

Abstract
Objective
To identify microRNAs (miRNAs) regulated by anti-α4 integrin monoclonal antibody therapy (natalizumab) in the peripheral blood of patients with relapsing-remitting (RR) multiple sclerosis (MS) and to confirm their role in experimental settings in vivo.
Methods
In a longitudinal study of 17 RR-MS patients, we investigated blood miRNA expression profiles at baseline and after 1 year of natalizumab therapy by microarray technique and quantitative PCR validation. We compared the baseline expression profiles of these patients to those of 18 age- and sex-matched healthy controls. We confirmed the contribution of resulting candidate miRNAs in an animal model of MS, experimental autoimmune encephalomyelitis (EAE) induced by adoptive transfer of proteolipid protein (PLP)139–151-activated lymphocytes in SJL/J mice or by active immunization of miR-106a∼363-deficient C57BL/6 mice (or wildtype litter mates) with myelin oligodendrocyte glycoprotein (MOG)35–55.
Results
Our longitudinal analysis revealed that miR-18a, miR-20b, miR-29a, and miR-103 were upregulated and predominantly expressed by CD4+ T cells, whereas miR-326 was downregulated upon natalizumab treatment. A comparison of untreated RR-MS patients at baseline with healthy controls revealed that the four natalizumab-upregulated targets were initially downregulated in MS. All confirmed targets showed disease-dependent expression in splenocytes of mice suffering from EAE. Genetic deletion of the miRNA cluster miR-106a∼363 (containing natalizumab-regulated miR-20b) resulted in a more severe EAE course and an in vivo upregulation of the miR-20b target genes rorgt, stat3, and vegfa.
Interpretation
Our study indicates that natalizumab restores dysregulated miRNA patterns in MS and reveals the contribution of miR-20b in autoimmune demyelination in vivo.
Introduction
Multiple sclerosis (MS) is the most common chronic inflammatory disease of the central nervous system (CNS) in the Western world and a major cause of sustained neurological disability among young adults.1 MS is considered an immune-mediated disease,2,3 characterized by the influx of activated immune cells into the CNS via crossing of the blood–brain barrier (BBB).4 Antibodies that bind α4-integrin and thus block the α4β1 dimer on T lymphocytes prevent this invasion process in experimental autoimmune encephalomyelitis (EAE), an animal model of MS.5,6 In line, monthly infusions of the humanized anti-α4 integrin monoclonal antibody natalizumab (Tysabri®) profoundly reduce disease activity in patients with relapsing MS, and therefore represent an important therapeutic option for MS.7–10 Although the accepted mode of action of natalizumab is the inhibition of lymphocyte transmigration,11 there may be additional hitherto unknown mechanisms that could contribute to both the therapeutic response and the adverse effects.12–16 Indeed, one recent study on messenger RNAs (mRNAs) found that natalizumab treatment regulated multiple genes relevant for the function and differentiation of lymphocytes.17 However, the mediators underlying this unexpectedly complex mode of action are unknown.
MicroRNAs (miRNAs) are small (20–25 nucleotides in length), noncoding, highly preserved RNA molecules that have recently emerged as key epigenetic regulators in cell biology.18 Utilizing a specialized molecular machinery, they bind complementarily to the 3′ UTR of a messenger RNA, thus preventing it from transcription and leading to its degradation and translational repression. It is estimated that at least one-third of all genes are regulated by miRNAs. Increasing evidence suggests that miRNAs play an indispensable role in immune function and autoimmunity.19 Indeed, cross-sectional studies investigating peripheral blood,20–32 inflammatory CNS lesions33,34 or cerebrospinal fluid (CSF)35 have revealed alterations of miRNA patterns in MS patients and reported different miRNA expression levels in MS patients treated with glatiramer acetate26 or interferon-beta.31
We thus set out to investigate the effect of natalizumab treatment on miRNA expression, using a combined longitudinal and cross-sectional approach with relapsing-remitting (RR-) MS patients and healthy controls (HCs), and confirming the resulting targets in experimental autoimmune demyelination.
Materials and Methods
Study population
MS patients were recruited to the Department of Neurology at Heinrich-Heine-University in Düsseldorf, Germany, diagnosed with definitive MS according to the 2005 revised McDonald criteria in accordance with the local ethics committee. All patients gave written informed consent. Samples were coded throughout the study, and sample handling was observer masked. If disease-modifying drugs had been used prior to natalizumab therapy, patients underwent a washout phase of at least 3 months. We included 17 patients with RR-MS with a female/male ratio of 3.3. At the initiation of natalizumab treatment, the mean age was 36.3 ± 7.7 years (SD), and the median expanded disability status scale (EDSS) score was 2.0 (range 1.0–6.5). Detailed demographic and clinical data are listed in Table S1. Patients donated two blood samples in Paxgene® RNA tubes (BD, Heidelberg, Germany). The first blood sample was collected immediately prior to the first natalizumab administration (before natalizumab). For samples obtained under therapy, blood was drawn in the moment before the regular natalizumab infusion was applied. The mean natalizumab treatment duration at the time of the second sample was 16.8 months (minimum 12 months, maximum 22 months). This collection schedule enabled an intraindividual, longitudinal approach. None of the patients experienced a relapse during blood withdrawal. The mean time between the last relapse and initiation of natalizumab was 6.5 months (minimum 3 months, maximum 12 months). The control group samples consisted of Paxgene® RNA tubes of blood from 18 age- and sex-matched healthy individuals with a mean age of 37.2 ± 12.5 years (SD) and a female/male ratio of 3.0.
MicroRNA microarray
Total RNA, including microRNA, was isolated from Paxgene® tubes using the miRNeasy® kit (Qiagen, Hilden, Germany). Samples were analyzed using a Geniom Realtime Analyzer (GRTA, CBC Comprehensive Biomarker Center, Heidelberg, Germany) with the Geniom Biochip miRNA Homo sapiens. Each array contained seven replicates of 866 miRNA and miRNA* sequences, as annotated in Sanger miRBase 12.0. Samples were labeled with biotin using microfluidic-based enzymatic on-chip miRNA labeling (MPEA), and further processed as described previously36 (see Data S1). Normal distribution of the data was verified with the Shapiro–Wilk test. We considered our microarray approach to represent an exploratory first step. Thus, to avoid a loss of sensitivity (as expected with other approaches), we based our analysis on the results of a separate parametric t-test (paired, two-tailed, cut-off P < 0.05) for each miRNA to detect miRNAs with different expression levels between the study groups. We then confirmed the resulting targets with quantitative PCR analyses (qPCR).
Quantitative PCR
For miRNA expression level quantification, the RNA samples used for the microarray analysis were subjected to qPCR by TaqMan microRNA assays (Applied Biosystems, Darmstadt, Germany). For human samples, RNU6B was used as a housekeeping gene, and snoRNA202 and snoRNA135 for mouse samples. Samples were measured in duplicate, and all resulting duplicate ΔCT values were analyzed using paired t-tests for human samples. For EAE miRNA expression levels were analyzed at three time points during disease course, groups were compared using analysis of variance (ANOVA) with post hoc analysis (Dunnett’s T test), and correlations between EAE disease scores and miRNA levels were determined using linear regression. For statistical analyses, P values <0.05 were considered significant, calculated with GraphPad Prism (La Jolla, CA) and SPSS (IBM, New York, NY).
In silico/systems biology analysis
In order to analyze the regulation of micro-RNAs via the α4β1 receptor we applied an overrepresentation analysis using our gene set analysis tool GeneTrail37 and the Transfac®-based P-Match tool (http://www.gene-regulation.com) for transcription factor binding site search by combining patterns and weight matrices.38 For computational prediction of gene targets of our miRNA candidates, we used the mirSVR scoring regression method39 provided by the http://www.microRNA.org information resource.
