

Abstract
Background and Purpose
Drug-induced arrhythmia due to blockade of the Kv11.1 channel (also known as the hERG K+ channel) is a frequent side effect. Previous studies have primarily focused on equilibrium parameters, i.e. affinity or potency, of drug candidates at the channel. The aim of this study was to determine the kinetics of the interaction with the channel for a number of known Kv11.1 blockers and to explore a possible correlation with the affinity or physicochemical properties of these compounds.
Experimental Approach
The affinity and kinetic parameters of 15 prototypical Kv11.1 inhibitors were evaluated in a number of [3H]-dofetilide binding assays. The lipophilicity (logKW-C8) and membrane partitioning (logKW-IAM) of these compounds were determined by means of HPLC analysis.
Key Results
A novel [3H]-dofetilide competition association assay was set up and validated, which allowed us to determine the binding kinetics of the Kv11.1 blockers used in this study. Interestingly, the compounds' affinities (Ki values) were correlated to their association rates rather than dissociation rates. Overall lipophilicity or membrane partitioning of the compounds were not correlated to their affinity or rate constants for the channel.
Conclusions and Implications
A compound's affinity for the Kv11.1 channel is determined by its rate of association with the channel, while overall lipophilicity and membrane affinity are not. In more general terms, our findings provide novel insights into the mechanism of action for a compound's activity at the Kv11.1 channel. This may help to elucidate how Kv11.1-induced cardiotoxicity is governed and how it can be circumvented in the future.
Tables of Links
| TARGETS |
|---|
| GPCRsa |
| β2-adrenoceptor |
| D2 receptor |
| OX2 receptor |
| P2Y12 receptor |
| Ion channelsb |
| Kv11.1 (hERG) |
| LIGANDS | ||
|---|---|---|
| Amiodarone | Clopidogrel | Ranolazine |
| Astemizole | Dofetilide | Sertindole |
| Chlorpromazine | E-4031 | Sotalol |
| Cisapride | Ibutilide | Terfenadine |
| Clofilium | Pimozide | Thioridazine |
| Clopidogrel | Ranolazine |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al.,2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b).
Introduction
The Kv11.1 channel, a voltage-gated potassium channel previously known as human ether-à-go-go related gene (hERG), encodes the pore-forming subunit of the rapid component of the delayed rectifier K+ channel, IKr, which contributes to phase 3 repolarization in cardiac action potentials (Doyle et al., 1998; Nerbonne, 2000; Vandenberg et al., 2001; 2012). Genetic defects or drug blockade of the Kv11.1 channel normally cause delayed repolarization of cardiac action potentials and Torsades de Pointes (TdP) (Hancox et al., 2008). In some cases, TdP can degenerate into ventricular fibrillation and lead to sudden death (Sanguinetti and Tristani-Firouzi, 2006; Rampe and Brown, 2013). Currently, a number of drugs, including astemizole and cisapride, have been withdrawn from the market due to their ability to block the Kv11.1 channel (Fitzgerald and Ackerman, 2005). In addition, either ‘black box’ warnings or significant restrictions have been rendered for several other drugs, such as droperidol and ibutilide (Fitzgerald and Ackerman, 2005). Withdrawing or restricting drugs due to their Kv11.1 liability in less than 1% of the patient population causes unpredictable and huge costs to the industry and brings troubles for patients as well, owing that some effective drugs would be beneficial for the vast majority of patients (Fitzgerald and Ackerman, 2005; Noble, 2008).
In the past decades, a series of techniques including whole-cell patch-clamp technologies, radioligand binding assays and ion flux assays have been developed and used to screen for Kv11.1 liability of compounds (Hancox et al., 2008; Heijman et al., 2014). From these, equilibrium radioligand displacement assays have been suggested as a low-cost and high-throughput technology alternative to other available methods and are widely employed to obtain the affinity of compounds at the Kv11.1 channel (Krohn, 2001; Chiu et al., 2004; Diaz et al., 2004). These studies provide equilibrium parameters, such as IC50 or Ki values. For instance, a 30-fold margin between Kv11.1 IC50 and maximum free plasma concentration (Cmax) has been recommended to assess drug safety with respect to arrhythmogenesis (Redfern et al., 2003). However, the kinetics of the interaction between a drug and the Kv11.1 channel may be equally, or even more, important. Knowledge of association and dissociation rate constants has already led to better insights in the action of drugs at other targets, such as enzymes and GPCRs (Copeland et al., 2006; Pan et al., 2013).
In the present study, we hypothesized that a slow association rate (kon) to and/or a fast dissociation rate constant (koff) from the channel may be beneficial to reduce or avoid Kv11.1-related cardiovascular side effects. Hence, a detailed understanding of a ligand's binding kinetics at the channel may provide clues to enable the optimization of its kinetic profiles and potentially ‘rescue’ it from removal of the drug discovery pipeline. In this context, we describe the development and validation of a [3H]-dofetilide competition association assay in HEK293 cell membranes stably expressing the Kv11.1 (hERG) channel (HEK293Kv11.1) to determine the kinetic binding parameters of 15 unlabelled reference compounds from different ‘Redfern’ categories (Redfern et al., 2003) (Table 1). We systematically investigated both equilibrium affinity and kinetic binding data of those compounds and compared these parameters with the compounds' physicochemical properties. Because the interaction of drugs with membrane phospholipids is not only driven by lipophilicity but also electrostatic interactions with both acidic and basic moieties on phosphate head groups (Taillardat-Bertschinger et al., 2003; Sykes et al., 2014), a regular column and an immobilized artificial membrane (IAM) column were used to derive two hydrophobic parameters for these compounds, i.e. logKW-C8 and logKW-IAM respectively. Finally, both hydrophobic parameters were compared with the compounds' affinity and kinetic data. Taken together, the present study provides a novel approach to study the kinetics of the interaction between a drug and the Kv11.1 channel, which may help to more precisely assess Kv11.1 liabilities of drug candidates in the future.
Table 1.
Chemical structures and information on the use of 15 Kv11.1 blockers examined in this study
| Name | Chemical structure | Therapeutic class | TdPa | Redfern categoryb | Status in the United Statesa |
|---|---|---|---|---|---|
| Dofetilide | 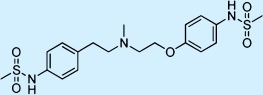 |
Antiarrhythmic | ++ | 1 | Restricted |
| Astemizole | 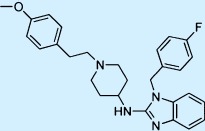 |
Antihistamine | ++ | 2 | Withdrawn |
| E-4031 | 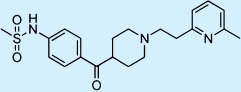 |
Antiarrhythmic | ++ | 1† | Not approved |
| Sertindole | 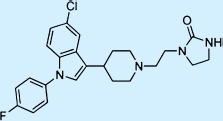 |
Antipsychotic | + | 2 | Not approved |
| Terfenadine | 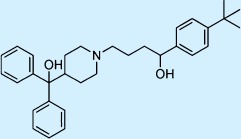 |
Antihistamine | ++ | 2 | Withdrawn |
| Moxifloxacin | 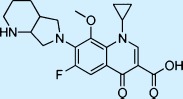 |
Antibacterial | ++ | 3† | Restricted |
| Amiodarone | 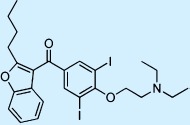 |
Antiarrhythmic | ++ | 1 | Restricted |
| Chlorpromazine | 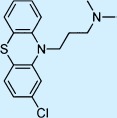 |
Antipsychotic | ++ | 3† | Restricted |
| Ibutilide | 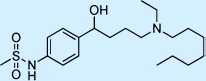 |
Antiarrhythmic | ++ | 1 | Restricted |
| Clofilium | 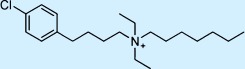 |
Antiarrhythmic | ++ | 1† | Not approved |
| Pimozide | 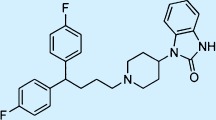 |
Antipsychotic | ++ | 3 | Restricted |
| Cisapride | 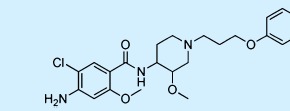 |
Gastroprokinetic | ++ | 2 | Withdrawn |
| Ranolazine |  |
Antianginal | + | 5† | Restricted |
| Sotalol |  |
Antiarrhythmic | ++ | 1 | Restricted |
| Thioridazine |  |
Antipsychotic | ++ | 3 | Restricted |
+, drugs with possible TdP risk and ++, drugs with known TdP risk. Information on these compounds was retrieved from CredibleMeds® available at: http://crediblemeds.org/ (accessed 10 April 2014).
Redfern categories for most compounds were derived from the literature (Redfern et al., 2003); they are 1: Class Ia and III antiarrythmics; 2: Withdrawn from market due to TdP; 3: Measurable incidence/numerous reports of TdP in humans; 4: Isolated reports of TdP in humans; 5: No reports of TdP in humans.
Redfern categories for these compounds were deduced according to the definition for different categories.
Methods
Cell culture and membrane preparation
HEK293Kv11.1 cells were cultured and membranes were prepared and stored as described previously (Yu et al., 2014).
