

Abstract
Background and Purpose
Mitochondrial dysfunction, triggered by mitochondria permeability transition (MPT), has been centrally implicated in the pathogenesis of podocytopathy and involves a multitude of cell signalling mechanisms, among which, glycogen synthase kinase (GSK) 3β has emerged as the integration point and plays a crucial role. This study aimed to examine the role of GSK3β in podocyte MPT and mitochondrial dysfunction.
Experimental Approach
The regulatory effect of GSK3β on MPT was examined in differentiated podocytes in culture and in a murine model of adriamycin-induced podocytopathy using 4-benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione (TDZD-8), a highly selective small-molecule inhibitor of GSK3β.
Key Results
TDZD-8 therapy prominently ameliorated the proteinuria and glomerular sclerosis in mice with adriamycin nephropathy; this was associated with a correction of GSK3β overactivity in the glomerulus and attenuation of podocyte injuries, including foot process effacement and podocyte death. Consistently, in adriamycin-injured podocytes, TDZD-8 treatment counteracted GSK3β overactivity, improved cell viability and prevented death, concomitant with diminished oxidative stress, improved mitochondrial dysfunction and desensitized MPT. Mechanistically, a discrete pool of GSK3β was found in podocyte mitochondria, which interacted with and phosphorylated clyclophilin F, a key structural component of the MPT pore. TDZD-8 treatment prevented the GSK3β-controlled phosphorylation and activation of cyclophilin F, desensitized MPT and alleviated the damage to mitochondria in podocytes induced by adriamycin in vivo and in vitro.
Conclusions and Implications
Our findings suggest that pharmacological targeting of GSK3β could represent a promising and feasible therapeutic strategy for protecting podocytes against mitochondrial dysfunction induced by oxidative injuries.
Tables of Links
| TARGETS |
|---|
| Caspase-3 |
| GSK3β |
| LIGANDS | |
|---|---|
| 2′Z, 3′E-6-bromoindirubin-3′-oxime | IFN-γ |
| Adriamycin | TDZD-8 |
| ATP |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Regardless of the original aetiology, podocyte dysfunction or injury is the common pathological pathway leading to glomerular proteinuria (Haraldsson et al., 2008). If podocyte injury persists, podocytopoenia ensues and non-selective nephrotic-range proteinuria develops, ultimately culminating in progressive glomerulosclerosis. Mitochondrial dysfunction is a fundamental mechanism central to cell death and plays an important role in podocyte injury (Papeta et al., 2010; Su et al., 2013). Stimulation of podocytes by numerous harmful stimuli, including hyperglycaemia, immune injury, vasoactive mediators like aldosterone and angiotensin II, as well as podocytotoxic agents like adriamycin, induces oxidative damages and the accumulation of cytotoxic reactive oxygen species (ROS) mostly in and around mitochondria. ROS can trigger the opening of the end-effector mitochondria permeability transition (MPT) pore, resulting in the immediate eradication of the mitochondrial membrane potential and permeability of the mitochondrial inner membrane allowing solutes with a molecular weight less than 1500 Da to leak from the mitochondria (Circu and Aw, 2010). The subsequent break-up of the mitochondrial outer membrane will lead to the release of pro-death proteins such as cytochrome c and other factors that play a critical role in cell death. The opening threshold of the MPT pore and the sensitivity of MPT to ROS are determined by the activation status of the mitochondrial membrane proteins like cyclophilin F (Cyp-F) (also known as mitochondrial cyclophilin D), which is regulated by a myriad of signalling pathways (Baines et al., 2005). The enzyme, glycogen synthase kinase (GSK) 3β, plays a crucial role in transferring regulatory signals downstream to modify the susceptibility to MPT (Nishihara et al., 2007).
GSK3β, originally identified as a kinase that mediates inhibitory phosphorylation of glycogen synthase, is a constitutively active and redox-sensitive multifunctional serine/threonine PK that is ubiquitously expressed in all cell types (Doble and Woodgett, 2003). In cell signalling networks, GSK3β resides at the centre of numerous pathways, including cell death, cytoskeletal organization, development control, insulin signalling, canonical wingless and NFκB (Ge et al., 2010). More recently, GSK3β was found to regulate MPT and mitochondria dysfunction in excitable cells including neurons and cardiomyocytes, and also in non-excitable cells, such as cancer cells and renal tubular epithelial cells (Plotnikov et al., 2007; Miura et al., 2009; Moreira et al., 2010; Rasola et al., 2010). Thus there is mounting evidence supporting the view that GSK3β is pivotal in determining renal tubular cell death and acute kidney injury. Nevertheless, its role in podocyte injury and in glomerular diseases remains unknown and is the subject of this study. The potential role of GSK3β in the regulation of MPT in adriamycin-induced podocyte mitochondria dysfunction and podocytopathy was examined using the selective inhibitor 4-benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione (TDZD-8), a novel, highly selective small-molecule inhibitor of GSK3β.
Methods
Animal experimental design
The animal experiments were approved by the Rhode Island Hospital Animal Care and Use Committee, and they conform to the United States Department of Agriculture regulations and the National Institutes of Health guidelines for humane care and use of laboratory animals. Male 8-week-old BALB/c mice weighing 20–25 g were randomly divided into five groups of 15 animals each as follows: control group (Ctrl) mice were injected i.p. with normal saline; TDZD group mice received an i.p. injection of 5 mg·kg−1 TDZD-8 (Sigma, St. Louis, MO, USA) (Bao et al., 2012; Wang et al., 2013); the adriamycin (ADR) group mice received an i.v. injection of vehicle 1 h before tail vein injection of 10 mg·kg−1 doxorubicin hydrochloride (adriamycin, Sigma) (Papeta et al., 2010; Su et al., 2013); T-L + ADR group mice received i.p. injections of 1 mg·kg−1 TDZD-8 (Bao et al., 2012; Wang et al., 2013) 1 h before tail vein injection of 10 mg·kg−1 adriamycin; and T + ADR group mice received i.p. injections of 5 mg·kg−1 TDZD-8 1 h before tail vein injection of 10 mg·kg−1 adriamycin. Urine was collected on post-injury days 3, 7 and 14. All mice were killed by an overdose of sodium thiobutabarbital (125 mg·kg−1, i.p.) 14 days after adriamycin injury and kidneys excised for further investigation. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Glomerular isolation
Mice were anaesthetized with an overdose of sodium thiobutabarbital (as above) and perfused by infusing the abdominal artery with 5 mL of PBS containing 8 × 107 Dynabeads M-450 (Dynal Biotech ASA, Oslo, Norway). After perfusion, the kidneys were removed, cut into 1 mm3 pieces, digested in collagenase A and the glomeruli containing Dynabeads were then gathered using a magnetic particle concentrator.
Histological studies
Formalin-fixed kidneys were embedded in paraffin, 3 μm thick sections were stained with periodic acid–Schiff (PAS) stain. Six coded slides from each group were examined by an observer blinded to the treatments. Glomerulosclerosis is defined as mesangial matrix expansion, capillary obliteration, hyalinosis and adhesion of the glomerular tuft to Bowman's capsule. If the glomerulus was normal, a score of 0 was given; if 0∼25% was affected, a score of 1 was given; 25∼50% was scored as 2; 50∼75% was scored as 3; and 75∼100% was scored as 4. A glomerulosclerosis index (GSI) was calculated by multiplying the number of glomeruli with a sclerosis score, and these values were summed and divided by the number of glomeruli assessed, including those with a sclerosis score of 0 (Han et al., 2013; Liu et al., 2013). The GSI for each kidney specimen was a sum of the points from 30 glomeruli.
Transmission electron microscopy was carried out according to published procedures (Ge et al., 2010). Ultra-thin sections (50∼60 nm) were prepared and examined using a Zeiss EM-10 electron microscope (Carl Zeiss AG, Oberkochen, Germany) and images were captured with an AMT Advantage 542 CCD camera system (Advanced Microscopy Techniques, Corp., Woburn, MA, USA).
Urine analysis
To discern the protein compositions of urine, equal amounts of urine samples were subjected to SDS/PAGE, followed by Coomassie blue (Sigma) staining. Urine albumin was measured using a mouse albumin elisa quantification kit (Bethyl Laboratories, Montgomery, TX, USA). Urine creatinine was measured by a creatinine assay kit (BioAssay Systems, Hayward, CA, USA).
Cell culture
The conditionally immortalized mouse podocyte cell line was kindly provided by Dr Stuart Shankland and cultured as previously described (Durvasula et al., 2004). Cells were grown on collagen-coated plastic Petri dishes at 33°C in RPMI-1640 (Life Technologies, Grand Island, NY, USA) medium supplemented with 10% FBS and recombinant IFN-γ (Life Technologies). To induce differentiation, podocytes were grown under non-permissive conditions at 37°C in the absence of IFN-γ. After being incubated in a serum-free medium (serum starvation) for 24 h, podocytes were pretreated with 5 μM TDZD-8 (Bao et al., 2012; Wang et al., 2013) or 2 mM N-acetyl-L-cysteine (NAC, Sigma), a free-radical scavenger (Chuang et al., 2007), or vehicle for 30 min before 0.25 μg·mL−1 adriamycin was added to the cultures. Cells were harvested at the indicated time points.
Cellular viability assay
Podocyte viability was measured using 3-4,5-dimethylthiazol-2,5-diphenyltetrazolium bromide (MTT) reduction as previously described (Wang et al., 2013). MTT (Sigma) was added (final concentration 0.5 mg·mL−1) to individual cultures 1 h before harvest, and tetrazolium released by DMSO. OD was determined with a spectrophotometer (570 nm), and data were normalized to solvent-treated cultures.