Ex vivo experiments with human samples
For ex vivo experiments, we randomly included nine patients (mean age 37 ± 10.0 years) out of the above mentioned longitudinal cohort of 17 patients treated with natalizumab for at least 1 year. Moreover, we included two additional patients that had developed natalizumab-associated progressive multifocal leukoencephalopathy (PML) and were diagnosed with PML few days after taking the blood sample (PML #1: 51-year old female, 3.1 years natalizumab treatment; PML #2: 32-year-old male, 5 years natalizumab treatment). Peripheral blood mononuclear cells (PBMC) were isolated from whole blood by Ficoll (Biochrom, Berlin, Germany) gradient and CD4+ cells were isolated by magnetic bead separation using STEMCELL EasySep Human CD4+ T Cell Enrichment Kit according to the manufacturer’s instructions. For generation of Th17 cells CD4+ cells were cultivated in RPMI (Roswell Park Memorial Institute) 1640 medium (Gibco, Carlsbad, CA) with 10% fetal calf serum (FCS, Gibco) and nonessential amino acids (Biochrom) in round-bottom 96-well plates coated with anti-CD3-antibody and supplemented with anti-CD28-antibody (both 1 μg/mL), IL-1 (10 ng/mL), IL-6 (10 ng/mL), IL-23 (10 ng/mL), and TGF-β (5 ng/mL, all eBioscience, Frankfurt, Germany). After 4 days, cells were harvested and miRNA was isolated for PCR as described above.
Experimental autoimmune encephalomyelitis
Disease induction and the clinical scoring of mice with EAE were performed as previously described.40 For confirmatory experiments regarding miRNA regulation in peripheral immune cells, female SJL/J mice (n = 5) were immunized subcutaneously with 200 μg proteolipid protein (PLP) peptide PLP139–151 (Pepceuticals, Leicestershire, UK) supplemented with 800 μg H37RA (Difco, Franklin Lakes, NJ) and emulsified in 100 μL complete Freund’s adjuvant (CFA, Sigma-Aldrich, Steinheim, Germany) with 100 μL PBS. On day 10 post immunization, spleens and lymph nodes were harvested, and the resulting single-cell suspension was restimulated with 10 μg/mL PLP139–151 in RPMI 1640 medium supplemented with 10% FCS, penicillin/streptomycin, glutamate, and 2-mercaptoethanol (Invitrogen, Karlsruhe, Germany). After 4 days in culture, cells were harvested and injected into naïve female SJL/J mice intraperitoneally (3 × 107 cells/mouse, n = 5 per time point). Control mice received a single intraperitoneal (i.p.) injection of PBS. Mice were euthanized either on day 13 or 50 post transfer. Spleens were harvested, total RNA including miRNA was isolated from single-cell suspensions and analyzed by qPCR. For experiments in transgenic animals, C57BL/6 mice (n = 5–7/group) deficient of miRNA cluster miR-106a∼363 (Jackson Laboratories, Bar Harbor, ME; stock number 008461)41 and wild-type litter mates were immunized subcutaneously with 200 μg of myelin oligodendrocyte glycoprotein (MOG) peptide MOG35–55 (Pepceuticals, Leicestershire, UK) supplemented with 800 μg H37RA and emulsified in 100 μL CFA with 100 μL PBS and received an i.p. injection of pertussis toxin (400 ng, List Biological Laboratories, Campbell, CA) with immunization and 2 days later. Mice were sacrificed on day 20 post immunization. Throughout all EAE experiments mice were housed specific pathogen-free, at a dark/light cycle of 12 h, stable temperature of 22–24°C and had unlimited access to food and water. All procedures were conducted according to protocols approved by the local animal welfare committee and comply with ARRIVE criteria.42
Immunohistochemistry
Mice were anaesthetized with isoflurane and cardially perfused with PBS. Brain and spinal cord were isolated and fixed in 4% paraformaldehyde and dehydrated in a 30% sucrose solution. Specimens were frozen at −80°C in Tissue Tek® (Sakura Fintek Europe, Zoeterwounde, The Netherlands), and cut with a cryostat (Leica, Wetzlar, Germany). The slices were permeabilized with 0.5% Triton and blocked using 5% normal goat serum (Invitrogen) and 1% bovine serum albumin. The following antibodies were used according to the manufacturer’s instructions: mouse anti-MBP (Upstate, New York, NY), rabbit anti-Iba1 (Wako Pure Chemical Industries, Osaka, Japan), Hoechst 33342 dye (Molecular Probes, Darmstadt, Germany). Visualization was performed by labeling with Cy2- and Cy3-conjugated secondary antibodies (Millipore, Billerica, MA). Primary antibodies (anti-mouse-Iba1 and anti-mouse MBP) were incubated over night at +4°C and the secondary antibody for 1 h at room temperature. After washing, Hoechst dye 33258 (Molecular Probes) was used to counterstain nuclei. Specimens were analyzed on an Olympus BX51 microscope and Photoshop 5.0 software (Adobe, San Jose, CA). For quantification, the mean number of lesions showing cell infiltration (Hoechst dye 33258, Sigma-Aldrich) and staining positive for microglia/macrophage-marker Iba1 from three sections per animal was calculated and statistics were calculated with 5 animals per group.
Results
Longitudinal analysis of miRNA expression by microarray and qPCR
In a first exploratory and unrestricted approach, we analyzed the expression of 866 miRNA sequences from Paxgene® blood samples of 17 patients with RR-MS intra-individually before natalizumab administration and after at least 1 year of continuous treatment. This analysis suggested that 14 miRNAs were regulated by natalizumab therapy (Table1). Of these, eight were upregulated after natalizumab (miR-18a, miR-29a, miR-20b, miR-103, miR-532-5p, miR-24, miR-342-3p, and miR-7-1*), and six miRNAs were downregulated (miR-411*, let-7d*, miR-380*, miR-2117, miR-2116*, and miR-1237). Interestingly, none of the downregulated miRNAs have been shown to be linked to specific functions, and some of these were miRNAs*, the passenger strand of the biologically active miRNA, which are thought to be biologically inactive.18
Table 1.
miRNA microarray data in the longitudinal study group (MS patients before and at least 12 months after natalizumab therapy)
| Upregulated | Downregulated | ||||||
|---|---|---|---|---|---|---|---|
| Fold change (log2) | P value | Fold change (log2) | P value | ||||
| 1 | hsa-miR-18a | 0.85 | <0.05 | 1 | hsa-miR-411* | −0.72 | <0.01 |
| 2 | hsa-miR-29a | 0.76 | <0.05 | 2 | hsa-let-7d* | −0.67 | <0.01 |
| 3 | hsa-miR-20b | 0.72 | <0.05 | 3 | hsa-miR-380* | −0.65 | <0.05 |
| 4 | hsa-miR-103 | 0.67 | <0.01 | 4 | hsa-miR-2117 | −0.64 | <0.05 |
| 5 | hsa-miR-532-5p | 0.65 | <0.05 | 5 | hsa-miR-2116* | −0.40 | <0.05 |
| 6 | hsa-miR-24 | 0.64 | <0.05 | 6 | hsa-miR-1237 | −0.30 | <0.05 |
| 7 | hsa-miR-342-3p | 0.49 | <0.05 | ||||
| 8 | hsa-miR-7-1* | 0.39 | <0.05 | ||||
MS, multiple sclerosis.
