

Abstract
Based upon evidence that the therapeutic properties of Cannabis preparations are not solely dependent upon the presence of Δ9-tetrahydrocannabinol (THC), pharmacological studies have been recently carried out with other plant cannabinoids (phytocannabinoids), particularly cannabidiol (CBD) and Δ9-tetrahydrocannabivarin (THCV). Results from some of these studies have fostered the view that CBD and THCV modulate the effects of THC via direct blockade of cannabinoid CB1 receptors, thus behaving like first-generation CB1 receptor inverse agonists, such as rimonabant. Here, we review in vitro and ex vivo mechanistic studies of CBD and THCV, and synthesize data from these studies in a meta-analysis. Synthesized data regarding mechanisms are then used to interpret results from recent pre-clinical animal studies and clinical trials. The evidence indicates that CBD and THCV are not rimonabant-like in their action and thus appear very unlikely to produce unwanted CNS effects. They exhibit markedly disparate pharmacological profiles particularly at CB1 receptors: CBD is a very low-affinity CB1 ligand that can nevertheless affect CB1 receptor activity in vivo in an indirect manner, while THCV is a high-affinity CB1 receptor ligand and potent antagonist in vitro and yet only occasionally produces effects in vivo resulting from CB1 receptor antagonism. THCV has also high affinity for CB2 receptors and signals as a partial agonist, differing from both CBD and rimonabant. These cannabinoids illustrate how in vitro mechanistic studies do not always predict in vivo pharmacology and underlie the necessity of testing compounds in vivo before drawing any conclusion on their functional activity at a given target.
Tables of Links
| TARGETS | |
|---|---|
| GPCRsa | MAGL, monoacylglycerol lipase |
| 5-HT1A receptors | PLA2 |
| Adenosine A2A receptors | Ion channelsc |
| CB1 receptors | TRPA1 |
| CB2 receptors | TRPM8 |
| Glutamate mGlu5 receptor | TRPV1 |
| GPR18 | TRPV2 |
| GPR55 | TRPV4 |
| Enzymesb | Transportersd |
| 5-lipoxygenase (5-LOX) | Adenosine transporter, (SLC29) |
| CYP1 | Glutamate transporter |
| CYP3A | Ligand-gated ion channelse |
| DAGLα, diacylglycerol lipase α | Glycine α3 receptors |
| DAGLβ, diacylglycerol lipase β | Nuclear hormone receptorsf |
| FAAH, fatty acid amide hydrolase | PARγ (NR1C3)"PPARγ (NR1C3) |
| iNOS, inducible NOS |
| LIGANDS | |
|---|---|
| 2-AG, 2-arachidonoylglycerol | |
| 5-HT | |
| AEA, anandamide | |
| AM251 | |
| CBD, cannabidiol | |
| CP55940 | |
| LTB4 | |
| NO | |
| Rimonabant, SR141716A | |
| THC, Δ9-tetrahydrocannabinol | |
| THCV, Δ9-tetrahydrocannabivarin | |
| WIN55212-2 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,c,d,e,fAlexander et al., 2013a,b,c,d,e,f,,,,,).
Introduction
Isolating and identifying the ‘primary active ingredient’ in Cannabis (the plant) and cannabis (the plant product) stymied chemists for over 150 years. Finally, Gaoni and Mechoulam (1964) isolated and defined Δ9-tetrahydrocannabinol (THC). THC and biosynthetically related and structurally similar plant cannabinoids are now called phytocannabinoids to distinguish them from structurally dissimilar but pharmacologically analogous endocannabinoids (see below) and synthetic cannabinoids (synthocannabinoids).
THC exerts most of its physiological actions via the endocannabinoid system. The endocannabinoid system consists of (i) GPCRs for THC, known as cannabinoid receptors; (ii) endogenous cannabinoid receptor ligands; and (iii) ligand metabolic enzymes. The salient homeostatic roles of the endocannabinoid system have been roughly portrayed as ‘relax, eat, sleep, forget, and protect’ (Di Marzo et al., 1998). When malfunctioning, the endocannabinoid system can contribute to pathological states (Russo, 2004; Di Marzo, 2008).
All vertebrate animals express at least two cannabinoid receptors. The CB1 receptor principally functions in the nervous system but is expressed in many cells throughout the body. CB2 receptors are primarily associated with cells governing immune function, such as splenocytes, macrophages, monocytes, microglia, and B- and T-cells. Recent evidence demonstrates the presence of CB2 receptors in other cells, often up-regulated under pathological conditions (reviewed in Pertwee et al., 2010).
The paradigmatic endocannabinoid ligands are N-arachidonylethanolamine (anandamide, AEA) and 2-arachidonoylglycerol (2-AG). One of AEA's key biosynthetic enzymes is N-acyl-phosphatidylethanolamine phospholipase D. The chief biosynthetic enzymes of 2-AG are two isoforms of diacylglycerol lipase: DAGLα and DAGLβ. The primary catabolic enzymes of AEA and 2-AG are fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) respectively. COX-2 can also catabolize AEA and 2-AG (Kozak et al., 2002).
Synthetic THC (dronabinol) became clinically available in the 1980s for indications including anorexia and weight loss in people with AIDS, and for nausea and vomiting associated with cancer chemotherapy. Off-label uses include migraine, multiple sclerosis, sleep disorders and chronic neuropathic pain. However, the therapeutic window of THC is narrowed by side effects. In clinical trials, dronabinol precipitated dysphoria, depersonalization, anxiety, panic reactions and paranoia (Cocchetto et al., 1981).
Psychological side effects occur more frequently with THC than with whole cannabis (Grinspoon and Bakalar, 1997). Only six years after Raphael Mechoulam successfully isolated THC, one of the authors of this article determined that THC did not act alone in cannabis (Gill et al., 1970). Other constituents in cannabis work in a paradoxical capacity of mitigating the side effects of THC, but improving the therapeutic activity of THC.
Cannabidiol (CBD) and Δ9-tetrahydrocannabivarin (THCV)
At last count, 108 phytocannabinoids have been characterized in various chemovars of the plant (Hanuš, 2008). The other phytocannabinoids of greatest clinical interest are CBD and THCV. THC and CBD are ‘sister’ molecules, biosynthesized by nearly identical enzymes in Cannabis – expressions of two alleles at a single gene locus (de Meijer et al., 2003). THC and CBD are C21 terpenophenols with pentyl alkyl tails, whereas THCV is a C19 propyl-tailed analogue of THC. Cannabis biosynthesizes these compounds as carboxylic acids, for example, THC-carboxylic acid (2-COOH-THC). When heated, dried or exposed to light, the parent compounds are decarboxylated.
Fundamentally, THC mimics AEA and 2-AG by acting as a partial agonist at CB1 and CB2 receptors (Mechoulam et al., 1998). But rather than simply substituting for AEA and 2-AG, cannabis and its many constituents work, in part, by ‘kick-starting’ the endocannabinoid system (McPartland and Guy, 2004). CBD, in particular, gained attention early in this regard. Several landmark studies published in the previous century have shown interactions between CBD and THC (see Box 2013a).
Box 1 Landmark 20th century studies regarding the effects of CBD upon THC.a.
Animal studies
CBD combined with isomeric tetrahydrocannabinols caused ‘synergistic hypnotic activity in the mouse’ – Loewe and Modell, 1941.
CBD inhibited THC effects on mouse catatonia, rat ambulation and rat aggression, but potentiated THC effects on mouse analgesia and rat rope climbing – Karniol and Carlini, 1973.
CBD decreased THC suppression of behaviour in rats and pigeons – Davis and Borgen, 1974.
CBD potentiated THC-induced changes in hepatic enzymes – Poddar et al., 1974.
CBD increased THC potentiation of hexobarbitone in rats – Fernandes et al., 1974.
CBD increased THC reduction of intestinal motility in mice – Anderson et al., 1974.
CBD reduced THC hypothermia and bradycardia – Borgen and Davis, 1974.
CBD blocked THC inhibition of pig brain monamine oxidase – Schurr and Livne, 1976.
CBD antagonized THC antinociceptive effects in mice – Welburn et al., 1976.
CBD prevented tonic and clonic convulsions induced by THC – Consroe et al., 1977.
CBD antagonized THC suppression of operant behaviour in monkeys – Brady and Balster, 1980.
CBD delayed THC discriminative effects – Zuardi et al., 1981.
CBD prolonged THC cue effects in rats – Hiltunen and Järbe, 1986.
CBD antagonized THC catalepsy in mice – Formukong et al., 1988a.
CBD increased THC analgesic activity and anti-erythema – Formukong et al., 1988b.
CBD prolonged and reduced the hydroxylation of THC – Bornheim et al., 1995, 1998.
Human clinical trials
CBD decreased anxiety caused by THC – Karniol et al., 1974.
CBD slightly increased time to onset, intensity and duration of THC intoxication – Hollister and Gillespie, 1975.
CBD attenuated THC euphoria – Dalton et al., 1976.
CBD reduced anxiety provoked by THC – Zuardi et al., 1982.
CBD improved sleep and decreased epilepsy – Cunha et al., 1980; Carlini and Cunha, 1981.
CBD decreased cortisol secretion and had sedative effects – Zuardi et al., 1993.
CBD provided antipsychotic benefits – Zuardi et al., 1995.
aFull citations appear in previous reviews (McPartland and Pruitt, 1999; McPartland and Russo, 2001; Russo and Guy, 2006).
Meta-analysis
Many narrative reviews have been written about THC, CBD and THCV, and cannabis as a ‘synergistic shotgun’ (McPartland and Pruitt, 1999; McPartland and Russo, 2001; Pertwee, 2004; 2005; 2008,,; Russo and Guy, 2006; Mechoulam et al., 2007; Zuardi, 2003; Izzo et al., 2009; Russo, 2011). CBD by itself has many therapeutic properties: anxiolytic, antidepressant, antipsychotic, anticonvulsant, anti-nausea, antioxidant, anti-inflammatory, anti-arthritic and antineoplastic.
The ability of CBD to antagonize THC has been the focus of many narrative reviews, as well as the studies listed in Box 2013a. This has led to the assumption that CBD exerts a direct pharmacodynamic blockade of THC. The pharmacological community tends to view CBD and THCV as negative modulators of CB1 receptor agonists. This view may be due to a superficial interpretation of the available pharmacological data. Hence, CBD and THCV would appear to mirror the mechanism of first-generation CB1 receptor inverse agonists known as cannABinoid ANTagonists (‘abants’), such as rimonabant, taranabant, otenabant and ibipinabant. Rimonabant was developed as an anti-obesity agent and marketed as an adjuvant to diet and exercise for weight loss in obese individuals. It was subsequently withdrawn from the market due to adverse psychiatric side effects (Bermudez-Silva et al., 2010).
We hypothesize that CBD and THCV may, under certain conditions, ‘antagonize’ the effects of THC via mechanisms other than direct CB1 receptor blockade. The purpose of this article is to review mechanistic studies (in vitro and ex vivo) of CBD and THCV. Rather than a narrative review, we will conduct a meta-analysis. The results of this synthesis of mechanistic studies will be applied to an emerging discussion: do CBD and THCV act as natural ‘abants’?
Methods
Meta-analysis uses an objective, transparent approach for research synthesis, with the aim of minimizing bias. A valid meta-analysis combines data from independent studies, identifies sources of heterogeneity among the studies and manages heterogeneity by placing defined limits upon data selection (Glass et al., 1981). This methodology usually focuses upon human clinical trials, but it can be applied to pre-clinical studies. We previously conducted a meta-analysis on CB1 receptor ligand binding affinity (McPartland et al., 2007). We follow the guidelines proposed by PRISMA, the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (Liberati et al., 2009). See Supporting Information Appendix S1.
Search strategy and study selection
We briefly describe the analysis here. For details regarding inclusion criteria, heterogeneity tests, subgroup analysis, quality assessment and publication bias, see Supporting Information Appendix S1.
