

Abstract
Lipid droplets (LDs) are intracellular structures that regulate neutral lipid homeostasis. In mammals, LD synthesis is inhibited by rapamycin, a known inhibitor of the mTORC1 pathway. In Saccharomyces cerevisiae, LD dynamics are modulated by the growth phase; however, the regulatory pathways involved are unknown. Therefore, we decided to study the role of the TORC1 pathway on LD metabolism in S. cerevisiae. Interestingly, rapamycin treatment resulted in a fast LD replenishment and growth inhibition. The discovery that osmotic stress (1 M sorbitol) also induced LD synthesis but not growth inhibition suggested that the induction of LDs in yeast is not a secondary response to reduced growth. The induction of LDs by rapamycin was due to increased triacylglycerol but not sterol ester synthesis. Induction was dependent on the TOR downstream effectors, the PP2A-related phosphatase Sit4p and the regulatory protein Tap42p. The TORC1-controlled transcriptional activators Gln3p, Gat1p, Rtg1p, and Rtg3p, but not Msn2p and Msn4p, were required for full induction of LDs by rapamycin. Furthermore, we show that the deletion of Gln3p and Gat1p transcription factors, which are activated in response to nitrogen availability, led to abnormal LD dynamics. These results reveal that the TORC1 pathway is involved in neutral lipid homeostasis in yeast.
INTRODUCTION
Lipid droplets (LDs) are intracellular structures formed by a core of neutral lipids, mainly triacylglycerols (TAG) and sterol esters (SE), which are delimited by a phospholipid monolayer embedded with proteins primarily related to lipid metabolism (1–3). At first, LDs were believed to be mere neutral lipid deposits, but later it became clear that LDs play other important roles in cellular physiology. The main function of LDs is to maintain cellular lipid homeostasis (4, 5), and it was shown that defects in the mobilization of neutral lipids from LDs are related to type 2 diabetes, inflammation, neurodegenerative disorders, and cancer (6–8). In addition to their importance in health, LDs also are studied in oleaginous yeast for their exploitation as cellular oil factories for biofuel production (9).
Although the metabolic steps of LD biogenesis are well known, the signals that govern LD dynamics are not clear yet. Because the signaling pathways are well conserved between yeast and mammals, Saccharomyces cerevisiae has been a model for studying LD dynamics. In S. cerevisiae, LDs follow a particular dynamic tightly linked to the growth phase and to the nutritional status of the cell (10, 11). When quiescent yeast cells encounter a rich medium containing glucose, cells must exit G0 in order to start duplication and cell growth (12). This start demands a large amount of sterols and fatty acids, leading to the strong mobilization of the neutral lipids stored in LDs. As a result, LDs are diminished in number and in size (10). After this strong lipolytic phase, yeast cells shift to a lipogenic phase to replenish the levels of LDs, reaching its maximum at early stationary phase (10, 11).
The regulatory mechanisms that orchestrate the correct expression and activity of lipolytic/lipogenic enzymes are not completely understood. However, some clues were given by the discovery that nitrogen limitation induces LD accumulation in microorganisms, such as the oleaginous yeast Yarrowia lipolytica, microalgae, and bacteria (3, 9, 13, 14). Thus, it is tempting to speculate that the TOR pathway, a well-established nitrogen-sensing and discriminating pathway (15, 16), coordinates lipid metabolism with growth.
While in mammals there is only a single TOR kinase that participates as part of both mammalian TOR complexes 1 and 2 (mTORC1 and mTORC2) (17), the yeast S. cerevisiae owns two different TOR kinases, Tor1p and Tor2p. Both isoforms can interchangeably form TOR complex 1 (TORC1), but only Tor2p can participate in TOR complex 2 (TORC2) (18, 19). Aside from this difference, the activities of TORC1, which regulates energetic metabolism and growth, and TORC2, which regulates cytoskeleton organization, are well conserved among organisms (20). In particular, only TORC1 is inhibited by rapamycin (19). In both yeast and mammals, TORC1/mTORC1 is activated when nutrients are available, promoting protein synthesis and growth. During nutrient starvation or rapamycin treatment, TORC1/mTORC1 is inhibited (17, 21). In yeast, TORC1 regulates the localization of several transcriptional factors coordinating transcriptional programs through two major distinct effectors: Sch9p, involved in ribosome biogenesis and translational regulation, and Tap42p-PP2A, involved in metabolic regulation (15, 20, 22, 23). The PP2A branch was the first to be discovered. When TORC1 is activated, Tap42p is phosphorylated and forms heterodimers with PP2A (Pph21p and Pph22p) and a PP2A-like protein phosphatase (Sit4p), preventing the activity of downstream transcription factors. Upon TORC1 inhibition (rapamycin treatment or nitrogen starvation), Tap42–PP2A/PP2A-like interaction is lost, and the transcription factors Gln3p and Gat1p are dephosphorylated and transiently localized to the nucleus (20, 23–25). TORC1 also regulates other outputs through the Tap42-PP2A branch, such as the retrograde pathway that coordinates mitochondrial function to changes in transcription, through Rtg1p and Rtg3p transcription factors, among others, and the environmental stress response, which coordinates a general transcriptional response to different stresses through the transcription factors Msn2p and Msn4p (20, 26, 27).
In mammals, there is evidence that mTORC1 must be active to allow the induction of lipid biosynthesis genes by growth factors (28). It is also known that LD formation caused by leptin treatment is mTORC1 dependent (29). Besides its lipogenic role, the activation of mTORC1 also results in the suppression of lipolysis in adipocytes (30). Although reports on the regulation of mammalian LD formation are increasing, the regulation of yeast lipid metabolism by TORC1 has not been studied.
In this work, we explored the role of the TORC1 pathway in the metabolism of LDs in S. cerevisiae, focusing on its Tap42p-Sit4p branch. Contrary to what has been shown in mammals, we discovered that treatment of cells with rapamycin, a specific TORC1 inhibitor, promotes the shift from the lipolytic to lipogenic phase by activating TAG but not SE synthesis. One of the proposed mechanisms is that the inhibition of the TOR pathway by rapamycin derepresses the nitrogen catabolite repression (NCR) pathway, since deletion of Gln3p and/or Gat1p blocks the rapamycin effect on LD dynamics.
MATERIALS AND METHODS
Yeast strains.
Saccharomyces cerevisiae strain BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0), the derived open reading frame knockout collection (MATa deletion library), and strains expressing proteins tagged with the TAP epitope (TAP collection) were obtained from Open Biosystems. The BY4742 (MATa his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0), dga1Δ lro1Δ (MATa his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 dga1::KanMX lro1::KanMX), and are1Δ are2Δ (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 are1::KanMX are2::KanMX) strains were kindly provided by Sepp D. Kohlwein (University of Graz, Austria). tor1-1 and tor2-1 mutants were derived from the JK9-3da (MATa leu2-3,112 ura3-52 rme1 trp1 his4 GAL+ HMLa) strain. Mutant gln3Δ gat1Δ (MATa leu2 ura3 rme1 trp1 his3Δ GAL+ HMLa gln3::KanMX gat1::HISMX) and tap42-11 (MATa leu2-3,112 ura3-52 rme1 GAL+ HMLa tap42::KanMX/YCplac111::tap42-11ts) strains, derived from the TB50a (MATa leu2 ura3 rme1 his3Δ GAL+ HMLa) strain, were kindly provided by M. N. Hall (University of Basel, Basel, Switzerland).
Reagents.
Rapamycin (Rap; Sigma-Aldrich, St. Louis, MO) was prepared at 1 mg/ml in dimethyl sulfoxide (DMSO). BODIPY 493/503 (referred to here as BODIPY) was purchased from Invitrogen, prepared as a 10 mM stock solution in DMSO, and kept at −80°C. All other reagents were obtained from Sigma-Aldrich (St. Louis, MO).