Following this exploratory microarray approach, results were validated by quantitative PCR as the gold standard for miRNA quantification. As most of the downregulated targets were miRNA* sequences or miRNAs that had not previously been ascribed to particular functions, we focused on the upregulated targets and confirmed the expression of the 4 most strongly upregulated targets miR-18a, miR-20b, miR-29a, and miR-103 by qPCR (Fig.1). In addition, we also analyzed the expression of miR-326 that has only recently been implicated in the pathogenesis of MS by three independent research groups.23,26,33 Indeed, although miR-326 was not identified in our microarray approach, the more sensitive qPCR analysis revealed its downregulation following natalizumab treatment in vivo (Fig.1).
Figure 1.
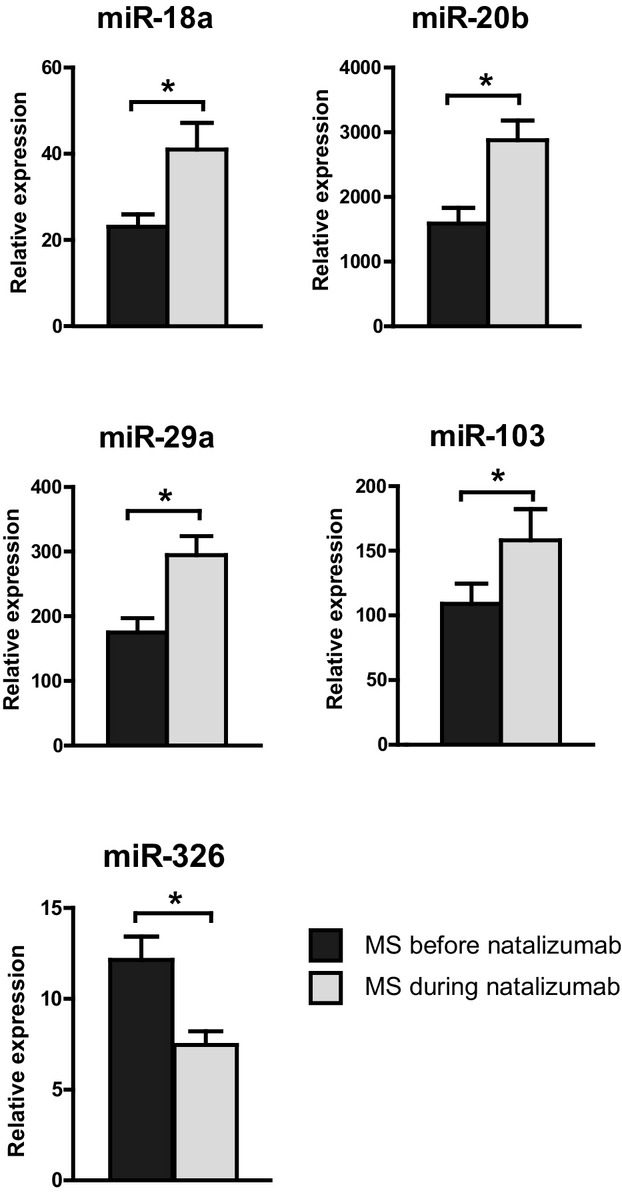
Regulation of miRNAs in the course of natalizumab therapy. Shown are quantitative PCR expression levels of selected miRNAs identified by exploratory array analysis (Table S1) and confirmed by TaqMan® quantitative PCR analysis. Data reflect fold changes in expression normalized to that of the housekeeping gene from 17 patients’ blood samples before and after at least 12 months of natalizumab treatment. Shown are mean + SEM from all ΔCT values measured in duplicate per patient (asterisks denominate statistical significance, P < 0.05). For samples obtained under therapy, blood was drawn in the moment before the regular natalizumab infusion was applied.
Disease relevance of the natalizumab-regulated miRNAs
To examine the disease relevance of the miRNA candidates identified in our longitudinal study, we performed a cross-sectional validation study. We conducted a microarray analysis to compare the miRNA profiles of our 17 patients at baseline (after a 3-month washout period and prior to receiving the first dose of natalizumab) as well as during natalizumab treatment to the profiles obtained from age- and sex-matched HCs. In order to cluster the three data sets, we applied a complete linkage hierarchical clustering of the miRNA patterns using the Euclidian distance as distance measure. To focus on the most informative miRNAs, the 100 with highest data variance have been selected (Fig.2A). In MS samples, 93 miRNAs were found to be significantly downregulated and 70 upregulated in comparison to the HC samples (Tables S2, S3). Remarkably, six of the eight miRNAs upregulated after natalizumab treatment were found to be downregulated in MS versus HCs. Of note, all miRNAs downregulated by natalizumab therapy were not found to be altered in untreated MS patients (Fig.2B).
Figure 2.
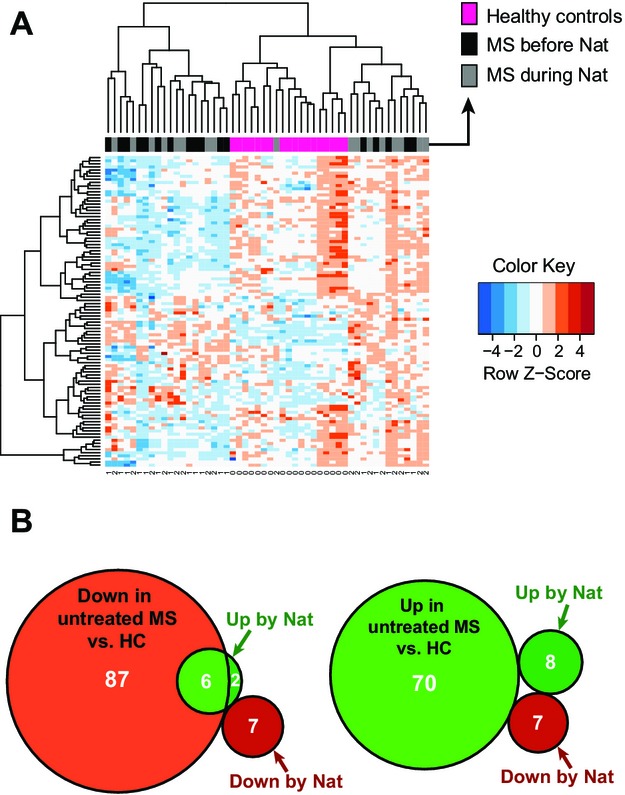
(A) Heat map using three data sets (healthy controls, multiple sclerosis [MS] before natalizumab, MS during natalizumab). In order to cluster the data we applied complete linkage hierarchical clustering of the miRNA patterns using the Euclidian distance as distance measure. To focus on the most informative miRNAs, the 100 with highest data variance have been selected. Nat = natalizumab treatment. B) Venn diagrams showing the overlapping miRNA targets for comparison of untreated MS subjects versus healthy controls (HC) and MS patients pre- versus postnatalizumab treatment. The left diagram shows the overlap of miRNAs downregulated in untreated MS with natalizumab-regulated miRNAs (Up by Nat: upregulated in natalizumab treatment; Down by Nat: downregulated in natalizumab treatment). The right diagram shows the overlap of miRNAs upregulated in untreated MS patients versus healthy controls (HC) with natalizumab-regulated miRNAs.
miRNA dysregulation and therapy response
Six of the 17 natalizumab-treated patients experienced an improvement with regard to the last available disability assessment, while the other 11 patients were either stable (n = 9) or showed disease activity (n = 2, EDSS worsening/relapses; see Table S1). We performed a subset analysis of the miRNA qPCR results in these subcohorts but were not able to detect a distinct miRNA regulation pattern (Fig. S1).