PubMed (www.ncbi.nlm.nih.gov/pubmed/) was searched from 1988 (beginning with Devane et al., 1988) through December 2013, using the following Boolean search string: (cannabidiol OR tetrahydrocannabivarin) AND (animal OR affinity OR efficacy) NOT (behavioral OR behavioural). References identified by the search strategy were scanned for inclusion and exclusion criteria by three independent reviewers who resolved disagreements by consensus.
Inclusion criteria included studies of CBD or THCV, their carboxylic acids (CBD-acid, THCV-acid), as well as CBD- or THCV-enriched plant extracts (‘botanical drug substances,’ CBD-BDS, THCV-BDS). Included studies reported in vitro or ex vivo mechanistic data (receptor affinity and efficacy assays), detailed in Supporting Information Appendix S1.
The rather broad search strategy retrieved many articles that were subsequently excluded as irrelevant. Excluded topics included (i) review articles or publications with duplicated data; (ii) animal studies or in vivo studies without mechanisms or an identified molecular target; (iii) studies of synthetic analogues, or metabolites of CBD or THCV; (iv) human clinical trials lacking mechanistic analysis; urinary metabolites of CBD and THCV and their use in drug testing; characterizations of cannabinoid drug delivery systems; and (v) other irrelevant topics (see Supporting Information Appendix S1 for elaboration).
Articles meeting inclusion and exclusion criteria were screened for supporting citations, and antecedent sources were retrieved. The search also included unpublished data communicated at research conferences, upon approval by the authors of the data. Lastly, we contacted world experts and asked them to contribute unpublished data (see Acknowledgements section).
Data extraction and synthesis
Extracted data included ligand (CBD or THCV), assay type, animal species, reported means, sample variance, sample size and methodological factors. Methodological factors (covariates) were extracted for use in subgroup analyses to test whether they exerted heterogeneous effects upon pooled means. Methodological factors were pre-specified, chosen in advance by a priori hypotheses based upon recognized methodological diversity among studies, and not undertaken after the results of the studies had been compiled (post hoc analyses).
Data were synthesized qualitatively (e.g. categorical data) or quantitatively [e.g. the continuous variables for affinity (Ki) and efficacy (EMAX)]. A quantitatively synthesized result is reported as a pooled mean ± SEM. Optimal quantitative synthesis would employ a weighted pooled mean, which adjusts each study's mean divided by its SEM, because larger studies with less variance should carry more ‘weight’ in a meta-analysis. Unfortunately, many studies omitted variance data or sample size.
To determine whether pooling was statistically appropriate, the coefficient of variation (CV) was determined for each pooled mean. The CV measures data dispersion of a probability distribution, defined simply as the ratio of the SD to the mean (Reed et al., 2002). We applied the Cochrane ‘skew test’ to the CV (Higgins and Green, 2005), where CV ≥ 1 (i.e. SD ≥ mean) identifies a skewed mean with excessive heterogeneity. Skewed mean was submitted to Grubb's test (www.graphpad.com/quickcalcs/). Data identified as significant outliers (P < 0.05) were reported in Supporting Information Appendix S2 and S3 and withdrawn from synthesis. The CV-skew test could not be used for sample sizes of n ≤ 3. In those cases, we used simple pooled means.
Results
The search algorithm identified 431 potentially relevant articles. Many of these publications were review articles or concerned topics irrelevant to this review. In addition to 174 articles that met the predefined selection criteria for relevance, we included 28 studies obtained from citation tracking or unpublished studies. See Figure 1 for a flowchart.
Figure 1.
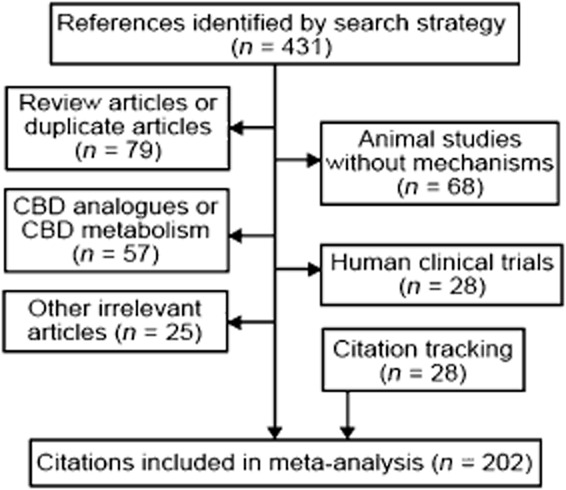
Flow diagram of article selection for meta-analysis.
The quality of statistical reporting has steadily improved between 1984 and 2013. For example, early studies that measured the Ki of CBD reported variance as SD, and sometimes omitted variance data or sample sizes (Supporting Information Appendix S2). Later studies reported variance as SEM. However, Ki values are not symmetric around a mean, and the best way to report non-parametric variance is the use of 95% confidence limits/intervals rather than SEM.
CBD has many molecular targets; we grouped them in three categories. The first category addresses the effects of CBD and THCV at CB1 receptors – directly and indirectly. The next category includes what some researchers consider an ‘expanded’ endocannabinoid system – other GPCRs (CB2, GPR55, GPR18), and transient receptor potential ion channels (e.g. TRPV1, TRPV2, TRPA1, TRPM8). The third category includes receptors and ligand enzymes beyond the expanded system, as well as the arachidonic acid (AA) cascade, nitric oxide signalling, cytokines, redox signalling and mechanisms involved in apoptosis.
CBD at CB1 receptors: direct and indirect interactions
The pooled mean affinity of CBD at CB1 receptors is Ki = 3245 ± 803 nM. The pooled mean was calculated from 1 human, 3 mice and 11 rat studies (Supporting Information Appendix S2). Species differences were not statistically different, including the human study (Ki = 1510 ± 100 nM). Several studies omitted quality measures such as statistical data (variance and/or sample size). Subgroup analysis of the five methodological factors produced surprising results. Only one factor – the use of crude brain homogenates – proved to be statistically relevant (Supporting Information Appendix S2). None of the other factors gave rise to data heterogeneity – species differences, class of tritiated ligand (agonist vs. antagonist) or even the use of non-specific probes (e.g. [3H]TMA-THC). One methodological factor could not be assessed – no studies used PMSF.
Eight studies tested CBD efficacy at CB1 receptors, assayed with forskolin-stimulated adenylate cyclase (FsAC) or [35S]GTPγS (Supporting Information Appendix S2). Six of these studies reported no measurable response or inconsistent dose–response curves hovering near zero. One study reported slight agonism, and one study reported slightly inverse agonism, both at high concentrations (≥10 μM).
Surprisingly, in some mechanistic studies, the effects of CBD could be reversed by CB1 receptor inverse agonists, or were absent in CB1 receptor knockout mice (n = 10 studies; Supporting Information Appendix S2). This suggests that CBD may exert ‘indirect agonism’ at CB1 receptors – either augmenting CB1 constitutional activity or augmenting endocannabinoid tone. Regarding the first mechanism, Howlett et al. (1989) presented thermodynamic data that suggest that CBD may alter CB1 receptor activity by increasing membrane fluidity. Sagredo et al. (2011) showed that Sativex® (a mix of THC and CBD; GW Pharmaceuticals, Salisbury, UK) can up-regulate CB1 receptor gene expression in a rat model of Huntington's disease. The second mechanism – augmenting endocannabinoid tone – is supported by studies showing that CBD or CBD-BDS inhibits AEA hydrolysis by FAAH (n = 5 studies; Supporting Information Appendix S2), with a pooled mean IC50 = 19.8 ± 4.77 μM. Four rodent studies show that CBD inhibits the putative AEA transporter, with a pooled mean IC50 = 10.2 ± 3.03 μM. Two studies reported CBD increasing 2-AG levels, 33 or 260% (Supporting Information Appendix S2).
CBD may affect the pharmacokinetics of THC when the two are co-administered (n = 17 studies; Supporting Information Appendix S2). One of these pharmacokinetic studies predates the isolation of pure THC – Loewe and Modell (1941) stated that CBD ‘synergized’ hypnotic action in mice induced by isomeric tetrahydrocannabinols. CBD may impair THC hydrolysis by CYP450 enzymes. The results of CYP studies vary due to species differences, timing (CBD pre-administration vs. co-administration) and specific CYP isoenzymes. Recent human studies show no pharmacokinetic interaction between THC and CBD at clinically relevant dosing (Supporting Information Appendix S2).
The effect of CBD on CYPs is important because these enzymes metabolize THC into 11-OH-THC. This metabolite may be up to four times more psychoactive than THC, according to a rat discriminative study (Browne and Weissman, 1981). The affinity and efficacy of 11-OH-THC at CB1 receptors has not been measured, although the affinity of 11-OH-Δ8-THC was Ki = 25.8 nM, displacing [3H]HU-243 from rCB1 COS cells (Rhee et al., 1997). In the same assay, Δ9-THC exhibited Ki = 80.3 nM (Rhee et al., 1997) or Ki = 39.5 nM (Bayewitch et al., 1996). The significance of CBD modulating the metabolism of THC into 11-OH-THC is an important variable that remains to be explored.
CBD may antagonize cannabinoid-induced effects. Six in vitro studies demonstrate that CBD can antagonize CP55,940- or WIN55212-2-induced efficacy (Supporting Information Appendix S2), with a pooled mean KB = 88.5 ± 18.46 nM. This unexpected KB is 37-fold more potent than the Ki of CBD in binding assays. This suggests an indirect mechanism, that is, mediated by (an)other target(s). The KB of CBD is 147-fold higher (less potent) than the KB of rimonabant, when this inverse agonist was run in parallel (n = 3 studies; Supporting Information Appendix S2). Note that this antagonism can be visualized by juxtaposing two log concentration–response curves; for example, the curve of CP55,940 for stimulation of [35S]GTPγS binding to CB1 receptors, compared to the curve of CP55,940 for such stimulation in the presence of CBD. A competitive antagonist would produce a parallel rightward shift in the curve of CP55,940. However, Petitet et al. (1998) found CBD to produce a parallel shift in the curve of CP55,940 that was downward rather than rightward in its direction, an effect that a competitive antagonist is not expected to produce.
CBD at targets in the ‘expanded endocannabinoid system’
The ability of CBD to antagonize a cannabinoid agonist in an efficacy assay may be confounded by the impact of CBD on other receptors or enzymes present in the assay. In Table 1, we summarize studies regarding CBD affinity and efficacy at other metabotropic receptors and ion channels in the ‘expanded endocannabinoid system.’
Table 1.
Affinity and efficacy of CBD at molecular targets in the ‘expanded endocannabinoid system,’ data pooled from Supporting Information Appendix S2
| Target | Assaya | Pooled means, number of studiesb |
|---|---|---|
| CB2 receptor | A: versus [3H]CP55,940 binding; human CB2 transfected cell cultures, or rat RBL-2H3 leukemia cells, centrifuged membranes | Ki = 3612 ± 1382 nM, n = 6 |
| CB2 receptor | E: [35S]GTPγS binding; human CB2-CHO cells | EMAX = −15% below basal at 10 μM EC50 = 503 ± 2080 nM; n = 1 |
| CB2 receptor | E: antagonism of CP55,940-induced [35S]GTPγS binding; human CB2-CHO cells | KB = 65 ± 54.1 nM, n = 1 |
| GPR55 | E: antagonism of agonist-induced signalling; human cell cultures or CB2-CHO cells | IC50 = 433 ± 42.6 nM, n = 3 |
| TRPV1 channels | A: versus [3H]-resiniferatoxin binding; human TRPV1-HEK293 cell membranes | Ki = 3600 ± 200 (SD) nM, n = 1 |
| TRPV1 channels | E: [Ca2+]i elevation in human TRPV1-HEK293 cells | EMAX 53.4 ± 5.03%; n = 4 EC50 = 1900 ± 802 nM |
| TRPA1 channels | E: [Ca2+]i elevation in rat TRPA1-HEK293 cell membranes | EMAX 98.6 ± 10.39% EC50 = 100 ± 10 nM; n = 3 |
| TRPV2 channels | E: [Ca2+]i elevation in rat or human TRPV2-HEK293 cell membranes | EMAX 100.2 ± 34.50% EC50 = 12.2 ± 9.77 μM; n = 3 |
| TRPM8 channels | E: [Ca2+]i elevation in rat TRPM8-HEK293 cell membranes | Signals as functional antagonist EC50 = 70.0 ± 14.1 nM; n = 2 |
aA, affinity; E, efficacy. Other abbreviations as defined in the manuscript.
bNumber of studies that met the CV-Cochrane ‘skew test’ (see Supporting Information Appendix S2).
