Hong ST, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash PE, Bieniek KF, Gendron TF, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Weng SM, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- Bendotti C, Atzori C, Piva R, et al. Activated p38MAPK is a novel component of the intracellular inclusions found in human amyotrophic lateral sclerosis and mutant SOD1 transgenic mice. J Neuropathol Exp Neurol. 2004;63:113–119. doi: 10.1093/jnen/63.2.113. [DOI] [PubMed] [Google Scholar]
- Kato T, Kurita K, Seino T, et al. Galectin-1 is a component of neurofilamentous lesions in sporadic and familial amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2001;282:166–172. doi: 10.1006/bbrc.2001.4556. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Ito H, Wate R, et al. Phosphorylated Smad2/3 immunoreactivity in sporadic and familial amyotrophic lateral sclerosis and its mouse model. Acta Neuropathol. 2008;115:327–334. doi: 10.1007/s00401-007-0337-z. [DOI] [PubMed] [Google Scholar]
- Collins M, Riascos D, Kovalik T, et al. The RNA-binding motif 45 (RBM45) protein accumulates in inclusion bodies in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) patients. Acta Neuropathol. 2012;124:717–732. doi: 10.1007/s00401-012-1045-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelkovnikova TA, Robinson HK, Troakes C, et al. Compromised paraspeckle formation as a pathogenic factor in FUSopathies. Hum Mol Genet. 2014;23:2298–2312. doi: 10.1093/hmg/ddt622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biere AL, Wood SJ, Wypych J, et al. Parkinson's disease-associated alpha-synuclein is more fibrillogenic than beta- and gamma-synuclein and cannot cross-seed its homologs. J Biol Chem. 2000;275:34574–34579. doi: 10.1074/jbc.M005514200. [DOI] [PubMed] [Google Scholar]
- Marsh JA, Singh VK, Jia Z, Forman-Kay JD. Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synuclein: implications for fibrillation. Protein Sci. 2006;15:2795–2804. doi: 10.1110/ps.062465306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninkina N, Peters O, Millership S, et al. Gamma-synucleinopathy: neurodegeneration associated with overexpression of the mouse protein. Hum Mol Genet. 2009;18:1779–1794. doi: 10.1093/hmg/ddp090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchman VL, Hunter HJ, Pinon LG, et al. Persyn, a member of the synuclein family, has a distinct pattern of expression in the developing nervous system. J Neurosci. 1998;18:9335–9341. doi: 10.1523/JNEUROSCI.18-22-09335.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchman VL, Adu J, Pinon LG, et al. Persyn, a member of the synuclein family, influences neurofilament network integrity. Nat Neurosci. 1998;1:101–103. doi: 10.1038/349. [DOI] [PubMed] [Google Scholar]
- Ninkina N, Papachroni K, Robertson DC, et al. Neurons expressing the highest levels of gamma-synuclein are unaffected by targeted inactivation of the gene. Mol Cell Biol. 2003;23:8233–8245. doi: 10.1128/MCB.23.22.8233-8245.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvin JE, Giasson B, Hurtig HI, et al. Neurodegeneration with brain iron accumulation, type 1 is characterized by alpha-, beta-, and gamma-synuclein neuropathology. Am J Pathol. 2000;157:361–368. doi: 10.1016/s0002-9440(10)64548-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvin JE, Uryu K, Lee VM, Trojanowski JQ. Axon pathology in Parkinson's disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc Natl Acad Sci USA. 1999;96:13450–13455. doi: 10.1073/pnas.96.23.13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surgucheva I, McMahan B, Ahmed F, et al. Synucleins in glaucoma: implication of gamma-synuclein in glaucomatous alterations in the optic nerve. J Neurosci Res. 2002;68:97–106. doi: 10.1002/jnr.10198. [DOI] [PubMed] [Google Scholar]
- Peters OM, Millership S, Shelkovnikova TA, et al. Selective pattern of motor system damage in gamma-synuclein transgenic mice mirrors the respective pathology in amyotrophic lateral sclerosis. Neurobiol Dis. 2012;48:124–131. doi: 10.1016/j.nbd.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninkina NN, Alimova-Kost MV, Paterson JW, et al. Organization, expression and polymorphism of the human persyn gene. Hum Mol Genet. 1998;7:1417–1424. doi: 10.1093/hmg/7.9.1417. [DOI] [PubMed] [Google Scholar]
- Ninkina N, Peters OM, Connor-Robson N, et al. Contrasting effects of alpha-synuclein and gamma-synuclein on the phenotype of cysteine string protein alpha (CSPalpha) null mutant mice suggest distinct function of these proteins in neuronal synapses. J Biol Chem. 2012;287:44471–44477. doi: 10.1074/jbc.M112.422402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen JV, Soto I, Kim KY, et al. Myelination transition zone astrocytes are constitutively phagocytic and have synuclein dependent reactivity in glaucoma. Proc Natl Acad Sci USA. 2011;108:1176–1181. doi: 10.1073/pnas.1013965108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JY, Afjehi-Sadat L, Asress S, et al. Galectin-3 is a candidate biomarker for amyotrophic lateral sclerosis: discovery by a proteomics approach. J Proteome Res. 2010;9:5133–5141. doi: 10.1021/pr100409r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Western blot analysis of γ-synuclein in protein fractions obtained by sequential extraction of the dorsolateral column area samples of sALS patients with (+ve) or without (−ve) γ-synuclein profiles and one of healthy controls as well as the motor cortex samples of a FTLD-MND and control patients. Fraction designations as in Figure 2. Note that for these Western blots PMS and HS samples were diluted 100 times comparing to those used for the blot shown in Figure 2.
Clinical characteristic ALS patients with γ-synuclein-positive inclusions detected in the cell bodies of spinal motor neurons