Growth conditions and rapamycin treatment.
Yeast cells were grown to stationary phase for 48 h in liquid rich medium (YPD; 1% yeast extract, 2% peptone, and 2% glucose) or in SD medium (2% glucose, 0.67% yeast nitrogen base, and the required supplements) by shaking at 30°C. At this time, the cells achieve a maximum of LD content. The cells then were seeded in fresh YPD medium at an optical density at 600 nm (OD600) of 0.25 and grown by shaking at 30°C. After 6 h, when the cultures reached the mid-log phase, the cultures were divided, and either rapamycin at a final concentration of 100 ng/ml or vehicle alone was added. The cultures were further incubated for 12 h with shaking at 30°C. Aliquots were taken for optical density (OD600) or LD index readings at the indicated time points.
Lipid droplet quantification by liquid fluorescence recovery (LFR) assay.
Lipid droplets were quantified by incubating formaldehyde-fixed cells with a solution containing the fluorescent dye BODIPY 493/503 quenched with potassium iodide. In this assay, BODIPY passively enters the cells, staining the LDs, as described previously (11). Briefly, aliquots (OD600 of 5) were withdrawn from the yeast cultures, harvested, and fixed with formaldehyde (final concentration of 3.7% [vol/vol]) for 15 min at room temperature. Fixed cells were collected by centrifugation (3,000 × g, 5 min) and washed once with distilled water. Pellets were resuspended in water and kept at 4°C until use. Readings were performed by adding fixed cells to a reading medium containing the neutral lipid probe BODIPY 493/503 (Invitrogen) (5 μM) and the fluorescence quencher potassium iodine (KI) (0.5 M). Fluorescence was read at 495/510 nm in a SpectraMax M5 plate reader (Molecular Devices) sequentially with absorbance at 600 nm (A600). The LD index, which reflects the cell neutral lipid content, is expressed as the ratio of fluorescence intensity to the A600 of the cells. To obtain the relative LD (rLD) index, results were normalized to the LD index of stationary-phase cells.
Confocal fluorescence microscopy.
LDs were visualized by microscopy. Yeast cells were fixed with formaldehyde, as described above, and resuspended in water. One μl of 10 mM BODIPY 493/503 and 3 μl of vector mounting medium for fluorescence (Vectashield) were added to 2 μl of fixed cells. Images were captured using an Olympus IX 81 microscope with a 100× oil immersion objective.
Lipid analysis.
Yeast cells were harvested by centrifugation at 3,000 × g for 5 min at room temperature and washed once with cold distilled water. Lipids were extracted based on a modified protocol described by Bourque and Titorenko using chloroform-methanol-water as solvents (31), and the final extract was dried under a stream of nitrogen and stored at 20°C. Lipids were resuspended in chloroform and applied to silica plates to perform thin-layer chromatography (TLC), employing triolein and cholesteryl oleate as standards (Sigma-Aldrich, St. Louis, MO). Neutral lipids were separated in an ascending manner by using a two-step separation system: light petroleum-diethyl ether-acetic acid (35:15:1, vol/vol) as a solvent system developed to 2/3 of the height of the plate, followed by a light petroleum-diethyl ether (49:1, vol/vol) solvent system developed to within 1 cm of the top (32). Lipids were revealed with iodine vapor, and spots were quantified by densitometry using Image Master TotalLab 1.11 (Amersham Pharmacia Biotech, England).
For the enzymatic determination of triacylglycerol content, cells were centrifuged and resuspended in 300 μl of extraction buffer (50 mM Tris-HCl, 0.3% Triton X-100, pH 7.5) and lysed with glass beads by vortexing for 5 cycles of 30 s each. Lysed cells were separated, and the glass beads were washed with 300 μl of extraction buffer. The total lysate was centrifuged at 3,000 rpm for 10 min. Neutral lipids were extracted from 200 μl of the supernatant as described by Bligh and Dyer (33). Triacylglycerols were measured, as previously described (11), using the triacylglycerol reagent kit (Doles, Brazil) according to the manufacturer's instructions against glycerol standards. Intracellular TAG was normalized by the protein concentration.
Preparation of protein homogenates and Western blotting.
Protein homogenates were prepared as previously described (34). Briefly, cells were centrifuged and the pellet was resuspended and incubated on ice for 10 min with 0.2 M NaOH and 0.2% of 2-mercaptoethanol. After the addition of 5% trichloroacetic acid, cells were further incubated for 10 min on ice. Total protein was collected by centrifugation, resuspended in Laemmli sample buffer, and immediately heated for 5 min at 80°C. Ten-microliter aliquots, corresponding to an OD of approximately 0.16, were separated in 6% SDS-acrylamide gel using the Mini-Protean II instrument (Bio-Rad) and electrotransferred to Immobilon-P for 30 min at 18 V in 25 mM Tris, 192 mM glycine, and 10% methanol, using a Transblot semidry cell (Bio-Rad). After transfer, membranes were treated with 5% nonfat dry milk solution in Tris-buffered saline with Tween 20 (TBS-T) buffer for 1 h at room temperature and then incubated overnight at 4°C with a 1:10,000 dilution of anti-TAP antibody (Open Biosystems). Blots were detected using the ECL Plus kit (GE Healthcare). For total protein quantification after anti-TAP Western blotting, membranes were incubated for 2 min in staining solution (0.1% Coomassie brilliant blue R in 50% methanol, 7% acetic acid) and then for 10 min in destaining solution (50% methanol, 7% acetic acid). Membranes were washed with distilled water and air dried.
qRT-PCR.
For quantitative real-time PCR (qRT-PCR), RNA was isolated as previously described (35). The cDNA was synthesized from extracted RNA using the high-capacity cDNA reverse transcription kit (Applied Biosystems) as indicated by the manufacturer and further used as a template for real-time PCR using SYBR green PCR master mix (Applied Biosystems, CA). Amplifications were performed in a StepOnePlus or 7500 real-time PCR system (Applied Biosystems, CA). PCR amplification was carried out under the following conditions: 95°C for 10 min, and then 40 cycles at 95°C for 15 s, 55°C for 30 s, and 72°C for 90 s. The threshold cycle (CT) for each gene of interest was calculated using StepOne software, v2.2, or 7500 software, v2.0.5. Normalization for each gene was performed against the mock-treated sample (treated with vehicle alone), and gene expression levels were based on the 2−ΔΔCT method (36, 37) by calculating the relative copy number (RCN) using ACT1 gene as an internal control. The primers used were the following: CIT2F (5′GCATTTGGTATTCTTGCTCAA3′), CIT2R (5′TGCTTTCAATGTTTTTGACCA3′), ACT1F (5′TTCCCAGGTATTGCCGAAA3′), and ACT1R (5′TTGTGGTGAACGATAGATGGA3′).
Statistical analysis.
The statistical significance of the results was evaluated using Student t tests. Analyses for differences in drug treatment and control during time courses were performed by two-way analysis of variance (ANOVA) with the repeated-measures test and the Bonferroni posttest using GraphPrism 5.0 software.
RESULTS
Inhibition of the TORC1 pathway by rapamycin induces LD accumulation in yeast.