CBD shows low affinity at CB2 receptors (Table 1). Its efficacy at CB2 receptors suggests weak inverse agonism at concentrations that may not be pharmacologically relevant. However, CBD antagonizes CP55,940 signalling at CB2 receptors with a KB potency not commensurate with its Ki. This suggests again an indirect mechanism of action. At GPR18, two studies suggest that CBD acts as a partial agonist and antagonizes THC (Supporting Information Appendix S2). The direct affinity and efficacy of CBD at GPR55 has not been measured, but it can antagonize GPR55 agonists (Supporting Information Appendix S2).
Table 1 highlights CBD signalling at transient receptor potential (TRP) channels, which have been characterized as ‘ionotropic cannabinoid receptors’ (Di Marzo et al., 2002). We find a species difference at TRPV1 channels and the pooled mean EMAX at human TRPV1 is 53.4% ± 5.03 (Table 1), whereas a single study of rat TRPV1 channels reports an EMAX of 21% (Qin et al., 2008). At TRPA1 channels, the pooled mean EMAX of CBD (Table 1) is considerably less than that of CBD-BDS (163.6 ± 11.9%; De Petrocellis et al., 2011). This suggests that terpenoids and other plant substances in the extract may enhance CBD activity at TRPA1 channels, either pharmacokinetically or pharmacodynamically. The same study showed that CBD-BDS also showed slightly greater efficacy than pure CBD at TRPV1 channels (Supporting Information Appendix S2). CBD acts as an antagonist at TRPM8, unlike its agonist activity at other TRP channels (Table 1).
CBD at other molecular targets
CBD is a promiscuous compound with activity at multiple targets. Promiscuity is governed by ligand structure, and cannabinoids exhibit the characteristics of promiscuous ligands: molecular mass >400 g·mol−1, and partition coefficient (CLogP) scores between 2 and 7 (Hopkins, 2008).
CBD inhibits adenosine uptake (pooled IC50 = 122 nM minus one outlier, n = 3 studies; Supporting Information Appendix S2) and this mechanism is likely to exert indirect agonism at adenosine receptors. Consistent with this, the beneficial effects of CBD in animal models of inflammation are blocked by adenosine A2A receptor antagonists (n = 8 studies). CBD may also regulate 5-HT levels, although six studies report disparate results. CBD has slight affinity for the 5-HT1A receptor (Ki ∼ 16 μM; Supporting Information Appendix S2), although its efficacy at 5-HT1A receptors is inconsistent. Several beneficial effects of CBD in vivo are blocked by 5-HT1A receptor antagonists (Supporting Information Appendix S2).
CBD exerts a positive allosteric modulation of α3 glycine receptors (pooled EC50 = 11.0 μM, n = 3 studies; Supporting Information Appendix S2). NMR analysis revealed a direct interaction between CBD and S296 in the third transmembrane domain of α3 glycine receptors. The cannabinoid-induced analgesic effect was absent in mice lacking the α3 glycine receptors (Xiong et al., 2011). CBD can go nuclear, as it affects PPARγ receptors (IC50 = 5 μM; Supporting Information Appendix S2), and its beneficial effects are blocked by PPARγ antagonists (n = 5 studies). A dozen studies indicate that CBD allosterically modulates other receptors: α1-adrenoceptors, dopamine D2, GABAA, μ- and δ-opioid receptors – some positively, some negatively, none with great efficacy (Supporting Information Appendix S2). CBD modulates intracellular calcium levels, via T-type and L-type voltage-regulated Ca2+ channels and mitochondrial Na+/Ca2+ exchange (n = 7 studies).
The effects on the AA cascade are complex. Three studies indicate that CBD mobilizes AA by stimulating PLA2 activity. The direction that AA goes from there is uncertain. CBD inhibits the metabolism of AA to LTB4 by 5-lipoxygenase – pooled IC50 = 3.1 ± 0.75 μM, n = 4 studies, although a fifth study reported no effect up to 80 μM (Supporting Information Appendix S2). Contradictory studies report CBD blocking the metabolism of AA to PGE1 or TxB2 (via COX-1) or PGE2 (via COX-2). Early studies did not identify which COX isoenzyme they tested. Recent studies indicate that CBD attenuates serum PGE2 in animal models of chronic pain, but this may or may not correlate with COX-2 protein expression (Supporting Information Appendix S2).
CBD clearly dampens NO production in animal models of acute and chronic inflammation – as measured by a reduction in nitrite levels or inducible NOS (iNOS) protein expression (n = 15 studies). A dozen studies show that CBD inhibits the expression of inflammatory cytokines and transcription factors (IL-1β, IL-2, IL-6, IL-8, TNF-α, IFN-γ, CCL3, CCL4, NF-κB).
CBD is a potent antioxidant; it reduces reactive oxygen species (ROS) induced by a variety of toxins and tissue insults (Supporting Information Appendix S2). However, one study suggests the opposite – that CBD hydroxyquinone, formed during hepatic microsomal metabolism of CBD, is capable of generating ROS and inducing cytotoxicity. Indeed, one mechanism by which CBD induces tumour cell apoptosis appears to be ROS generation (Supporting Information Appendix S2). In general, it appears that CBD can induce ROS in cancer cells and reduce ROS in healthy cells stimulated by agents that induce ROS formation. In fact, even in cancer cells, CBD inhibits ROS formation induced by H2O2 (Ligresti et al., 2006).
THCV at CB1 and CB2
Affinity studies show that THCV binds to CB1 receptors with significant potency. There may be species differences – at human CB1 receptors transfected into CHO cells, mean Ki = 5.47 ± 4.02 nM (n = 2 studies); at mouse brain membranes, mean Ki = 61.0 ± 14.40 nM (n = 2); and at rat brain membranes (n = 1), mean Ki = 286 ± 43 nM. The rat study used [3H]SR141716A, whereas others used [3H]CP55,940 (Supporting Information Appendix S3).
Four studies tested the efficacy of THCV at CB1 receptors. THCV did not inhibit or stimulate [35S]GTPγS binding to mouse whole brain membranes (n = 3 studies) or to rat membranes (cortical, cerebellar or piriform cortical membranes) at concentrations up to 10 μM. This suggests that THCV targets the CB1 receptor as a neutral antagonist rather than as an antagonist/inverse agonist. The single study that tested FsAC in human CB1 receptors in CHO cells produced signs of inverse agonism at concentrations of 10, 100 and 1000 nM (Supporting Information Appendix S3).
In the presence of other cannabinoids, THCV binds to CB1, in vitro, in a manner that gives rise to competitive antagonism rather than to agonism. More specifically, THCV produces significant parallel rightward shifts in the log concentration–response curves of established CB1 receptor agonists such as CP55,940 or WIN55212-2, in [35S]GTPγS binding to mouse whole brain membranes, and in the inhibition of electrically evoked contractions of mouse isolated vasa deferentia. Pooling five studies (Supporting Information Appendix S3) results in a KB = 64.2 ± 14.14 nM. Thus, the KB of THCV is in the same range of concentrations as its Ki.
The five studies hint at ‘probe dependence’. That is, THCV has greater potency when antagonizing WIN55212-2 than when antagonizing CP55,940, although the difference falls short of statistical significance (Supporting Information Appendix S3). Two studies of THCV as an antagonist in the vas deferens assay also hint at probe dependence – the potency of THCV is not the same for all CB1 receptor agonists. In rank order: AEA (KB = 1.2 nM), methAEA (KB = 2.6 nM), WIN55212-2 (KB = 3.15 nM), CP55,940 (KB = 10.3 nM), THC (KB = 96.7 nM) (Supporting Information Appendix S3). THCV also reverses WIN55212-2-induced decreases of miniature inhibitory postsynaptic current frequency at mouse cerebellar interneuron-Purkinje cell synapses, albeit with much less potency than the CB1 receptor inverse agonist AM251, which was tested in parallel (Supporting Information Appendix S3).
At human CB2 receptors transfected into CHO cells, THCV has significant affinity, with pooled mean Ki = 124.7 ± 64.55 nM (n = 3 studies; Supporting Information Appendix S3). THCV acts as a partial agonist at these receptors as measured by [35S]GTPγS binding or FsAC assays, pooled EMAX = 56.7 from basal at 1–10 μM, EC50 = 74.2 ± 34.4 nM (n = 3 studies; Supporting Information Appendix S3).
THCV may exert ‘indirect agonism’ at CB1 and CB2 receptors by augmenting endocannabinoid tone. It does this by inhibiting the putative AEA transporter, inhibiting FAAH activity and inhibiting MAGL activity, albeit at 25–100 μM (n = 3 studies; Supporting Information Appendix S3). Whether or not THCV exerts ‘indirect agonism’ to an extent that can overcome its surmountable CB1 receptor antagonism requires further experiments.
THCV at other targets
Unlike CBD, much less evidence exists for non-cannabinoid receptor-mediated effects of THCV, and this evidence has to do mostly with the capability of THCV to interact with ‘thermo-TRP’ channels. One study demonstrates that THCV acts as an agonist at rat TRPA1, human TRPV1 and rat TRPV2-4 channels and a potent antagonist at rat TRPM8 channels (Supporting Information Appendix S3). Data regarding its effects at GPR55 are controversial, and the potentiation of 5-HT1A receptors was observed only at the concentration of 100 nM (Supporting Information Appendix S3).
Discussion
Our previous meta-analysis of receptor affinity of several synthetic and natural cannabinoid ligands (McPartland et al., 2007) involved more studies than this current analysis of CBD and THCV. Subgroup analysis suggests that methodological nuances employed in the larger group of studies (such as PMSF usage) were absent in studies of CBD and THCV.
The low affinity of CBD at CB1 and CB2 receptors dampened the signal-to-noise ratio to such an extent that some nuances were rendered unnecessary – subgroup analysis showed no differences arising from methodological factors that proved important in our previous meta-analysis, such as species differences and the quality of tritiated ligand. THCV, on the contrary, with its moderate to high potency in displacement assays for CB1 and CB2 receptors, hints at species differences in its affinity at CB1 receptors, as well as ‘probe dependence’ in its ability to antagonize other cannabinoids.
Pre-clinical animal studies demonstrated that CBD expands the therapeutic window of THC. CBD may accomplish this by enhancing the efficacy of THC as well as by mitigating the ‘central’ side effects of THC. These effects have been known for some time (Box 2013a). In particular, our discussion below indicates that CBD enhances the efficacy of THC in preclinical models of multiple sclerosis, muscle spasticity, epilepsy, chronic pain and inflammation, anorexia and nausea, diabetes and metabolic syndrome. CBD mitigates the side effects of THC in animal models of psychosis, anxiety and depression-anhedonia.
CBD at CB1 receptors: direct and indirect interactions
Let us return to our original question: does CBD act as a natural ‘abant'? Data clearly show that CBD does not act directly at CB1 receptors. The affinity of CBD at CB1 receptors (Ki = 3245 nM) is at least three orders of magnitude less than that of rimonabant (Kd = 1.0 nM in rat, 2.9 nM in human; McPartland et al., 2007). Several efficacy studies made direct comparisons of [35S]GTPγS binding: CBD showed no measurable activity, whereas rimonabant elicited strong inverse agonism with a mean IMAX = −35.5% (Supporting Information Appendix S2).