miRNA regulation in EAE
As these results suggest a link between natalizumab treatment and the miRNA profile of patients with chronic autoimmune neuroinflammation, we next evaluated the miRNA patterns in experimental autoimmune demyelination. Peripheral leukocytes were isolated from the spleens of mice at different stages of EAE, and miRNAs previously identified in MS patients were investigated by qPCR. Because an active immunization with a local inflammation at the site of antigen inoculation may potentially confound the interpretation of neuroinflammation-specific miRNA alterations, we opted for utilizing an adoptive transfer EAE model in SJL/J mice. At the peak of disease, the expression of miR-18a, miR-20b, miR-29a, and miR-103 was significantly downregulated, whereas that of miR-326 was upregulated in comparison to controls (Fig.3). At late-stage disease, these changes were reversed for miR-18a, miR-20b, miR-29a, but not miR-103 and miR-326. A linear regression analysis revealed that the expression levels of miR-18a, miR-20b, miR-29a, and miR-103 are negatively correlated with disease severity. In contrast, the expression of miR-326 appeared to be positively correlated with disease severity (Fig.4).
Figure 3.
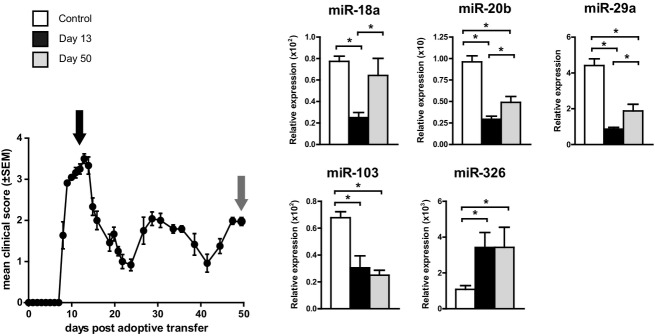
Regulation of natalizumab-associated human miRNA targets in a preclinical model of MS. Naïve SJL/J mice received activated encephalitogenic T lymphocytes (stimulated with PLP139–151) to induce adoptive transfer experimental autoimmune encephalomyelitis (EAE) or PBS only (controls). RNA from splenocytes was isolated at different time points during disease, and data show mean fold changes in expression normalized to that of the housekeeping genes +SEM for duplicate ΔCT values per mouse (n = 3 for control group; n = 5 for day 13 and day 50 groups; asterisks denominate statistical significance, P < 0.05). MS, multiple sclerosis; PLP, proteolipid protein.
Figure 4.
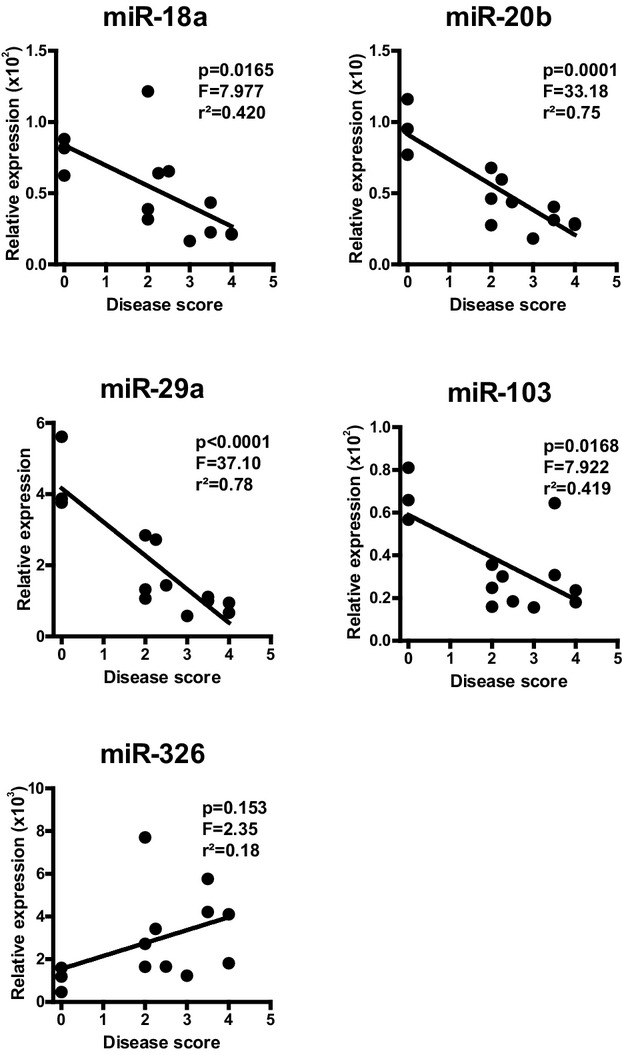
Disease activity-dependent miRNA regulation in experimental autoimmune encephalomyelitis (EAE). Splenocyte miRNA levels determined in the course of EAE from Figure3 were correlated to disease scores of the corresponding animals by linear regression analysis.
EAE in miR-106a∼363 deficient mice
Three of the above described miRNA targets have already been linked to the pathogenesis of MS: miR-18a,43 miR-29a,44 as well as miR-326.23 However, extending these previous findings, we were able to identify miR-20b, a member of miR-106a∼363 cluster and miR-17∼93 family, as a novel target. Thus, we further explored the contribution of this cluster to autoimmune demyelination by inducing active EAE in mice with a homozygous deficiency of the miRNA cluster miR-106a∼363 (miR-106a∼363−/−) or their direct wild-type litter mates. Of note, an in vitro analysis of Th17 differentiation patterns did not show a deviation in this strain (Fig. S2). However, regarding in vivo EAE manifestation, we found that miR-106a∼363−/− mice had an earlier onset of symptoms and a more severe disease course (Fig.5A). Histological analysis of CNS tissue for EAE lesions correlated to clinical signs and showed significantly more lesions with infiltrating immune cells in miR-106a∼363−/− than in miR-106a∼363+/+ (Fig.5B and C). Finally, quantitative PCR from preonset peripheral splenocytes (day 7 post immunization, Fig.5D) and spinal cord tissue from day 20 post immunization (Fig.5E) revealed an upregulation of Th17-related proinflammatory target genes rorgt and stat3, as well as vegfa, all computationally predicted targets of miR-20b (Table S4) that recently have been validated experimentally.45–47
Figure 5.

EAE in miR-106a∼363-deficient mice. (A) Active EAE was induced in miR-106a∼363-deficient C57BL/6 mice using MOG35–55 peptide (n = 6–7). (B) Representative images of CNS lesions with mononuclear cell infiltration and microglia/macrophage activation. (C) Quantification of number of lesions per slice (n = 5). (D) Quantitative PCR from splenocytes extracted from animals at day 20 post immunization. The different panels represent targets associated to (from left to right) Th1 and Th17/GMCSF differentiation, as well as the macrophage lineage, all previously validated targets of miR-20b. (E) Quantitative PCR from spinal cord tissue at day 20 post immunization. Statistical tests were performed using t-test. EAE, experimental autoimmune encephalomyelitis; CNS, central nervous system.