Radioligand saturation assay
Membrane aliquots containing 20 μg of protein were incubated in a total volume of 100 μL of incubation buffer (10 mM HEPES, 130 mM NaCl, 60 mM KCl, 0.8 mM MgCl2, 1 mM EGTA, 10 mM glucose, 0.1% BSA, pH 7.4) at 25°C for 120 min to ensure that the equilibrium was reached at all concentrations of radioligand. Total binding was determined at a range of concentrations (0.2∼22 nM) of [3H]-dofetilide, whereas non-specific binding was determined at three different concentrations of radioligand in the presence of 10 μM astemizole and analysed by linear regression. Incubations were terminated by dilution with ice-cold wash buffer (25 mM Tris-HCl, 130 mM NaCl, 60 mM KCl, 0.8 mM MgCl2, 0.05 mM CaCl2, 0.05% BSA, pH 7.4). Separation of bound from free radioligand was performed by rapid filtration through Whatman GF/B filters (GE Healthcare, Buckinghamshire, UK) using a Brandel harvester (Brandel, MD, USA). Filters were subsequently washed six times with 2 mL ice-cold wash buffer. Filter-bound radioactivity was determined by scintillation spectrometry using a liquid Scintillation Analyzer (Tri-Carb 2900TR, PerkinElmer, Groningen, The Netherlands) after addition of 3.5 mL of Packard Emulsifier-Safe (PerkinElmer) and 2 h extraction.
Radioligand association and dissociation assay
Kinetic association experiments were performed by incubating membrane aliquots containing 20 μg of protein in a total volume of 100 μL incubation buffer at 25°C for 120 min with 16 different concentrations (0.7∼16 nM) of [3H]-dofetilide. The amount of radioligand bound to the receptor was measured at various time intervals during the incubation. Incubations were terminated and samples were obtained and analysed as described in radioligand saturation assay. Further traditional association and dissociation assays were performed as described previously (Yu et al., 2014).
Radioligand displacement assay
The [3H]-dofetilide binding assay for the Kv11.1 channel was performed as described previously (Yu et al., 2014). In short, membrane aliquots containing 20 μg protein were incubated in a total volume of 100 μL incubation buffer at 25°C for 60 min. Radioligand displacement experiments were conducted using 11 concentrations of the competing ligand in the presence of 5 nM [3H]-dofetilide. At this concentration, total radioligand binding did not exceed 10% of the radioligand added to prevent ligand depletion. Non-specific binding was determined in the presence of 10 μM astemizole and represented approximately 15% of the total binding. [3H]-dofetilide did not bind to membranes prepared from empty HEK293 cells lacking the Kv11.1 channel (data not shown). Total binding was determined in the presence of incubation buffer and was set at 100% in all experiments, whereas non-specific binding was set at 0%. Incubations were terminated by dilution with ice-cold wash buffer. Separation of bound from free radioligand was performed by rapid filtration through a 96-well GF/B filter plate using a PerkinElmer Filtermate-harvester (PerkinElmer). Filters were subsequently washed 12 times with ice-cold wash buffer. The filter-bound radioactivity was determined by scintillation spectrometry using the P-E 1450 Microbeta Wallac Trilux scintillation counter (PerkinElmer) after addition of 25 μL Microscint (PerkinElmer) and 2 h extraction.
Radioligand competition association assay
The binding kinetics of unlabelled reference compounds were determined at 25°C using the competition association assay according to a previously published method (Motulsky and Mahan, 1984). In a standard assay, three different concentrations (0.3-, one- and threefold of their Ki values) of unlabelled dofetilide, astemizole and E-4031 were tested. We assessed the binding kinetics of all other unlabelled reference compounds in a simplified one-concentration competition association assay based on Guo et al. (2012). The experiments were initiated by incubating membrane aliquots containing 20 μg of protein in a total volume of 100 μL of incubation buffer in the absence (control) or presence of a certain concentration of unlabelled ligands at 25°C for 120 min with 5 nM [3H]-dofetilide. The amounts of radioligand bound to the receptor were measured at various time intervals during the incubation. Incubations were terminated and samples were obtained and analysed as described in radioligand displacement assay.
Determination of logKW-C8 and logKW-IAM parameters by HPLC
LogKW-C8 values were measured on a Supelcosil LC-ABZ, 5 cm × 4.6 mm, 5 μm column (Supelco, Bellefonte, PA, USA) according to a methodology described previously (Lombardo et al., 2001; Heitman et al., 2009). In short, retention times of the compounds were determined at three different methanol percentages. These retention times were converted to k values by using the formula k = (tR − t0) / tR in which tR is the retention time and t0 is the retention time of a ‘non-delayed’ compound (pure methanol). The calculated logk values were plotted against the methanol concentrations and extrapolated to a 0% methanol situation yielding the logKW-C8 values for 15 reference compounds (intercept of Y axis).
An isocratic method was applied to measure the logKW-IAM values of all tested compounds on a 10 cm × 4.6 mm, 10 μm Regis IAM PC DD2 column (Regis, Morton Grove, IL, USA) (Valko et al., 2000). Retention times of the compounds were determined at three different concentrations of acetonitrile. The kIAM values were calculated by the equation kIAM = (tR − t0) / tR in which tR represents retention times of tested compounds, whereas t0 is determined by injecting a sodium nitrate solution in the HPLC system. The logkIAM values for a compound were plotted against the applied acetonitrile concentrations. The intercept with the Y axis of the straight line through these data points yielded the extrapolated logKW-IAM values for the 15 reference compounds.
Data analysis
All data of radioligand binding assays were analysed using the non-linear regression curve fitting program Prism v. 5.1 (GraphPad, San Diego, CA, USA). KD and Bmax values of [3H]-dofetilide at HEK293Kv11.1 membranes were obtained by computational analysis of saturation curves. Apparent inhibitory binding constants (Ki values) were derived from the IC50 values according to the Cheng and Prusoff equation Ki = IC50 / (1 + [L*] / KD), where [L*] was the concentration of radioligand and KD was its dissociation constant from the saturation assay (Cheng and Prusoff, 1973). In the kinetic association experiments, the on- and off-rates were derived from the linear regression analysis using the equation kobs = kon[L*] + koff, where the kobs value was obtained by computer analysis of the exponential association of [3H]-dofetilide bound to the receptor with [L*] being the concentration of radioligand. The association and dissociation rates were used to calculate the kinetic KD value using the following equation KD = koff / kon. The association and dissociation rates for unlabelled compounds were calculated by fitting the data into the competition association model using ‘kinetics of competitive binding’ (Motulsky and Mahan, 1984):
Where X is the time (min), Y the specific binding of [3H]-dofetilide, k1 and k2 are the kon (M−1·min−1) and koff (min−1) of [3H]-dofetilide obtained from the traditional association and dissociation assay, L the concentration of [3H]-dofetilide (nM), Bmax the maximum specific binding (dpm) and I the concentration of the unlabelled compound (nM). Fixing these parameters allowed the following parameters to be calculated: k3, which is the kon value (M−1·min−1) of the unlabelled compound and k4, which is the koff value (min−1) of the unlabelled compound. LogKW-C8 and logKW-IAM values were derived from linear regression analysis as mentioned earlier. The MW, logP and pKa values were calculated using a structure-based calculation plug-in provided by ChemAxon (Budapest, Hungary). All values obtained from radioligand binding assays in this study are means of at least three independent experiments performed in duplicate. Statistical analysis was performed with Student's two-tailed unpaired t-test.
Materials
Astemizole, sertindole, terfenadine, moxifloxacin, amiodarone, chlorpromazine, ibutilide, clofilium, pimozide, cisapride, ranolazine, sotalol, thioridazine and all the solutes for HPLC determinations were purchased from Sigma Aldrich (Zwijndrecht, The Netherlands). Dofetilide and E-4031 were synthesized in our own laboratory (Shagufta et al., 2009; Vilums et al., 2012). Tritium-labelled dofetilide (specific activity 65∼87 Ci·mmol−1) was purchased from PerkinElmer (Groningen, The Netherlands). BSA (fraction V) was purchased from Sigma (St. Louis, MO, USA). G418 was obtained from Stratagene (Cedar Creek, TX, USA). The chemicals for HPLC were of HPLC grade; all the other chemicals were of analytical grade and achieved from standard commercial sources. HEK293Kv11.1 cells were kindly provided by Dr Eckhard Ficker (University of Cleveland, Cleveland, OH, USA). The molecular target nomenclature (Kv11.1) conforms to ‘The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels' (Alexander et al., 2013b).
Results
[3H]-dofetilide saturation assay
The binding of [3H]-dofetilide to HEK293Kv11.1 cell membranes was saturable and best described by a one-site binding model. A representative saturation curve and the averaged data of three independent experiments performed in duplicate are shown in Figure 1 and Table 2 respectively. The KD and Bmax values obtained from this assay were 2.4 ± 0.1 nM and 1.6 ± 0.1 pmol·mg−1 protein respectively. The KD value for [3H]-dofetilide from this assay was used to calculate Ki rather than IC50 values from the displacement assay for 15 reference compounds.