Transient transfection
Transient transfection of podocyte was carried out using Lipofectamine 2000 (Life Technologies) according to the manufacture's instruction. The expression vectors encoding the haemagglutinin (HA) conjugated constitutively active mutant (S9A-GSK3β-HA/pcDNA3; S9A) and kinase-dead (KD) GSK-3β (KD-GSK3β-HA/pcDNA3) were employed as previously described (Gong et al., 2008b; Bao et al., 2012), the pcDNA3 empty vector (EV) was used as a negative control. After transfection with equal amounts of expression plasmid or EV, immunofluorescent staining revealed that >75% of the cells expressed the HA-tagged constructs 24 h after transfection. Cells were then subjected to various treatments as indicated.
Fluorescent staining
Cells were fixed with 4% paraformaldehyde and permeabilized. Following serum blocking for 30 min, cells were incubated with the primary antibody to GSK3β, Cyp-F (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or preimmune IgG and then the Alexa fluorophore-conjugated secondary antibody (Invitrogen). For mitochondria staining, live cells were incubated with MitoTracker green (Life Technologies, Grand Island, NY, USA) for 30 min before fixation (Dagda et al., 2011; Bao et al., 2012; Wang et al., 2013). Finally, cells were mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA) and visualized using a fluorescence microscope. The 3 μm thick frozen cryostat sections of mice kidney cortex were stained with antibodies (Santa Cruz Biotechnology) against podocin and Wilms tumour 1 (WT-1). The confocal images were acquired with a Zeiss LSM510 Meta confocal microscope (Carl Zeiss AG). For dual-colour staining, images were acquired sequentially to avoid dye interference. The NIH ImageJ software (National Institutes of Health, Bethesda, MD, USA) was used for post-processing of the images, for example scaling, merging and co-localization analysis.
Detection of ROS generation by fluorescence
The production of ROS in cultured podocytes and in vivo in the kidney was evaluated by detecting the fluorescence intensity of 2′, 7′-dichlorofluorescein-diacetate (DCF-DA; Sigma) (Koya et al., 2003; Lee et al., 2010; Zhu et al., 2011). Briefly, cells were loaded with 20 μM DCF-DA for 0.5 h before termination of the treatments and were subsequently washed and scraped into ice-cold PBS. Cell suspensions were then subjected to fluorometric analysis for DCF fluorescence intensity. To examine the production of ROS in vivo in the kidney, fresh kidney cryostat sections were incubated with 10 μM DCF-DA in a light-protected humidified chamber at 37°C for 30 min as described previously (Takimoto et al., 2005; Zhu et al., 2011), subsequently washed twice with PBS for 5 min, mounted with mounting medium and visualized with a fluorescence microscope.
GSK3β activity assay
Activity of GSK3β in the homogenates of glomeruli isolated from differently treated mice was measured using a GENMED GSK3β activity assay kit (Genmed Scientifics, Inc., Arlington, MA, USA) according to manufacturer's instructions and expressed as fold induction over the control group.
Western immunoblot analysis and immunoprecipitation
Cultured podocytes were lysed and isolated glomeruli homogenized in RIPA buffer (1% Nonidet P-40, 0.1% SDS, 100 μg·mL−1 phenylmethanesulfonyl fluoride, 0.5% sodium deoxycholate, 1 mM sodium orthovanadate, 2 μg·mL−1 aprotin, 2 μg·mL−1 leupeptin, 5 mM EDTA in PBS) supplemented with protease inhibitors. Mitochondrial fractions were prepared from cultured cells or isolated glomeruli using the mitochondria isolation kit (Pierce, Rockford, IL USA). Samples with equal amounts of total protein (50 mg·mL−1) were processed for immunoblot as described previously (Gong et al., 2006). The antibodies against GSK3β, p-GSK3β, GAPDH and Cyp-F were purchased from Santa Cruz Biotechnology. The antibody against cleaved caspase-3 was purchased from Cell Signaling Technology (Beverly, MA, USA).
For detection of phosphorylated Cyp-F and to examine potential physical interactions between GSK3β and Cyp-F, Cyp-F was immunoprecipitated and the antibody against phosphorylated serine (Abcam, Cambridge, MA, USA) or GSK3β was used to probe the immunoprecipitates by immunoblot analysis.
MPT assay
The respiratory chain in mitochondria is a major source of ROS, and increased ROS generation may lead to a catastrophic cycle of mitochondrial dysfunction (Ide et al., 2001; Tsutsui et al., 2009). To measure MPT, mitochondria were isolated from cultured cells or isolated glomeruli by differential centrifugation as previously described (Enrique Guerrero-Beltran et al., 2010; Szeto et al., 2011; Silva and Oliveira, 2012). The protein concentration was determined by bicinchoninic acid protein assay. MPT, reflected by susceptibility to mitochondrial swelling, was estimated on the basis of the decrease in the absorbance of mitochondria (1.0 mg protein) at 540 nm in 1 mL of a medium containing 125 mmol·L−1 sucrose, 65 mM KCl, 5 mM succinate, 5 μM rotenone, 20 μM CaCl2, and 10 mM HEPES-KOH, pH 7.2, at 30°C.
Apoptosis analysis
TUNEL staining was performed on fixed tissue sections or cell cultures with a cell apoptosis detection kit (Roche Applied Science, Indianapolis, IN, USA) according to the manufacturer's instructions. Apoptosis was also analysed by flow cytometry using a commercially available kit (Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit for flow cytometry, Life Technologies) according to the manufacturer's instructions. Briefly, cells were harvested following the indicated treatments, washed in cold PBS and resuspend the cells in 1× annexin-binding buffer to about 1 × 106 cells·mL−1. Cells were then labelled with Alexa Fluor 488 annexin V and propidium iodide (PI) and rinsed. The stained cells were analysed by flow cytometry; the fluorescence emission was measured at 499 and 617 nm using 488 and 535 nm excitation respectively. Flow cytometry results were confirmed by viewing the cells under a fluorescence microscope.
Statistical analyses
All quantitative data are expressed as mean ± SD, and qualitative data presented as percentages. Statistical analysis of the data from multiple groups was performed by one-way anova followed by the Student–Newman–Kuels test. Data from two groups were compared using Student's t-test. A value of P < 0.05 was considered statistically significant. All statistical analysis was performed using SPSS v.18.0 (IBM, Armonk, NY, USA).
Results
TDZD-8 treatment ameliorates proteinuria and glomerular sclerosis in mice with adriamycin nephropathy
The murine model of adriamycin nephropathy is a standard model of acute podocytopathy, which replicates key features of nephrosis and focal segmental glomerulosclerosis in humans, including podocyte injury, massive proteinuria and progressive glomerulosclerosis (Haraldsson et al., 2008). Indeed, mice injured with an i.v. injection of adriamycin (10 mg·kg−1) developed massive proteinuria that peaked on day 7 and slightly subsided by day 14, as estimated by SDS/PAGE coupled with Coomassie brilliant blue stain and as quantified by urine albumin levels, adjusted to urine creatinine concentrations (Figure 1A, B).
Figure 1.
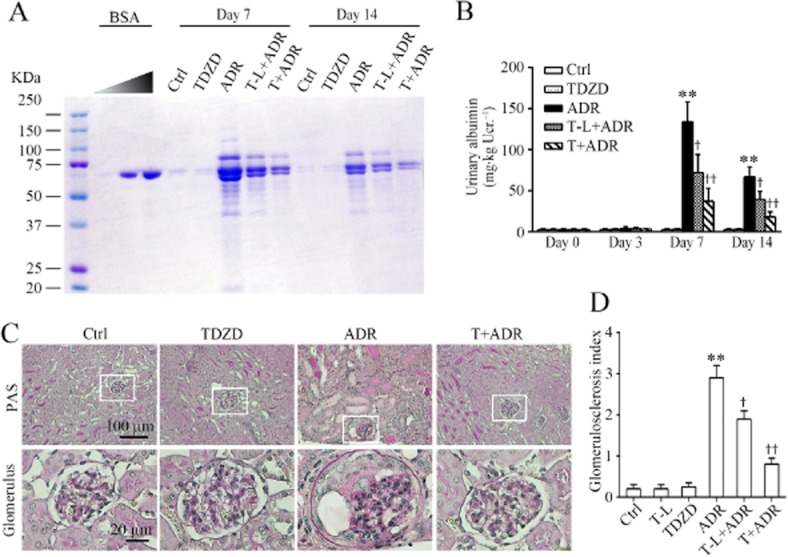
Inhibition of GSK3β by TDZD-8 protects against proteinuria and glomerular sclerosis mice with adriamycin nephropathy. (A) Mice were injected with adriamycin (ADR, i.v.) 1 h following a single i.p. injection of a regular (5 mg·kg−1; T) or low (1 mg·kg−1; T-L) dose of TDZD-8, a highly selective small-molecule inhibitor of GSK3β, or an equal volume of vehicle. Ctrl mice were injected with vehicle instead of adriamycin. Urine was collected at the indicated time points and subjected to SDS/PAGE and staining with Coomassie brilliant blue. BSA of 0, 1 and 4 μg, served as standard controls. Urine samples (2 μL) were collected on the post-injury days indicated from each group. (B) Quantification of urine albumin levels adjusted with urine creatinine concentrations. **P < 0.01 versus Ctrl; †P < 0.05, ††P < 0.01 versus ADR; (n = 15). (C) Representative micrographs demonstrate kidney sections stained with PAS (original magnification, ×200; ×400 for inserts). (D) Semi-quantitative morphometric estimation of glomerulosclerosis on PAS-stained mouse kidney sections. **P < 0.01 versus Ctrl; †P < 0.05, ††P < 0.01 versus ADR; (n = 15).