Target miRNA expression patterns in immune cell subsets
In order to identify the cellular source of the here identified miRNA targets, we analyzed the expression corresponding to immune cell subsets. For this purpose, we purified PBMC from natalizumab-treated patients (n = 9; randomly selected subcohort from 17 patients of our initial longitudinal patient cohort), isolated CD4+ T helper cells and generated Th17 cells from the same patients. Figure6A shows that the expression of miR-20b expression was significantly higher in CD4+ T cells as compared to the CD4− subpopulation. Neither T-cell activation without addition of cytokines nor T-cell activation with Th17-skewing conditions (IL-1, IL-6, IL-23, and TGF-β) resulted in a relevant regulation of miR-20b (Fig.6B). A similar pattern indicating CD4+ T cells as the major source of our observations was evident for the other here described miRNA targets (Fig. S3A for miR-18a, miR-29a, miR-103, and miR-326) which were mostly stable during T-cell differentiation (Fig. S3B).
Figure 6.
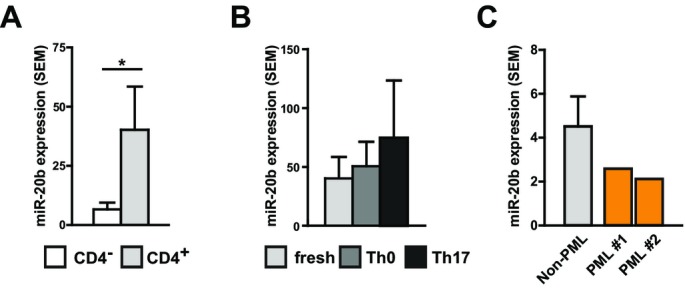
miR-20b expression in T cells and in PBMC from natalizumab-associated progressive multifocal leukoencephalopathy (PML). (A) PBMC were isolated using a Ficoll gradient and CD4+ T cells were sorted with magnetic beads (n = 9; randomly selected patients of our initial longitudinal cohort consisting of 17 patients). miR-20b expression (qPCR) was measured in the CD4+ and CD4-negative fraction and compared using t-test. (B) CD4+ cells were activated by plate-bound anti-CD3-antibody and soluble anti-CD28-antibody without further cytokines, termed “Th0”, and with IL-1 (10 ng/mL), IL-6 (10 ng/mL), IL-23 (10 ng/mL) and TGF-β (5 ng/mL), termed “Th17”, as well as freshly isolated CD4+ T cells, termed “fresh”. miR-20b levels were measured by qPCR. (C) miR-20b levels in PBMC from natalizumab-treated patients (n = 9, left bar) were compared to miR-20b levels in two PBMC samples from patients with natalizumab-associated PML (each patient one bar).
miR-20b expression in samples from patients with natalizumab-associated PML
Considering opportunistic JC-virus-mediated progressive multifocal leukoencephalopathy (PML) a major challenge in natalizumab therapy, we further investigated PBMC samples from two patients with natalizumab-associated PML. A comparison of miRNA expression patterns in these two patients with the corresponding patterns of natalizumab-treated patients without PML showed no relevant regulation for our miRNA targets, neither miR-20b (Fig.6C) nor miR-18a, miR-29a, miR-103, and miR-326 (Fig. S3C).
In silico analysis of α4β1/miRNA-associated signaling cascades
In order to understand the potential regulation of miR-20b via α4β1 and its downstream signaling pathways we performed a systems biology pathway analysis. An initial overrepresentation analysis with GeneTrail37 focusing on integrin-mediated outside-in signaling showed that miR-20b is regulated by the extracellular matrix (ECM)-receptor interaction and especially the MAPK pathway. Moreover, we applied the Transfac®-based P-Match tool for transcription factor binding site search by combining patterns and weight matrices.38 We found that all miRNAs regulated by α4β1 engagement contain putative NF-kB regulatory elements in their promoter region (Table S5).
Discussion
We report the effects of natalizumab treatment on miRNA expression patterns in vivo, as determined by a longitudinal follow-up analysis of RR-MS patients during the course of at least 1 year of continuous therapy. Using a stepwise approach starting with microarray analysis and subsequent qPCR-based verification, we found that five microRNAs (miR-18a, miR-20b, miR-29a, miR-103, and miR-326) were regulated by natalizumab. Remarkably, in a cross-sectional study comparing our MS patients prior to natalizumab therapy with HCs, all of the natalizumab-upregulated miRNAs (miR-18a, miR-29a, miR-20b, and miR-103) were downregulated. Thus, natalizumab treatment appears to restore an altered expression of miRNAs in vivo. Moreover, we were able to confirm the involvement of all five newly identified natalizumab-regulated miRNAs in experimental autoimmune demyelination, as they were associated with disease severity. Induction of EAE in animals deficient for miRNA cluster miR-106∼363, which contains one of the natalizumab-regulated miRNAs (miR-20b) resulted in a more severe phenotype with histologically more CNS lesions and an in vivo upregulation of immunological targets of miR-20b. CD4+ T cells were confirmed to be the main source of miR-20b and of most of the other here identified miRNA targets in natalizumab-treated patients.
It is widely assumed that disturbed immunity is key to the pathogenesis of MS. The majority of MS-associated genes identified in a recent large genome-wide association study have immunological functions.48 Epigenetic mechanisms responsible for altered gene expression, such as microRNAs, have recently been shown to act as major modulators of many physiological functions in health and disease, including the immune system18,19 and MS.49 MicroRNAs are of particular interest because only a limited number exists, and each miRNA regulates several genes through partially complementary sequences in the target mRNA. Therefore, understanding the effects of miRNAs on an immune-mediated disease such as MS may not only increase the general understanding of the pathogenic mechanisms but may also lead to the development of biomarkers for drug response monitoring or the discovery of therapeutic miRNA targets. Indeed, tools for taking miRNA modulation into therapy have already been developed.50
In our study, two miRNAs of the miR-17∼92 family were shown to be upregulated due to natalizumab therapy (miR-18a of the miR-17∼92 cluster and miR-20b of the miR-106a∼363 cluster). Indeed, members of this family have repeatedly been reported to be associated with immune dysfunction in MS20–22,24–26 and even in natalizumab treatment.51 However, the precise expression patterns of members of this miRNA family are complex and depend on the specific miRNA as well as the compartment being investigated. Moreover, members of the miR-17∼92 family have been implicated in a plethora of different processes, including cell cycle progression, angiogenesis, cancer, TGF-β-signaling, and lymphoproliferative disorders.52 Although miR-17∼92 family members have been shown to be regulated in all published miRNA microarray analyses of MS patients, it remains unknown if this is an epiphenomenon or if some of these miRNAs play a causative role. Our study implicates miR-20b (a member of miR-106a-363 cluster) to contribute to disease pathogenesis: EAE in miR-106a∼363-deficient mice is more severe and two previously experimentally validated targets of miR-20b were significantly upregulated in inflamed CNS tissue in vivo, that is, stat3 and vegfa.45,46 VEGF (vascular endothelial growth factor) has recently been shown to regulate autoimmune inflammation and BBB breakdown.53 STAT3 plays an important role in Th17 differentiation,54 which in turn is crucial for the development of autoimmune neuroinflammation. A very recent experimental study showed that lentiviral overexpression of miR-20b inhibits Th17 differentiation and suppresses EAE by inhibition of the Th17 transcription factor RORγt.47 In line, we found that RORγt was upregulated in miR-106a∼363-deficient CNS tissue in EAE. Furthermore, miR-20b has been shown to be regulated in MS peripheral blood,21,24,32 as well as MS brain lesions,33 and we identified CD4+ T helper cells as the main source for miR-20b in PBMC of natalizumab-treated patients. Our data add a direct pathogenic link of miR-20b to autoimmune inflammation and suggest miR-20b-related processes as a possible therapeutic target in MS. Regarding such processes, our in silico promoter analysis indicates that the here identified candidate including miR-20b may be sensitive to regulation by the NF-kB pathway known as a central downstream target of α4β1 signaling.55
Regarding consistency with previous reports, our qPCR analysis was able to confirm two miRNAs, miR-326 and miR-29a, recently linked to MS and experimental autoimmune demyelination. For miR-326, we observed a downregulation in patients upon natalizumab treatment. This finding is in line with observations on upregulation of miR-326 in the peripheral blood of MS patients and the contribution of this miRNA to an encephalitogenic Th17 response in EAE.23,26 Furthermore, miR-326 was found to be upregulated in acute MS brain lesions.33 A similar pattern is obvious for miR-29a, recently shown to be regulated in MS patients with RR disease course, as compared to HCs,28 and shown to regulate differentiation of proinflammatory Th1 immune responses.44 Apparently, natalizumab therapy is associated with the normalization of two previously identified disease-relevant encephalitogenic miRNAs, thus exerting effects beyond pure blockage of adhesion molecule-mediated immune cell infiltration.