The inactivity of CBD in vitro is confirmed by in vivo studies. CBD does not elicit the classic CB1-mediated tetrad of hypolocomotion, analgesia, catalepsy and hypothermia (Long et al., 2010). In drug discrimination studies, CBD failed to generalize with THC (Järbe et al., 1977; Zuardi et al., 1981) and did not alter the discriminative stimulus effects of THC (Vann et al., 2008). Its lack of cannabimimetic effects is well known in humans (Hollister, 1973).
CBD exerts CB1 receptor agonist-like activity in some in vitro functional assays at high concentrations (>10 μM), which can be reversed by CB1 receptor inverse agonists, and is absent in CB1 receptor knockouts. This probably occurs by CBD augmenting endocannabinoid tone. Pooled rodent studies show moderate inhibition of FAAH and the putative AEA transporter. CBD may also augment endocannabinoid tone by activating PLA2, thus mobilizing AA – the feedstock for AEA and 2-AG synthesis.
On the contrary, CBD inhibits adenosine uptake, thereby acting as an indirect agonist at A2A receptors. Agonism of these receptors in post-synaptic cells prevents glutamate mGlu5 receptor-mediated release of AEA and 2-AG through A2A/mGlu5 heteromers (Lerner et al., 2010). These opposing effects – CBD augmenting endocannabinoid tone and augmenting adenosine tone – often strike a balance in favour of the former: Robust rodent studies indicate that CBD augments endocannabinoid tone (e.g. Campos et al., 2013,2013). One clinical study showed that AEA levels increased in schizophrenic patients given CBD 800 mg·day−1 (Leweke et al., 2012). Future in vitro studies of CBD affinity and efficacy in the presence or absence of PMSF or MAFP would be instructive (these are FAAH inhibitors that prevent the synthesis of AEA). Knocking down tonic adenosine A2A receptor signalling may also shed light on this complex situation (Savinainen et al., 2003).
Our second paradoxical finding is the in vitro capacity of CBD to ‘functionally antagonize’ cannabinoid-induced activity at CB1 receptors. However, the KB of CBD (88.5 ± 18.46 nM) is far higher (implying less potency) than that of rimonabant (mean KB of 0.6 ± 0.41 nM when tested in parallel in the same studies; Supporting Information Appendix S2). Indeed, CBD in vivo mostly behaved in a different way from that of rimonabant. For example, CBD produced no effect on food intake, energy expenditure or insulin sensitivity in obese mice (Cawthorne et al., 2008). Its anxiolytic, antidepressant and anti-nausea effects are all opposite to those reported for rimonabant (Pertwee, 2008) (Box 2013b).
Box 2 Landmark 21st century in vivo studies of CBD functional antagonism of THC.
Animal studies
CBD antagonized THC-induced spatial memory – Fadda et al., 2004.
CBD reversed THC-induced conditioned place aversion – Vann et al., 2008.
CBD reversed THC-induced decrease in social interaction – Malone et al., 2009.
CBD increased hippocampal cell survival and neurogenesis, whereas THC has the opposite effect; the CBD response is absent in CB1−/− knockout mice – Wolf et al., 2010.
Human clinical trials and epidemiology studies
CBD reduced THC intoxication and impairment in binocular depth perception (a model of psychosis) – Leweke et al., 2000.
Sativexa compared to THC alone reduced adverse effects in patients with multiple sclerosis – Wade et al., 2003; Zijicek et al., 2003.
CBD counteracted THC somnolence and morning-after memory deficits – Nicholson et al., 2004.
High-THC cannabis with higher dose of CBD caused less anxiety than high-THC cannabis with lower dose of CBD – Ilan et al., 2005.
No difference in appetite and quality of life (QOL) scores between cannabis extract and THC alone – Strasser et al., 2006.
Increased CBD-to-THC ratios in chronic cannabis users inversely correlated with expression of psychotic symptoms – Morgan and Curran, 2008.
CBD reduced anxiety, skin conductance response and amygdala activity – the opposite of THC effects – Fusar-Poli et al., 2009.
CBD reduced ‘psychotic scores’ of THC – Bhattacharyya et al., 2010.
CBD attenuated the appetitive effects of THC – Morgan et al., 2010a.
Increased CBD-to-THC ratios in chronic cannabis users correlated with a reduction of cognitive and memory deficits – Morgan et al., 2010a.
Increased CBD-to-THC ratios in chronic cannabis users inversely correlated with liking for drug-related stimuli including food – Morgan et al., 2010b.
Increased CBD-to-THC ratio is associated with lower degrees of negative psychiatric symptoms – Schubart et al., 2011.
No difference in side effect profile between cannabis extract and THC alone – Karschner et al., 2011b.
Increased CBD-to-THC ratios in chronic cannabis users inversely correlated with volume loss in the hippocampus – Demirakca et al., 2011.
CBD inhibited THC-elicited paranoid symptoms and hippocampal-dependent memory impairment – Englund et al., 2013.
aSativex® contains THC and CBD in a 1:1 ratio, with minor cannabinoids (5–6%), terpenoids (6–7%), sterols (6%), triglycerides, alkanes, squalene, tocopherol, carotenoids and other minor components derived from the plant material (Guy and Stott,2005).
Non-CB1 receptor mechanisms of CBD antagonism of THC
Earlier we proposed that the functional antagonism of THC by CBD may be mediated by a non-CB1 receptor mechanism of action. Pharmacodynamic antagonism arises when one drug diminishes the effect of another drug by targeting different receptors or enzymes. For example, dry mouth caused by a sympathomimetic drug is antagonized by a cholinergic drug.
We propose several non-CB1 receptor mechanisms of action:
CBD augments AEA levels. Both AEA and CBD have affinity for TRPV1 channels and signal as agonists (Table 1). Pre-synaptic TRPV1 channel activation enhances glutamate release in the spinal cord and brain. This may counteract or antagonize the inhibitory action of pre-synaptic CB1 receptors co-localizing on glutamatergic neurons (Campos and Guimaraes, 2009; Di Marzo, 2010).
CBD inhibits adenosine uptake and therefore acts as an indirect agonist at adenosine receptors. Functionally CBD does the opposite of caffeine, an adenosine receptor antagonist (El Yacoubi et al., 2000). There are four adenosine receptor subtypes; cannabinoid research has focused on A2A receptors (Table 1). Pre-synaptic A2A receptors form heteromers with CB1 receptors on glutamatergic neurons, and A2A receptor agonism inhibits CB1 receptor-mediated effects in rat striatum (Martire et al., 2011). Post-synaptic A2A receptor signalling is a different story, as we mentioned earlier. On the contrary, some effects of CBD are reversed by the A1 receptor-selective antagonist DPCPX (Castillo et al., 2010; Maione et al., 2011). Possible indirect agonism of A1 receptors would place CBD in the company of benzodiazepines, which target A1, as well as GABAA, receptors and, like benzodiazepines, might also strengthen, and not only oppose, CBD indirect activation of CB1 receptors in a tissue-specific manner (see Maione et al., 2011 for an example).
CBD acts as a direct and indirect agonist at 5-HT1A receptors and facilitates 5-HT1A-mediated anxiolytic effects (e.g. Campos and Guimaraes, 2008; Gomes et al., 2011; Campos et al., 2013,2013). CBD reduces WIN55,212-2-induced catalepsy via 5-HT1A receptors (Gomes et al., 2013). It is tempting to attribute this again to CBD indirect agonism via AEA – Palazzo et al. (2006) demonstrated that sciatic nerve chronic constriction injury (CCI) elevates AEA in the dorsal raphe, resulting in 5-HT-mediated hyperactivity. The study also showed that CCI increased extracellular 5-HT levels, a mechanism likely to be shared by CBD administration, which suppresses enzymatic depletion of tryptophan, the precursor of 5-HT (Supporting Information Appendix S2).
Non-CB1 receptor mechanisms of CBD potentiation of THC
CBD potentiates some effects of THC in an additive or synergistic fashion. Williamson (2001) has reviewed the mathematical definitions of synergy. Wilkinson et al. (2005) provided examples of synergy within polypharmaceutical cannabis extracts. CBD may potentiate the behavioural effects of THC via pharmacokinetic mechanisms, for example, increasing the area under the curve of THC in blood and brain (Klein et al., 2011). Pharmacodynamic interactions arise when CBD and THC act at separate but interrelated receptor sites. Landmark studies demonstrating the potentiation of THC by CBD or by CBD-rich cannabis extracts are summarized in Box 2013a and Box 2013c.
Box 3 Landmark 21st century studies of CBD potentiating the effects of THC.
Animal studies
CBD potentiated THC antinociception – Varvel et al., 2006.
CBD enhanced THC tetrad effects – Hayakawa et al., 2008.
CBD turned an ineffective THC dose into an effective one in colitis – Jamontt et al., 2010.
CBD altered THC pharmacokinetics and augments some THC behavioural effects – Klein et al., 2011.
Sativex compared to THC alone enhanced antinociception in a rat model of neuropathic pain – Comelli et al., 2008.
Human clinical trials and epidemiology studies
CBD plus THC imparted synergistic inhibition of human glioblastoma cancer cell growth and apoptosis – Marcu et al., 2010.
Sativexa compared to THC alone provides greater pain relief and improvement in sleep – Notcutt et al., 2004.
Sativexa compared to THC extract reduced cancer-related pain – Johnson et al., 2010.
Sativexa compared to THC alone reduced abnormalities in psychomotor performance associated with schizophrenia – Roser et al., 2009.
aSativex® contains THC and CBD in a 1:1 ratio, with minor cannabinoids (5–6%), terpenoids (6–7%), sterols (6%), triglycerides, alkanes, squalene, tocopherol, carotenoids and other minor components derived from the plant material (Guy and Stott, 2005).
We propose eight non-CB1 mechanism of action by which CBD may potentiate THC:
CBD inhibition of FAAH and consequential increase in AEA may synergize with THC CB1 receptor agonism in peripheral injury: AEA suppresses pain through a peripheral mechanism (Clapper et al., 2010), whereas THC works through central CB1 receptor-mediated mechanisms. CBD, like cannabichromene, another cannabinoid capable of inhibiting the putative AEA transporter, when injected into the periaqueductal grey area increased 2-AG levels by 2.6-fold and elicited antinociception in the tail-flick test. The antinociceptive effect was blunted by antagonists of CB1, adenosine A1 receptors and TRPA1, but not TRPV1 channels (Maione et al., 2011).
CBD reduces peripheral hyperalgesia via TRPV1 channels (Costa et al., 2004). This peripheral mechanism may hypothetically potentiate THC central mechanisms via CB1. Sativex provided better antinociception than THC given alone, and the difference was likely to be mediated by TRPV1 channels (Comelli et al., 2008).
CBD reduces inflammation and inflammatory cytokines (e.g. TNF-α, IL-6, IL-1β) through TRPV1-, A2A- and PPARγ-mediated mechanisms (Supporting Information Appendix S2). THC reduces inflammation through separate CB1- and CB2 receptor-mediated mechanisms.
CBD acts as a positive allosteric modulator of glycine receptors, which contributes to cannabis-induced analgesia (Xiong et al., 2011).
CBD is an antioxidant and ROS scavenger, more potent than ascorbate or α-tocopherol – thus, it controls free radical-associated diseases (Hampson et al., 1998). The U.S. Department of Health patented this discovery (U.S. Patent No. 6630507), despite its classification of cannabis as a Schedule I substance having ‘no currently accepted medical use.’ Antioxidants limit neurological damage following stroke, ethanol poisoning or trauma, as well as animal models of multiple sclerosis, Alzheimer's, Parkinson's and Huntington's disease (Supporting Information Appendix S2).