A single injection of TDZD-8 (5 mg·kg−1) 1 h before adriamycin was given to mice based on previous experience (Bao et al., 2012; Wang et al., 2013) and this decreased the proteinuria by more than 70%. Even at a low dose (1 mg·kg−1), TDZD-8 also attenuated the adriamycin-induced urine protein and albumin excretion by approximately 50%. Morphologically, the adriamycin-injured kidney exhibited prominent glomerular sclerosis and extensive protein casts in renal tubules as evidenced by PAS staining (Figure 1C). These lesions were significantly ameliorated after TDZD-8 treatment with both the regular dose and low dose. These morphological findings were further corroborated by the semi-quantitative morphometric analysis of GSI (Figure 1D). TDZD-8 alone-treated mice did not produce any noticeable side effects or any evidence of kidney injury.
TDZD-8 prevents GSK3β overactivity and attenuates podocyte injury in adriamycin-injured kidney
Podocyte dysfunction has been regarded as the common pathological pathway that leads to massive proteinuria. To determine if the TDZD–8-induced reduction in proteinuria in adriamycin nephropathy is attributable to an improvement in podocyte injury, the podocyte integrity was examined by probing specific podocyte markers and by electron microscopy. As shown in Figure 2A, glomerular staining of podocyte markers, including podocin and WT-1 was substantially blunted following adriamycin injury. The expression levels of these molecules in glomeruli were measured by immunoblot analysis of homogenates of glomeruli that were isolated from the excised kidney (Figure 2B) by the Dynabeads-based approach. Adriamycin exposure consistently diminished protein expression levels of podocin and WT-1, suggesting podocyte dysfunction and depletion. Furthermore, electron microscopy of glomerular tufts indicated massive podocyte foot process effacement and podocytopoenia in adriamycin-injured kidneys (Figure 2A). Adriamycin is a quintessential pro-oxidant and elicits prominent oxidative stress, which could alter the activity of redox-sensitive kinases in target organs, including the kidneys. The adriamycin-induced podocyte injury was followed by overactivity of the redox-sensitive GSK3β in the glomerulus, as assessed by the GSK3β kinase activity assay of the homogenates of the isolated glomeruli (Figure 2C). As a highly selective small-molecule inhibitor of GSK3β, TDZD-8 lessened the adriamycin-elicited GSK3β overactivity and, in parallel, restored the expression of podocin and WT-1 in glomerulus, and ameliorated the adriamycin-induced ultrastructural injuries in podocytes, indicating a protective effect on the podocytes.
Figure 2.
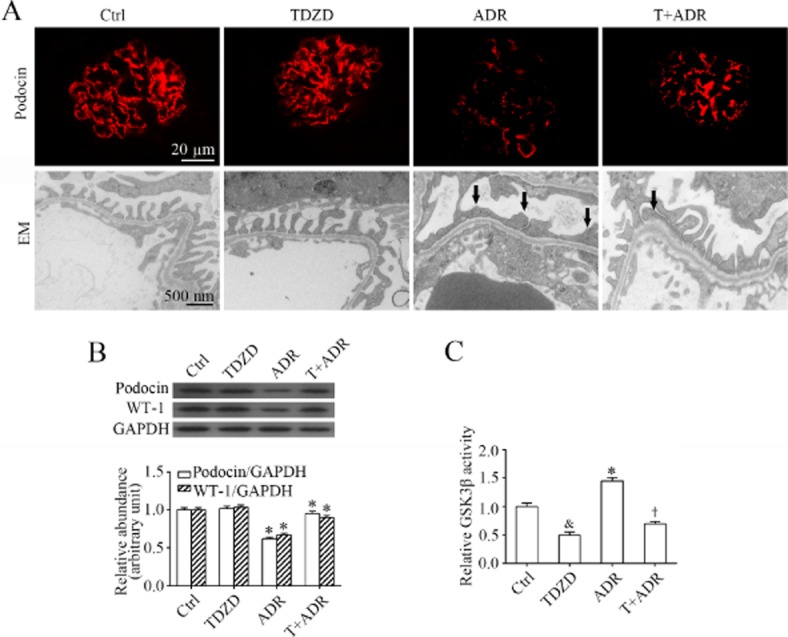
TDZD-8 prevents GSK3β overactivity and attenuates podocyte injury in adriamycin-injured kidney. (A) Kidney tissues obtained on day 14 from the mice after their different treatments, as described in Figure 1, were prepared for fluorescent immunohistochemistry staining for podocin (red signals) and electron microscopy. Adriamycin induced severe podocyte injury in mice, as indicated by the markedly reduced expression of podocin and extensive podocyte foot process effacement (indicated by arrows), and this effect was significantly attenuated by TDZD-8. (B) Glomeruli were isolated from kidneys excised on day 14 by the magnetic beads-based approach and homogenized for immunoblot analysis for GAPDH and podocyte markers including podocin and WT-1. Densitometric analysis of immunoblots estimated the relative abundance of podocin and WT-1 in glomeruli from the differently-treated mice. *P < 0.05 versus Ctrl; †P < 0.05 versus ADR; (n = 15). (C) Homogenates of isolated glomeruli were subjected to a GSK3β kinase assay to verify the GSK3β inhibitory effect of TDZD-8 in the kidney. &,*P < 0.05 versus Ctrl; †P < 0.05 versus ADR; (n = 15).
Inhibition of GSK3β by TDZD-8 diminishes oxidative stress and podocyte death in adriamycin-injured kidneys
Oxidative stress-induced podocyte injury and loss has been implicated in the pathogenesis of podocytopathy, proteinuria and glomerular sclerosis. To further understand the mechanism accounting for the podocyte protective action conferred by TDZD-8, oxidative stress and apoptosis were next examined in the kidneys. As a potent pro-oxidant, adriamycin caused robust oxidative stress in both glomerulus and renal tubules, as indicated by the green fluorescence of the ROS marker, DCF (Figure 3A). Computerized morphometric analysis of DCF-stained sections demonstrated an approximate threefold increase in the DCF fluorescence intensity in glomerulus (Figure 3B). In line with the causative role of oxidative stress in apoptotic cell death, the adriamycin-induced ROS accumulation in glomerulus was accompanied by more podocyte death, as assessed by dual-colour staining of cells positive for both WT-1 and TUNEL in the glomerulus (Figure 3A), as well as a reduced number of WT–1-positive podocytes in each glomerulus. The disparity between the drastic depletion of WT–1-positive podocytes and a more modest increase in TUNEL-positive apoptotic cells per glomerulus following adriamycin injury is consistent with other potential mechanisms for podocytopoenia besides apoptosis, including necrosis and autophagic cell death. The absolute count of apoptotic podocytes in each glomerulus indicated that the number of podocytes that were undergoing apoptosis in the adriamycin-injured kidney ranged between one and two cells per glomerulus (Figure 3C). TDZD-8 treatment diminished the number of TUNEL-positive podocytes to 0 to 1 cell per glomerulus and prevented podocyte loss, as indicated by the lack of a depletion in the number of WT–1-positive podocytes in each glomerulus, contingent on an abolished DCF fluorescence and oxidative stress in glomerulus in adriamycin-injured kidneys.
Figure 3.
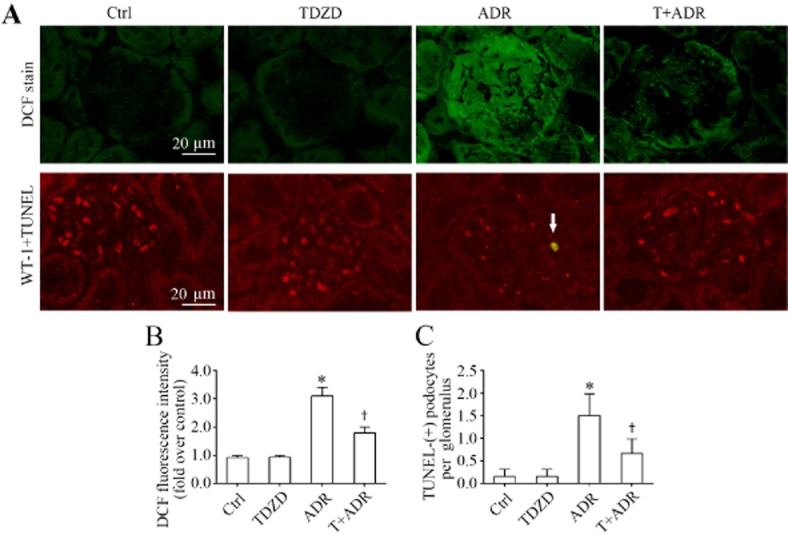
Inhibition of GSK3β attenuates oxidative stress and podocyte death in adriamycin-injured kidneys. (A) Fresh renal cortical tissues were obtained from the differently-treated animals on day 7 and prepared as frozen cryostat sections for staining of DCF (green), a marker of ROS as well as dual-colour staining for WT-1 (red) and TUNEL (green). The apoptotic podocyte that was positive for both WT-1 and TUNEL is indicated by an arrow. (B) Computerized morphometric analysis evaluated ROS production in the glomerulus in different groups as the relative integrated pixel density of DCF fluorescence in the glomerulus expressed as fold induction over the control group. *P < 0.05 versus Ctrl; †P < 0.05 versus ADR; (n = 15). (C) Absolute count of apoptotic podocytes that were positive for both WT-1 and TUNEL in the glomerulus and expressed as cells per glomerulus. *P < 0.05 versus Ctrl; †P < 0.05 versus ADR; (n = 15).