We investigated the effect of rapamycin, a TORC1 inhibitor, on LD dynamics in yeast by using a liquid fluorescence recovery (LFR) assay previously described by our group (11). Stationary cells were seeded at a low density in fresh rich medium and incubated for 6 h at 30°C before the addition of rapamycin. We have chosen the 6-h time point to add rapamycin, because this is the moment when the LD index reaches the lowest value under this experimental condition. We observed that rapamycin induced a faster increase in LD replenishment than nontreated yeast cells (Fig. 1A). Fluorescence microscopy showed that rapamycin-treated cells stained more intensely with BODIPY than the nontreated yeast cells (Fig. 1C and D), reinforcing LFR assay results. We have also tested the effect of the addition of rapamycin just after the seeding in fresh rich medium and observed that in this case, rapamycin blocks the lipolytic phase and slightly induces LD accumulation (Fig. 1B).
FIG 1.

Rapamycin induces rapid LD replenishment. Wild-type (BY4741) yeast cells were pregrown for 48 h to stationary phase and then inoculated into fresh YPD medium at low density (OD600 of 0.25). Rapamycin (100 ng/ml) (•) or vehicle alone (○) was added after 6 h of growth (A) or at the time of seeding (B). The LD indexes were measured using the LFR assay and normalized to that obtained in pregrown stationary-phase cells (relative LD index, or rLD) (upper). Growth was determined by absorbance at 600 nm (Abs600nm) (lower). (C) Aliquots from the control culture or rapamycin-treated cells after 2 h of rapamycin treatment, from the experiment depicted in panel A, were analyzed by confocal fluorescence microscopy. Cells were stained with BODIPY (green) and 4′,6-diamidino-2-phenylindole (DAPI) (blue) (scale bar, 2 μm). (D) Intensity of fluorescence was determined as described in Materials and Methods. Data are from three independent experiments ± standard deviations (*, P < 0.05; **, P < 0.01; ***, P < 0.001 for the control versus rapamycin treatment).
While mammalian cells present a unique TOR kinase, named mTOR, the yeast S. cerevisiae presents two TOR isoforms, Tor1p and Tor2p (17, 18, 38). In yeast, both isoforms can participate with other proteins in the formation of TOR complex 1 (TORC1), but only Tor2p can participate in TOR complex 2 (TORC2) (19). Despite this difference, like in mammalian cells, only TORC1 is affected by rapamycin treatment (19). Rapamycin acts by forming a complex with FKBP12, which interacts with Tor1/2p kinases, inhibiting TORC1 activity. The tor1-1 and tor2-1 rapamycin-resistant mutants, first reported by Heitman et al. (18), present mutations that disrupt the FKBP-rapamycin binding site in Tor1/2, avoiding TORC1 inhibition (39). The resistance to rapamycin conferred by these alleles is dominant, as one TORC1-resistant complex is enough to guarantee TORC1 function upon rapamycin treatment (18, 39).
In order to investigate whether the rapamycin effect is indeed caused by the specific inhibition of TORC1, we studied the effect of rapamycin on the LD dynamics of tor1-1 and tor2-1 mutants. We observed that these mutations do not affect LD dynamics compared to the isogenic WT strain under normal growth conditions, but both mutants fail to respond to rapamycin (Fig. 2A to C).
FIG 2.

Rapamycin effect on LDs is due to the inhibition of the TORC1 pathway. tor1-1 (A), tor2-1 (B), and WT (JK9-3da) (C) strains were treated with rapamycin (100 ng/ml) (•) or vehicle alone (○) as described in the legend to Fig. 1A. The LD indexes were measured and normalized to that obtained in pregrown stationary-phase cells (rLD index) (upper), and growth was determined by absorbance at 600 nm (lower). ***, P < 0.001. (D) Rapamycin strongly induces CIT2 expression. The WT (BY4741) strain was treated with vehicle alone (Control) or rapamycin (100 ng/ml) (Rap) for 30 min up to 2 h, and the relative copy number (RCN) of CIT2 transcript was determined by real-time PCR. The ACT1 gene was used for normalization. Data are from three independent experiments ± standard deviations (*, P < 0.05; **, P < 0.01). (E) Cit2p-TAP protein levels were analyzed by Western blotting using the anti-TAP antibody (upper). Densitometry is from three independent experiments (***, P < 0.001 between rapamycin and the control).
Previous studies show that CIT2, encoding the peroxisomal form of citrate synthase (40), is highly induced by rapamycin treatment (27, 41). In order to confirm that the TORC1 pathway is being activated under our experimental conditions, we determined the effect of rapamycin on the CIT2 mRNA and protein levels by quantitative RT-PCR and immunoblot assays, respectively (Fig. 2D and E). We found that CIT2 expression is highly induced by rapamycin treatment at both the mRNA and protein levels. Taken together, these results confirm that the TORC1 pathway is inhibited by rapamycin under the conditions tested and that the effects of rapamycin on the LDs are a consequence of the inhibition of TORC1, suggesting that TORC1 is involved in the regulation of LD homeostasis in yeast.
Growth inhibition does not necessarily correlate with LD accumulation and vice versa.
It has been described that in different microorganisms, such as in microalgae and the yeast Yarrowia lipolytica, lipid accumulation could be a consequence of lower growth rates (9, 42, 43). To investigate if the lipogenic effect of rapamycin was related to the inhibition of growth, we subjected cells to different types of stress, such as osmotic stress (1 M sorbitol), saline stress (1 M NaCl), and nitrogen starvation (Fig. 3). Osmotic stress reduced growth but did not affect LD homeostasis after 2 h of treatment. To confirm this effect, we extended the incubation to 24 h, and no effect on LD dynamics was observed compared to that of nontreated yeast cells (Fig. 3A). On the other hand, saline stress reduced growth and induced LD synthesis in short-time incubations (2 h of treatment) (Fig. 3B). Nitrogen starvation did not reduce growth in the first 2 h of treatment but induced the LD levels within this period (Fig. 3C). As a control for the nitrogen starvation experiment, we tested the effect of rapamycin under the same experimental conditions (SD medium). We observed that both the induction of LD replenishment and the inhibition of growth were similar, as observed in complete medium (Fig. 3D). These results show that the LD accumulation observed in different stress conditions, including rapamycin treatment, are not necessarily related to the decrease in growth rate.
FIG 3.

LD dynamics after saline stress, osmotic stress, and nitrogen starvation. (A and B) Wild-type (BY4741) yeast cells were inoculated into fresh YPD medium, and after reaching exponential phase (5 h), they were left untreated (○) or were treated with 1 M sorbitol or 1 M NaCl (●). For nitrogen starvation (C), the wild-type (BY4741) yeast cells were inoculated in complete SD (0.17% yeast nitrogen base, 0.5% NH2SO4, 2% glucose, and supplements), and after reaching exponential phase, the culture was filtered through a 0.22-μm Millipore filter and reseeded in complete SD (○) or SD without NH2SO4 (•). The effect of nitrogen starvation was compared to that of rapamycin treatment (100 ng/ml) (•) or the addition of vehicle alone (○) in cells grown in complete SD (D). The rLD index (upper) and growth (lower) were determined. Data are from three independent experiments ± standard deviations (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
LD replenishment induced by rapamycin depends on induction of TAG but not SE synthesis.
The core of LD is formed by TAG and SE. The LD index analyzed by the LFR assay might reflect changes in both neutral lipids, so we investigated whether the TAG and/or the SE levels were increased by rapamycin. Analysis of neutral lipids by TLC showed that only the TAG content was induced after 2 h of rapamycin treatment (Fig. 4A), which led to an increase in the TAG-to-SE ratio (Fig. 4B). Enzymatic determination of TAG content corroborated the TLC results, showing a TAG content 40% higher in rapamycin-treated cells than in nontreated cells (62.0 ± 7.0 nmol TAG · mg protein−1 in rapamycin-treated cells, in contrast to 43.0 ± 12 nmol TAG · mg protein−1 in control cells; n = 3, P < 0.05). The specific effect of rapamycin on TAG synthesis was reinforced by the results showing that rapamycin does not affect LD in the strain deleted of the main DAG acyltransferases (dga1Δ lro1Δ strain), but it does affect LD in the strain deleted of the main sterol acyltransferases (are1Δ are2Δ strain) (Fig. 4E to G).