CBD suppression of ROS, TNF-α and IL-1β predictably reduces NF-κB, which is induced by these stimuli (Supporting Information Appendix S2). Elevation of NF-κB occurs in many inflammatory diseases, such as arthritis, inflammatory bowel disease, gastritis, asthma, atherosclerosis and possibly schizophrenia. THC may dampen NF-κB through a CB2-mediated mechanism (Jeon et al., 1996). THC-mediated mechanisms may diverge from CBD-mediated mechanisms via other pathways (Kozela et al., 2010). CBD suppression of ROS, pro-inflammatory cytokines and NF-κB also predictably reduces iNOS, and consequent NO and peroxynitrite formation (Supporting Information Appendix S2). Inhibiting iNOS helps control chronic neurological diseases as well as cardiovascular disease. THC probably reduces iNOS through a different mechanism (Jeon et al., 1996).
CBD modulation of cytosolic Ca2+ levels via several mechanisms (voltage-gated Ca2+ channels, mitochondrial Na+/Ca2+ exchange, adenosine A2A and 5-HT1A receptor agonism) is likely to contribute to its anticonvulsant benefits, particularly for partial or generalized seizures (Jones et al., 2012).
CBD inhibits cancer growth and induces apoptosis. CBD generates ROS and up-regulates caspase proteases (Supporting Information Appendix S2). The capacity of CBD to selectively generate ROS in cancer cells likely works through superoxide-generating NADPH oxidases or by inducing endoplasmic reticulum and mitochondrial stress. THC inhibition of cancer works through CB1- and CB2 receptor-mediated MAPK/ERK pathways and ceramide accumulation. Combining these mechanisms by using THC and CBD together, produces synergistic inhibition of cancer cell growth and apoptosis (Marcu et al., 2010).
THCV at CB1 receptors
As discussed previously, meta-analysis indicates that THCV acts as a neutral CB1 and CB2 receptor antagonist, at least in most in vitro studies. Indeed, evidence has also emerged that THCV can block CB1 receptors in vivo, as indicated by its ability to oppose (i) anti-nociception induced by THC in the mouse tail-flick test and by CP55,940 in the rat hot plate test; (ii) hypothermia induced in mice by THC; and (iii) CP55,940-induced inhibition of rat locomotor activity (Pertwee et al., 2007; García et al., 2011).
Results from other in vivo experiments have shown that THCV can suppress food consumption and body weight in non-fasted mice, like AM251 (Riedel et al., 2009), and like rimonabant, THCV can reduce signs of motor inhibition in rats caused by 6-hydroxydopamine (García et al., 2011). It remains to be established whether THCV produced these effects through CB1 receptor inverse agonism or because it was competitively antagonizing CB1 receptor-mediated effects of one or more endogenously released endocannabinoids.
However, it is noteworthy that, like the established neutral CB1 receptor antagonists, THCV does not share the ability (i) of SR141716A or AM251 to produce signs of nausea in rats (Rock et al., 2013) or (ii) of SR141716A to produce an anxiogenic-like reaction in the rat light–dark immersion model of anxiety-like behaviour, or a suppression of saccharin hedonic reactions of rats in the taste reactivity test of palatability processing (O'Brien et al., 2013). Furthermore, in some in vivo settings where neutral antagonists produce effects, THCV does not. For example, THCV did not reduce food intake and body weight in obese mice, even though it did improve insulin resistance in these animals (Wargent et al., 2013). Furthermore, THCV did not reduce food deprivation-induced food intake in mice (Izzo et al., 2013).
THCV has anti-epileptiform actions in the rat pentylenetetrazole seizure model (Hill et al., 2010). This is in contrast to rimonabant safety data, where seizures were occasionally observed (Bermudez-Silva et al., 2010). Rimonabant significantly increased seizure duration and seizure frequency in the rat pilocarpine model of epilepsy (Wallace et al., 2003). Pro-convulsant effects were also observed with AM251 in a model of generalized seizures (Shafaroodi et al., 2004).
Also, when given in vivo at doses well above those at which it can block CB1 receptors, THCV produces signs of CB1 receptor activation. For example, in experiments performed with mice, THCV has been shown to induce (i) immobility in the Pertwee ring test, albeit with a potency 4.8 times less than that of THC, and (ii) anti-nociception in the tail-flick test (Gill et al., 1970; Pertwee et al., 2007). Importantly, although SR141716A was found to antagonize the antinociceptive effect of THCV, it did not give rise to any significant antagonism of THCV-induced hypothermia or ring immobility (Pertwee et al., 2007). THCV also decreased pain behaviour in the formalin test; this effect appeared to be CB1 and CB2 receptor-mediated (Bolognini et al., 2010). It remains to be established how THCV produces apparent CB1 receptor activation in vivo but not in vitro at doses above those at which it can block CB1 receptors both in vivo and in vitro. One possibility is the ability of THCV to inhibit endocannabinoid re-uptake (see previous discussion), with indirect activation of cannabinoid receptors. Models of intestinal transit are often used as assays of CB1 receptor activation and THCV inhibits upper intestinal transit in a dose-dependent manner (Izzo et al., 2013). However, this effect was not antagonized by a dose of rimonabant inactive per se. This study, together with the hypothermia and immobility assay, indicates that THCV can have cannabimimetic effects, which appear to be independent of the CB1 receptor.
The ‘abants’ were withdrawn from the market because of their worsening of anxiety and depression in obese patients. In contrast, THCV has very little effect in animal models of anxiety and depression. For example, the dose of THCV required to impart anxiogenic effects in the elevated plus maze is 30-fold higher than that of rimonabant (Izzo et al., 2013).
The observed similarities or differences between THCV and rimonabant (Box 2013d) may depend upon several factors. These include (i) the initial physiopathological status of the cell/tissue/organ, and the degree of expression therein of functional CB1 receptors, which might affect the activity of different ligands of the same receptor in different manners; (ii) the presence or absence in the cell/tissue/organ of non-CB1, non-CB2 receptor targets for THCV – in particular, TRP channels and CB2 receptors, particularly when THCV is administered at doses higher than those required for CB1 receptor antagonism, might counteract some of the CB1 receptor antagonism-mediated effects of THCV, but not others; (iii) the fact that at higher doses, THCV might start behaving as a CB1 receptor agonist, thus clearly counteracting some of the effects due to CB1 receptor neutral antagonism; (iv) different pharmacokinetic properties of the two compounds, particularly, but not limited to, under pathological conditions that may alter their tissue distribution and catabolism.
Box 4 Landmark 21st century in vivo studies concerning THCV.
THCV induced anti-nociception in the tail-flick test – Pertwee et al., 2007.
THCV suppressed food consumption and body weight in non-fasted mice – Riedel et al., 2009.
THCV antagonized the antinociceptive effects of THC in the rat acetic acid model of visceral nociception – Booker et al., 2009.
THCV exerted anticonvulsant actions in the PTZ model of seizure – Hill et al., 2010.
THCV reduced pain behaviour in the formalin test – Bolognini et al., 2010.
THCV imparted neuroprotective effects and relieves symptoms in rat models of Parkinson's disease – García et al., 2011.
THCV did not produce nausea or a suppression of saccharin hedonic reactions of rats in the taste reactivity test of palatability processing – Rock et al., 2013.
THCV did not reduce food intake and body weight in obese mice – Wargent et al., 2013.
THCV did not produce an anxiogenic-like reaction in the rat light–dark immersion model of anxiety-like behaviour – O'Brien et al., 2013.
Conclusions
Based upon the meta-analysis of in vitro data, and in vivo data cited in the Discussion section, we conclude that the pharmacology of neither CBD nor THCV has much in common with that of the ‘abants’ and of rimonabant in particular. While CBD does counteract some of the actions of THC, particularly in the brain, it potentiates other actions of THC. The capacity of CBD to modulate several signalling systems and to enhance endocannabinoid levels might produce varying effects on the endocannabinoid system and CB1 receptors. This may be organ/tissue/cell-dependent, and unlikely due to direct molecular interactions with CB1 receptors.
Although THCV undoubtedly behaves as a CB1 (and CB2) receptor orthosteric ligand in binding assays, and as a neutral CB1 receptor antagonist in functional assays, its pharmacological profile in vivo overlaps only in part with that of rimonabant and other CB1 receptor inverse agonists, or even with that of synthetic CB1 receptor neutral antagonists.
Thus, CBD and THCV represent the two opposing ends of activity at CB1 receptors: a low-affinity CB1 receptor ligand, which can affect CB1 receptor activity in vivo in an indirect manner, and a high-affinity CB1 receptor ligand and potent antagonist in vitro, which instead produces CB1 receptor antagonism-mediated effects in vivo only in a few instances. These paradoxical examples of how in vivo pharmacology is not always predicted from in vitro mechanistic studies underlie the necessity of testing compounds in vivo before drawing any conclusion on their functional activity at a given target. This is especially true regarding compounds that have complex pharmacology, such as the phytocannabinoids.
However, it must be remembered that whether or not in vitro mechanistic studies predict in vivo pharmacology depends, in part, upon pharmacokinetic data – particularly on the ability of CBD and THCV to reach concentrations in plasma and tissues similar to the concentrations shown in vitro to be necessary to interact with certain targets. In mice, the plasma Cmax of THCV, given at a dose of 30 mg·kg−1 either p.o. or i.p., was 0.24 and 0.88 mg·L−1, and thus lower than that of a higher dose of CBD (120 mg·kg−1), which was 2.2 and 14.3 mg·L−1 (Deiana et al., 2012). A similar scenario was described by the same authors also for the brain, although in this case the difference between THCV and CBD was less striking, with the Cmax of the former being 0.43 and 1.69 μm·g−1 and that of CBD being 1.3 and 6.9 μm·g−1.
In rats, two different vehicles were compared for CBD (120 mg·kg−1), and the plasma Cmax values were 2 and 2.6 mg·L−1 after p.o. and i.p. administration, respectively, for cremophor, and 3.2 and 2.4 mg·L−1 for solutol. Brain Cmax values were 8.6 and 6.8 μm·g−1 after p.o. and i.p. administration, respectively, for cremophor, and 12.6 and 5.2 μm·g−1 for solutol. Rat pharmacokinetic values for THCV (30 mg·kg−1) were Cmax 0.21 and 0.4 mg mL−1 for p.o. and i.p. administration, respectively, in plasma, and 0.3 and 1.62 μm·g−1 for oral and i.p. administration, respectively, in the brain. Thus, there are only small differences between the two species, for the two compounds.
No human studies have been published regarding THCV pharmacokinetics. Human subjects given Sativex as a buccal spray (THC 16.2 mg and CBD 15.0 mg) averaged a CBD Cmax plasma concentration of 6.7 μg·L−1, range 2.0–20.5 μg·L−1 (Karschner et al., 2011a). The metabolism of THC administered as Sativex was slightly altered, as they observed lower 11-OH-THC Cmax after Sativex in relation to 15 mg p.o. THC (falling short of significance, P = 0.09). Subjects given eight buccal sprays of Sativex per day (= 20 mg CBD daily) over 9 days averaged CBD plasma concentration of 3.2 ng·mL−1 (Stott et al., 2013).
Thus, given these low Cmax values, many in vitro studies reporting effects in the micromolar range, especially for CBD, might be regarded as irrelevant. Yet, in the clinic, both CBD and THCV, like rimonabant, are likely to be administered chronically and their tissue concentrations are therefore likely to accumulate. Furthermore, it must be emphasized that a proper comparison between the pharmacokinetics and bioavailability of CBD or THCV with those of rimonabant and other ‘abants’ cannot be made as data for the latter are scant and mostly unpublished possibly because these compounds are still proprietary and their development has been interrupted.