TDZD-8 prevents GSK3β overactivity, diminishes cell death and improves viability in adriamycin-injured podocytes
To explore whether TDZD-8 has a direct protective effect on podocytes, we next employed differentiated mouse podocytes in culture. Similar to the in vivo findings described earlier, podocytes exposed to adriamycin elicited a robust increase in ROS that was detected as early as 1 h by fluorometric analysis of the DCF green fluorescence in cell lysates (Figure 4A), followed by a continuous accumulation of ROS for at least 24 h. Cellular viability measured by the tetrazolium assay was drastically impaired (Figure 4B), and podocyte death defined by both TUNEL staining (Figure 4C, 4D) and immunoblot analysis for caspase-3 cleavage, an essential signalling event involved in apoptosis (Figure 4E), was markedly amplified. GSK3β appears to be a redox-sensitive signalling transducer and its activity could be facilitated upon oxidative stress. Accordingly, the adriamycin-elicited oxidative stress resulted in enhanced GSK3β activity in podocytes (Figure 4F), as indicated by the decreased inhibitory phosphorylation of GSK3β at the serine 9 residue. In contrast, treatment with the GSK3β inhibitor TDZD-8 1 h before adriamycin prevented the ROS accumulation, improved podocyte viability, abolished the pro-apoptotic effect of adriamycin, overrode the decreased inhibitory phosphorylation of GSK3β and counteracted GSK3β overactivity in adriamycin-injured podocytes, similar to the effect of NAC (Figure 4A), a quintessential antioxidant and ROS scavenger.
Figure 4.
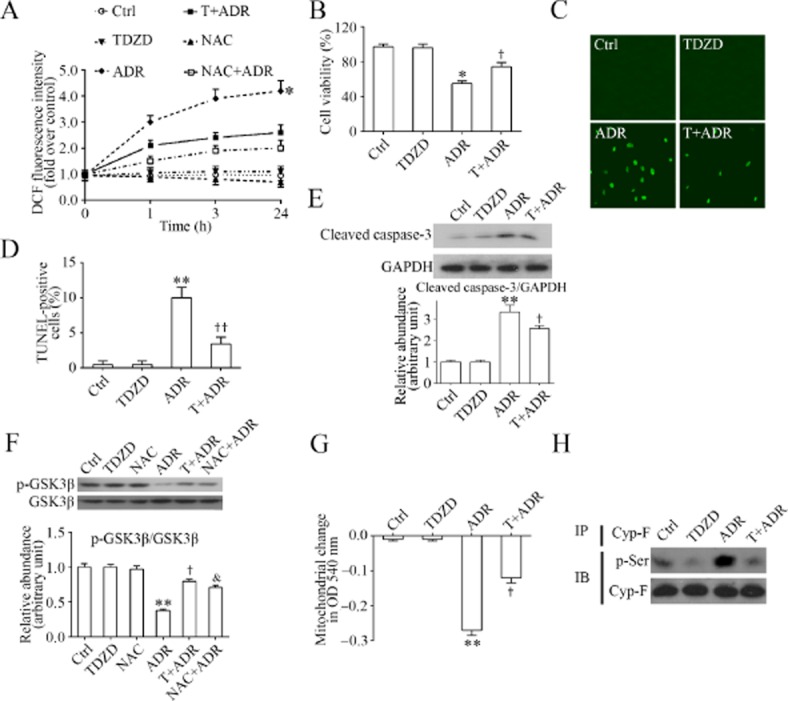
Inhibition of GSK3β by TDZD-8 reduces the oxidative stress, improves mitochondrial dysfunction and prevents GSK3β overactivity in adriamycin-injured podocytes. (A) Differentiated podocytes in culture were administered adriamycin (0.25 μg·mL−1) or vehicle (Ctrl) 1 h following pretreatment with TDZD-8 (TDZD or T, 5 μM), vehicle, or NAC (2 mM), a quintessential antioxidant and ROS scavenger. Before termination of the treatments at the indicated time points, podocytes were incubated with 10 μM DCF-DA for 30 min and the amount of reactive oxygen species was measured by fluorometric analysis of the fluorescence intensity of DCF in cell suspensions and expressed as fold induction over the value at time 0. *P < 0.05 versus other groups (n = 6). (B) Differentiated podocytes in culture were treated as described in (A) for 24 h. Cell viability was estimated by the MTT assay. *P < 0.05 versus Ctrl; †P < 0.05 versus ADR; (n = 6). (C) Representative fluorescent micrographs demonstrating TUNEL staining of cells that were treated as described in (A) for 24 h; (D) TUNEL-positive cells were counted and expressed as % of 100 cells that were labelled with DAPI. **P < 0.01 versus Ctrl; ††P < 0.01 versus ADR; (n = 6). (E) Cell lysates were prepared from cells that were differently treated for 24 h as described in (A). Cell lysates were subjected to immunoblot analysis for cleaved of caspase-3 and GAPDH. Arbitrary units of cleaved caspase-3/GAPDH ratios are expressed as immunoblot densitometric ratios of the molecules as fold of Ctrl. **P < 0.01 versus Ctrl; †P < 0.05 versus ADR; (n = 4). (F) Cell lysates were prepared from cells that were differently treated for 24 h as described in (A). Cell lysates were subjected to immunoblot analysis for inhibitory phosphorylation of GSK3β (p-GSK3β) and total GSK3β. Arbitrary units of p-GSK3β/GSK3β ratios are expressed as immunoblot densitometric ratios of the molecules as fold of Ctrl. Both TDZD-8 and NAC effectively prevented the inhibitory effect of adriamycin on the phosphorylation of GSK3β. **P < 0.01 versus Ctrl; †,&P < 0.05 versus ADR; (n = 6). (G) Mitochondria were isolated from cells that were differently treated for 24 h as described in (A). Mitochondrial dysfunction, reflected by the propensity of mitochondria to swell when incubated with 20 mM CaCl2 for 20 min, was spectrophotometrically monitored at 540 nm. **P < 0.01 versus Ctrl; †P < 0.05 versus adriamycin; (n = 6). (H) Cell lysates were prepared from cells that were differently treated for 24 h as described in (A). These lysates were then subjected to immunoprecipitation for Cyp-F and immunoprecipitates were processed for immunoblot analysis for phosphorylated serine (p-Ser).
TDZD-8 ameliorates mitochondria dysfunction in adriamycin-injured podocytes
The mitochondrial respiratory chain is a major source of ROS production. Mitochondria dysfunction, characterized by MPT, plays a key role in oxidative burst and ROS accumulation. Reciprocally, ROS overproduction could further exacerbate mitochondria dysfunction. To assess whether the TDZD–8-attenuated oxidative stress in adriamycin-injured podocytes was ascribed to an improvement in mitochondria dysfunction, mitochondria were isolated from podocytes following different treatments and MPT, as measured by calcium-induced mitochondrial swelling, was examined as a decrease in mitochondrial spectrophotometric absorbance at 540 nm upon calcium challenge (Figure 4G). In isolated mitochondria, the capacity to tolerate a calcium challenge is inversely correlated with mitochondria dysfunction and the susceptibility to the MPT. As shown in Figure 4G, mitochondria derived from the adriamycin-injured podocytes displayed a greater reduction in absorbance at 540 nm, denoting a marked mitochondrial dysfunction and sensitized MPT. TDZD-8 ameliorated this effect, inferring a mitochondria protective effect. MPT results from the opening of an MPT pore that is structurally composed of many interacting molecules, including Cyp-F (Circu and Aw, 2010). To examine if the TDZD–8-attenuated mitochondria dysfunction was associated with changes in the activity of MPT pore proteins, Cyp-F was immunoprecipitated from podocyte lysates and subjected to immunoblot analysis for phosphorylated serine. As shown in Figure 4H, adriamycin injury elicited evident hyperphosphorylation of Cyp-F in cultured podocytes and this effect was reduced by TDZD-8 treatment.
Cyp-F, a key regulator of MPT, physically interacts with GSK3β in podocytes and serves as its cognate substrate
The results suggested that GSK3β is involved in MPT, and this prompted us to further explore the mechanism underlying the mitochondria protective effect of TDZD-8 in podocytes. Cyp-F was immunoprecipitated from total cell lysates, and samples were probed for GSK3β. As shown in Figure 5A, Cyp-F co-precipitated with GSK3β in podocyte lysates, indicating that GSK3β physically interacts with Cyp-F in podocytes. To validate this observation, dual-colour fluorescent staining for GSK3β and mitochondria was carried out. As shown in Figure 5B, an abundant expression of GSK3β was observed in the cytoplasm of podocytes with a discrete pool co-localizing with mitochondria, which were highlighted with MitoTracker green, suggesting that a discrete pool of GSK3β is located in mitochondria. Frequency scatterplots of ImageJ co-localization analysis demonstrated that GSK3β staining and MitoTracker staining had a Spearman's correlation coefficient of 0.812 (Figure 5B), suggestive of a significant co-localization. To further define the specific localization of GSK3β in the mitochondria, GSK3β was co-stained with multiple mitochondrial molecules and was found to co-localize with key structural components of the MPT pore, including Cyp-F. As shown in Figure 5C, a high-powered view of podocytes by confocal microscopy demonstrated a close spatial association and co-localization between a discrete pool of GSK3β and Cyp-F on both XY and Z planes. The co-localization level (Figure 5C), determined by ImageJ co-localization analysis and expressed as a percentage of GSK3β staining co-localized with Cyp-F staining or Cyp-F staining co-localized with GSK3β staining, indicated that GSK3β was partially co-localized with mitochondria Cyp-F. This finding is consistent with the fact that GSK3β is widely distributed in the cytoplasm in addition to subcellular organelles, including mitochondria, centrosome, rough endoplasmic reticulum and the Golgi apparatus (Doble and Woodgett, 2003).