Our data imply that certain miRNAs might serve as therapy response markers. As natalizumab is generally a very effective therapy, the number of patients in our cohort who did not respond to the drug was obviously too small to draw final conclusions, thus requiring a confirmation in larger patient cohorts. Notably, the expression of the five miRNA targets reverted by natalizumab therapy was also altered in peripheral immune cells during EAE and negatively correlated with disease severity. While it is clear that uncritical extrapolation from EAE to MS has several limitations,4,56 the close correlation of miRNA expression patterns in MS and EAE in our study supports the role of common miRNA-dependent pathways in autoimmune neuroinflammation.34
There are obvious restrictions to our study. The sample size is small, and the analysis is based on exploratory approaches. However, despite these limitations, we found a coherent set of confirmed miRNAs reverted by natalizumab therapy, suggesting that an effective MS therapy can have a considerable impact on this epigenetic regulation pathway. Furthermore, we were able to detect sustained alterations of specific microRNAs in different scenarios, including longitudinal and cross-sectional expression analyses as well as in animal studies. This consistent pattern suggests these miRNAs to be relevant for the regulation of immune-mediated demyelination. In light of the high efficacy and the risk profile of selective immunointervention therapy approaches, such markers may contribute to the assessment of a patient’s individual treatment response and may help to better determine personalized risk-benefit profiles.
Acknowledgments
We thank the patients and healthy subjects who donated blood samples for their support. The Düsseldorf MS center at the Department of Neurology of the Heinrich-Heine-Universität in Düsseldorf is supported in part by the Walter-and-Ilse-Rose-Stiftung. This work was supported by grants from the German Ministry for Education and Research (BMBF, “German Competence Network Multiple Sclerosis” (KKNMS), Natalizumab-Pharmakovigilanzstudie, 01GI1002 to B. C. K.). We thank Carsten Berndt for valuable discussions.
Author Contributions
J. Ingwersen designed and conceptualized the study, collected, analyzed, and interpreted the data and drafted and revised the manuscript. T. Menge conceptualized the study, interpreted the data, and drafted and revised the manuscript. B. Wingerath, D. Kaya, J. Graf, and T. Dehmel collected and analyzed the data and revised the manuscript. J T. Prozorovski and A. Keller collected, analyzed, and interpreted the data and revised the manuscript. C. Backes, M. Beier, and M. Scheffler analyzed and interpreted the data.B. K. Kieseier, H. P. Hartung, and P. Küry conceptualized the study, interpreted the data, and revised the manuscript.. O. Aktas designed and conceptualized the study, analyzed, and interpreted the data and drafted and revised the manuscript.
Conflict of Interest
J. Ingwersen, B. Wingerath ,D. Kaya, J. Graf, C. Backes, T. Prozorovski, Dehmel report no disclosures. T. Menge received honoraria for lectures and travel/accommodations/meeting expenses from BiogenIdec, Teva, MerckSerono, and Bayer. , A. Keller, M. Beier, M. Scheffler were employees of febit biomed GmbH (now CBC Comprehensive Biomarker Center GmbH) and hold patents.T.. B. Kieseier received honoraria for consultancy and travel/accommodations/meeting expenses by Bayer, BiogenIdec, CSLBehring, MerckSerono, Novartis, Sanofi, TEVA. H.-P. Hartung received grants by Walter-and-Ilse-Rose-Stiftung, Eugène Devic European Network (EU-FP7) and the German Ministry for Education and Research, received honoraria for consultancy by Bayer HealthCare, BiogenIdec, Genzyme, Novartis, Teva, SanofiAventis, Hoffman LaRoche and holds patents. P. Küry received grants and travel/accommodations/meeting expenses by German Research Foundation (DFG), Novartis and Baxter and holds patents. O. Aktas received grants by German Research Foundation (DFG), Eugène Devic European Network (EU-FP7), German Ministry for Education and Research, Schaufler Foundation, honoraria for lectures by Novartis, BayerSchering, Teva, BiogenIdec, holds patents and received travel/accommodations/meeting expenses by Novartis, BayerSchering, and MerckSerono.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Data S1. Supplementary methods.
Figure S1. Disease response analysis. Shown are ratios of quantitative PCR expression levels of selected miRNAs from patients before (baseline) and during natalizumab therapy. A) Comparison of patients that improved in EDSS (n = 6) and that were stable or deteriorated (n = 11). B) Comparison of patients that improved or were stable (n = 15) and that deteriorated (n = 2). See also Table S1.
Figure S2. Magnetic bead isolated mouse CD4+ cells were incubated with anti-CD3-antibody (1 μg/mL), IL-6 (20 ng/mL), and TGF-beta (2 ng/mL) for 4 days in the presence of irradiated APC. Analysis was performed using BD Leukocyte Activation Cocktail, intracellular staining using BD Cytofix/Cytoperm buffers (Becton Dickinson), and subsequent FACS analysis. Pooled data from 4 experiments (each n = 5/per group).
Figure S3. (A) PBMC were isolated using a ficoll gradient and CD4+ T cells were sorted with magnetic beads (n = 9). miRNA expression was measured in CD4+ cells and CD4- PBMC by qPCR. Significance was calculated using t-test. (B) PBMC were isolated using a ficoll gradient and CD4+ T cells were sorted with magnetic beads (n = 9). miRNA expression was measured in freshly isolated CD4+ cells (“fresh), CD4+ cells that were activated by plate-bound anti-CD3-antibody and soluble anti-CD28-antibody without further cytokines, (termed “Th0”) and with IL-1 (10 ng/mL), IL-6 (10 ng/mL), IL23 (10 ng/mL), and TGB-beta (5 ng/mL), termed “Th17”) by qPCR. Significance was calculated using t-test. (C) miRNA levels in PBMC from natalizumab-treated patients (n = 9, left bar) were compared to two PBMC samples from patients with natalizumab-associated PML (each patient one bar). Due to small sample size statistical tests were not performed.
Table S1. Demographical and clinical data for the longitudinal study population.
Table S2. Data from the miRNA microarray of MS baseline samples versus healthy control samples: Downregulated miRNAs with P values <0.05 (corrected P value, limma P value) were sorted by P value.
Table S3. Data from the miRNA microarray of MS baseline samples versus healthy control samples: Upregulated miRNAs with P values <0.05 (corrected P value, limma P value) were sorted by P value.
Table S4. Data show computationally predicted targets of miR-20b (mirSVR scoring regression method by the http://www.microRNA.org information resource).
Table S5. Data show analysis of promoters of miRNA targets with regard to regulation by α4β1-receptor engagement investigated by the Transfac®-based P-Match tool (http://www.gene-regulation.com) for transcription factor binding site search by combining patterns and weight matrices.