At any rate, much of the confusion about CBD as a ‘rimonabant-like drug’ might have been avoided if one had followed the algorithm shown in Figure 2 to decide whether, based upon in vitro pharmacological data, a substance can be described as a CB1 receptor orthosteric ligand first and then as agonist, inverse agonist, neutral antagonist or functionally inactive (Figure 2). Indeed, based upon this algorithm, one might even question the necessity of testing compounds in functional assays of a given receptor in vitro if they do not exhibit high affinity in radioligand displacement assays for that receptor. An exception to this rule might be allosteric ligands, which cannot always be identified by the binding assays normally used to evaluate the affinity of compounds for GPCRs (May et al., 2007).
Figure 2.

Algorithm for investigating cannabinoids in mechanistic studies. Dashed arrow: Note that in some cases compounds that do not enhance the binding of high affinity ligands to their receptors might still be allosteric modulators (May et al., 2007).
Our results may have important effects on the future clinical development of CBD as an antipsychotic, anti-inflammatory and anti-epileptic drug, and of THCV for the treatment of type 2 diabetes. Meta-analysis suggests that these two phytocannabinoids are very unlikely to produce the central adverse events typical of THC, as well as the central adverse events of the ‘abants’.
Acknowledgments
This work was partially supported by an unrestricted grant from GW Pharmaceuticals, Salisbury, UK. We thank all the scientists who responded to our queries regarding their unpublished results.
Glossary
- 2-AG
sn-2 arachidonoyl glycerol
- AEA
anandamide
- CBD
cannabidiol
- CV
coefficient of variation
- DAGL
diacylglycerol lipase
- FAAH
fatty acid amide hydrolase
- FsAC
forskolin-stimulated adenylate cyclase
- MAFP
methylarachidonoyl fluorophosphonate
- MAGL
monoacylglycerol lipase
- PMSF
phenylmethyl sulfonyl fluoride
- THCV
Δ9-tetrahydrocannabivarin
Conflict of interest
M. D. is an employee of GW Pharmaceuticals, UK. J. M. M., V. D. and R. P. are consultants for, and receive research funds from, GW Pharmaceuticals, UK.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Appendix S1 PRISMA 2009 Checklist and extended methods section for meta-analysis.
Appendix S2 The CBD data extraction table, followed by subgroup meta-regression tests.
Appendix S3 The THCV data extraction table, followed by subgroup meta-regression tests.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013c;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013d;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ligand-Gated Ion Channels. Br J Pharmacol. 2013e;170:1582–1606. doi: 10.1111/bph.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Nuclear Hormone Receptors. Br J Pharmacol. 2013f;170:1652–1675. doi: 10.1111/bph.12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayewitch M, Rhee MH, Avidor-Reiss T, Breuer A, Mechoulam R, Vogel Z. (-)-delta9-tetrahydrocannabinol antagonizes the peripheral cannabinoid receptor-mediated inhibition of adenylyl cyclase. J Biol Chem. 1996;271:9902–9905. doi: 10.1074/jbc.271.17.9902. [DOI] [PubMed] [Google Scholar]
- Bermudez-Silva FJ, Viveros MP, McPartland JM, Rodriguez de Fonseca F. The endocannabinoid system, eating behavior and energy homeostasis: the end or a new beginning? Pharmacol Biochem Behav. 2010;95:375–382. doi: 10.1016/j.pbb.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Morrison PD, Fusar-Poli P, Martin-Santos R, Borgwardt S, Winton-Brown T, et al. Opposite effects of delta-9-tetrahydrocannabinol and cannabidiol on human brain function and psychopathology. Neuropsychopharmacology. 2010;35:764–774. doi: 10.1038/npp.2009.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognini D, Costa B, Maione S, Comelli F, Marini P, Di Marzo V, et al. The plant cannabinoid Δ9-tetrahydrocannabivarin can decrease signs of inflammation and inflammatory pain in mice. Br J Pharmacol. 2010;160:677–687. doi: 10.1111/j.1476-5381.2010.00756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker L, Naidu PS, Razdan RK, Mahadevan A, Lichtman AH. Evaluation of prevalent phytocannabinoids in the acetic acid model of visceral nociception. Drug Alcohol Depend. 2009;105:42–47. doi: 10.1016/j.drugalcdep.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne RG, Weissman A. Discriminative stimulus properties of delta 9-tetrahydrocannabinol: mechanistic studies. J Clin Pharmacol. 1981;21:227S–234S. doi: 10.1002/j.1552-4604.1981.tb02599.x. [DOI] [PubMed] [Google Scholar]
- Campos AC, Guimaraes FS. Involvement of 5HT1A receptors in the anxiolytic-like effects of cannabidiol injected into the dorsolateral periaqueductal gray of rats. Psychopharmacology (Berl) 2008;199:223–230. doi: 10.1007/s00213-008-1168-x. [DOI] [PubMed] [Google Scholar]
- Campos AC, Guimaraes FS. Evidence for a potential role for TRPV1 receptors in the dorsolateral periaqueductal gray in the attenuation of the anxiolytic effects of cannabinoids. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:1517–1521. doi: 10.1016/j.pnpbp.2009.08.017. [DOI] [PubMed] [Google Scholar]
- Campos AC, de Paula Soares V, Carvalho MC, Ferreira FR, Vicente MA, Brandão ML, et al. Involvement of serotonin-mediated neurotransmission in the dorsal periaqueductal gray matter on cannabidiol chronic effects in panic-like responses in rats. Psychopharmacology (Berl) 2013;226:13–24. doi: 10.1007/s00213-012-2878-7. [DOI] [PubMed] [Google Scholar]
- Campos AC, Ortega Z, Palazuelos J, Fogaça MV, Aguiar DC, Díaz-Alonso J, et al. The anxiolytic effect of cannabidiol on chronically stressed mice depends on hippocampal neurogenesis: involvement of the endocannabinoid system. Int J Neuropsychopharmacol. 2013;16:1407–1419. doi: 10.1017/S1461145712001502. [DOI] [PubMed] [Google Scholar]
- Castillo A, Tolón MR, Fernández-Ruiz J, Romero J, Martinez-Orgado J. The neuroprotective effect of cannabidiol in an in vitro model of newborn hypoxic-ischemic brain damage in mice is mediated by CB(2) and adenosine receptors. Neurobiol Dis. 2010;37:434–440. doi: 10.1016/j.nbd.2009.10.023. [DOI] [PubMed] [Google Scholar]
- Cawthorne M, Stott C, Wright S, Guy G, Zaibi M, Wargent E. 2008. The metabolic effects of tetrahydrocannabivarin (THCV) and cannabidiol (CBD). Proceedings of the 18th Annual Symposium on the Cannabinoids. International Cannabinoid Research Society, Burlington, VT, p. 42.
- Clapper JR, Moreno-Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A, et al. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat Neurosci. 2010;13:1265–1270. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchetto DM, Cook LF, Cato AE. A critical review of the safety and antiemetic efficacy of delta-9-tetrahydrocannabinol. Drug Intell Clin Pharm. 1981;15:867–875. doi: 10.1177/106002808101501104. [DOI] [PubMed] [Google Scholar]
- Comelli F, Giagnoni G, Bettoni I, Colleoni M, Costa B. Antihyperalgesic effect of a Cannabis sativa extract in a rat model of neuropathic pain: mechanisms involved. Phytother Res. 2008;22:1017–1024. doi: 10.1002/ptr.2401. [DOI] [PubMed] [Google Scholar]
- Costa B, Giagnoni G, Franke C, Trovato AE, Colleoni M. Vanilloid TRPV1 receptor mediates the antihyperalgesic effect of the nonpsychoactive cannabinoid, cannabidiol, in a rat model of acute inflammation. Br J Pharmacol. 2004;143:247–250. doi: 10.1038/sj.bjp.0705920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Ligresti A, Moriello AS, Allarà M, Bisogno T, Petrosino S, et al. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol. 2011;163:1479–1494. doi: 10.1111/j.1476-5381.2010.01166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiana S, Watanabe A, Yamasaki Y, Amada N, Arthur M, Fleming S, et al. Plasma and brain pharmacokinetic profile of cannabidiol (CBD), cannabidivarine (CBDV), Δ9-tetrahydrocannabivarin (THCV) and cannabigerol (CBG) in rats and mice following oral and intraperitoneal administration and CBD action on obsessive-compulsive behaviour. Psychopharmacology (Berl) 2012;219:859–873. doi: 10.1007/s00213-011-2415-0. [DOI] [PubMed] [Google Scholar]
- Demirakca T, Sartorius A, Ende G, Meyer N, Welzel H, Skopp G, et al. Diminished gray matter in the hippocampus of cannabis users: possible protective effects of cannabidiol. Drug Alcohol Depend. 2011;114:242–245. doi: 10.1016/j.drugalcdep.2010.09.020. [DOI] [PubMed] [Google Scholar]
- Devane WA, Dysarz FA, 3rd, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- Di Marzo V. Targeting the endocannabinoid system: to enhance or reduce? Nat Rev Drug Discov. 2008;7:438–455. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. Anandamide serves two masters in the brain. Nat Neurosci. 2010;13:1446–1448. doi: 10.1038/nn1210-1446. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Melck D, Bisogno T, De Petrocellis L. Endocannabinoids: endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998;21:521–528. doi: 10.1016/s0166-2236(98)01283-1. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, De Petrocellis L, Fezza F, Ligresti A, Bisogno T. Anandamide receptors. Prostaglandins Leukot Essent Fatty Acids. 2002;66:377–391. doi: 10.1054/plef.2001.0349. [DOI] [PubMed] [Google Scholar]
- El Yacoubi M, Ledent C, Ménard JF, Parmentier M, Costentin J, Vaugeois JM. The stimulant effects of caffeine on locomotor behaviour in mice are mediated through its blockade of adenosine A(2A) receptors. Br J Pharmacol. 2000;129:1465–1473. doi: 10.1038/sj.bjp.0703170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund A, Morrison PD, Nottage J, Hague D, Kane F, Bonaccorso S, et al. Cannabidiol inhibits THC-elicited paranoid symptoms and hippocampal-dependent memory impairment. J Psychopharmacol. 2013;27:19–27. doi: 10.1177/0269881112460109. [DOI] [PubMed] [Google Scholar]
- Fadda P, Robinson L, Fratta W, Pertwee RG, Riedel G. Differential effects of THC- or CBD-rich cannabis extracts on working memory in rats. Neuropharmacology. 2004;47:1170–1179. doi: 10.1016/j.neuropharm.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Fusar-Poli P, Crippa JA, Bhattacharyya S, Borgwardt SJ, Allen P, Martin-Santos R, et al. Distinct effects of delta-9-tetrahydrocannabinol and cannabidiol on neural activation during emotional processing. Arch Gen Psychiatry. 2009;66:95–105. doi: 10.1001/archgenpsychiatry.2008.519. [DOI] [PubMed] [Google Scholar]
- Gaoni Y, Mechoulam R. Isolation, structure, and partial synthesis of an active constituent of hashish. J Am Chem Soc. 1964;86:1646–1647. [Google Scholar]
- García C, Palomo-Garo C, Garcia-Arencibia M, Ramos JA, Pertwee RG, Fernandez-Ruiz J. Symptom-relieving and neuroprotective effects of the phytocannabinoid Δ9-THCV in animal models of Parkinson's disease. Br J Pharmacol. 2011;163:1495–1506. doi: 10.1111/j.1476-5381.2011.01278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill EW, Paton WDM, Pertwee RG. Preliminary experiments on the chemistry and pharmacology of Cannabis. Nature. 1970;228:134–136. doi: 10.1038/228134a0. [DOI] [PubMed] [Google Scholar]
- Glass GV, McGaw B, Smith ML. Meta-Analysis in Social Research. Beverly Hills, CA: SAGE Publications; 1981. [Google Scholar]
- Gomes FV, Resstel LB, Guimarães FS. The anxiolytic-like effects of cannabidiol injected into the bed nucleus of the stria terminalis are mediated by 5-HT1A receptors. Psychopharmacology (Berl) 2011;213:465–473. doi: 10.1007/s00213-010-2036-z. [DOI] [PubMed] [Google Scholar]
- Gomes FV, Del Bel EA, Guimarães FS. Cannabidiol attenuates catalepsy induced by distinct pharmacological mechanisms via 5-HT1A receptor activation in mice. Prog Neuropsychopharmacol Biol Psychiatry. 2013;46:43–47. doi: 10.1016/j.pnpbp.2013.06.005. [DOI] [PubMed] [Google Scholar]
- Grinspoon L, Bakalar JB. Marihuana, the Forbidden Medicine. New Haven, CT: Yale University Press; 1997. [Google Scholar]
- Guy GW, Stott CG. The development of Sativex – a natural cannabis-based medicine. In: Mechoulam R, editor. Cannabinoids as Therapeutics. Basel: Birkhäuser Verlag; 2005. pp. 231–263. . In: (ed.). [Google Scholar]
- Hampson AJ, Grimaldi M, Axelrod J, Wink D. Cannabidiol and (-)Delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci U S A. 1998;95:8268–8273. doi: 10.1073/pnas.95.14.8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanuš L. Pharmacological and therapeutic secrets of plant and brain (endo)cannabinoids. Med Res Rev. 2008;29:213–271. doi: 10.1002/med.20135. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Mishima K, Hazekawa M, Sano K, Irie K, Orito K, et al. Cannabidiol potentiates pharmacological effects of Delta(9)-tetrahydrocannabinol via CB(1) receptor-dependent mechanism. Brain Res. 2008;1188:157–164. doi: 10.1016/j.brainres.2007.09.090. [DOI] [PubMed] [Google Scholar]
- Higgins JPT, Green S. Cochrane Handbook for Systematic Reviews of Interventions 4.2.5. Chichester, UK: John Wiley & Sons; 2005. [Google Scholar]
- Hill AJ, Weston SE, Jones NA, Smith I, Bevan SA, Williamson EM, et al. Δ9-Tetrahydrocannabivarin suppresses in vitro epileptiform and in vivo seizure activity in adult rats. Epilepsia. 2010;51:1522–1532. doi: 10.1111/j.1528-1167.2010.02523.x. [DOI] [PubMed] [Google Scholar]
- Hollister LE. Cannabidiol and cannabinol in man. Experientia. 1973;29:825–826. doi: 10.1007/BF01946311. [DOI] [PubMed] [Google Scholar]
- Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4:682–690. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Scott DK, Wilken GH. Regulation of adenylate cyclase by cannabinoid drugs. Insights based n thermodynamic studies. Biochem Pharmacol. 1989;38:3297–3304. doi: 10.1016/0006-2952(89)90628-x. [DOI] [PubMed] [Google Scholar]
- Ilan AB, Gevins A, Coleman M, ElSohly MA, de Wit H. Neurophysiological and subjective profile of marijuana with varying concentrations of cannabinoids. Behav Pharmacol. 2005;16:487–496. doi: 10.1097/00008877-200509000-00023. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Borrelli F, Capasso R, Di Marzo V, Mechoulam R. Non-psychotropic plant cannabinoids: new therapeutic opportunities from an ancient herb. Trends Pharmacol Sci. 2009;30:515–527. doi: 10.1016/j.tips.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Zaibi MS, Wargent E, Capasso R, Arbuckle C, Duncan M, et al. 2013. The effect of Δ9-tetrahydrocannabivarin on food deprivation-induced food intake and upper gastrointestinal motility: differences from rimonabant. Proceedings of the 7th Conference on Cannabinoids in Medicine. International Association for Cannabis as Medicine, Köln, p. 57.