FIG 4.

(A and B) Rapamycin-induced LD accumulation depends on TAG synthesis. WT (BY4741) cells were treated, as described in the legend to Fig. 1A, with rapamycin (100 ng/ml) (gray bars) or vehicle alone (white bars). After 2 h of treatment, cells were harvested and lipids were extracted and analyzed by TLC. (A) The sterol ester (SE) and triacylglycerol (TAG) contents after rapamycin treatment were estimated relative to those of samples treated with vehicle alone. (B) The TAG/SE ratio was calculated in control yeast cells and rapamycin-treated cells. Data are from three independent experiments ± standard deviations (*, P < 0.05; ns, nonsignificant). (C and D) Phosphorylation of Tgl4p is not induced by rapamycin. (C) The Tgl4-TAP strain was grown as described in the legend to Fig. 1A and treated with rapamycin (100 ng/ml) or vehicle alone after 6 h of seeding. Total protein was extracted, and after separation of proteins in 6% SDS-PAGE, Tgl4p-TAP was revealed by Western blotting using anti-TAP (1:10,000) antibody. (D) The ratio of phosphorylated Tgl4p to nonphosphorylated Tgl4p was quantified by densitometry. (E to G) Rapamycin-induced LD replenishment is dependent on TAG but not SE synthesis. The WT (BY4742) (E), the are1Δ are2Δ strain, deficient in SE synthesis (F), and the dga1Δ lro1Δ strain, deficient in TAG synthesis (G), were treated with rapamycin (○) or vehicle alone (•) as described in the legend to Fig. 1A. The rLD indexes (upper) and culture growth (lower) were recorded. Data are from three independent experiments ± standard deviations (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
It is believed that the LD levels reflect an equilibrium state between continuous synthesis and hydrolysis of neutral lipids (10). Thus, the rapamycin effect observed could be due to the activation of neutral lipid synthesis and/or to inhibition of neutral lipid mobilization. Kurat and coworkers (34) have shown that the onset of the lipolytic phase depends on the activation of Tgl4p lipase by phosphorylation. Thus, we tested whether rapamycin decreased the phosphorylation level of Tgl4p. We observed that the ratio between phosphorylated and nonphosphorylated forms of Tgl4p was not changed by rapamycin treatment (Fig. 4C and D), although we observed a reduction in total Tgl4p level at 2 h of rapamycin treatment. The latter effect could be related to general rapamycin-induced protein degradation (44). This result suggests that rapamycin reduces the level of Tgl4p, altering the balance between lipolysis and lipogenesis, favoring the latter.
Involvement of the Tap42-PP2A branch of the TOR pathway on the LD induction by rapamycin.
It has become clear that in yeast, TORC1 monitors nutrients and stress signals and then transduces these signals through two main signaling branches: the Sch9p branch, which regulates protein and ribosome synthesis genes, and the Tap42-PP2A branch, which participates in the regulation, mostly of metabolism genes during nitrogen starvation and rapamycin treatment (20, 22, 23). In the next experiments, we studied the participation of the PP2A branch in the modulation of LD homeostasis by rapamycin. We chose to study this branch because we have shown that, just like rapamycin, nitrogen starvation enhances LD accumulation (Fig. 3C). Also, we have shown previously that the deletion of the PP2A-like Ser/Thr protein phosphatase Sit4 leads to lower levels of LDs in a growing culture via modulation of AMP-activated kinase (AMPK)/Snf1p phosphorylation (11). This kinase is known to regulate lipid metabolism by controlling the activity of acetyl coenzyme A (acetyl-CoA) carboxylase (ACCase) (45, 46). First, we tested the effect of rapamycin on LD levels in a sit4Δ strain. We observed that the deletion of SIT4 partially suppresses the induction of LD accumulation caused by rapamycin (Fig. 5A). The role of Sit4p in the response to rapamycin treatment probably is not related to its known role in regulating Snf1p (11), because rapamycin can induce LD levels in an snf1Δ strain (Fig. 5B). Furthermore, we could not detect any effect of rapamycin on the phosphorylation state of Thr210-Snf1p under this experimental condition (data not shown).
FIG 5.
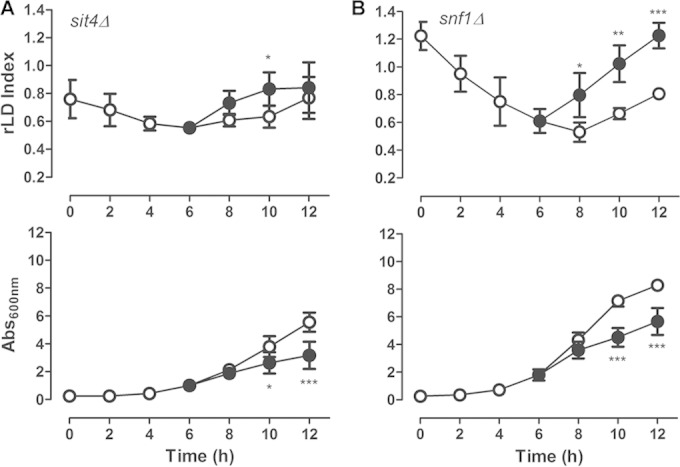
Rapamycin-induced LD replenishment is dependent on SIT4. sit4Δ (A) and snf1Δ (B) mutant strains were treated with rapamycin (100 ng/ml) (•) or vehicle alone (○), as described in the legend to Fig. 1A. The relative LD index was normalized to the LD index obtained in WT (BY4741) cells pregrown to stationary phase (upper), and culture growth was determined (lower). Data are from three independent experiments ± standard deviations (*, P < 0.05; ***, P < 0.001).
TORC1 signaling through the protein phosphatase Sit4 requires its regulatory protein, Tap42p (23, 26). Tap42p is essential to cell viability, so we tested the effect of rapamycin on a strain carrying the temperature-sensitive tap42-11 allele, which is partially resistant to rapamycin at 25°C. We observed that this mutant was unresponsive to rapamycin and LD levels were not increased within 2 h of treatment (Fig. 6). Sit4-Tap42 effectors include the GATA-family transcription factors Gln3p and Gat1p (15, 47), the heterodimeric transcriptional activators Rtg1p and Rtg3p (20, 41), and the Zn finger transcription factors Msn2p and Msn4p (26, 48). These factors regulate the nitrogen catabolic response, the retrograde pathway, and the general stress response, respectively. We next explored the involvement of these PP2A branch effectors. We observed that LD induction by rapamycin was significantly diminished in the gln3Δ, gat1Δ, rtg1Δ, and rtg3Δ strains but not in the msn2Δ and msn4Δ mutant strains (Fig. 7). Failure to reduce the LD induction in msn2Δ and msn4Δ strains could be due to the overlapping function of Msn2p and Msn4p; however, the double-deletion msn2Δ msn4Δ strain still responded to rapamycin (data not shown), ruling out this possibility.
FIG 6.