Figure 1.
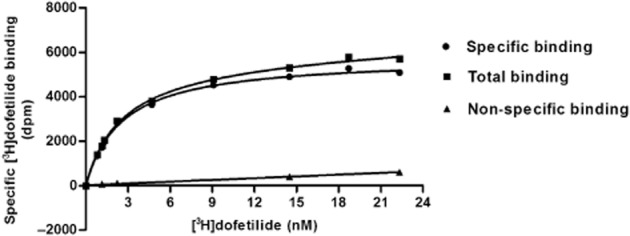
Saturation of [3H]-dofetilide binding to HEK293Kv11.1 membranes. Total binding was determined at increasing concentrations of [3H]-dofetilide. Non-specific binding was determined at three concentrations of [3H]-dofetilide and non-specific binding at other concentrations of radioligand was extrapolated by linear regression. Specific binding was calculated as the difference between the total and non-specific binding. The KD value was 2.4 ± 0.1 nM and the Bmax value was 1.6 ± 0.1 pmol·mg−1 protein. Data shown are representative results from a single experiment performed in duplicate.
Table 2.
Binding parameters of dofetilide from different equilibrium and kinetic binding assays
| Binding parameters | Saturation assay | Kinetic association assaya | Traditional association and dissociation assayb | Displacement assay | Competition association assay |
|---|---|---|---|---|---|
| kon (nM−1·min−1) | – | 0.017 | 0.032 ± 0.003 | – | 0.048 ± 0.011 |
| koff (min−1) | – | 0.12 | 0.20 ± 0.03 | – | 0.13 ± 0.02 |
| KD (nM) | 2.4 ± 0.1 | 7.1c | 6.4 ± 1.3c | – | 2.7 ± 0.3c |
| Ki (nM) | – | – | – | 5.4 ± 0.8 | – |
Values are means (±SEM) of three independent assays performed in duplicate.
Data were derived from linear regression of one independent association assay of [3H]-dofetilide at different concentrations.
Data from our previous study (Yu et al., 2014).
Kinetic KD = koff / kon.
[3H]-dofetilide association and dissociation assay
Initial experiments were performed to fully characterize the association and dissociation rates of [3H]-dofetilide to and from HEK293Kv11.1 membranes respectively. As the association rate of a ligand is dependent upon the concentration used, kinetic association experiments with a range of [3H]-dofetilide concentrations were conducted. In Figure 2A, curves are shown for four of such concentrations (0.76, 2.7, 3.8 and 7.1 nM). A plot of the kobs values against more, including higher, concentrations of [3H]-dofetilide (Figure 2B) was consistent with a linear correlation (r2 = 0.86, P < 0.0001), indicating that the binding of [3H]-dofetilide to the Kv11.1 channel followed the law of mass action for a simple bimolecular interaction and that the equation kon = (kobs − koff) / [L*] was applicable in this study. The kon and koff values obtained from this plot were 0.017 nM−1·min−1 and 0.12 min−1 respectively (Table 3). When koff was divided by kon, a kinetically derived KD value of 7.1 nM was obtained. These values were in agreement with values for apparent on- and off-rates and kinetic KD of [3H]-dofetilide assessed at one (5 nM) concentration (kon = 0.032 ± 0.003 nM−1·min−1, koff = 0.20 ± 0.03 min−1 and KD = 6.4 ± 1.3 nM) derived from the traditional association and dissociation assays published previously (Yu et al., 2014).
Figure 2.
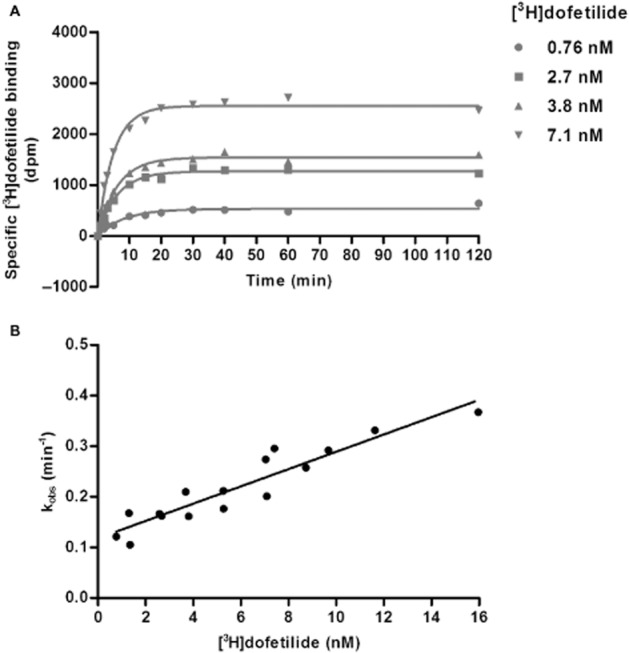
Characterization of the association and dissociation rates of [3H]-dofetilide to HEK293Kv11.1 membranes in the kinetic association assay. (A) Representative association curves of [3H]-dofetilide at four different concentrations. (B) A plot of kobs values versus the concentration of [3H]-dofetilide. Data shown are representative results from a single experiment performed in duplicate.
Table 3.
The affinity constants and kinetic parameters of 15 compounds at the Kv11.1 channel obtained from the [3H]-dofetilide displacement and competition association assay
| Compound | Ki (nM)a | kon (nM−1·min−1)b | koff (min−1)b | KD (nM)c |
|---|---|---|---|---|
| Dofetilide | 5.4 ± 0.8 | 0.048 ± 0.011 | 0.13 ± 0.02 | 2.7 ± 0.3 |
| Astemizole | 2.5 ± 0.2 | 0.17 ± 0.03 | 0.083 ± 0.003 | 0.53 ± 0.07 |
| E-4031 | 13 ± 0.7 | 0.026 ± 0.003 | 0.27 ± 0.02 | 10 ± 1 |
| Sertindole | 34 ± 5 | 0.048 ± 0.007 | 0.86 ± 0.17 | 18 ± 1 |
| Terfenadine | 63 ± 4 | 0.0071 ± 0.0025 | 0.25 ± 0.03 | 39 ± 7 |
| Moxifloxacin | 252 347 ± 120 995 | (4.7 ± 1.0) × 10−6 | 0.28 ± 0.06 | 64 531 ± 10 276 |
| Amiodarone | 308 ± 33 | (6.0 ± 0.7) × 10−4 | 0.23 ± 0.02 | 387 ± 37 |
| Chlorpromazine | 2518 ± 301 | (1.3 ± 0.2) × 10−4 | 0.36 ± 0.05 | 2 714 ± 154 |
| Ibutilide | 5.1 ± 0.4 | 0.046 ± 0.006 | 0.20 ± 0.02 | 4.6 ± 1.1 |
| Clofilium | 0.55 ± 0.09 | 0.23 ± 0.07 | 0.10 ± 0.01 | 0.54 ± 0.17 |
| Pimozide | 28 ± 7 | 0.071 ± 0.020 | 0.22 ± 0.07 | 3.2 ± 0.4 |
| Cisapride | 54 ± 10 | 0.031 ± 0.009 | 0.59 ± 0.14 | 20 ± 2 |
| Ranolazine | 21 379 ± 5 776 | (1.5 ± 0.4) × 10−5 | 0.23 ± 0.04 | 16 672 ± 1 820 |
| Sotalol | 24 663 ± 1 379 | (1.7 ± 0.3) × 10−5 | 0.32 ± 0.03 | 19 740 ± 1 591 |
| Thioridazine | 1 065 ± 41 | (2.3 ± 0.4) × 10−4 | 0.24 ± 0.05 | 1 050 ± 48 |
Values are means (±SEM) of three independent assays performed in duplicate.
Ki values were derived from the [3H]dofetilide displacement assay.
kon (k3) and koff (k4) values of unlabelled compounds were determined in the [3H]-dofetilide competition association assay.
Kinetic KD = koff / kon.
[3H]-dofetilide displacement assay
Competition binding assays were performed to generate Ki values for 15 reference compounds. Compounds were selected based on structural diversity and having a wide range of Kv11.1 binding affinities, and included both anti-arrhythmic drugs and drugs for other therapeutic areas (e.g. astemizole and terfenadine for the treatment of allergic conditions). All compounds produced a concentration-dependent inhibition of specific [3H]-dofetilide binding and their displacement curves were best described by a one-site competition model (Figure 3). All Ki values are listed in Table 3. Among the 15 compounds, clofilium had the highest affinity to the Kv11.1 channel, displacing [3H]-dofetilide with a Ki value of 0.55 ± 0.09 nM, whereas moxifloxacin exhibited the lowest affinity of 252 ± 121 μM. Ranolazine and sotalol showed similar and relatively weak inhibition of the channel with Ki values of 21 ± 6 and 25 ± 1 μM respectively. Additionally, amiodarone, thioridazine and chlorpromazine displayed modest Kv11.1 blockade with Ki values from 0.3 to 3 μM. All other compounds demonstrated relatively high affinity to the Kv11.1 channel, between 2.5 ± 0.2 nM (astemizole) and 63 ± 4 nM (terfenadine).