- Jamontt JM, Molleman A, Pertwee RG, Parsons ME. The effects of Delta-tetrahydrocannabinol and cannabidiol alone and in combination on damage, inflammation and in vitro motility disturbances in rat colitis. Br J Pharmacol. 2010;160:712–723. doi: 10.1111/j.1476-5381.2010.00791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järbe TU, Henriksson BG, Ohlin GC. Delta9-THC as a discriminative cue in pigeons: effects of delta8-THC, CBD, and CBN. Arch Int Pharmacodyn Ther. 1977;228:68–72. [PubMed] [Google Scholar]
- Jeon YJ, Yang KH, Pulaski JT, Kaminski NE. Attenuation of inducible nitric oxide synthase gene expression by delta 9-tetrahydrocannabinol is mediated through the inhibition of nuclear factor- kappa B/Rel activation. Mol Pharmacol. 1996;50:334–341. [PubMed] [Google Scholar]
- Johnson JR, Burnell-Nugent M, Lossignol D, Ganae-Motan ED, Potts R, Fallon MT. Multicenter, double-blind, randomized, placebo-controlled, parallel-group study of the efficacy, safety, and tolerability of THC:CBD extract and THC extract in patients with intractable cancer-related pain. J Pain Symptom Manage. 2010;39:167–179. doi: 10.1016/j.jpainsymman.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Jones NA, Glyn SE, Akiyama S, Hill TD, Hill AJ, Weston SE, et al. Cannabidiol exerts anti-convulsant effects in animal models of temporal lobe and partial seizures. Seizure. 2012;21:344–352. doi: 10.1016/j.seizure.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Karschner EL, Darwin WD, Goodwin RS, Wright S, Huestis MA. Plasma cannabinoid pharmacokinetics following controlled oral delta9-tetrahydrocannabinol and oromucosal cannabis extract administration. Clin Chem. 2011a;57:66–75. doi: 10.1373/clinchem.2010.152439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karschner EL, Darwin WD, McMahon RP, Liu F, Wright S, Goodwin RS, et al. Subjective and physiological effects after controlled Sativex and oral THC administration. Clin Pharmacol Ther. 2011b;89:400–407. doi: 10.1038/clpt.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Karanges E, Spiro A, Wong A, Spencer J, Huynh T, et al. Cannabidiol potentiates Δ9-tetrahydrocannabinol (THC) behavioural effects and alters THC pharmacokinetics during acute and chronic treatment in adolescent rats. Psychopharmacology (Berl) 2011;218:443–457. doi: 10.1007/s00213-011-2342-0. [DOI] [PubMed] [Google Scholar]
- Kozak KR, Crews BC, Morrow JD, Wang LH, Ma YH, Weinander R, et al. Metabolism of the endocannabinoids, 2-arachidonylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol esters and ethanolamides. J Biol Chem. 2002;277:44877–44885. doi: 10.1074/jbc.M206788200. [DOI] [PubMed] [Google Scholar]
- Kozela E, Pietr M, Juknat A, Rimmerman N, Levy R, Vogel Z. Cannabinoids delta(9)-tetrahydrocannabinol and cannabidiol differentially inhibit the lipopolysaccharide-activated NF-kappaB and interferon-beta/STAT proinflammatory pathways in BV-2 microglial cells. J Biol Chem. 2010;285:1616–1626. doi: 10.1074/jbc.M109.069294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner TN, Horne EA, Stella N, Kreitzer AC. Endocannabinoid signaling mediates psychomotor activation by adenosine A2A antagonists. J Neurosci. 2010;30:2160–2164. doi: 10.1523/JNEUROSCI.5844-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leweke FM, Schneider U, Radwan M, Schmidt E, Emrich HM. Different effects of nabilone and cannabidiol on binocular depth inversion in man. Pharmacol Biochem Behav. 2000;66:175–181. doi: 10.1016/s0091-3057(00)00201-x. [DOI] [PubMed] [Google Scholar]
- Leweke FM, Piomelli D, Pahlisch F, Muhl D, Gerth CW, Hoyer C, et al. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry. 2012;2:e94. doi: 10.1038/tp.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gotzsche PC, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. PLoS Med. 2009;6:e1000100. doi: 10.1371/journal.pmed.1000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligresti A, Schiano Moriello A, Starowicz K, Matias I, Pisanti S, De Petrocellis L, et al. Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J Pharmcol Exp Ther. 2006;318:1375–1387. doi: 10.1124/jpet.106.105247. [DOI] [PubMed] [Google Scholar]
- Loewe WS, Modell W. The action of chemical components of cannabis extracts. J Pharmacol Exp Ther. 1941;72:27. [Google Scholar]
- Long LE, Chesworth R, Huang XF, McGregor IS, Arnold JC, Karl T. A behavioural comparison of acute and chronic D9-tetrahydrocannabinol and cannabidiol in C57BL/6JArc mice. Int J Neuropsychopharmacol. 2010;13:861–876. doi: 10.1017/S1461145709990605. [DOI] [PubMed] [Google Scholar]
- Maione S, Piscitelli F, Gatta L, Vita D, De Petrocellis L, Palazzo E, et al. Non-psychoactive cannabinoids modulate the descending pathway of antinociception in anaesthetized rats through several mechanisms of action. Br J Pharmacol. 2011;162:584–596. doi: 10.1111/j.1476-5381.2010.01063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone DT, Jongejan D, Taylor DA. Cannabidiol reverses the reduction in social interaction produced by low dose Delta(9)-tetrahydrocannabinol in rats. Pharmacol Biochem Behav. 2009;93:91–96. doi: 10.1016/j.pbb.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Marcu JP, Christian RT, Lau D, Zielinski AJ, Horowitz MP, Lee J, et al. Cannabidiol enhances the inhibitory effects of delta9-tetrahydrocannabinol on human glioblastoma cell proliferation and survival. Mol Cancer Ther. 2010;9:180–189. doi: 10.1158/1535-7163.MCT-09-0407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martire A, Tebano MT, Chiodi V, Ferreira SG, Cunha RA, Köfalvi A, et al. Pre-synaptic adenosine A2A receptors control cannabinoid CB1 receptor-mediated inhibition of striatal glutamatergic neurotransmission. J Neurochem. 2011;116:273–280. doi: 10.1111/j.1471-4159.2010.07101.x. [DOI] [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulous A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- McPartland JM, Guy G. The evolution of Cannabis and coevolution with the cannabinoid receptor – a hypothesis. In: Guy G, Robson R, Strong K, Whittle B, editors. The Medicinal Use of Cannabis. London: Royal Society of Pharmacists; 2004. pp. 71–102. . In: (eds). [Google Scholar]
- McPartland JM, Pruitt PL. Side effects of pharmaceuticals not elicited by comparable herbal medicines: the case of tetrahydrocannabinol and marijuana. Altern Ther Health Med. 1999;5:57–62. [PubMed] [Google Scholar]
- McPartland JM, Russo EB. Cannabis and cannabis extracts: greater than the sum of their parts? J Cannabis Therapeutics. 2001;1:103–132. [Google Scholar]
- McPartland JM, Glass M, Pertwee RG. Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: interspecies differences. Br J Pharmacol. 2007;152:583–593. doi: 10.1038/sj.bjp.0707399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Fride E, Di Marzo V. Endocannabinoids. Eur J Pharmacol. 1998;359:1–18. doi: 10.1016/s0014-2999(98)00649-9. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Peters M, Murillo-Rodriguez E, Hanuš LO. Cannabidiol – recent advances. Chem Biodivers. 2007;4:1678–1692. doi: 10.1002/cbdv.200790147. [DOI] [PubMed] [Google Scholar]
- de Meijer EPM, Bagatta M, Carboni A, Crucitti P, Cristiana Moliterni VM, Ranalli P, et al. The inheritance of chemical phenotype in Cannabis sativa L. Genetics. 2003;163:335–346. doi: 10.1093/genetics/163.1.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan CJ, Curran HV. Effects of cannabidiol on schizophrenia-like symptoms in people who use cannabis. Br J Psychiatry. 2008;192:306–307. doi: 10.1192/bjp.bp.107.046649. [DOI] [PubMed] [Google Scholar]
- Morgan CJ, Freeman TP, Schafer GL, Curran HV. Cannabidiol attenuates the appetitive effects of delta 9-tetrahydrocannabinol in humans smoking their chosen cannabis. Neuropsychopharmacology. 2010a;35:1879–1885. doi: 10.1038/npp.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan CJ, Schafer G, Freeman TP, Curran HV. Impact of cannabidiol on the acute memory and psychotomimetic effects of smoked cannabis: naturalistic study. Br J Psychiatry. 2010b;197:285–290. doi: 10.1192/bjp.bp.110.077503. [DOI] [PubMed] [Google Scholar]
- Nicholson AN, Turner C, Stone BM, Robson PJ. Effect of delta-9-tetrahydrocannabinol and cannabidiol on nocturnal sleep and early-morning behavior in young adults. J Clin Psychopharmacol. 2004;24:305–313. doi: 10.1097/01.jcp.0000125688.05091.8f. [DOI] [PubMed] [Google Scholar]
- Notcutt W, Price M, Miller R, Newport S, Phillips C, Simmons S, et al. Initial experiences with medicinal extracts of cannabis for chronic pain: results from 34 ‘N of 1’ studies. Anaesthesia. 2004;59:440–452. doi: 10.1111/j.1365-2044.2004.03674.x. [DOI] [PubMed] [Google Scholar]
- O'Brien LD, Wills KL, Segsworth B, Dashney B, Rock EM, Limebeer CL, et al. Effect of chronic exposure to rimonabant and phytocannabinoids on anxiety-like behavior and saccharin palatability. Pharmacol Biochem Behav. 2013;103:597–602. doi: 10.1016/j.pbb.2012.10.008. [DOI] [PubMed] [Google Scholar]