References
- 1.Compston A, Coles A. Multiple sclerosis. Lancet. 2002;9313:1221–1231. doi: 10.1016/S0140-6736(02)08220-X. [DOI] [PubMed] [Google Scholar]
- 2.Hohlfeld R, Wekerle H. Autoimmune concepts of multiple sclerosis as a basis for selective immunotherapy: from pipe dreams to (therapeutic) pipelines. Proc Natl Acad Sci USA. 2004;101:14599–14606. doi: 10.1073/pnas.0404874101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 4.Engelhardt B, Ransohoff RM. The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol. 2005;9:485–495. doi: 10.1016/j.it.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 5.Yednock TA, Cannon C, Fritz LC, et al. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature. 1992;6364:63–66. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- 6.Baron JL. Surface expression of alpha 4 integrin by CD4 T cells is required for their entry into brain parenchyma. J Exp Med. 1993;1:57–68. doi: 10.1084/jem.177.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;9:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- 8.Phillips JT, Giovannoni G, Lublin FD, et al. Sustained improvement in Expanded Disability Status Scale as a new efficacy measure of neurological change in multiple sclerosis: treatment effects with natalizumab in patients with relapsing multiple sclerosis. Mult Scler. 2011;8:970–979. doi: 10.1177/1352458511399611. [DOI] [PubMed] [Google Scholar]
- 9.Weinstock-Guttman B, Galetta SL, Giovannoni G, et al. Additional efficacy endpoints from pivotal natalizumab trials in relapsing-remitting MS. J Neurol. 2012;5:898–905. doi: 10.1007/s00415-011-6275-7. [DOI] [PubMed] [Google Scholar]
- 10.Aktas O. Natalizumab in clinical practice: managing the risks, enjoying the benefits. J Neurol Neurosurg Psychiatry. 2014;85:1181. doi: 10.1136/jnnp-2013-307355. [DOI] [PubMed] [Google Scholar]
- 11.Stüve O, Bennett JL. Pharmacological properties, toxicology and scientific rationale for the use of natalizumab (Tysabri) in inflammatory diseases. CNS Drug Rev. 2007;1:79–95. doi: 10.1111/j.1527-3458.2007.00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Theien BE, Vanderlugt CL, Eagar TN, et al. Discordant effects of anti-VLA-4 treatment before and after onset of relapsing experimental autoimmune encephalomyelitis. J Clin Invest. 2001;8:995–1006. doi: 10.1172/JCI11717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Theien BE, Vanderlugt CL, Nickerson-Nutter C, et al. Differential effects of treatment with a small-molecule VLA-4 antagonist before and after onset of relapsing EAE. Blood. 2003;13:4464–4471. doi: 10.1182/blood-2003-03-0974. [DOI] [PubMed] [Google Scholar]
- 14.Mittelbrunn M, Molina A, Escribese MM, et al. VLA-4 integrin concentrates at the peripheral supramolecular activation complex of the immune synapse and drives T helper 1 responses. Proc Natl Acad Sci USA. 2004;30:11058–11063. doi: 10.1073/pnas.0307927101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niino M, Bodner C, Simard M, et al. Natalizumab effects on immune cell responses in multiple sclerosis. Ann Neurol. 2006;5:748–754. doi: 10.1002/ana.20859. [DOI] [PubMed] [Google Scholar]
- 16.Warnke C, Mausberg AK, Stettner M, et al. Natalizumab affects the T-cell receptor repertoire in patients with multiple sclerosis. Neurology. 2013;16:1400–1408. doi: 10.1212/WNL.0b013e3182a84101. [DOI] [PubMed] [Google Scholar]
- 17.Lindberg RLP, Achtnichts L, Hoffmann F, et al. Natalizumab alters transcriptional expression profiles of blood cell subpopulations of multiple sclerosis patients. J Neuroimmunol. 2008;194:153–164. doi: 10.1016/j.jneuroim.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Bartel D. MicroRNAs genomics, biogenesis, mechanism, and function. Cell. 2004;2:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 19.Baltimore D, Boldin MP, O’Connell RM, et al. MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008;8:839–845. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- 20.Otaegui D, Baranzini SE, Armañanzas R, et al. Differential micro RNA expression in PBMC from multiple sclerosis patients. PLoS One. 2009;7:e6309. doi: 10.1371/journal.pone.0006309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keller A, Leidinger P, Lange J, et al. Multiple sclerosis: microRNA expression profiles accurately differentiate patients with relapsing-remitting disease from healthy controls. PLoS One. 2009;10:e7440. doi: 10.1371/journal.pone.0007440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindberg RLP, Hoffmann F, Mehling M, et al. Altered expression of miR-17-5p in CD4+ lymphocytes of relapsing-remitting multiple sclerosis patients. Eur J Immunol. 2010;3:888–898. doi: 10.1002/eji.200940032. [DOI] [PubMed] [Google Scholar]
- 23.Du C, Liu C, Kang J, et al. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol. 2009;12:1252–1259. doi: 10.1038/ni.1798. [DOI] [PubMed] [Google Scholar]
- 24.Cox MB, Cairns MJ, Gandhi KS, et al. MicroRNAs miR-17 and miR-20a inhibit T cell activation genes and are under-expressed in MS whole blood. PLoS One. 2010;8:e12132. doi: 10.1371/journal.pone.0012132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Santis G, Ferracin M, Biondani A, et al. Altered miRNA expression in T regulatory cells in course of multiple sclerosis. J Neuroimmunol. 2010;1–2:165–171. doi: 10.1016/j.jneuroim.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 26.Waschbisch A, Atiya M, Linker RA, et al. Glatiramer acetate treatment normalizes deregulated microRNA expression in relapsing remitting multiple sclerosis. PLoS One. 2011;9:e24604. doi: 10.1371/journal.pone.0024604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guerau-de-Arellano M, Smith KM, Godlewski J, et al. Micro-RNA dysregulation in multiple sclerosis favours pro-inflammatory T-cell-mediated autoimmunity. Brain. 2011;12:3578–3589. doi: 10.1093/brain/awr262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gandhi R, Healy B, Gholipour T, et al. Circulating microRNAs as biomarkers for disease staging in multiple sclerosis. Ann Neurol. 2013;6:729–740. doi: 10.1002/ana.23880. [DOI] [PubMed] [Google Scholar]
- 29.Fenoglio C, Ridolfi E, Cantoni C, et al. Decreased circulating miRNA levels in patients with primary progressive multiple sclerosis. Mult Scler. 2013;14:1938–1942. doi: 10.1177/1352458513485654. [DOI] [PubMed] [Google Scholar]
- 30.Sondergaard HB, Hesse D, Krakauer M, et al. Differential microRNA expression in blood in multiple sclerosis. Mult Scler. 2013;14:1849–1857. doi: 10.1177/1352458513490542. [DOI] [PubMed] [Google Scholar]
- 31.Hecker M, Thamilarasan M, Koczan D, et al. MicroRNA expression changes during interferon-beta treatment in the peripheral blood of multiple sclerosis patients. Int J Mol Sci. 2013;8:16087–16110. doi: 10.3390/ijms140816087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keller A, Leidinger P, Steinmeyer F, et al. Comprehensive analysis of microRNA profiles in multiple sclerosis including next-generation sequencing. Mult Scler. 2014;3:295–303. doi: 10.1177/1352458513496343. [DOI] [PubMed] [Google Scholar]
- 33.Junker A, Krumbholz M, Eisele S, et al. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain. 2009;12:3342–3352. doi: 10.1093/brain/awp300. [DOI] [PubMed] [Google Scholar]