Figure 3.
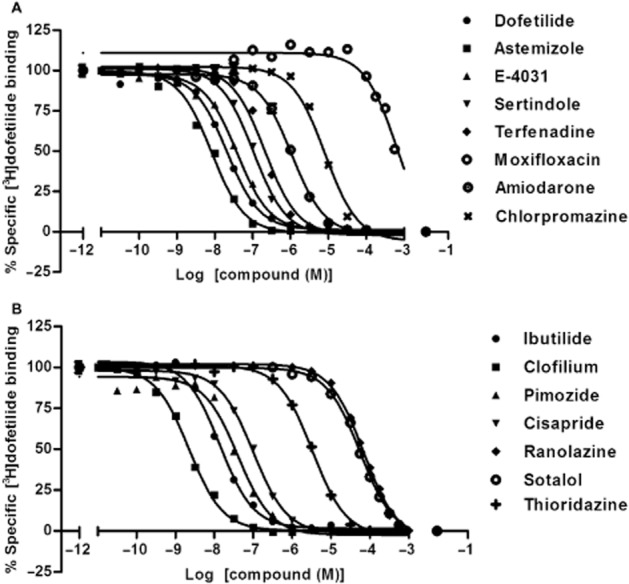
Displacement curves of [3H]-dofetilide from HEK293Kv11.1 membranes by different known Kv11.1 channel blockers. (A) Dofetilide, astemizole, E-4031, sertindole, terfenadine, moxifloxacin, amiodarone and chlorpromazine; (B) ibutilide, clofilium, pimozide, cisapride, ranolazine, sotalol and thioridazine. Data shown are representative results from a single experiment performed in duplicate.
[3H]-dofetilide competition association assay
With the kon (k1) and koff (k2) values of [3H]-dofetilide obtained from the traditional association and dissociation assays, it was possible to determine the kon (k3) and koff (k4) values of unlabelled compounds by performing the so-called competition association experiments. Firstly, we validated the competition association assay at the Kv11.1 channel using three concentrations of ‘cold’ dofetilide equivalent to 0.3-, one- and threefold of its Ki value. A representative experiment is shown in Figure 4. The kon (k3) and koff (k4) values for dofetilide determined in this assay were 0.048 ± 0.011 nM−1·min−1 and 0.13 ± 0.02 min−1, respectively, and in good agreement with the kinetic parameters determined in the traditional association and dissociation assays, as shown in Table 4. Furthermore, the kinetic KD value (2.7 ± 0.3 nM) derived from this assay was similar to the KD value (2.4 ± 0.1 nM) obtained from the [3H]-dofetilide saturation assay. In addition, these KD values were in the same range as dofetilide's affinity constant that stemmed from the displacement assay (Ki = 5.4 ± 0.8 nM) and the kinetically derived KD values from kinetic association assay and traditional kinetic experiments (7.1 nM or 6.4 ± 1.3 nM respectively). Overall, the results presented here demonstrated that the competition association assay could be applied to determine the association and dissociation rates of other unlabelled ligands at the Kv11.1 channel. It is noteworthy that a good experimental window was achieved using a concentration of dofetilide at onefold of its Ki value in the competition association assay, as displayed in Figure 4. Reassuringly, when astemizole and E-4031 were tested in this standard three-concentration assay, similar findings were observed as well (data not shown). Thus, the other unlabelled ligands were only tested at onefold of their Ki values rather than three different concentrations in the further competition association experiments in order to improve the throughput of this method.
Figure 4.
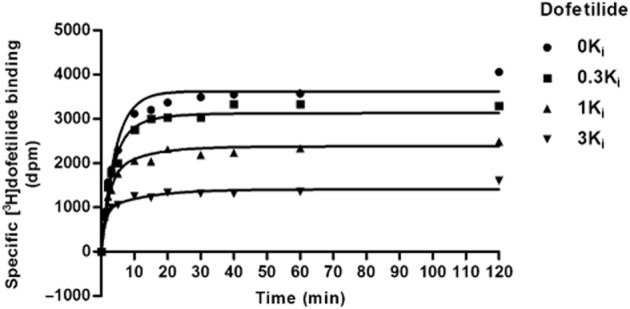
Competition association assay of [3H]-dofetilide in the absence (control) or presence of 0.3-, one- and threefold of unlabelled dofetilide's Ki value. Data shown are representative results from a single experiment performed in duplicate.
Table 4.
The lipophilicity, membrane partition coefficients and other physicochemical properties of 15 Kv11.1 blockers
| Compound | LogKW-C8a | LogKW-IAMb | MWc | LogPc | pKac |
|---|---|---|---|---|---|
| Dofetilide | 0.84 | 2.08 | 441.57 | 0.59 | 8.99 |
| Astemizole | 3.52 | 3.40 | 458.57 | 5.39 | 8.75 |
| E-4031 | 1.29 | 1.98 | 401.52 | 1.73 | 8.01 |
| Sertindole | 3.97 | 3.38 | 440.94 | 3.77 | 8.59 |
| Terfenadine | 4.05 | 4.01 | 471.67 | 6.48 | 9.02 |
| Moxifloxacin | 1.12 | 1.57 | 401.43 | 1.97 | 9.42 |
| Amiodarone | 5.52 | 3.30 | 645.31 | 7.64 | 8.47 |
| Chlorpromazine | 3.39 | 3.36 | 318.86 | 4.54 | 9.20 |
| Ibutilide | 0.90 | 2.53 | 384.58 | 3.25 | 10.40 |
| Clofilium | 2.00 | −0.35 | 338.98 | 2.91 | nad |
| Pimozide | 4.69 | 3.80 | 461.55 | 5.83 | 8.38 |
| Cisapride | 3.12 | 2.66 | 465.95 | 2.49 | 8.24 |
| Ranolazine | 2.17 | 2.39 | 427.54 | 2.83 | 7.17 |
| Sotalol | 0.56 | 0.66 | 272.36 | 0.05 | 9.43 |
| Thioridazine | 3.53 | 3.80 | 370.58 | 5.47 | 8.93 |
LogKW-C8 values were derived from HPLC experiments on a Supelcosil LC-ABZ, 5 cm × 4.6 mm, 5 μm column.
LogKW-IAM values were derived from HPLC experiments on a 10 cm × 4.6 mm, 10 μm Regis IAM PC DD2 column.
Values were derived from the structure-based calculation plug-in by ChemAxon.
na, not applicable; this compound is permanently charged.
Subsequently, kinetic parameters of the 12 other known Kv11.1 blockers were evaluated and representative normalized curves for several compounds are depicted in Figure 5. The on- and off-rates of all compounds determined by these experiments are shown in Table 4. The association rates for all the compounds were quite distinct with kon values ranging from (4.7 ± 1.0) × 10−6 nM−1·min−1 (moxifloxacin) to 0.23 ± 0.07 nM−1·min−1 (clofilium), i.e. an almost 50 000-fold difference between the fastest and slowest associating compounds. On the other hand, the dissociation rates of these 15 compounds were more similar, with the highest value of 0.86 ± 0.17 min−1 for sertindole and lowest koff of 0.083 ± 0.003 min−1 for astemizole, i.e. only a 10-fold difference. Considering the kinetically derived KD values shown in Table 4, clofilium was the most potent inhibitor to the Kv11.1 channel with a kinetic KD value of 0.54 ± 0.17 nM, while moxifloxacin had the lowest affinity (KD = 65 ± 10 μM) to the channel. These results were in the same rank order as the Ki values derived from the equilibrium displacement assay.
Figure 5.
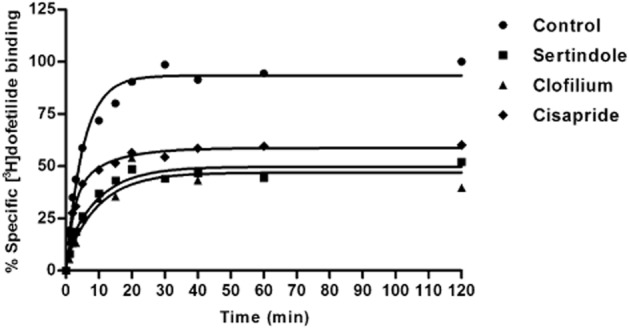
Representative competition association curves for [3H]-dofetilide in the absence (control) or presence of unlabelled sertindole, clofilium and cisapride at a concentration of onefold their Ki values. Data shown are representative results from a single experiment performed in duplicate.
Correlations of equilibrium Ki with kinetic KD, kon and koff values
A plot of the logarithms of kinetic KD values (i.e. koff/kon) derived from the [3H]-dofetilide competition association assays and the logarithms of equilibrium Ki values obtained from the displacement experiments was made and a significant correlation (Figure 6A) was observed. This showed an excellent consistency of the results from two different methods and indicated a high reliability of the [3H]-dofetilide competition association assay. More interestingly, a significant inverse relationship was also found between pkon and pKi values for unlabelled compounds in Figure 6B. In contrast, there was no significant linear relationship between pkoff and pKi values (r2 = 0.15, P = 0.15, data not shown). Together, this suggested that the [3H]-dofetilide competition association assay was successfully validated for assessing the kinetics of other unlabelled competitive compounds and that the affinity of these compounds at the Kv11.1 channel was mainly controlled by their on-rates rather than off-rates.