- Palazzo E, de Novellis V, Petrosino S, Marabese I, Vita D, Giordano C, et al. Neuropathic pain and the endocannabinoid system in the dorsal raphe: pharmacological treatment and interactions with the serotonergic system. Eur J Neurosci. 2006;24:2011–2020. doi: 10.1111/j.1460-9568.2006.05086.x. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. The pharmacology and therapeutic potential of cannabidiol. In: Di Marzo V, editor. Cannabinoids. New York: Kluwer Academic/Plenum Publishers; 2004. pp. 32–83. . In: (ed.). [Google Scholar]
- Pertwee RG. The therapeutic potential of drugs that target cannabinoid receptors or modulate the tissue levels or actions of endocannabinoids. AAPS J. 2005;7:E625–E654. doi: 10.1208/aapsj070364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153:199–215. doi: 10.1038/sj.bjp.0707442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Thomas A, Stevenson LA, Ross RA, Varvel SA, Lichtman AH, et al. The psychoactive plant cannabinoid, Δ9-tetrahydrocannabinol, is antagonized by Δ8- and Δ9-tetrahydrocannabivarin in mice in vivo. Br J Pharmacol. 2007;150:586–594. doi: 10.1038/sj.bjp.0707124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol Rev. 2010;62:588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitet F, Jeantaud B, Reibaud M, Imperato A, Dubroeucq MC. Complex pharmacology of natural cannabinoids: evidence for partial agonist activity of delta9-tetrahydrocannabinol and antagonist activity of cannabidiol on rat brain cannabinoid receptors. Life Sci. 1998;63:PL1–PL6. doi: 10.1016/s0024-3205(98)00238-0. [DOI] [PubMed] [Google Scholar]
- Qin N, Neeper MP, Liu Y, Hutchinson TL, Lubin ML, Flores CM. TRPV2 is activated by cannabidiol and mediates CGRP release in cultured rat dorsal root ganglion neurons. J Neuroscience. 2008;28:6231–6238. doi: 10.1523/JNEUROSCI.0504-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed GF, Lynn F, Meade BD. Use of coefficient of variation in assessing variability of quantitative assays. Clin Diagn Lab Immunol. 2002;9:1235–1239. doi: 10.1128/CDLI.9.6.1235-1239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee MH, Vogel Z, Barg J, Bayewitch M, Levy R, Hanus L, et al. Cannabinol derivatives: binding to cannabinoid receptors and inhibition of adenylylcyclase. J Med Chem. 1997;40:3228–3233. doi: 10.1021/jm970126f. [DOI] [PubMed] [Google Scholar]
- Riedel G, Fadda P, McKillop-Smith S, Pertwee RG, Platt B, Robinson L. Synthetic and plant-derived cannabinoid receptor antagonists show hypophagic properties in fasted and non-fasted mice. Br J Pharmacol. 2009;156:1154–1166. doi: 10.1111/j.1476-5381.2008.00107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock EM, Sticht MA, Duncan M, Stott C, Parker LA. Evaluation of the potential of the phytocannabinoids, cannabidivarin (CBDV) and Δ9-tetrahydrocannabivarin (THCV), to produce CB1 receptor inverse agonism symptoms of nausea in rats. Br J Pharmacol. 2013;170:671–678. doi: 10.1111/bph.12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roser P, Gallinat J, Weinberg G, Juckel G, Gorynia I, Stadelmann AM. Psychomotor performance in relation to acute oral administration of Delta9-tetrahydrocannabinol and standardized cannabis extract in healthy human subjects. Eur Arch Psychiatry Clin Neurosci. 2009;259:284–292. doi: 10.1007/s00406-009-0868-5. [DOI] [PubMed] [Google Scholar]
- Russo E, Guy GW. A tale of two cannabinoids: the therapeutic rationale for combining tetrahydrocannabinol and cannabidiol. Med Hypotheses. 2006;66:234–246. doi: 10.1016/j.mehy.2005.08.026. [DOI] [PubMed] [Google Scholar]
- Russo EB. Clinical endocannabinoid deficiency (CECD): can this concept explain therapeutic benefits of cannabis in migraine, fibromyalgia, irritable bowel syndrome and other treatment-resistant conditions? Neuro Endocrinol Lett. 2004;25:31–39. [PubMed] [Google Scholar]
- Russo EB. Taming THC: potential cannabis synergy and phytocannabinoid-pterpenoid entourage effects. Br J Pharmacol. 2011;163:1344–1364. doi: 10.1111/j.1476-5381.2011.01238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagredo O, Pazos MR, Satta V, Ramos JA, Pertwee RG, Fernández-Ruiz J. Neuroprotective effects of phytocannabinoid-based medicines in experimental models of Huntington's disease. J Neurosci Res. 2011;89:1509–1518. doi: 10.1002/jnr.22682. [DOI] [PubMed] [Google Scholar]
- Savinainen JR, Saario SM, Niemi R, Järvinen T, Laitinen JT. An optimized approach to study endocannabinoid signaling: evidence against constitutive activity of rat brain adenosine A1 and cannabinoid CB1 receptors. Br J Pharmacol. 2003;140:1451–1459. doi: 10.1038/sj.bjp.0705577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubart CD, Sommer IE, van Gastel WA, Goetgebuer RL, Kahn RS, Boks MP. Cannabis with high cannabidiol content is associated with fewer psychotic experiences. Schizophr Res. 2011;130:216–221. doi: 10.1016/j.schres.2011.04.017. [DOI] [PubMed] [Google Scholar]
- Shafaroodi H, Samini M, Moezi L, Homayoun H, Sadeghipour H, Tavakoli S, et al. The interaction of cannabinoids and opioids on pentylenetetrazole-induced seizure threshold in mice. Neuropharmacology. 2004;47:390–400. doi: 10.1016/j.neuropharm.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Stott CG, White L, Wright S, Wilbraham D, Guy GW. A phase I study to assess the single and multiple dose pharmacokinetics of THC/CBD oromucosal spray. Eur J Clin Pharmacol. 2013;69:1135–1147. doi: 10.1007/s00228-012-1441-0. [DOI] [PubMed] [Google Scholar]
- Strasser F, Luftner D, Possinger K, Ernst G, Ruhstaller T, Meissner W, et al. Comparison of orally administered cannabis extract and delta-9-tetrahydrocannabinol in treating patients with cancer-related anorexia-cachexia syndrome: a multicenter, phase III, randomized, double-blind, placebo-controlled clinical trial from the Cannabis-In-Cachexia-Study-Group. J Clin Oncol. 2006;24:3394–3400. doi: 10.1200/JCO.2005.05.1847. [DOI] [PubMed] [Google Scholar]
- Vann RE, Gamage TF, Warner JA, Marshall EM, Taylor NL, Martin BR, et al. Divergent effects of cannabidiol on the discriminative stimulus and place conditioning effects of Delta(9)-tetrahydrocannabinol. Drug Alcohol Depend. 2008;94:191–198. doi: 10.1016/j.drugalcdep.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varvel SA, Wiley JL, Yang R, Bridgen DT, Long K, Lichtman AH, et al. Interactions between THC and cannabidiol in mouse models of cannabinoid activity. Psychopharmacology (Berl) 2006;186:226–234. doi: 10.1007/s00213-006-0356-9. [DOI] [PubMed] [Google Scholar]
- Wade DT, Robson P, House H, Makela P, Aram J. A preliminary controlled study to determine whether whole-plant cannabis extracts can improve intractable neurogenic symptoms. Clin Rehabil. 2003;17:21–29. doi: 10.1191/0269215503cr581oa. [DOI] [PubMed] [Google Scholar]
- Wallace MJ, Blair RE, Falenski KW, Martin BR, DeLorenzo RJ. The endogenous cannabinoid system regulates seizure frequency and duration in a model of temporal lobe epilepsy. J Pharmacol Exp Ther. 2003;307:129–137. doi: 10.1124/jpet.103.051920. [DOI] [PubMed] [Google Scholar]
- Wargent ET, Zaibi MS, Silvestri C, Hislop DC, Stocker CJ, Stott CG, et al. The cannabinoid Δ(9)-tetrahydrocannabivarin (THCV) ameliorates insulin sensitivity in two mouse models of obesity. Nutr Diabetes. 2013;3:e68. doi: 10.1038/nutd.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson JD, Whalley BJ, Baker D, Pryce G, Constanti A, Gibbons S, et al. Medicinal cannabis: is delta9-tetrahydrocannabinol necessary for all its effects? J Pharm Pharmacol. 2005;55:1687–1694. doi: 10.1211/0022357022304. [DOI] [PubMed] [Google Scholar]
- Williamson EM. Synergy and other interactions in phytomedicines. Phytomedicine. 2001;8:401–409. doi: 10.1078/0944-7113-00060. [DOI] [PubMed] [Google Scholar]
- Wolf SA, Bick-Sander A, Fabel K, Leal-Galicia P, Tauber S, Ramirez-Rodriguez G, et al. Cannabinoid receptor CB1 mediates baseline and activity-induced survival of new neurons in adult hippocampal neurogenesis. Cell Commun Signal. 2010;8:12. doi: 10.1186/1478-811X-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Cheng K, Cui T, Godlewski G, Rice KC, Xu Y, et al. Cannabinoid potentiation of glycine receptors contributes to cannabis-induced analgesia. Nat Chem Biol. 2011;7:296–303. doi: 10.1038/nchembio.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zijicek J, Fox P, Sanders H, Wright D, Vickery J, Nunn A, et al. Cannabinoids for treatment of spasticity and other symptoms related to multiple sclerosis (CAMS study): multicentre randomised placebo-controlled trial. Lancet. 2003;362:1517–1526. doi: 10.1016/S0140-6736(03)14738-1. [DOI] [PubMed] [Google Scholar]
- Zuardi AW. Cannabidiol: from an inactive cannabinoid to a drug with wide spectrum of action. Rev Bras Psiquiatr. 2008;30:271–280. doi: 10.1590/s1516-44462008000300015. [DOI] [PubMed] [Google Scholar]
- Zuardi AW, Finkelfarb E, Bueno OF, Musty RE, Karniol IG. Characteristics of the stimulus produced by the mixture of cannabidiol with delta 9-tetrahydrocannabinol. Arch Int Pharmacodyn Ther. 1981;249:137–146. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 PRISMA 2009 Checklist and extended methods section for meta-analysis.
Appendix S2 The CBD data extraction table, followed by subgroup meta-regression tests.
Appendix S3 The THCV data extraction table, followed by subgroup meta-regression tests.