Figure 5.
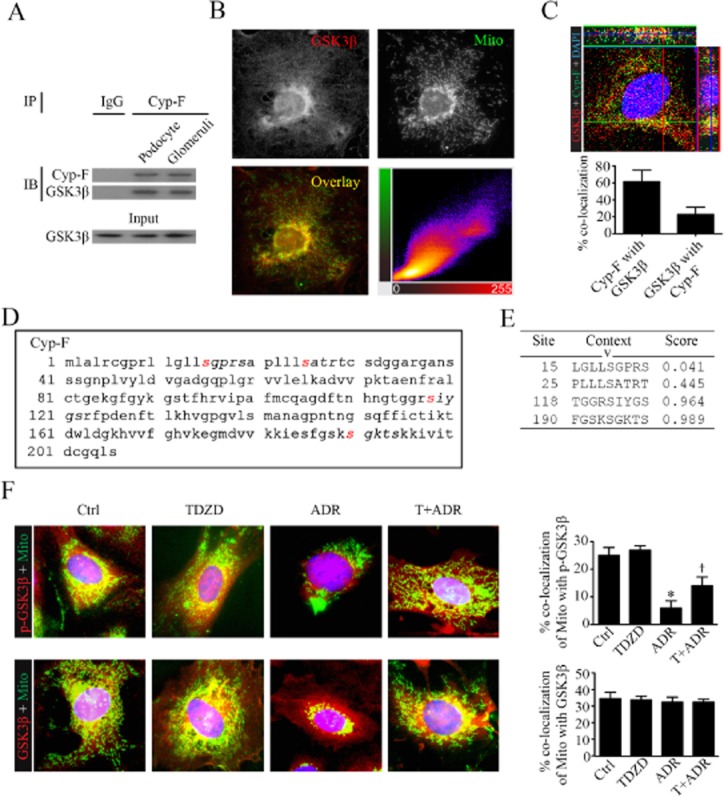
GSK3β is a mitochondrial molecule in podocytes and interacts with Cyp-F, a key regulator of MPT. (A) Lysates of cultured podocyte cells and homogenates of isolated glomeruli were subjected to immunoprecipitation using an anti-Cyp-F antibody or a preimmune IgG. Immunoprecipitates were processed for immunoblot analysis for Cyp-F and GSK3β. Equal amounts of glomerular lysates served as input control for immunoblot analysis. (B) Representative fluorescent micrographs of dual-colour florescence staining for GSK3β (red) and mitochondria (Mito, MitoTracker green) in differentiated podocytes demonstrated that a discrete pool of cytoplasmic GSK3β co-localized with mitochondria. Cells were counterstained with DAPI (blue colour); frequency scatterplots of ImageJ co-localization analysis demonstrated that GSK3β staining and MitoTracker green staining had a Spearman's correlation coefficient of 0.812, suggestive of a significant co-localization. (C) Representative high-powered view of podocytes by confocal microscopy demonstrated a close spatial association and co-localization between a discrete pool of GSK3β (red) and Cyp-F (green) on both XY and Z planes. Cells were counterstained with DAPI; The co-localization level, presented as % of GSK3β staining co-localized with Cyp-F staining or Cyp-F staining co-localized with GSK3β staining, was determined by ImageJ co-localization analysis and indicated that GSK3β partially co-localized with mitochondria Cyp-F (n = 6). (D) In silico analysis demonstrated that multiple serine residuals of Cyp-F resided in the consensus motifs for phosphorylation by GSK3β, denoting Cyp-F as a cognate substrate for GSK3β. (E) Characteristics of consensus GSK3β phosphorylation motifs in Cyp-F, as estimated by in silico analysis, including the predicted phosphorylation sites, prediction confidence scores and sequence context. Note that Ser118 and Ser190 were statistically significant and possessed prediction scores that were higher than 0.5, denoting high-confidence matches to GSK3β phosphorylation motifs. (F) Differentiated podocytes in culture were treated with adriamycin (0.25 μg·mL−1) or vehicle (Ctrl) 1 h following pretreatment with TDZD-8 (5 μM) or vehicle. Cells were processed for dual-colour fluorescent staining for mitochondria by MitoTracker green (Mito) and GSK3β or phosphorylated GSK3β (p-GSK3β). The % of MitoTracker green co-localized with GSK3β staining in podocytes as measured by ImageJ co-localization analysis was barely altered upon different treatments, suggesting mitochondrial trafficking of GSK3β was probably unaffected. Whereas, the % of MitoTracker green co-localized with p-GSK3β staining was markedly inhibited in adriamycin-treated podocytes and this effect was significantly attenuated by TDZD-8 treatment, inferring that TDZD-8 restores the phosphorylation of GSK3β in mitochondria upon injury. *P < 0.05 versus Ctrl; †P < 0.05 versus ADR; (n = 6).
To further understand the functionality of this physical interaction between GSK3β and Cyp-F, the amino acid sequence of murine Cyp-F (NCBI Reference Sequence: NP_598845.1) was analysed (Figure 5D). In silico analysis (http://scansite.mit.edu/motifscan_seq.phtml) revealed multiple serine residues of Cyp-F (Figure 5D) reside in the consensus motifs for phosphorylation by GSK3β (S/T-XXX-S/T). Among these sites, residues Ser118 and Ser190 possessed statistically significant prediction scores that were higher than 0.5 (Figure 5E), denoting high-confidence matches to GSK3β phosphorylation motifs. Taken together, these data are consistent with Cyp-F serving as a putative cognate substrate for GSK3β.
To understand how TDZD-8 affects GSK3β phosphorylation and translocation in mitochondria, podocytes were injured with adriamycin in the presence or absence of TDZD-8 for 24 h followed by dual-colour fluorescence staining for GSK3β or phosphorylated GSK3β and mitochondria. As shown in Figure 5F, the percentage of MitoTracker green co-localized with GSK3β staining in podocytes, as measured by ImageJ co-localization analysis, was barely altered by the different treatments, suggesting that mitochondrial trafficking of GSK3β was probably unaffected. In contrast, the percentage of MitoTracker green co-localized with phosphorylated GSK3β staining was markedly attenuated in adriamycin-injured podocytes and this effect was significantly diminished by TDZD-8 treatment, inferring that TDZD-8 restores GSK3β phosphorylation in the injured mitochondria.
GSK3β directly controls Cyp-F activation, regulates MPT and cellular viability in podocytes
To further ascertain the role of GSK3β in the regulation of Cyp-F in podocytes, the activity of GSK3β was specifically manipulated in cultured podocytes by transient transfection of vectors encoding the HA-conjugated KD mutant or S9A. As shown in Figure 6A, the immunoblot for native and HA-conjugated GSK3β demonstrated a satisfactory transfection efficiency. Forced expression of S9A considerably enhanced, whereas ectopic expression of KD significantly abolished, the adriamycin-elicited serine phosphorylation of Cyp-F (Figure 6A). In parallel, the adriamycin-induced MPT, as measured by the calcium overload-induced mitochondrial swelling as a decrease in spectrophotometric absorbance at 540 nm, was markedly sensitized in S9A-expressing podocytes, but substantially blunted in KD-expressing podocytes (Figure 6B). In agreement with the function of mitochondria to reduce tetrazolium to formazan, S9A expression aggravated, while KD expression attenuated the adriamycin-impaired podocyte viability as measured by the MTT assay (Figure 6C). Moreover, to a lesser extent, adriamycin elicited podocyte death as determined by the flow cytometry-based annexin V/PI apoptosis assay, and this effect was potentiated in S9A-expressing podocytes, but was abolished in KD-expressing podocytes, (Figure 6D). The discrepancy between the degree of impairment of podocyte viability as measured by the MTT assay and the extent of apoptosis suggests that an alternative mode of podocyte death might be involved, such as programmed necrosis (Baines, 2010).
Figure 6.