Figure 6.
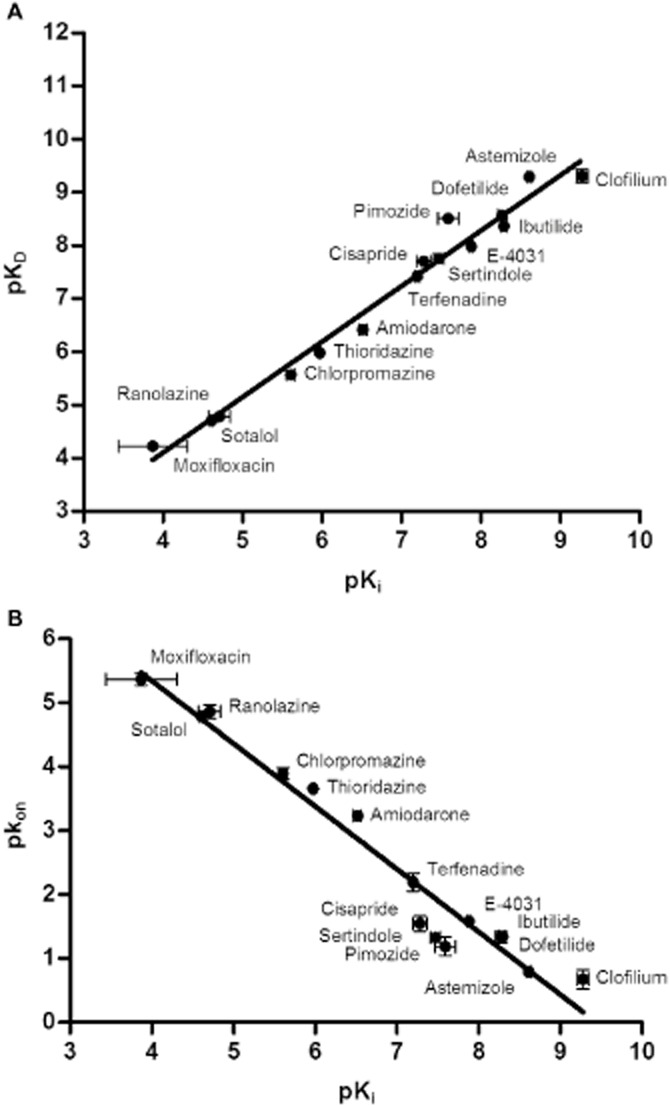
Correlations between the affinity constant (Ki) and (A) the kinetically derived equilibrium dissociation constants, KD (r2 = 0.98, P < 0.0001) and (B) the association rates, kon, for unlabelled ligands at the Kv11.1 channel (r2 = 0.95, P < 0.0001). Ki, KD and kon values are listed in Table 4.
Lipophilicity (logKW-C8) and membrane partition coefficient (logKW-IAM) of Kv11.1 blockers
The isocratical logKW-C8 values (‘lipophilicity’) were evaluated at pH 7.4 and are detailed in Table 4. The lipophilicity of the 15 reference compounds covered a wide numerical range, varying from 0.56 (sotalol) to 5.52 (amiodarone). We also calculated logP values as a measure for lipophilicity and plotted these against the logKW-C8 data (Table 4). A significant correlation was found between them (r2 = 0.81, P < 0.0001), which implied that for this series of compounds, calculated logP values can be used interchangeably with the experimentally determined values.
To mimic the interactions of our ligands with membrane phospholipids, an IAM HPLC column that is a reflection of the lipid environment of a fluid cell membrane on a solid matrix was used to determine membrane partition coefficients (logKW-IAM) for all reference compounds. Their logKW-IAM values were measured at pH 7.4 and are summarized in Table 1973. Terfenadine had the highest logKW-IAM value of 4.01, indicating that this compound possessed the highest affinity for membrane phospholipids. On the contrary, the logKW-IAM for clofilium was only −0.35, most likely due to its quaternary ammonium moiety, which demonstrated that this compound hardly interacted with phospholipid membranes.
Subsequently, the possible correlation between logKW-C8 and logKW-IAM was studied and the result is shown in Figure 7. A significant linear relationship (r2 = 0.52, P < 0.0024) was observed for these two parameters even when including the outlier clofilium. Obviously, a similar significant correlation was also found between calculated logP and logKW-IAM values (r2 = 0.52, P = 0.0022, data not shown). Next, the relationship between affinity constants or kinetic rate constants of the 15 Kv11.1 inhibitors and their membrane interactions were investigated, as shown in Figure 8A–C. Apparently, no relationship was found for any of them (P > 0.05), demonstrating that membrane interactions did not affect affinity and binding kinetics of these ligands at the Kv11.1 channel. Similarly, there were no correlations between logKW-C8 and pKi, pkon or pkoff values (data not shown).
Figure 7.
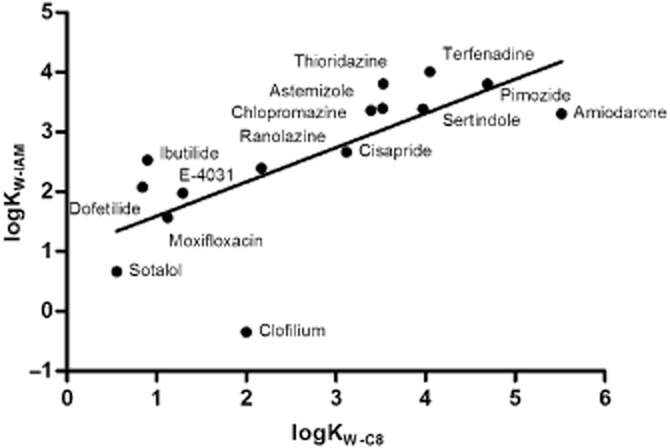
Correlation between logKW-C8 and logKW-IAM values at pH 7.4 for 15 Kv11.1 inhibitors (r2 = 0.52, P < 0.0024). LogKW-C8 and logKW-IAM values are listed in Table 1973.
Figure 8.
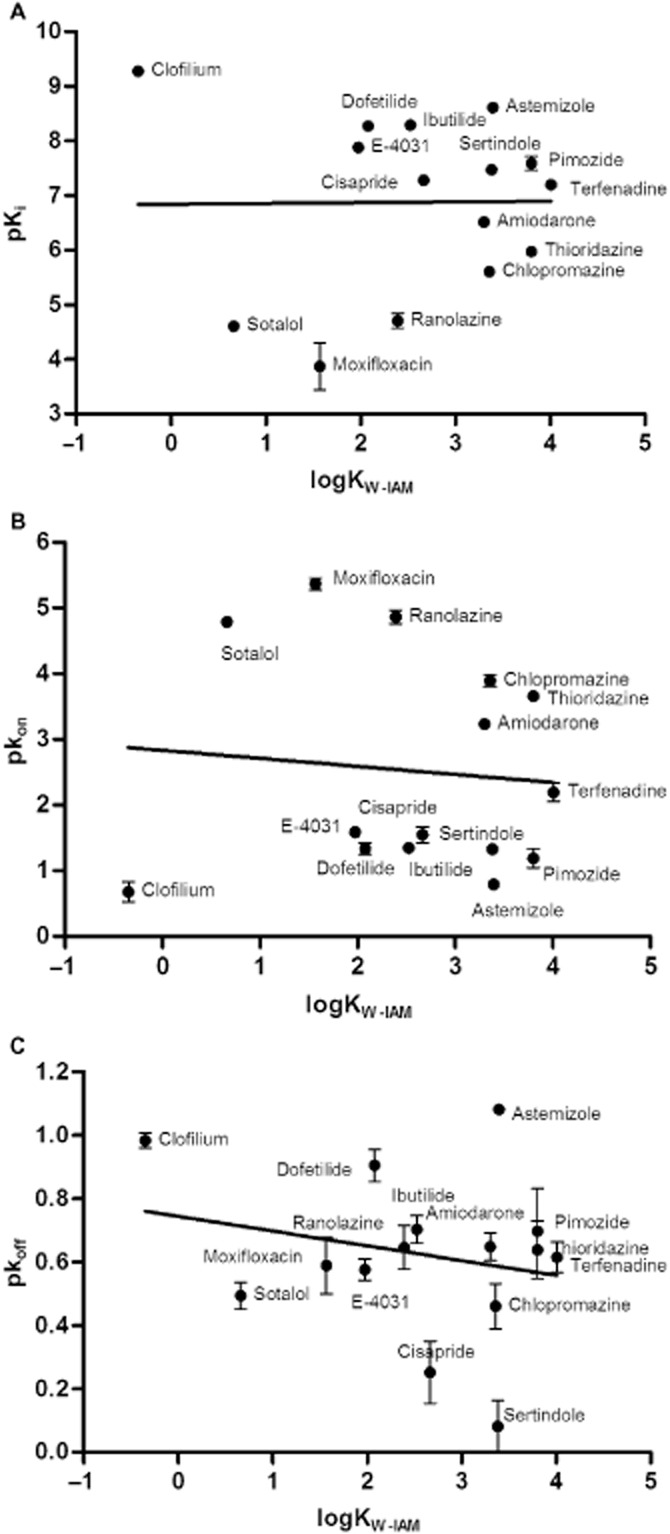
Correlation of logKW-IAM values and binding parameters of 15 Kv11.1 inhibitors. No significant relationship between logKW-IAM and values of (A) pKi (r2 = 0.00011, P = 0.97), (B) pkon (r2 = 0.0087, P = 0.74) or (C) pkoff was observed (r2 = 0.051, P = 0.42). All data used in these plots are listed in Tables 2014 and 1973.