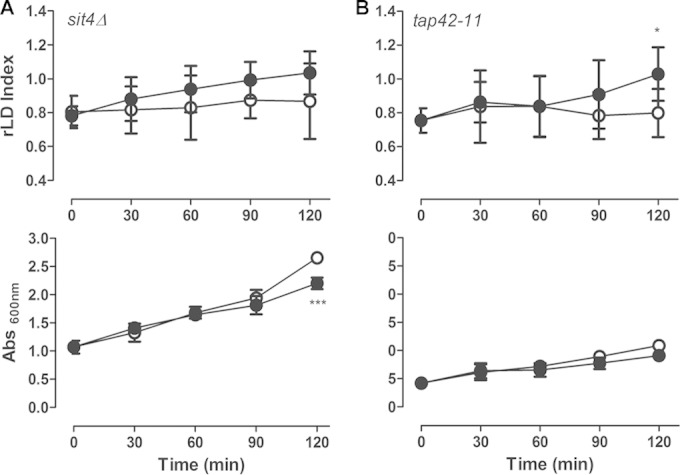
Tap42p is important to mediate LD replenishment by rapamycin. The sit4Δ strain (A) and the temperature-sensitive tap42-11 mutant strains (B) were treated with rapamycin (100 ng/ml) (•) or vehicle alone (○), as described in the legend to Fig. 1, for 2 h. The temperature-sensitive tap42-11 strain was grown at 25°C, where it is partially resistant to rapamycin. The LD indexes were measured and normalized to that obtained in WT (BY4741 and TB50-a, respectively) pregrown stationary cells using the LFR assay (upper), and culture growth was determined (lower). Data are from three independent experiments ± standard deviations (*, P < 0.05; ***, P < 0.001).
FIG 7.
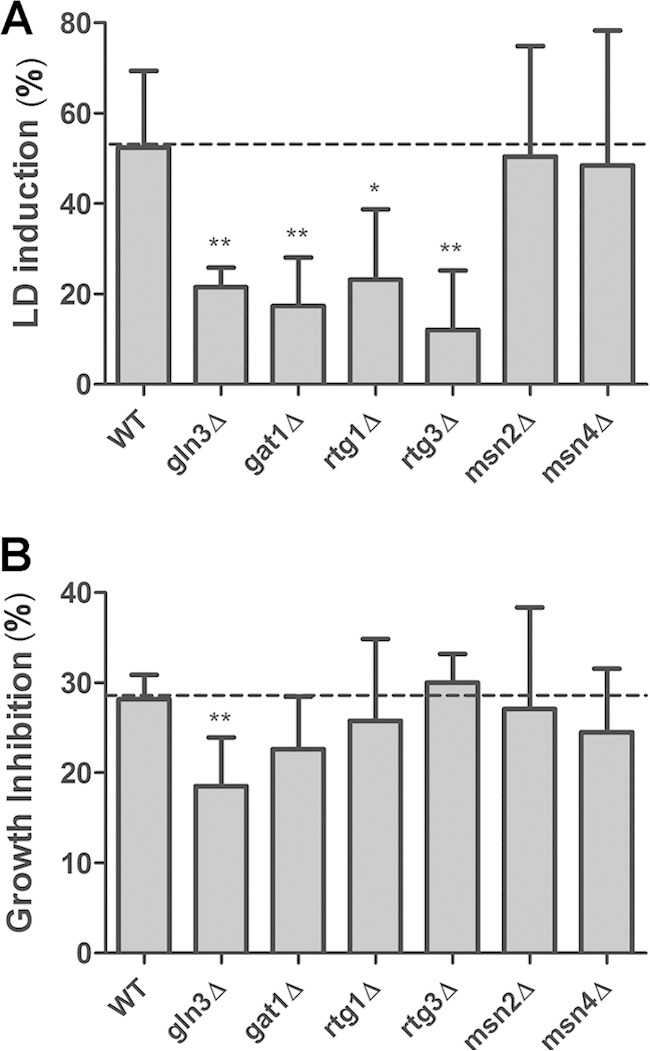
TORC1-PP2A downstream effectors participate in LD replenishment induced by rapamycin. The indicated deletion strains were treated with rapamycin (100 ng/ml) or vehicle alone, as described in the legend to Fig. 1A, after 2 h of treatment. LD induction (A) and growth inhibition (B) caused by rapamycin treatment were calculated. Data are from three independent experiments ± standard deviations (*, P < 0.05; **, P < 0.01).
Nitrogen starvation and lipid droplet synthesis in S. cerevisiae.
The transcription factors Gln3p and Gat1p regulate the expression of genes involved in nitrogen catabolism. Upon nitrogen limitation, Gln3p and Gat1p are activated and promote the transcription of genes important for the utilization of alternative nitrogen sources. It is well known that Gln3p/Gat1p activation and nuclear translocation are dependent on TORC1 inhibition (16, 49, 50). We studied the LD profile of gln3Δ, gat1Δ, and double-mutant gln3Δ gat1Δ strains. Interestingly, we found that these strains had an aberrant LD profile in which the lipolytic and lipogenic phases were not clearly identified (Fig. 8A and B).
FIG 8.

Disruption of Gln3p and Gat1p transcription factors affect LD dynamics. gln3Δ, gat1Δ, and gln3 Δgat1Δ strains and the isogenic strains were grown in YPD, and the LD dynamics and growth were monitored. (A) gln3Δ (○) and gat1Δ (△) strains and the isogenic WT (BY4741) strain (gray). (B) The gln3Δ gat1Δ strain (black) and its isogenic WT (TB50-a) strain (gray). Data are from three independent experiments ± standard deviations (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
DISCUSSION
Rapamycin or nitrogen starvation induces LD accumulation in S. cerevisiae.
We investigated the role of the TORC1 pathway in the regulation of LD dynamics in the yeast S. cerevisiae. Rapamycin, a TORC1 inhibitor that mimics several outputs of nitrogen starvation, was added to yeast cultures. As expected, the addition of rapamycin (Fig. 1A and 3D) and nitrogen starvation (Fig. 3C) readily promoted a shift to a lipogenic phase when cells are treated at the lipolytic phase. The rapid accumulation of LDs was accompanied by an increase in TAG but not SE levels (Fig. 4A and B). We could not observe differences in the phosphorylation state of the major TAG lipase in the first hour of rapamycin treatment (Fig. 4C and D), although Tgl4p was partially degraded at longer incubation times (2 h). These results suggest that the induction of LD synthesis is due in part to the induction of TAG synthesis and reduction of lipolysis.
Different metabolic pathways could be used by TORC1 to induce TAG synthesis as de novo fatty acid synthesis by the glyceroneogenesis pathway or the use of the free amino acid pool as a result of rapamycin-induced autophagy. From these we have discarded de novo synthesis of fatty acids by glyceroneogenesis, since key enzymes in this pathway, such as phosphoenolpyruvate carboxykinase, were not induced by rapamycin (data not shown). We also did not detect the incorporation of labeled 14C from [14C]pyruvate into TAG after rapamycin treatment (data not shown). TORC1 inhibition already was demonstrated to promote autophagy (20), which might provide blocks for TAG synthesis. It has been shown that hepatocytes and cardiac myocytes accumulate LDs after short periods of starvation, which is dependent on the autophagic protein Atg7 (51). Additional experiments are needed to study if, in yeast, the induction of TAG synthesis by rapamycin is indeed linked to autophagy.
Growth impairment is not necessarily linked to LD accumulation.
A main concern about our results was that TORC1 is known for its role as a coordinator of cellular growth in response to nutrient availability. Since TORC1 inhibition leads to diminished growth rates and LD dynamics are profoundly affected by growth phase, we questioned if the replenishment of LDs induced by rapamycin was a nonspecific effect of growth impairment. If this was true, we would expect that inhibition of growth by other stresses would affect LD dynamics. Our evidence showed that LD accumulation was not always connected to growth inhibition. First, osmotic stress impaired growth without affecting LD dynamics (Fig. 3A). Second, if reduced growth and LD synthesis were linked phenomena, the sit4Δ mutant would break this rule, as it shows slow growth but lower levels of LDs (11).
TORC1 is implicated in lipid homeostasis.