- 34.Lescher J, Paap F, Schultz V, et al. MicroRNA regulation in experimental autoimmune encephalomyelitis in mice and marmosets resembles regulation in human multiple sclerosis lesions. J Neuroimmunol. 2012;1–2:27–33. doi: 10.1016/j.jneuroim.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 35.Haghikia A, Haghikia A, Hellwig K, et al. Regulated microRNAs in the CSF of patients with multiple sclerosis: a case-control study. Neurology. 2012;22:2166–2170. doi: 10.1212/WNL.0b013e3182759621. [DOI] [PubMed] [Google Scholar]
- 36.Vorwerk S, Ganter K, Cheng Y, et al. Microfluidic-based enzymatic on-chip labeling of miRNAs. N Biotechnol. 2008;25:142–149. doi: 10.1016/j.nbt.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 37.Backes C, Keller A, Kuentzer J, et al. GeneTrail – advanced gene set enrichment analysis. Nucleic Acids Res. 2007;35:W186–W192. doi: 10.1093/nar/gkm323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chekmenev DS, Haid C, Kel AE. P-Match: transcription factor binding site search by combining patterns and weight matrices. Nucleic Acids Res. 2005;33(suppl 2):W432–W437. doi: 10.1093/nar/gki441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Betel D, Koppal A, Agius P, et al. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010;8:R90. doi: 10.1186/gb-2010-11-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huehnchen P, Prozorovski T, Klaissle P, et al. Modulation of adult hippocampal neurogenesis during myelin-directed autoimmune neuroinflammation. Glia. 2011;1:132–142. doi: 10.1002/glia.21082. [DOI] [PubMed] [Google Scholar]
- 41.Ventura A, Young AG, Winslow MM, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;5:875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baker D, Amor S. Publication guidelines for refereeing and reporting on animal use in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2012;1–2:78–83. doi: 10.1016/j.jneuroim.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 43.de Kouchkovsky D, Esensten JH, Rosenthal WL, et al. microRNA-17-92 regulates IL-10 production by regulatory T cells and control of experimental autoimmune encephalomyelitis. J Immunol. 2013;4:1594–1605. doi: 10.4049/jimmunol.1203567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith KM, Guerau-de-Arellano M, Costinean S, et al. miR-29ab1 deficiency identifies a negative feedback loop controlling Th1 bias that is dysregulated in multiple sclerosis. J Immunol. 2012;4:1567–1576. doi: 10.4049/jimmunol.1103171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cascio S, D’Andrea A, Ferla R, et al. miR-20b modulates VEGF expression by targeting HIF-1 alpha and STAT3 in MCF-7 breast cancer cells. J Cell Physiol. 2010;1:242–249. doi: 10.1002/jcp.22126. [DOI] [PubMed] [Google Scholar]
- 46.Lei Z, Li B, Yang Z, et al. Regulation of HIF-1alpha and VEGF by miR-20b tunes tumor cells to adapt to the alteration of oxygen concentration. PLoS One. 2009;10:e7629. doi: 10.1371/journal.pone.0007629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu E, Wang X, Zheng B, et al. miR-20b suppresses Th17 differentiation and the pathogenesis of experimental autoimmune encephalomyelitis by targeting RORγt and STAT3. J Immunol. 2014;192:5599–5609. doi: 10.4049/jimmunol.1303488. [DOI] [PubMed] [Google Scholar]
- 48.Sawcer S, Hellenthal G, Pirinen M, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;7359:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koch MW, Metz LM, Kovalchuk O. Epigenetic changes in patients with multiple sclerosis. Nat Rev Neurol. 2013;1:35–43. doi: 10.1038/nrneurol.2012.226. [DOI] [PubMed] [Google Scholar]
- 50.Krützfeldt J, Rajewsky N, Braich R, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;7068:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 51.Sievers C, Meira M, Hoffmann F, et al. Altered microRNA expression in B lymphocytes in multiple sclerosis: towards a better understanding of treatment effects. Clin Immunol. 2012;1:70–79. doi: 10.1016/j.clim.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 52.Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008;2:217–222. doi: 10.1016/j.cell.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Argaw AT, Gurfein BT, Zhang Y, et al. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci USA. 2009;6:1977–1982. doi: 10.1073/pnas.0808698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harris TJ, Grosso JF, Yen H, et al. Cutting edge: an in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007;7:4313–4317. doi: 10.4049/jimmunol.179.7.4313. [DOI] [PubMed] [Google Scholar]
- 55.Nguyen K, Sylvain NR, Bunnell SC. T cell costimulation via the integrin VLA-4 inhibits the actin-dependent centralization of signaling microclusters containing the adaptor SLP-76. Immunity. 2008;6:810–821. doi: 10.1016/j.immuni.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 56.Steinman L, Zamvil SS. Virtues and pitfalls of EAE for the development of therapies for multiple sclerosis. Trends Immunol. 2005;11:565–571. doi: 10.1016/j.it.2005.08.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplementary methods.
Figure S1. Disease response analysis. Shown are ratios of quantitative PCR expression levels of selected miRNAs from patients before (baseline) and during natalizumab therapy. A) Comparison of patients that improved in EDSS (n = 6) and that were stable or deteriorated (n = 11). B) Comparison of patients that improved or were stable (n = 15) and that deteriorated (n = 2). See also Table S1.
Figure S2. Magnetic bead isolated mouse CD4+ cells were incubated with anti-CD3-antibody (1 μg/mL), IL-6 (20 ng/mL), and TGF-beta (2 ng/mL) for 4 days in the presence of irradiated APC. Analysis was performed using BD Leukocyte Activation Cocktail, intracellular staining using BD Cytofix/Cytoperm buffers (Becton Dickinson), and subsequent FACS analysis. Pooled data from 4 experiments (each n = 5/per group).
Figure S3. (A) PBMC were isolated using a ficoll gradient and CD4+ T cells were sorted with magnetic beads (n = 9). miRNA expression was measured in CD4+ cells and CD4- PBMC by qPCR. Significance was calculated using t-test. (B) PBMC were isolated using a ficoll gradient and CD4+ T cells were sorted with magnetic beads (n = 9). miRNA expression was measured in freshly isolated CD4+ cells (“fresh), CD4+ cells that were activated by plate-bound anti-CD3-antibody and soluble anti-CD28-antibody without further cytokines, (termed “Th0”) and with IL-1 (10 ng/mL), IL-6 (10 ng/mL), IL23 (10 ng/mL), and TGB-beta (5 ng/mL), termed “Th17”) by qPCR. Significance was calculated using t-test. (C) miRNA levels in PBMC from natalizumab-treated patients (n = 9, left bar) were compared to two PBMC samples from patients with natalizumab-associated PML (each patient one bar). Due to small sample size statistical tests were not performed.
Table S1. Demographical and clinical data for the longitudinal study population.
Table S2. Data from the miRNA microarray of MS baseline samples versus healthy control samples: Downregulated miRNAs with P values <0.05 (corrected P value, limma P value) were sorted by P value.
Table S3. Data from the miRNA microarray of MS baseline samples versus healthy control samples: Upregulated miRNAs with P values <0.05 (corrected P value, limma P value) were sorted by P value.
Table S4. Data show computationally predicted targets of miR-20b (mirSVR scoring regression method by the http://www.microRNA.org information resource).
Table S5. Data show analysis of promoters of miRNA targets with regard to regulation by α4β1-receptor engagement investigated by the Transfac®-based P-Match tool (http://www.gene-regulation.com) for transcription factor binding site search by combining patterns and weight matrices.