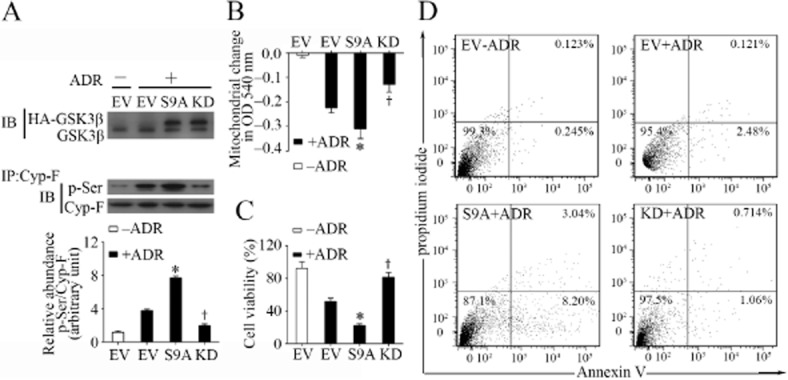
GSK3β controls Cyp-F phosphorylation and regulates MPT in podocytes upon adriamycin injury. (A) Differentiated podocytes were transfected with an EV or vectors encoding the HA-conjugated dominant negative KD mutant, or constitutively active mutant (S9A) of GSK3β for 48 h. Cells were then treated with adriamycin (0.25 μg·mL−1) or vehicle for 24 h before cell lysates were prepared. Cell lysates were subjected to immunoblot analysis for GSK3β or immunoprecipitation using an anti-Cyp-F antibody, and immunoprecipitates were probed for Cyp-F and phosphorylated serine (p-Ser). Densitometric analysis of immunoblots demonstrated the relative abundance of phosphorylated Cyp-F measured as densitometric ratios of phosphorylated serine and Cyp-F and expressed as fold of the vehicle treated (−ADR) EV group. *,†P < 0.05 versus EV + ADR; (n = 6). (B) Cells were transfected and treated as described in (A) and following different treatments cells were collected and mitochondria extracted. MPT was assessed by the decrease in spectrophotometric absorbance of calcium-challenged mitochondria at 540 nm. *,†P < 0.05 versus EV + adriamycin; (n = 6). (C) Cells were transfected and treated as described in (A) and following different treatments podocyte viability was estimated by the MTT assay. *,†P < 0.05 versus EV + ADR; (n = 6). (D) Podocytes were transfected and treated as described in (A) and following the different treatments cells were prepared for staining of Alexa Fluor-annexin V and PI. Subsequently, podocytes apoptosis was determined by flow cytometry.
Inhibition of GSK3β by TDZD-8 prevents Cyp-F overactivation, desensitizes MPT and prevents podocyte mitochondrial damage in adriamycin nephropathy
To validate the GSK3β-directed control of Cyp-F activity and MPT in vivo, glomeruli isolated from differently treated mice (Figure 7A) were analysed. Adriamycin injury reduced the inhibitory phosphorylation of GSK3β, inferring GSK3β overactivity (Figure 7B). This was associated with a prominently increased phosphorylation and activation of Cyp-F (Figure 7C). In contrast, TDZD-8 abolished GSK3β overactivity and blocked the adriamycin-amplified Cyp-F activation. Moreover, glomerular mitochondria derived from adriamycin-injured kidneys exhibited a greater reduction in absorbance at 540 nm upon calcium overload as compared with glomerular mitochondria derived from control mice (Figure 7D), denoting a potentiated MPT and prominent mitochondrial dysfunction. This effect was largely attenuated in glomerular mitochondria prepared from TDZD-8 pretreated mice, indicating a mitochondrial protective effect of TDZD-8. Furthermore, renal cortical specimens were subjected to electron microscopy to evaluate the effect on mitochondrial morphology. Adriamycin injury resulted in prominent mitochondrial damage in podocytes in the glomerulus (Figure 7E), defined as decreased total mitochondrial numbers, mitochondrial swelling and fragmentation, destruction of the inner mitochondrial membrane, and disruption of mitochondrial cristae. This injurious effect on mitochondria was attenuated by TDZD-8 treatment.
Figure 7.
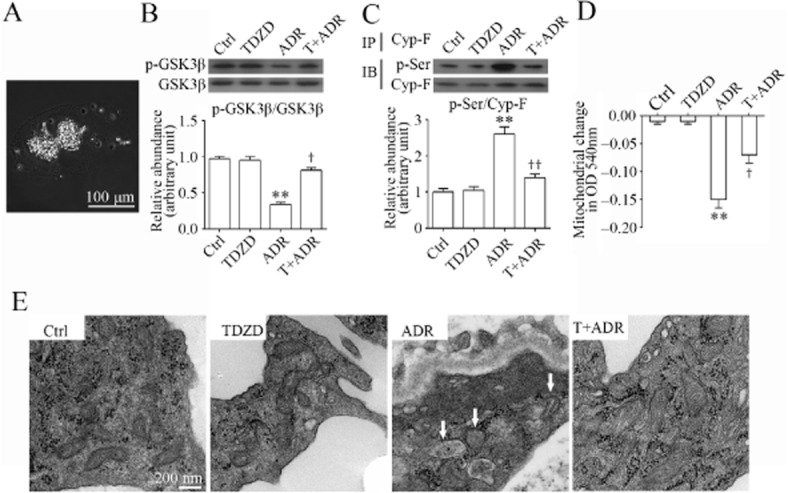
TDZD-8 counteracts GSK3β overactivity, prevents Cyp-F hyperphosphorylation and attenuates MPT and mitochondrial damages in podocytes in adriamycin-injured kidneys. (A) A representative micrograph showing glomeruli, isolated from kidneys by use of the magnetic beads-based approach. (B) Homogenates of glomeruli isolated from the differently-treated animals on day 14 were subjected to immunoblot analysis of phosphorylated GSK3β and total GSK3β. Densitometric analysis of immunoblots estimated the relative abundance of phosphorylated GSK3β in glomeruli from the differently-treated mice. **P < 0.01 versus Ctrl; †P < 0.05 versus ADR; (n = 15). (C) Homogenates of glomeruli isolated from the differently-treated animals on day 14 were subjected to immunoprecipitation by an anti-Cyp-F antibody, and immunoprecipitates were probed for Cyp-F or p-serine (p-Ser). Densitometric analysis of immunoblots demonstrated the relative abundance of phosphorylated Cyp-F in glomeruli from the differently- treated mice measured as densitometric ratios of phosphorylated serine and Cyp-F and expressed as fold of the control group. **P < 0.01 versus Ctrl; ††P < 0.01 versus adriamycin; (n = 15). (D) Mitochondria were extracted from glomeruli freshly isolated from the differently-treated animals on day 14. MPT was assessed by the decrease in spectrophotometric absorbance of calcium-challenged mitochondria at 540 nm. **P < 0.01 versus Ctrl; †P < 0.05 versus ADR; (n = 15). (E) Kidney cortex tissues were obtained from the differently-treated animals on day 14 and prepared for electron microscopy. Representative electron micrographs of glomerulus demonstrated severe mitochondrial damage in podocytes in adriamycin-injured mice kidneys (indicated by arrows), defined as decreased total mitochondrial numbers, mitochondrial swelling and fragmentation, destruction of the inner mitochondrial membrane, and disruption of mitochondrial cristae. This ADR-induced mitochondrial injury was significantly attenuated by TDZD-8 treatment.
Discussion
Despite advances in immunosuppression and blockade of the renin–angiotensin system, debilitating podocytopathies including minimal change disease and focal segmental glomerulosclerosis, which are clinically manifested with massive proteinuria and glomerulosclerosis, continue to be a therapeutic challenge (Kuhn et al., 2006; Gong, 2011). In an effort to develop a novel podocyte protective treatment, numerous potential therapeutic targets in podocytes have been tested (Durvasula and Shankland, 2006; Geroldi et al., 2006). To the best of our knowledge, this study is the first attempt to test the effect of GSK3β inhibition on mitochondria dysfunction and the ensuing cellular injury in podocytes. Our data demonstrated that TDZD-8, a highly selective small-molecule inhibitor of GSK3β-desensitized MPT, attenuated the adriamycin-elicited mitochondria dysfunction in podocytes in vitro and in vivo, and thereby improved podocyte viability, prevented podocyte death, improved podocyte injury, and ameliorated proteinuria and glomerular sclerosis.
Podocytes are a core structural constituent of the glomerular filtration barrier (Kawachi et al., 2006). Through their constant motility driven by intricate cytoskeletal machinery, glomerular podocytes play a pivotal role in withstanding the pulsating hydrostatic pressure and maintaining the structural and functional integrity of this filtration barrier. In addition, podocytes are actively involved in other energy-intensive cellular events, including membrane trafficking and recycling and the transcytosis of the primary filtrate (Swiatecka-Urban, 2013). All of these high energy-consuming cellular processes are driven by ATP and powered primarily by mitochondria in podocytes. Therefore, mitochondrial homeostasis is essential for podocyte health. There is accumulating evidence supporting the view that mitochondrial dysfunction is centrally involved in podocyte injury and progressive glomerular sclerosis (Papeta et al., 2010; Su et al., 2013). Patients with congenital mitochondrial disorders, including defects in respiratory chain complexes, such as mutations in the COQ2 genes, frequently present as steroid-resistant nephrotic syndrome and podocytopathies (Goldenberg et al., 2005; Diomedi-Camassei et al., 2007; Machuca et al., 2010). Conversely, coenzyme Q10 supplementation induced proteinuria remission and an improvement in renal disease in patients with aarF domain containing kinase 4 gene mutations, which disrupt coenzyme Q10 biosynthesis and reduce mitochondrial respiratory enzyme activity (Ashraf et al., 2013; Malaga-Dieguez and Susztak, 2013; Ozaltin, 2014). In non-congenital podocytopathies induced by various harmful stimuli, such as aldosterone, hyperglycaemia and toxins like adriamycin, mitochondrial dysfunction has also been implicated as a major pathogenic mechanism (Mathieson, 2009). Accordingly, correction of this mitochondrial dysfunction by up-regulation of PPAR-γ coactivator 1α or by supplementation with free-radical scavengers like NAC substantially protected against podocyte injury and ameliorated proteinuria and glomerular sclerosis (Zheng et al., 2008; Yuan et al., 2012). MPT is a hallmark of mitochondria dysfunction, and the MPT threshold is regulated by the activation status of the structural components of the MPT pore, including Cyp-F (Baines et al., 2005). An elevation of the MPT threshold, subsequent to inhibition of the activity of Cyp-F, has been found to prevent mitochondrial dysfunction and reduce cell death (Crompton, 1999). In the present study, we demonstrated that GSK3β-controlled phosphorylation and activation of Cyp-F mediated adriamycin injury-induced oxidative stress, enhanced the activity of the redox-sensitive GSK3β, and resulted in hyperphosphorylation and overactivation of Cyp-F in glomerular podocytes. All these events were associated with a more sensitized MPT and aggravated mitochondrial dysfunction. In contrast, TDZD-8 blocked GSK3β activity, prevented Cyp-F overactivation and this was accompanied by a desensitized MPT and improved mitochondrial dysfunction, resulting in a diminished podocyte death, a reduction in glomerular podocyte injury, and amelioration of proteinuria and glomerulosclerosis.