Role of other calculated physicochemical properties in ligand-receptor binding kinetics
Lastly, two other physicochemical properties of the unlabelled compounds, MW and acid/base constant (pKa) (Table 1973), were compared with their on-/off-rates and dissociation constants. As depicted in Figure 9, no significant correlations were observed between the logarithms of on-rates of the compounds in this study and their molecular properties. Moreover, there were no obvious relationships between the logarithms of off-rates or equilibrium Ki values and these physicochemical properties either (P > 0.05, data not shown). These results implied that the binding kinetics of compounds at the Kv11.1 channel was not governed by their overall, macroscopic, physicochemical properties.
Figure 9.
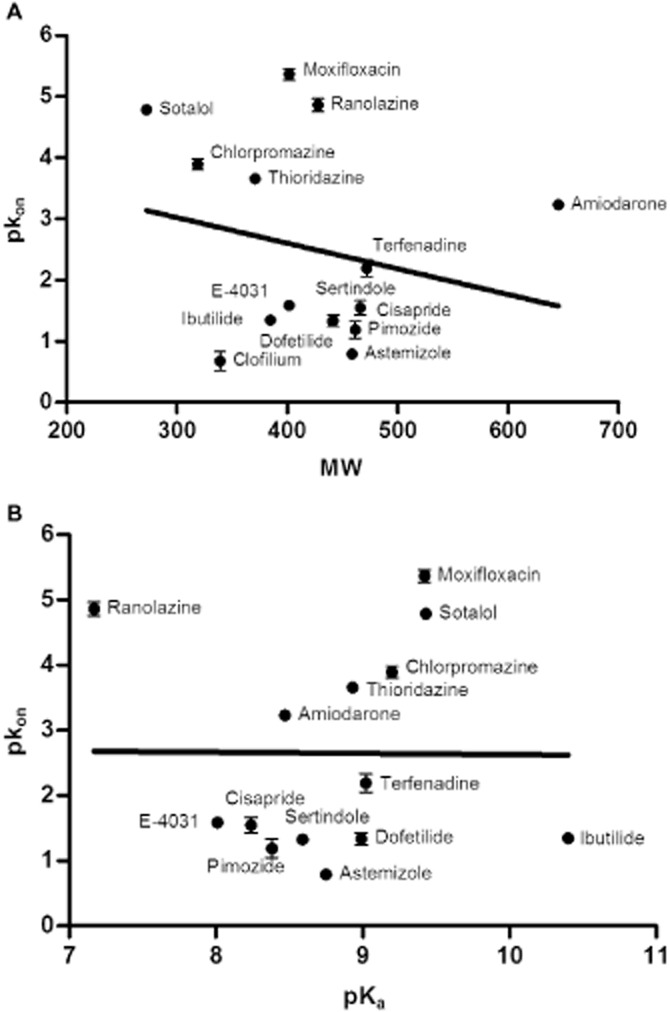
Lack of correlation between the association rates (pkon values) of selected compounds and their physicochemical properties. (A) No significant correlation was observed with MW (r2 = 0.049, P = 0.43); (B) no significant correlation was observed with pKa (r2 = 0.000076, P = 0.98). MW and pKa values are shown in Table 1973.
Discussion and conclusions
Since the introduction of the competition association assay (Motulsky and Mahan, 1984), more and more researchers have utilized this method to study the binding kinetics of unlabelled ligands at their targets, such as muscarinic receptors and the adenosine A2A receptor (Schreiber et al., 1985; Dowling and Charlton, 2006; Guo et al., 2012). In addition, a few examples in the literature have been reported to apply this technique for the determination of ligand binding kinetics at ligand-gated ion channels (Hawkinson and Casida, 1992). To the best of our knowledge, the current study is the first to assess binding kinetics of ligands interacting with voltage-gated ion channels, in particular the Kv11.1 channel.
It might be argued that our study has some limitations. The stably transfected HEK293Kv11.1 cells lack the (co-)expression of two ancillary β-subunits, minK and MiRP1 (Vandenberg et al., 2001). Discrepancies could also exist between membrane binding assays and whole-cell experiments, which are more relevant to the channel's natural orientation. For instance, the kinetics of the drug–Kv11.1 interaction have been reported to be use- and frequency-dependent in whole-cell patch-clamp assays due to special gating kinetics of the channel (Stork et al., 2007; Windisch et al., 2011). In our membrane binding assays, we speculate that the channels maintain the same configurations and thus the kinetic parameters in this investigation should resemble the actual binding process between Kv11.1 blockers and the channel. To perform straightforward and accurate kinetic determinations, experiments were carried out at 25°C with membrane preparations of stably transfected HEK293Kv11.1 cells. We reason that all parameters obtained at 25°C in this investigation lead to a similar compound rank order as those at physiological temperature. In fact, this has been shown for another target based on van't Hoff and Eyring equations (Mondal et al., 2013).
The linear plot of kobs values obtained at increasing concentrations of [3H]-dofetilide (Figure 2B) strongly supported a pseudo-first-order kinetic behaviour of its binding to the Kv11.1 channel, which provided the theoretical foundation for all kinetic analyses in our present study. Other mechanisms of action such as induced-fit or conformational selection would have resulted in deviations from linearity; in such cases, the Motulsky and Mahan model and equations would not have been applicable (Tummino and Copeland, 2008; Vogt and Di Cera, 2012). All 15 reference compounds had a pseudo-Hill coefficient close to unity in the [3H]-dofetilide displacement assay (data not shown), which indicated a competitive mode of inhibition with regard to the radioligand. This is another indication of the simple bimolecular interaction model and corroborated a pseudo-first-order kinetic behaviour of the binding to the Kv11.1 channel. This finding was in accordance with previous studies describing the presence of a single affinity state for Kv11.1 inhibitors at the channel (Finlayson et al., 2001a,b). Subsequently, a newly developed [3H]-dofetilide competition association assay was applied to study the binding kinetics of 15 unlabelled reference compounds with diverse chemical structures at the Kv11.1 channel (Table 2013a). The kon, koff and KD values of dofetilide from this assay were comparable with the values derived from saturation and kinetic association experiments and traditional association and dissociation assays (Yu et al., 2014), indicative of the accuracy and reliability of this new assay. Moreover, the excellent linear correlation between these kinetically derived KD values of 15 reference compounds and their Ki values from the equilibrium displacement assay further supported the latter (Figure 6A). Taken together, this led us to conclude that a novel [3H]-dofetilide competition association assay was successfully developed and validated.
Correlations between the affinity data (Ki) and the kinetic parameters (on- and off-rates) were also investigated in the present study. Surprisingly, there was a significant inverse correlation between pKi and pkon values for reference compounds (Figure 6B), whereas no relationship was found between pKi and pkoff values (data not shown). Although this correlation between affinity and association rates has been shown to be the case for β2-adrenoceptor agonists (Sykes and Charlton, 2012) and OX2-receptor antagonists (Mould et al., 2014), this phenomenon is supposed to be unusual and counterintuitive. Dogma has it that the collision theory sets the maximum value for kon as the diffusion limit for a ligand and a target, which is about 108∼109 M−1·s−1 (Smith, 2009). In this view, there can only be small variations in the on-rate constants and thus the equilibrium affinity changes are mainly dictated by the off-rates of ligands (Copeland et al., 2006; Smith, 2009; Lu and Tonge, 2010). In contrast to these classical assumptions, association rates of our selected structurally diverse compounds varied around 50 000-fold, whereas their dissociation rates differed only 10-fold. This clearly indicated that apart from off-rates, on-rates also play a pivotal role in regulating affinity of ligands to their targeted receptors, at least for the Kv11.1 channel.