LD dynamics are linked to the progression of the cell cycle, as evidenced by the activation of Tgl4p lipase by the cyclin-dependent kinase 1, Cdk1/Cdc28, and the acute lipolytic activity observed in prelogarithmic growth (10, 34, 52). We speculate that TORC1 is part of the cross talk between regulators of cell cycle and LD dynamics. Here, we provide evidence that the TORC1 pathway is involved in LD homeostasis, since the deletion of downstream effectors, such as the Ser-Thr phosphatase Sit4p (Fig. 5A) and the transcription factors Gln3p and Gat1p, led to abnormal LD dynamics (Fig. 8).
Besides TORC1, AMPK (or Snf1p) in yeast cells is also a hub that senses the energetic status of the cell and triggers the cellular response to a variety of stresses (53, 54). The existence of cross talk between TOR and Snf1p/AMPK pathways still is controversial in yeast. In mammals, AMPK and mTORC1 pathways are linked, since AMPK is able to inhibit mTORC1 through the phosphorylation of its upstream regulators, TSC2 and Raptor, when the cellular energy level is low (55, 56). In yeast, such regulation is not yet well established. Evidence suggests that TORC1 and Snf1p/AMPK converge in the regulation of fatty acid metabolism and other outputs independently (57). A recent work provided strong evidence that Snf1p/AMPK participates as a possible upstream regulator of the TORC1 pathway under glucose-limiting conditions (58).
In fact, previous studies demonstrated that Sit4p and Reg1p-Glc7 phosphatase are negative regulators of Snf1p/AMPK activity (11, 59, 60). In sit4Δ and reg1Δ mutants, Snf1p/AMPK is hyperactivated, which might inhibit acetyl-CoA carboxylase activity, a key enzyme in the synthesis of fatty acids (11, 59). Preliminarily, we hypothesized that the effect of rapamycin on LD replenishment was via Snf1p/AMPK. Thus, the activation of the TORC1 pathway would have a negative effect on the accumulation of LD. We had to discard this hypothesis, because the deletion of SNF1 did not affect the cellular response to rapamycin (Fig. 5B).
By which mechanism did the inhibition of TORC1 induce the shift to the lipogenic phase? Summarizing our data, we believe that the modulation of LD homeostasis by TORC1 requires the PP2A-like phosphatase, Sit4p, as well as the Gln3p and Gat1p transcriptional complex (Fig. 9). This assumption is based on the observation that strains deleted of the corresponding genes have aberrant LD dynamics during growth, where the transition between the lipolytic and lipogenic phase was not clearly observed (Fig. 8). Furthermore, sit4Δ, gln3Δ, and gat1Δ mutants did not respond to rapamycin by inducing LD replenishment in the first 2 h of treatment. Thus, rapamycin-induced NCR genes might be involved in the regulation of lipid homeostasis in batch cultures.
FIG 9.
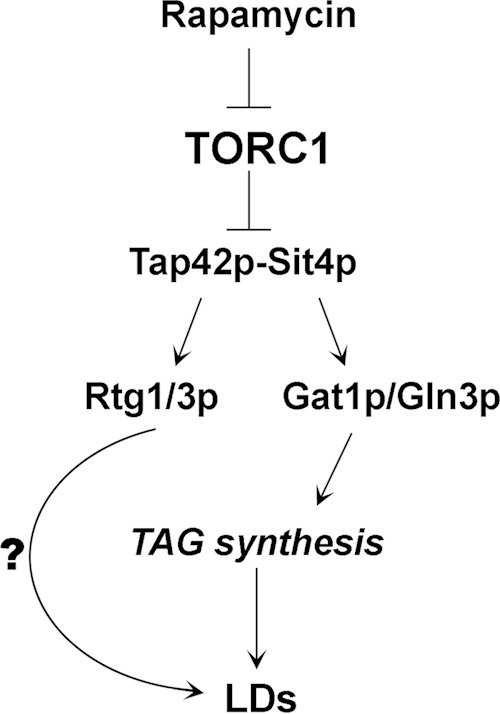
Upon rapamycin treatment or nutrient starvation, TORC1 is inhibited, freeing Tap42p-Sit4p to act on downstream effectors, such as Rtg1/3p and Gat1p/Gln3p. Gln3p/Gat1p are translocated to the nucleus and induce early TAG synthesis, which is reflected in increased LDs. The effect of rapamycin on LD replenishment also is dependent on Rtg1/3p, but the role of these transcription factors has yet to be investigated.
TORC1 inhibition by rapamycin has different outcomes in yeast and mammalian cells.
Our results showing that TORC1 inhibition in yeast leads to LD synthesis do not agree with the role of the mTOR pathway in mammals, where it is well established that the activation of the TORC1 pathway is essential to promote lipogenesis (28, 61).
One possible reason for the different outcomes of TORC1 activation/inhibition on LD homeostasis between yeast and mammals is the different roles of LDs in those organisms. Although LDs have common roles, such as lipid storage and protection against lipotoxicity, in both organisms (4, 62, 63), in mammals, LDs are involved in other biological processes, such as immune response (64) and even viral replication (65). The role of LDs in those differential processes could explain the different regulatory role of TORC1 in LD homeostasis.
Another possible explanation for the different outcomes regarding LD homeostasis of the activation/inactivation of TORC1 in yeast and mammals is the different strategies used by these organisms during nutrient limitation conditions. For example, contrary to what is expected for most mammalian cells, yeast cells consume glycogen when they are presented with proper growth conditions (e.g., fresh medium) which allow rapid growth, and they accumulate glycogen when the nutrients start to become limited (66). As unicellular organisms, yeast cells cannot rely on other cells to offer them nutrients under nutrient starvation conditions. When nutrients start to become limiting, they slow down division and start to accumulate nutrients to handle the rough times ahead (12). If we apply this same reasoning to lipids, it does makes sense that yeast cells utilize lipids when they are presented proper growth conditions (e.g., fresh medium), and evidence shows that they use these lipids for rapid membrane synthesis (10, 52). When cells sense a stress or nutrient limitation conditions, they accumulate neutral lipids. In this scenario, it does make sense that a signaling pathway that coordinates growth with nutrient availability in yeast, such as the TORC1 pathway (20), would activate lipolysis under proper growth conditions to provide building blocks and energy for proliferation and would induce neutral lipid storage and growth arrest under nutrient-limiting conditions. It will be interesting to understand evolutionarily how a well-conserved signaling cascade, such as the TORC1 pathway, diverged to completely opposite outputs to maintain lipid homeostasis in different organisms. Further work needs to be performed to address this question.
ACKNOWLEDGMENTS
This work was supported by grants from Conselho Nacional de Ciência e Tecnologia (CNPq), a masters degree and Ph.D. degree fellowship to J.B.M. (CNPq), postdoctoral fellowship CAPES-PNPD Institucional to B.L.B.-M., the Fundação de Amparo a Pesquisa do Rio de Janeiro (FAPERJ-Cientistas do Nosso Estado to M.M.-L.), and an Iniciação Científica fellowship to G.S.M. (CNPq).
We gratefully acknowledge Suelene Francisca Bispo, Andressa Piedade Motta, and Jens Rietdorf (CAPES/CDTS) for their technical assistance.