GSK3β is a well-conserved, ubiquitously expressed serine/threonine PK that participates in numerous cellular processes, including proliferation, apoptosis, and necrosis, and has an important role in the pathophysiology of a number of diseases, including kidney diseases (Gong et al., 2008b; Ge et al., 2010). Indeed, recent evidence suggests that GSK3β plays a detrimental role in acute kidney injury. Inhibition of GSK3β by small-molecule inhibitors or genetic knockout strikingly prevented acute renal histological injury induced by ischaemia reperfusion and nephrotoxic substances, such as non-steroidal anti-inflammatory drugs, mercury and the pro-oxidant paraquat (Plotnikov et al., 2007; Bao et al., 2012; Wang et al., 2013). The role of GSK3β in podocyte injury, however, remains uncertain. We recently found that the expression of GSK3β is aberrantly up-regulated in diseased human kidneys, in both tubules and glomeruli (Gong et al., 2008a). Waters and Koziell (2009) also noted an up-regulation of GSK3β in human podocytes in association with NPHS1 mutations, suggesting that GSK3β deregulation might be involved in podocyte dysfunction. In line with these observations, inhibition of GSK3β by a selective small-molecule inhibitor 2′Z, 3′E-6-bromoindirubin-3′-oxime (BIO) at a low dose dramatically ameliorated proteinuria and attenuated glomerular injury in rat models of diabetic nephropathy, even though hyperglycaemia was not affected, suggesting a direct anti-proteinuric and renoprotective action (Lin et al., 2006). However, in stark contrast, Matsui et al. found that a high-dose of BIO exacerbated proteinuria and loss of glomerular nephrin in puromycin-injured rats (Matsui et al., 2007). Also, another study Dai et al. (2009) reported that transient proteiuria was induced by an ultrahigh dose of lithium in normal mice, but immediately receded to normal levels within 24 h. Both studies concluded that inhibition of GSK3β worsens podocytopathy. These data sharply contradict those obtained in gene-targeted knock-in mice with a mutated GSK3 that cannot be inhibited, which developed spontaneous albuminuria and podocyte injury (Boini et al., 2008), suggesting a detrimental role of GSK3β in podocyte injury. As mentioned earlier, Matsui et al. used high-dose BIO, which is about 10 times higher than that used in the earlier study of diabetic nephropathy (Matsui et al., 2007) and Dai et al. used an ultrahigh dose of lithium chloride (16 mmol·kg−1), which is almost 2 times the median lethal dose of lithium chloride in mice (Dai et al., 2009). At ultrahigh doses, both lithium and BIO could have non-selective off-target effects, and could thus induce cytotoxic and even lethal actions. Collectively, it seems that low-dose GSK3β inhibitors have a beneficial effect that protects podocytes from injury, and ameliorates proteinura and glomerulopathy, whereas ultrahigh doses of GSK3β inhibitors have a toxic effect on podocytes and worsen glomerular injury. In support of this viewpoint, the results from the present study indicate that inhibition of GSK3β by both a regular dose and a low dose of TDZD-8 prevents podocyte injury and partially attenuates proteinuria and glomerular sclerosis. Based on these observations, the efficacy of multi-dose TDZD-8 treatment merits further exploration in animal models of proteinuria and podocytopathy.
In summary, TDZD-8, a highly selective small-molecule inhibitor of GSK3β, counteracted the adriamycin-induced overactivity of the redox-sensitive GSK3β, inhibited the GSK3β-controlled phosphorylation and activation of Cyp-F, desensitized MPT in podocytes, and prevented adriamycin-induced proteinuria and glomerular sclerosis. Our findings suggest that pharmacological targeting of GSK3β might represent a novel, promising and feasible therapeutic strategy to desensitize MPT in podocytes and to protect podocytes against mitochondrial dysfunction induced by oxidative injuries.
Acknowledgments
Z. W. and H. B. are International Society of Nephrology (ISN) Fellows and recipients of the ISN fellowship. This work was made possible, in part, by the support from the U.S. National Institutes of Health grant R01DK092485, the Natural Science Foundation of China (Program Nos. 81101414/H1503, 81171792/H1503, 81270136/H0111) and the ISN Sister Renal Center Trio Program.
Glossary
- BIO
2′Z, 3′E-6-bromoindirubin-3′-oxime
- Cyp-F
cyclophilin F
- DCF-DA
2′, 7′-dichlorofluorescein-diacetate
- EV
empty vector
- GSK3β
glycogen synthase kinase 3β
- HA
haemagglutinin
- KD
kinase dead
- MPT
mitochondria permeability transition
- MTT
3-4,5-dimethylthiazol-2,5-diphenyltetrazolium bromide
- NAC
N-acetyl-L-cysteine
- PAS
periodic acid–Schiff
- PI
propidium iodide
- ROS
reactive oxygen species
- S9A
constitutively active mutant of GSK3β
- TDZD-8
4-benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione
- WT-1
Wilms tumour 1
Author contributions
R. G. and Z. W. conceived the project. Z. W., Y. G., H. B. and R. G. carried out most of the experiments. Z. W. conducted flow cytometry and analysis. Z. W., Y. G. and H. B. performed the animal experiment, glomerular isolation and histology analyses. Z. W. and H. B. performed transient transfection, cellular staining, TUNEL assay and Western blot analysis. Z. W. and Y. G. carried out mitochondria preparation and performed MPT assay with the advice of R. G., S. Z. and A. P. Z. W. and R. G. performed data interpretation. Z. W and R. G. analysed the data and wrote the paper with the help of all authors.
Conflict of interest
None of the authors has any potential financial conflict of interest related to this manuscript.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest. 2013;123:5179–5189. doi: 10.1172/JCI69000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines CP. Role of the mitochondrion in programmed necrosis. Front Physiol. 2010;29:156. doi: 10.3389/fphys.2010.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Bao H, Ge Y, Zhuang S, Dworkin LD, Liu Z, Gong R. Inhibition of glycogen synthase kinase-3beta prevents NSAID-induced acute kidney injury. Kidney Int. 2012;81:662–673. doi: 10.1038/ki.2011.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boini KM, Amann K, Kempe D, Alessi DR, Lang F. Proteinuria in mice expressing PKB/SGK-resistant GSK3. Am J Physiol Renal Physiol. 2008;296:F153–F159. doi: 10.1152/ajprenal.90398.2008. [DOI] [PubMed] [Google Scholar]
- Chuang P, Yu Q, Fang W, Uribarri J, He J. Advanced glycation endproducts induce podocyte apoptosis by activation of the FOXO4 transcription factor. Kidney Int. 2007;72:965–976. doi: 10.1038/sj.ki.5002456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48:749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, Gusdon AM, Pien I, Strack S, Green S, Li C, et al. Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson's disease. Cell Death Differ. 2011;18:1914–1923. doi: 10.1038/cdd.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Stolz DB, Kiss LP, Monga SP, Holzman LB, Liu Y. Wnt/beta-catenin signaling promotes podocyte dysfunction and albuminuria. J Am Soc Nephrol. 2009;20:1997–2008. doi: 10.1681/ASN.2009010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diomedi-Camassei F, Di Giandomenico S, Santorelli FM, Caridi G, Piemonte F, Montini G, et al. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol. 2007;18:2773–2780. doi: 10.1681/ASN.2006080833. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durvasula RV, Shankland SJ. Podocyte injury and targeting therapy: an update. Curr Opin Nephrol Hypertens. 2006;15:1–7. doi: 10.1097/01.mnh.0000199012.79670.0b. [DOI] [PubMed] [Google Scholar]
- Durvasula RV, Petermann AT, Hiromura K, Blonski M, Pippin J, Mundel P, et al. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int. 2004;65:30–39. doi: 10.1111/j.1523-1755.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- Enrique Guerrero-Beltran C, Calderon-Oliver M, Martinez-Abundis E, Tapia E, Zarco-Marquez G, Zazueta C, et al. Protective effect of sulforaphane against cisplatin-induced mitochondrial alterations and impairment in the activity of NAD(P)H: quinone oxidoreductase 1 and gamma glutamyl cysteine ligase: studies in mitochondria isolated from rat kidney and in LLC-PK1 cells. Toxicol Lett. 2010;199:80–92. doi: 10.1016/j.toxlet.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Ge Y, Si J, Tian L, Zhuang S, Dworkin LD, Gong R. Conditional ablation of glycogen synthase kinase 3β in postnatal mouse kidney. Lab Invest. 2010;91:85–96. doi: 10.1038/labinvest.2010.142. [DOI] [PubMed] [Google Scholar]