Recently, it has been reported that drug-receptor association rates and corresponding affinity were enhanced due to concentrating effects and lateral diffusion of membrane associated β2-adrenoceptor ligands and kon values were positively correlated to their lipophilicity and membrane interactions (Hanson et al., 2012; Sykes et al., 2014). Furthermore, Sykes et al. (2014) observed that dissociation rates were much more correlated to the corrected ‘true’ affinity, which considers drug concentration gradients in the local environment due to membrane affinity than the apparent affinity. Additionally, it was recommended that any prediction of pharmacodynamic properties should take membrane interactions as well as lipophilicity into account (Taillardat-Bertschinger et al., 2003). Hence, the potential influence of membrane affinity on ligand binding to the Kv11.1 channel was investigated by the determination of both logKW-C8 and logKW-IAM values. However, when pKi and pkon were compared with logKW-C8 and logKW-IAM, no significant relationships were observed in our case (Figure 8). This indicated that membrane interactions of our ligands did not influence their association rates and binding affinity to the Kv11.1 channel and that the apparent affinity of these reference compounds is their ‘true’ affinity. Interestingly, when logKW-C8 and logKW-IAM were compared with each other, clofilium was observed to deviate from the significant linear correlation (Figure 7). Herein, we hypothesized that the lipophilic alkyl chains of clofilium compensated for its hydrophilic quaternary ammonium group and thus dominated its lipophilicity in the octanol-water system, while the positive charge at the nitrogen played a more critical role in the IAM system and weakened its membrane interactions. With regard to dissociation rates, no correlations were found to either logKW-C8 or logKW-IAM values as well (data not shown). This was in accordance with previous findings (Mason et al., 1991; Sykes et al., 2014) and demonstrated that the dissociation rate of a drug was not affected by its local concentrations and thus independent of membrane affinity. Of note, typical Kv11.1 inhibitors are known to bind to the inner cavity in the intracellular part of the channel (Vandenberg et al., 2001). However, all experiments in the present study were performed with HEK293Kv11.1 membranes instead of intact cells and were thus independent of transmembrane transport of ligands. From Figure 6B, it follows that a significant relationship between affinity and association rates for 15 Kv11.1 inhibitors was found, which showed that ligand-Kv11.1 rather than ligand-membrane association controlled their affinity. Apparently, an aqueous entry pathway predominates the binding of Kv11.1 blockers to the channel and an additional compartment induced by the lipid membranes does not play a significant role.
We questioned whether it would be possible to correlate other general physicochemical properties of these molecules to their biological profile (Figure 9). This was not the case, as typical features such as MW and the basicity of the Kv11.1 blockers were not correlated to any of the equilibrium or kinetic binding parameters tested. However, it has been stated previously that physicochemical properties of drugs are relevant for their kinetic parameters (Shaikh et al., 2007; Smith, 2009). For instance, Miller et al. (2012) reported that MW was one of the most important factors to affect the dissociation kinetics of ligands from their biological targets including enzymes (kinases) and GPCRs. From the present study, it follows that the effects of general molecular properties on the binding kinetics of a ligand to Kv11.1 channel are negligible compared with other targets.
Previous studies have also suggested that koff values, or drug-target residence times (RTs, the reciprocal of koff), play an important role in the duration of pharmacological effects (Copeland et al., 2006; Lu and Tonge, 2010; Guo et al., 2012). In other words, an increased drug-target RT could elicit an improved or prolonged drug effect (Dahl and Åkerud, 2013). Nevertheless, when adverse effects result directly from drug occupancy at the pharmacological target, such as chlorpromazine binding to D2 receptor or clopidogrel to P2Y12 receptor, a long RT would result in so-called ‘on-target’ drug toxicity, and thus a short RT is favoured (Copeland, 2010). In our hands, however, the dissociation rates and hence RTs of selected 15 Kv11.1 inhibitors were very similar (Table 2014). In contrast, association rates of our reference compounds varied widely and are therefore potentially more important to predict the safety of Kv11.1 inhibitors.
Previously, Redfern et al. (2003) have assigned drugs into five categories of torsadogenic propensity and proposed a ‘30-fold safety margin’ between Cmax and Kv11.1 IC50 values as a marker to predict TdP. However, it is known that TdP are not only induced by inhibition of the Kv11.1 channel but also regulated by other potassium, sodium and calcium channels (Redfern et al., 2003; Sager et al., 2014). Therefore, a comprehensive in vitro pro-arrhythmia assay has recently been introduced to assess drug-induced pro-arrhythmic risk more efficiently and accurately (Cavero, 2014; Sager et al., 2014). The kinetics of drug block and unblock were suggested to be incorporated together with drug potency at ion channels in arrhythmia evaluation during the interpretation of this paradigm (Sager et al., 2014). Although the present study focuses on the Kv11.1 channel only, it provides a new medium throughput method to determine the association and dissociation rates of Kv11.1 blockers, which can be used subsequently for other ion channels as well, if a radioligand is available. Interestingly, Veroli et al. (2014) derived from mathematical models that fast binding Kv11.1 blockers in the untrapped configuration and trapped blockers induced greater action potential and QT prolongation. This strengthens the application of our method in the future and suggests that using slow association and/or fast dissociation characteristics as a ‘novel marker’ might be beneficial for reducing Kv11.1 cardiotoxicity of drug candidates.
In conclusion, a novel [3H]-dofetilide competition association assay has been successfully developed and validated to characterize the kinetic binding parameters of unlabelled compounds at the Kv11.1 channel. Importantly, association rates of Kv11.1 blockers were divergent, i.e. not diffusion limited, and excellently correlated to their affinity values. In addition, membrane interactions and other molecular properties do not influence the affinity and kinetic binding parameters of ligands at the Kv11.1 channel. Altogether, this is quite unlike the mechanisms of interaction proposed for other drug target classes, such as kinases and GPCRs. Hence, we postulate that association rates can be used to assess a compound's Kv11.1 liability, apart from its affinity. However, further studies involving compounds with a wide range of koff values are required to assess the effect of RT on drug-induced Kv11.1 cardiotoxicity. Overall, we believe that this research provides novel insights into the kinetic study of ion channels, which can hopefully help to avoid Kv11.1-induced cardiotoxicity of drug candidates in the future.
Acknowledgments
Z. Y. is supported by a scholarship from the Chinese Scholarship Council. We thank Dr Steven Charlton (Novartis, UK) for helpful discussions, particularly about the IAM column.
Glossary
- Cmax
maximum free plasma concentration
- HEK293Kv11.1
HEK293 cells stably expressing the Kv11.1 (hERG) channel
- hERG
human ether-à-go-go related gene
- IAM
immobilized artificial membrane
- MW
molecular weight
- TdP
Torsade de Pointes
Author contributions
Z. Y. designed and conducted the experiments, analysed the data and wrote the manuscript. A. P. I. designed the experiments, assessed the results and wrote the manuscript. L. H. H. designed the experiments, assessed the results and wrote the manuscript.
Conflict of interest
None.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The concise guide to PHARMACOLOGY 2013/14: ion channels. Br J Pharmacol. 2013b;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavero I. 13th Annual Meeting of the Safety Pharmacology Society: focus on novel technologies and safety pharmacology frontiers. Expert Opin Drug Saf. 2014;13:1271–1281. doi: 10.1517/14740338.2014.940310. [DOI] [PubMed] [Google Scholar]
- Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Chiu PJ, Marcoe KF, Bounds SE, Lin C-H, Feng J-J, Lin A, et al. Validation of a [3H] astemizole binding assay in HEK293 cells expressing HERG K+ channels. J Pharmacol Sci. 2004;95:311–319. doi: 10.1254/jphs.fpe0040101. [DOI] [PubMed] [Google Scholar]
- Copeland RA. The dynamics of drug-target interactions: drug-target residence time and its impact on efficacy and safety. Expert Opin Drug Discov. 2010;5:305–310. doi: 10.1517/17460441003677725. [DOI] [PubMed] [Google Scholar]
- Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- Dahl G, Åkerud T. Pharmacokinetics and the drug-target residence time concept. Drug Discov Today. 2013;18:697–707. doi: 10.1016/j.drudis.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Diaz GJ, Daniell K, Leitza ST, Martin RL, Su Z, McDermott JS, et al. The [3H]dofetilide binding assay is a predictive screening tool for hERG blockade and proarrhythmia: comparison of intact cell and membrane preparations and effects of altering [K+]o. J Pharmacol Toxicol Methods. 2004;50:187–199. doi: 10.1016/j.vascn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Dowling MR, Charlton SJ. Quantifying the association and dissociation rates of unlabelled antagonists at the muscarinic M3 receptor. Br J Pharmacol. 2006;148:927–937. doi: 10.1038/sj.bjp.0706819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Finlayson K, Pennington AJ, Kelly JS. [3H]dofetilide binding in SHSY5Y and HEK293 cells expressing a HERG-like K+ channel? Eur J Pharmacol. 2001a;412:203–212. doi: 10.1016/s0014-2999(01)00731-2. [DOI] [PubMed] [Google Scholar]