REFERENCES
- 1.Clausen MK, Christiansen K, Jensen PK, Behnke O. 1974. Isolation of lipid particles from baker's yeast. FEBS Lett 43:176–179. doi: 10.1016/0014-5793(74)80994-4. [DOI] [PubMed] [Google Scholar]
- 2.Leber R, Zinser E, Paltauf F, Daum G, Zellnig G. 1994. Characterization of lipid particles of the yeast, Saccharomyces cerevisiae. Yeast 10:1421–1428. doi: 10.1002/yea.320101105. [DOI] [PubMed] [Google Scholar]
- 3.Murphy DJ. 2001. The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog Lipid Res 40:325–438. doi: 10.1016/S0163-7827(01)00013-3. [DOI] [PubMed] [Google Scholar]
- 4.Fujimoto T, Ohsaki Y, Cheng J, Suzuki M, Shinohara Y. 2008. Lipid droplets: a classic organelle with new outfits. Histochem Cell Biol 130:263–279. doi: 10.1007/s00418-008-0449-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beller M, Sztalryd C, Southall N, Bell M. 2008. COPI complex is a regulator of lipid homeostasis. PLoS Biol 6:e292. doi: 10.1371/journal.pbio.0060292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krahmer N, Farese RV, Walther TC. 2013. Balancing the fat: lipid droplets and human disease. EMBO Mol Med 5:905–915. doi: 10.1002/emmm.201100671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bozza PT, Viola JPB. 2010. Lipid droplets in inflammation and cancer. Prostaglandins Leukot Essent Fatty Acids 82:243–250. doi: 10.1016/j.plefa.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 8.Cole NB, Murphy DD, Grider T, Rueter S, Brasaemle D, Nussbaum RL. 2002. Lipid droplet binding and oligomerization properties of the Parkinson's disease protein alpha-synuclein. J Biol Chem 277:6344–6352. doi: 10.1074/jbc.M108414200. [DOI] [PubMed] [Google Scholar]
- 9.Beopoulos A, Cescut J, Haddouche R, Uribelarrea J-L, Molina-Jouve C, Nicaud J-M. 2009. Yarrowia lipolytica as a model for bio-oil production. Prog Lipid Res 48:375–387. doi: 10.1016/j.plipres.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 10.Kurat CF, Natter K, Petschnigg J, Wolinski H, Scheuringer K, Scholz H, Zimmermann R, Leber R, Zechner R, Kohlwein SD. 2006. Obese yeast: triglyceride lipolysis is functionally conserved from mammals to yeast. J Biol Chem 281:491–500. doi: 10.1074/jbc.M508414200. [DOI] [PubMed] [Google Scholar]
- 11.Bozaquel-Morais BL, Madeira JB, Maya-Monteiro CM, Masuda CA, Montero-Lomeli M. 2010. A new fluorescence-based method identifies protein phosphatases regulating lipid droplet metabolism. PLoS One 5:e13692. doi: 10.1371/journal.pone.0013692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gray JV, Petsko GA, Johnston GC, Ringe D, Singer RA, Werner-Washburne M. 2004. “Sleeping beauty”: quiescence in Saccharomyces cerevisiae. Microbiol Mol Biol 68:187–206. doi: 10.1128/MMBR.68.2.187-206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ratledge C, Wynn JP. 2002. The biochemistry and molecular biology of lipid accumulation in oleaginous microorganisms. Adv Appl Microbiol 51:1–51. doi: 10.1016/S0065-2164(02)51000-5. [DOI] [PubMed] [Google Scholar]
- 14.Richardson B, Orcutt DM, Schwertner HA, Martinez CL, Wickline HE. 1969. Effects of nitrogen limitation on the growth and composition of unicellular algae in continuous culture. Appl Microbiol 18:245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beck T, Hall M. 1999. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 3:689–692. [DOI] [PubMed] [Google Scholar]
- 16.Cardenas ME, Cutler NS, Lorenz MC, Di Como CJ, Heitman J. 1999. The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev 13:3271–3279. doi: 10.1101/gad.13.24.3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Virgilio C, Loewith R. 2006. The TOR signalling network from yeast to man. Int J Biochem Cell Biol 38:1476–1481. doi: 10.1016/j.biocel.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Heitman J, Movva NR, Hall MN. 1991. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 19.Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. 2002. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 10:457–468. doi: 10.1016/S1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 20.Loewith R, Hall MN. 2011. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics 189:1177–1201. doi: 10.1534/genetics.111.133363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wullschleger S, Loewith R, Hall MN. 2006. TOR signaling in growth and metabolism. Cell 124:4714–4784. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 22.Urban J, Soulard A, Huber A, Lippman S, Mukhopadhyay D, Deloche O, Wanke V, Anrather D, Ammerer G, Riezman H, Broach JR, De Virgilio C, Hall MN, Loewith R. 2007. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol Cell 26:663–674. doi: 10.1016/j.molcel.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 23.Di Como CJ, Arndt KT. 1996. Nutrients, via the Tor proteins, stimulate the association of Tap42 with type 2A phosphatases. Genes Dev 10:1904–1916. doi: 10.1101/gad.10.15.1904. [DOI] [PubMed] [Google Scholar]
- 24.Yan G, Shen X, Jiang Y. 2006. Rapamycin activates Tap42-associated phosphatases by abrogating their association with Tor complex 1. EMBO J 25:3546–3555. doi: 10.1038/sj.emboj.7601239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tate JJ, Georis I, Dubois E, Cooper TG. 2010. Distinct phosphatase requirements and GATA factor responses to nitrogen catabolite repression and rapamycin treatment in Saccharomyces cerevisiae. J Biol Chem 285:17880–17895. doi: 10.1074/jbc.M109.085712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Düvel K, Santhanam A, Garrett S, Schneper L, Broach JR. 2003. Multiple roles of Tap42 in mediating rapamycin-induced transcriptional changes in yeast. Mol Cell 11:1467–1478. doi: 10.1016/S1097-2765(03)00228-4. [DOI] [PubMed] [Google Scholar]
- 27.Crespo L, Powers T, Fowler B, Hall MN. 2002. The TOR-controlled transcription activators GLN3, RTG1, and RTG3 are regulated in response to intracellular levels of glutamine. Proc Natl Acad Sci U S A 99:6784–6789. doi: 10.1073/pnas.102687599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laplante M, Sabatini DM. 2009. An emerging role of mTOR in lipid biosynthesis. Curr Biol 19:R1046–R1052. doi: 10.1016/j.cub.2009.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maya-Monteiro CM, Almeida PE, D'Avila H, Martins AS, Rezende AP, Castro-Faria-Neto H, Bozza PT. 2008. Leptin induces macrophage lipid body formation by a phosphatidylinositol 3-kinase- and mammalian target of rapamycin-dependent mechanism. J Biol Chem 283:2203–2210. doi: 10.1074/jbc.M706706200. [DOI] [PubMed] [Google Scholar]
- 30.Chakrabarti P, English T, Shi J, Smas CM, Kandror KV. 2010. Mammalian target of rapamycin complex 1 suppresses lipolysis, stimulates lipogenesis, and promotes fat storage. Diabetes 59:775–781. doi: 10.2337/db09-1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bourque SD, Titorenko VI. 2009. A quantitative assessment of the yeast lipidome using electrospray ionization mass spectrometry. J Vis Exp 2009:1513. doi: 10.3791/1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidt C, Ploier B, Koch B, Daum G. 2013. Analysis of yeast lipid droplet proteome and lipidome. Methods Cell Biol 116:15–37. doi: 10.1016/B978-0-12-408051-5.00002-4. [DOI] [PubMed] [Google Scholar]