- Geroldi D, Falcone C, Emanuele E. Soluble receptor for advanced glycation end products: from disease marker to potential therapeutic target. Curr Med Chem. 2006;13:1971–1978. doi: 10.2174/092986706777585013. [DOI] [PubMed] [Google Scholar]
- Goldenberg A, Ngoc LH, Thouret M-C, Cormier-Daire V, Gagnadoux M-F, Chrétien D, et al. Respiratory chain deficiency presenting as congenital nephrotic syndrome. Pediatr Nephrol. 2005;20:465–469. doi: 10.1007/s00467-004-1725-4. [DOI] [PubMed] [Google Scholar]
- Gong R. The renaissance of corticotropin therapy in proteinuric nephropathies. Nat Rev Nephrol. 2011;8:122–128. doi: 10.1038/nrneph.2011.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong R, Rifai A, Dworkin LD. Hepatocyte growth factor suppresses acute renal inflammation by inhibition of endothelial E-selectin. Kidney Int. 2006;69:1166–1174. doi: 10.1038/sj.ki.5000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong R, Ge Y, Chen S, Liang E, Esparza A, Sabo E, et al. Glycogen synthase kinase 3β: a novel marker and modulator of inflammatory injury in chronic renal allograft disease. Am J Transplant. 2008a;8:1852–1863. doi: 10.1111/j.1600-6143.2008.02319.x. [DOI] [PubMed] [Google Scholar]
- Gong R, Rifai A, Ge Y, Chen S, Dworkin LD. Hepatocyte growth factor suppresses proinflammatory NF kappa B activation through GSK3 beta inactivation in renal tubular epithelial cells. J Biol Chem. 2008b;283:7401–7410. doi: 10.1074/jbc.M710396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P, Sun H, Xu Y, Zeng Y, Yi W, Wu J, et al. Lisinopril protects against the adriamycin nephropathy and reverses the renalase reduction: potential role of renalase in adriamycin nephropathy. Kidney Blood Press Res. 2013;37:295–304. doi: 10.1159/000350157. [DOI] [PubMed] [Google Scholar]
- Haraldsson B, Nystroem J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008;88:451–487. doi: 10.1152/physrev.00055.2006. [DOI] [PubMed] [Google Scholar]
- Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K-I, et al. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–535. doi: 10.1161/01.res.88.5.529. [DOI] [PubMed] [Google Scholar]
- Kawachi H, Miyauchi N, Suzuki K, Han GD, Orikasa M, Shimizu F. Role of podocyte slit diaphragm as a filtration barrier (review article) Nephrology (Carlton) 2006;11:274–281. doi: 10.1111/j.1440-1797.2006.00583.x. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koya D, Hayashi K, Kitada M, Kashiwagi A, Kikkawa R, Haneda M. Effects of antioxidants in diabetes-induced oxidative stress in the glomeruli of diabetic rats. J Am Soc Nephrol. 2003;14(Suppl. 3):S250–S253. doi: 10.1097/01.asn.0000077412.07578.44. [DOI] [PubMed] [Google Scholar]
- Kuhn C, Kuhn A, Markau S, Kästner U, Osten B. Effect of immunoadsorption on refractory idiopathic focal and segmental glomerulosclerosis. J Clin Apher. 2006;21:266–270. doi: 10.1002/jca.20104. [DOI] [PubMed] [Google Scholar]
- Lee SH, Nam BY, Kang EW, Han SH, Li JJ, Kim SH, et al. Effects of an oral adsorbent on oxidative stress and fibronectin expression in experimental diabetic nephropathy. Nephrol Dial Transplant. 2010;25:2134–2141. doi: 10.1093/ndt/gfq063. [DOI] [PubMed] [Google Scholar]
- Lin C-L, Wang J-Y, Huang Y-T, Kuo Y-H, Surendran K, Wang F-S. Wnt/β-catenin signaling modulates survival of high glucose-stressed mesangial cells. J Am Soc Nephrol. 2006;17:2812–2820. doi: 10.1681/ASN.2005121355. [DOI] [PubMed] [Google Scholar]
- Liu S, Jia Z, Zhou L, Liu Y, Ling H, Zhou S-F, et al. Nitro-oleic acid protects against adriamycin-induced nephropathy in mice. Am J Physiol Renal Physiol. 2013;305:F1533–F1541. doi: 10.1152/ajprenal.00656.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machuca E, Benoit G, Nevo F, Tête M-J, Gribouval O, Pawtowski A, et al. Genotype–phenotype correlations in non-Finnish congenital nephrotic syndrome. J Am Soc Nephrol. 2010;21:1209–1217. doi: 10.1681/ASN.2009121309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaga-Dieguez L, Susztak K. ADCK4 ‘reenergizes’ nephrotic syndrome. J Clin Invest. 2013;123:4996–4999. doi: 10.1172/JCI73168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson PW. Update on the podocyte. Curr Opin Nephrol Hypertens. 2009;18:206–211. doi: 10.1097/mnh.0b013e328326f3ca. [DOI] [PubMed] [Google Scholar]
- Matsui I, Ito T, Kurihara H, Imai E, Ogihara T, Hori M. Snail, a transcriptional regulator, represses nephrin expression in glomerular epithelial cells of nephrotic rats. Lab Invest. 2007;87:273–283. doi: 10.1038/labinvest.3700518. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura T, Nishihara M, Miki T. Drug development targeting the glycogen synthase kinase-3β (GSK-3β)-mediated signal transduction pathway: role of GSK-3β in myocardial protection against ischemia/reperfusion injury. J Pharmacol Sci. 2009;109:162–167. doi: 10.1254/jphs.08r27fm. [DOI] [PubMed] [Google Scholar]
- Moreira PI, Zhu X, Wang X, Lee H-G, Nunomura A, Petersen RB, et al. Mitochondria: a therapeutic target in neurodegeneration. Biochim Biophys Acta. 2010;1802:212–220. doi: 10.1016/j.bbadis.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihara M, Miura T, Miki T, Tanno M, Yano T, Naitoh K, et al. Modulation of the mitochondrial permeability transition pore complex in GSK-3beta-mediated myocardial protection. J Mol Cell Cardiol. 2007;43:564–570. doi: 10.1016/j.yjmcc.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Ozaltin F. Primary coenzyme Q10 (CoQ10) deficiencies and related nephropathies. Pediatr Nephrol. 2014;29:961–969. doi: 10.1007/s00467-013-2482-z. [DOI] [PubMed] [Google Scholar]
- Papeta N, Zheng Z, Schon EA, Brosel S, Altintas MM, Nasr SH, et al. Prkdc participates in mitochondrial genome maintenance and prevents Adriamycin-induced nephropathy in mice. J Clin Invest. 2010;120:4055–4064. doi: 10.1172/JCI43721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikov E, Kazachenko A, Vyssokikh MY, Vasileva A, Tcvirkun D, Isaev N, et al. The role of mitochondria in oxidative and nitrosative stress during ischemia/reperfusion in the rat kidney. Kidney Int. 2007;72:1493–1502. doi: 10.1038/sj.ki.5002568. [DOI] [PubMed] [Google Scholar]
- Rasola A, Sciacovelli M, Chiara F, Pantic B, Brusilow WS, Bernardi P. Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc Natl Acad Sci U S A. 2010;107:726–731. doi: 10.1073/pnas.0912742107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AM, Oliveira PJ. Evaluation of respiration with clark type electrode in isolated mitochondria and permeabilized animal cells. Methods Mol Biol. 2012;810:7–24. doi: 10.1007/978-1-61779-382-0_2. [DOI] [PubMed] [Google Scholar]
- Su M, Dhoopun A-R, Yuan Y, Huang S, Zhu C, Ding G, et al. Mitochondrial dysfunction is an early event in aldosterone-induced podocyte injury. Am J Physiol Renal Physiol. 2013;305:F520–F531. doi: 10.1152/ajprenal.00570.2012. [DOI] [PubMed] [Google Scholar]
- Swiatecka-Urban A. Membrane trafficking in podocyte health and disease. Pediatr Nephrol. 2013;28:1723–1737. doi: 10.1007/s00467-012-2281-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, et al. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol. 2011;22:1041–1052. doi: 10.1681/ASN.2010080808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, et al. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221–1231. doi: 10.1172/JCI21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc Res. 2009;81:449–456. doi: 10.1093/cvr/cvn280. [DOI] [PubMed] [Google Scholar]
- Wang Z, Ge Y, Bao H, Dworkin L, Peng A, Gong R. Redox-sensitive glycogen synthase kinase 3β-directed control of mitochondrial permeability transition: rheostatic regulation of acute kidney injury. Free Radic Biol Med. 2013;65:849–858. doi: 10.1016/j.freeradbiomed.2013.08.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters A, Koziell A. Activation of canonical Wnt signaling meets with podocytopathy. J Am Soc Nephrol. 2009;20:1864–1866. doi: 10.1681/ASN.2009070762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Huang S, Wang W, Wang Y, Zhang P, Zhu C, et al. Activation of peroxisome proliferator-activated receptor-γ coactivator 1α ameliorates mitochondrial dysfunction and protects podocytes from aldosterone-induced injury. Kidney Int. 2012;82:771–789. doi: 10.1038/ki.2012.188. [DOI] [PubMed] [Google Scholar]
- Zheng C-X, Chen Z-H, Zeng C-H, Qin W-S, Li L-S, Liu Z-H. Triptolide protects podocytes from puromycin aminonucleoside induced injury in vivo and in vitro. Kidney Int. 2008;74:596–612. doi: 10.1038/ki.2008.203. [DOI] [PubMed] [Google Scholar]
- Zhu C, Huang S, Yuan Y, Ding G, Chen R, Liu B, et al. Mitochondrial dysfunction mediates aldosterone-induced podocyte damage a therapeutic target of PPAR gamma. Am J Pathol. 2011;178:2020–2031. doi: 10.1016/j.ajpath.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]