- Finlayson K, Turnbull L, January CT, Sharkey J, Kelly JS. [3H]dofetilide binding to HERG transfected membranes: a potential high throughput preclinical screen. Eur J Pharmacol. 2001b;430:147–148. doi: 10.1016/s0014-2999(01)01362-0. [DOI] [PubMed] [Google Scholar]
- Fitzgerald PT, Ackerman MJ. Drug-induced torsades de pointes: the evolving role of pharmacogenetics. Heart Rhythm. 2005;2:S30–S37. doi: 10.1016/j.hrthm.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Guo D, Mulder-Krieger T, IJzerman AP, Heitman LH. Functional efficacy of adenosine A2A receptor agonists is positively correlated to their receptor residence time. Br J Pharmacol. 2012;166:1846–1859. doi: 10.1111/j.1476-5381.2012.01897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancox JC, McPate MJ, El Harchi A, Zhang YH. The hERG potassium channel and hERG screening for drug-induced torsades de pointes. Pharmacol Ther. 2008;119:118–132. doi: 10.1016/j.pharmthera.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, et al. Crystal structure of a lipid G protein-coupled receptor. Science. 2012;335:851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkinson J, Casida JE. Binding kinetics of gamma-aminobutyric acidA receptor noncompetitive antagonists: trioxabicyclooctane, dithiane, and cyclodiene insecticide-induced slow transition to blocked chloride channel conformation. Mol Pharmacol. 1992;42:1069–1076. [PubMed] [Google Scholar]
- Heijman J, Voigt N, Carlsson LG, Dobrev D. Cardiac safety assays. Curr Opin Pharmacol. 2014;15:16–21. doi: 10.1016/j.coph.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Heitman LH, Narlawar R, de Vries H, Willemsen MN, Wolfram D, Brussee J, et al. Substituted terphenyl compounds as the first class of low molecular weight allosteric inhibitors of the luteinizing hormone receptor. J Med Chem. 2009;52:2036–2042. doi: 10.1021/jm801561h. [DOI] [PubMed] [Google Scholar]
- Krohn KA. The physical chemistry of ligand-receptor binding identifies some limitations to the analysis of receptor images. Nucl Med Biol. 2001;28:477–483. doi: 10.1016/s0969-8051(01)00216-5. [DOI] [PubMed] [Google Scholar]
- Lombardo F, Shalaeva MY, Tupper KA, Gao F. ElogDoct: a tool for lipophilicity determination in drug discovery. 2. Basic and neutral compounds. J Med Chem. 2001;44:2490–2497. doi: 10.1021/jm0100990. [DOI] [PubMed] [Google Scholar]
- Lu H, Tonge PJ. Drug–target residence time: critical information for lead optimization. Curr Opin Chem Biol. 2010;14:467–474. doi: 10.1016/j.cbpa.2010.06.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason RP, Rhodes DG, Herbette LG. Reevaluating equilibrium and kinetic binding parameters for lipophilic drugs based on a structural model for drug interactions with biological membranes. J Med Chem. 1991;34:869–877. doi: 10.1021/jm00107a001. [DOI] [PubMed] [Google Scholar]
- Miller DC, Lunn G, Jones P, Sabnis Y, Davies NL, Driscoll P. Investigation of the effect of molecular properties on the binding kinetics of a ligand to its biological target. Medchemcomm. 2012;3:449–452. [Google Scholar]
- Mondal K, Regnstrom K, Morishige W, Barbour R, Probst G, Xu Y-Z, et al. Thermodynamic and kinetic characterization of hydroxyethylamine β-secretase-1 inhibitors. Biochem Biophys Res Commun. 2013;441:291–296. doi: 10.1016/j.bbrc.2013.09.081. [DOI] [PubMed] [Google Scholar]
- Motulsky HJ, Mahan L. The kinetics of competitive radioligand binding predicted by the law of mass action. Mol Pharmacol. 1984;25:1–9. [PubMed] [Google Scholar]
- Mould R, Brown J, Marshall FH, Langmead C. Binding kinetics differentiates functional antagonism of orexin-2 receptor ligands. Br J Pharmacol. 2014;171:351–363. doi: 10.1111/bph.12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM. Molecular basis of functional voltage-gated K+ channel diversity in the mammalian myocardium. J Physiol. 2000;525:285–298. doi: 10.1111/j.1469-7793.2000.t01-1-00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble D. Computational models of the heart and their use in assessing the actions of drugs. J Pharmacol Sci. 2008;107:107–117. doi: 10.1254/jphs.cr0070042. [DOI] [PubMed] [Google Scholar]
- Pan AC, Borhani DW, Dror RO, Shaw DE. Molecular determinants of drug–receptor binding kinetics. Drug Discov Today. 2013;18:667–673. doi: 10.1016/j.drudis.2013.02.007. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampe D, Brown AM. A history of the role of the hERG channel in cardiac risk assessment. J Pharmacol Toxicol Methods. 2013;68:13–22. doi: 10.1016/j.vascn.2013.03.005. [DOI] [PubMed] [Google Scholar]
- Redfern W, Carlsson L, Davis A, Lynch W, MacKenzie I, Palethorpe S, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- Sager PT, Gintant G, Turner JR, Pettit S, Stockbridge N. Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium. Am Heart J. 2014;167:292–300. doi: 10.1016/j.ahj.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- Schreiber G, Henis YI, Sokolovsky M. Analysis of ligand binding to receptors by competition kinetics. Application to muscarinic antagonists in rat brain cortex. J Biol Chem. 1985;260:8789–8794. [PubMed] [Google Scholar]
- Shagufta, Guo D, Klaasse E, de Vries H, Brussee J, Nalos L, et al. Exploring chemical substructures essential for hERG K+ channel blockade by synthesis and biological evaluation of dofetilide analogues. ChemMedChem. 2009;4:1722–1732. doi: 10.1002/cmdc.200900203. [DOI] [PubMed] [Google Scholar]
- Shaikh SA, Jain T, Sandhu G, Latha N, Jayaram B. From drug target to leads-sketching a physicochemical pathway for lead molecule design in silico. Curr Pharm Des. 2007;13:3454–3470. doi: 10.2174/138161207782794220. [DOI] [PubMed] [Google Scholar]
- Smith GF. Medicinal chemistry by the numbers: the physicochemistry, thermodynamics and kinetics of modern drug design. Prog Med Chem. 2009;48:1–29. doi: 10.1016/s0079-6468(09)04801-2. [DOI] [PubMed] [Google Scholar]
- Stork D, Timin E, Berjukow S, Huber C, Hohaus A, Auer M, et al. State dependent dissociation of HERG channel inhibitors. Br J Pharmacol. 2007;151:1368–1376. doi: 10.1038/sj.bjp.0707356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes D, Parry C, Reilly J, Wright P, Fairhurst R, Charlton SJ. Observed drug-receptor association rates are governed by membrane affinity: the importance of establishing ‘micro PK/PD relationships’ at the β2-adrenoceptor. Mol Pharmacol. 2014;85:608–617. doi: 10.1124/mol.113.090209. [DOI] [PubMed] [Google Scholar]
- Sykes DA, Charlton SJ. Slow receptor dissociation is not a key factor in the duration of action of inhaled long-acting β2-adrenoceptor agonists. Br J Pharmacol. 2012;165:2672–2683. doi: 10.1111/j.1476-5381.2011.01639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taillardat-Bertschinger A, Carrupt P-A, Barbato F, Testa B. Immobilized artificial membrane HPLC in drug research. J Med Chem. 2003;46:655–665. doi: 10.1021/jm020265j. [DOI] [PubMed] [Google Scholar]
- Tummino PJ, Copeland RA. Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry. 2008;47:5481–5492. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- Valko K, Du CM, Bevan CD, Reynolds DP, Abraham MH. Rapid-gradient HPLC method for measuring drug interactions with immobilized artificial membrane: comparison with other lipophilicity measures. J Pharm Sci. 2000;89:1085–1096. doi: 10.1002/1520-6017(200008)89:8<1085::aid-jps13>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Vandenberg JI, Walker BD, Campbell TJ. HERG K+ channels: friend and foe. Trends Pharmacol Sci. 2001;22:240–246. doi: 10.1016/s0165-6147(00)01662-x. [DOI] [PubMed] [Google Scholar]
- Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. hERG K+ channels: structure, function, and clinical significance. Physiol Rev. 2012;92:1393–1478. doi: 10.1152/physrev.00036.2011. [DOI] [PubMed] [Google Scholar]
- Veroli GYD, Davies MR, Zhang H, Abi-Gerges N, Boyett MR. hERG inhibitors with similar potency but different binding kinetics do not pose the same proarrhythmic risk: implications for drug safety assessment. J Cardiovasc Electrophysiol. 2014;25:197–207. doi: 10.1111/jce.12289. [DOI] [PubMed] [Google Scholar]
- Vilums M, Overman J, Klaasse E, Scheel O, Brussee J, IJzerman AP. Understanding of molecular substructures that contribute to hERG K+ channel blockade: synthesis and biological evaluation of E-4031 analogues. ChemMedChem. 2012;7:107–113. doi: 10.1002/cmdc.201100366. [DOI] [PubMed] [Google Scholar]
- Vogt AD, Di Cera E. Conformational selection or induced fit? A critical appraisal of the kinetic mechanism. Biochemistry. 2012;51:5894–5902. doi: 10.1021/bi3006913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windisch A, Timin E, Schwarz T, Stork-Riedler D, Erker T, Ecker G, et al. Trapping and dissociation of propafenone derivatives in HERG channels. Br J Pharmacol. 2011;162:1542–1552. doi: 10.1111/j.1476-5381.2010.01159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Klaasse E, Heitman LH, Ijzerman AP. Allosteric modulators of the hERG K+ channel: radioligand binding assays reveal allosteric characteristics of dofetilide analogs. Toxicol Appl Pharmacol. 2014;274:78–86. doi: 10.1016/j.taap.2013.10.024. [DOI] [PubMed] [Google Scholar]