- 33.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 34.Kurat CF, Wolinski H, Petschnigg J, Kaluarachchi S, Andrews B, Natter K, Kohlwein SD. 2009. Cdk1/Cdc28-dependent activation of the major triacylglycerol lipase Tgl4 in yeast links lipolysis to cell-cycle progression. Mol Cell 33:53–63. doi: 10.1016/j.molcel.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 35.Schmitt ME, Brown TA, Trumpower BL. 1990. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res 18:3091–3092. doi: 10.1093/nar/18.10.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conceição TM, El-Bacha T, Villas-Bôas CSA, Coello G, Ramírez J, Montero-Lomeli M, Da Poian AT. 2010. Gene expression analysis during dengue virus infection in HepG2 cells reveals virus control of innate immune response. J Infect 60:65–75. doi: 10.1016/j.jinf.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 37.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 38.Helliwell SB, Wagner P, Kunz J, Deuter-Reinhard M, Henriquez R, Hall MN. 1994. TOR1 and TOR2 are structurally and functionally similar but not identical phosphatidylinositol kinase homologues in yeast. Mol Biol Cell 5:105–118. doi: 10.1091/mbc.5.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lorenz MC, Heitman J. 1995. TOR mutations confer rapamycin resistance by preventing interaction with FKBP12-rapamycin. J Biol Chem 270:27531–275317. doi: 10.1074/jbc.270.46.27531. [DOI] [PubMed] [Google Scholar]
- 40.Lewin A, Hines V, Small G. 1990. Citrate synthase encoded by the CIT2 gene of Saccharomyces cerevisiae is peroxisomal. Mol Cell Biol 10:1399–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Komeili A, Wedaman K. 2000. Mechanism of metabolic control target of rapamycin signaling links nitrogen quality to the activity of the Rtg1 and Rtg3 transcription factors. J Cell Biol 151:863–878. doi: 10.1083/jcb.151.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beopoulos A, Nicaud J-M, Gaillardin C. 2011. An overview of lipid metabolism in yeasts and its impact on biotechnological processes. Appl Microbiol Biotechnol 90:1193–1206. doi: 10.1007/s00253-011-3212-8. [DOI] [PubMed] [Google Scholar]
- 43.Courchesne NMD, Parisien A, Wang B, Lan CQ. 2009. Enhancement of lipid production using biochemical, genetic and transcription factor engineering approaches. J Biotechnol 141:31–41. doi: 10.1016/j.jbiotec.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 44.Barbet NC, Schneider U, Helliwell SB, Stansfield I, Tuite MF, Hall MN. 1996. TOR controls translation initiation and early G1 progression in yeast. Mol Biol Cell 7:25–42. doi: 10.1091/mbc.7.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Woods A, Mundays MR, Scott J, Yango X, Carlson M, Carling D. 1994. Yeast SNF1 is functionally related to mammalian AMP-activated protein kinase and regulates acetyl-CoA carboxylase in vivo. J Biol Chem 269:19509–19515. [PubMed] [Google Scholar]
- 46.Tehlivets O, Scheuringer K, Kohlwein SD. 2007. Fatty acid synthesis and elongation in yeast. Biochim Biophys Acta 1771:255–270. doi: 10.1016/j.bbalip.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 47.Hofman-Bang J. 1999. Nitrogen catabolite repression in Saccharomyces cerevisiae. Mol Biotechnol 12:35–39. doi: 10.1385/MB:12:1:35. [DOI] [PubMed] [Google Scholar]
- 48.Santhanam A, Hartley A, Düvel K, James R, Garrett S, Du K, Broach JR. 2004. PP2A phosphatase activity is required for stress and Tor kinase regulation of yeast stress response factor Msn2p. Eukaryot Cell 3:1261–1271. doi: 10.1128/EC.3.5.1261-1271.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coffman JA, Rai R, Cooper TG. 1995. Genetic evidence for Gln3p-independent, nitrogen catabolite repression-sensitive gene expression in Saccharomyces cerevisiae. J Bacteriol 177:6910–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coffman JA, Rai R, Cunningham T, Svetlov V, Cooper TG. 1996. Gat1p, a GATA family protein whose production is sensitive to nitrogen catabolite repression, participates in transcriptional activation of nitrogen-catabolic genes in Saccharomyces cerevisiae. Mol Cell Biol 16:847–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shibata M, Yoshimura K, Tamura H, Ueno T, Nishimura T, Inoue T, Sasaki M, Koike M, Arai H, Kominami E, Uchiyama Y. 2010. LC3, a microtubule-associated protein1A/B light chain3, is involved in cytoplasmic lipid droplet formation. Biochem Biophys Res Commun 393:274–279. doi: 10.1016/j.bbrc.2010.01.121. [DOI] [PubMed] [Google Scholar]
- 52.Zanghellini J, Natter K, Jungreuthmayer C, Thalhammer A, Kurat CF, Gogg-Fassolter G, Kohlwein SD, von Grünberg H-H. 2008. Quantitative modeling of triacylglycerol homeostasis in yeast–metabolic requirement for lipolysis to promote membrane lipid synthesis and cellular growth. FEBS J 275:5552–5563. doi: 10.1111/j.1742-4658.2008.06681.x. [DOI] [PubMed] [Google Scholar]
- 53.Hardie DG. 2007. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev 8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 54.Hardie DG. 2011. Sensing of energy and nutrients by AMP-activated protein kinase. Am J Clin Nutr 93:891–896. doi: 10.3945/ajcn.110.001925. [DOI] [PubMed] [Google Scholar]
- 55.Inoki K, Zhu T, Guan K. 2003. TSC2 mediates cellular energy response to control cell growth and survival. Cell 115:577–590. doi: 10.1016/S0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 56.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. 2008. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang J, Vaga S, Chumnanpuen P, Kumar R, Vemuri GN, Aebersold R, Nielsen J. 2011. Mapping the interaction of Snf1 with TORC1 in Saccharomyces cerevisiae. Mol Syst Biol 7:1–11. doi: 10.1038/msb.2011.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hallett J, Luo X, Capaldi A. 2014. State transitions in the TORC1 signaling pathway and information processing in Saccharomyces cerevisiae. Genetics 198:773–786. doi: 10.1534/genetics.114.168369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ruiz A, Xu X, Carlson M. 2011. Roles of two protein phosphatases, Reg1-Glc7 and Sit4, and glycogen synthesis in regulation of SNF1 protein kinase. Proc Natl Acad Sci U S A 108:6349–6354. doi: 10.1073/pnas.1102758108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruiz A, Liu Y, Xu X, Carlson M. 2012. Heterotrimer-independent regulation of activation-loop phosphorylation of Snf1 protein kinase involves two protein phosphatases. Proc Natl Acad Sci U S A 109:8652–8657. doi: 10.1073/pnas.1206280109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Soliman GA, Acosta-Jaquez HA, Fingar DC. 2010. mTORC1 inhibition via rapamycin promotes triacylglycerol lipolysis and release of free fatty acids in 3T3-L1 adipocytes. Lipids 45:1089–1100. doi: 10.1007/s11745-010-3488-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Petschnigg J, Wolinski H, Kolb D, Zellnig G, Kurat CF, Natter K, Kohlwein SD. 2009. Good fat, essential cellular requirements for triacylglycerol synthesis to maintain membrane homeostasis in yeast. J Biol Chem 284:30981–30993. doi: 10.1074/jbc.M109.024752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Ory DS, Schaffer JE. 2003. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A 100:3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saka HA, Valdivia R. 2012. Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu Rev Cell Dev Biol 28:411–437. doi: 10.1146/annurev-cellbio-092910-153958. [DOI] [PubMed] [Google Scholar]
- 65.Ogawa K, Hishiki T, Shimizu Y, Funami K, Sugiyama K, Miyanari Y, Shimotohno K. 2009. Hepatitis C virus utilizes lipid droplet for production of infectious virus. Proc Jpn Acad 85:217–228. doi: 10.2183/pjab.85.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.François J, Parrou JL. 2006. Reserve carbohydrates metabolism in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev 25:125–145. doi: 10.1111/j.1574-6976.2001.tb00574.x. [DOI] [PubMed] [Google Scholar]