

Abstract
Interferons regulate immunity by inducing DNA binding of the transcription factor STAT1 through Y701 phosphorylation. Transcription by STAT1 needs to be restricted to minimize the adverse effects of prolonged immune responses. It remains unclear how STAT1 inactivation is regulated such that the transcription output is adequate. Here we show that efficient STAT1 inactivation in macrophages is coupled with processive transcription. Ongoing transcription feeds back to reduce the promoter occupancy of STAT1 and, consequently, the transcriptional output. Once released from the promoter, STAT1 is ultimately inactivated by Y701 dephosphorylation. We observe similar regulation for STAT2 and STAT3, suggesting a conserved inactivation mechanism among STATs. These findings reveal that STAT1 promoter occupancy in macrophages is regulated such that it decreases only after initiation of the transcription cycle. This feedback control ensures the fidelity of cytokine responses and provides options for pharmacological intervention.
INTRODUCTION
Cytokines perform their functions as key regulators of immune responses through activation of the JAK-STAT signaling pathway (1). Inadequate (either low or exacerbated) cytokine signaling may result in diseases such as immunodeficiency, autoimmunity, or cancer (2, 3). The strength of cytokine responses is regulated by various positive and negative feedback mechanisms that act at all steps of the signaling pathway: the cytokine receptors, JAKs, and STAT transcription factors. Yet it remains unclear how the process of cytokine-induced transcription is controlled once the transcription machinery has been turned on by activated STAT.
STAT1 is indispensable for the biological function of interferons (IFNs), which are crucial cytokines for antiviral and antibacterial immunity. STAT1 nuclear translocation and DNA binding are activated by JAK-mediated phosphorylation of Y701. Other modifications that tune STAT1 function in IFN signaling include CDK8-mediated S727 phosphorylation, IκB kinase ε (IKKε)-mediated S708 phosphorylation, and K703 sumoylation (4–6). Y701-phosphorylated STAT1 binds to target gene promoters in two major forms: (i) STAT1 homodimers induced by type I, II, and III IFNs bind to gamma interferon-activated sequence (GAS) elements, and (ii) the trimeric interferon-stimulated gene factor 3 (ISGF3; composed of STAT1, STAT2, and IRF9) induced by type I and III IFNs binds to interferon-stimulated response elements (ISRE) (1, 7). The principal mechanism of STAT1 inactivation is Y701 dephosphorylation, which causes both STAT1 homodimers and ISGF3 to lose their DNA-binding activity and to relocate to the cytoplasm. The nuclear T-cell protein tyrosine phosphatase (TC-PTP) is the major Y701-directed phosphatase (8). STAT1 acetylation was reported to facilitate dephosphorylation by TC-PTP (9), but this issue has been controversial (10). The access of phosphatase to phosphorylated Y701 appears to be restricted, since DNA-bound STAT1 is protected from Y701 dephosphorylation (11, 12). For type II IFN (IFN-γ) responses, it has been proposed that the DNA-bound STAT1 homodimers at some point switch their conformation from parallel to antiparallel, thereby becoming accessible for Y701 dephosphorylation (12). How this fundamental process of STAT1 homodimer inactivation is regulated and how the ISGF3 complex is disabled remain unknown.
Using genetic and biochemical approaches, we demonstrate that in the responses of primary murine macrophages to IFNs, the promoter occupancy of Y701-phosphorylated STAT1 gradually decreases as a result of processive transcription. Both STAT1 homodimers and the ISGF3 complex are controlled in this way. Once released from the chromatin, STAT1 is Y701 dephosphorylated, revealing that the regulated step of STAT1 inactivation is its dissociation from the promoter, not Y701 dephosphorylation. Macrophages expressing solely the less transcriptionally active STAT1β isoform exhibit longer STAT1 promoter occupancy than wild-type (WT) cells. Blockade of transcription also results in impaired inactivation of STAT2 and STAT3, suggesting that coupling of promoter dissociation with ongoing transcription is conserved among STATs. Such feedback control ensures that after successfully launching the transcriptional process, STAT1 quickly relocates to the IFN receptor to monitor the activation status of the receptor. This mechanism ensures that the IFN-induced transcriptional output is instantly adjusted to the cytokine levels and that excessive gene expression resulting from biological indolence is prevented.
MATERIALS AND METHODS
Cell culture.
All cell lines used were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS) and penicillin-streptomycin. Immortalized mouse embryonic fibroblasts (MEFs) expressing STAT1 with the K336A mutation have been described previously (13). Control immortalized MEFs expressing WT STAT1 were generated by transfection of STAT1 into immortalized STAT1−/− MEFs using TurboFect (Thermo Scientific). Cells stably expressing WT STAT1 were selected using 100 μg/ml Zeocin (InvivoGen) and limited dilution cloning. For the generation of bone marrow-derived macrophages (BMDMs), bone marrow of 6- to 10-week-old C57BL/6N mice or Irf9−/− (14), STAT1β/β (15), or p53−/− (Trp53−/−) (stock no. 002101; The Jackson Laboratory) (16) mice was isolated from the femur and tibia. Macrophages were differentiated in DMEM supplemented with 10% FCS in the presence of L929 cell-derived colony-stimulating factor 1 (CSF-1) as described previously (17).
Quantitation of RNA by qRT-PCR.
To measure mRNA expression, total RNA was reverse transcribed using an oligo(dT18) primer and Moloney murine leukemia virus (M-MuLV) reverse transcriptase (Fermentas). cDNA was amplified with GoTaq qPCR master mix (Promega) using mRNA primers shown below. Reverse transcription-quantitative PCR (qRT-PCR) was performed using a Mastercycler ep realplex 2 system (Eppendorf). To measure the abundance of precursor mRNA (pre-mRNA), RNA was digested with Turbo DNase (Ambion) and was reverse transcribed using random hexamer primers (N6). To measure the expression of microRNAs (miRNAs), isolation was performed using a miRNeasy minikit (Qiagen). Reverse transcription was carried out using a miScript II RT kit (Qiagen). miRNA was amplified with a miScript SYBR green PCR kit (Qiagen) using miRNA/snRNA primers. qPCR primers for mRNAs were CCGAAGACCTTATGAAGCTCTTTG (forward [FWD]) and GCAAGTATCCCTTGCCATCG (reverse [Rev]) for Irf1, CCAGTTCCTCTCAGTCCCAAGATT (Fwd) and TACTGGATGATCAAGGGAACGTGG (Rev) for Mx2, QuantiTect primer assays (Qiagen) for Socs1 (catalog no. QT01059268) and Ifit1 (catalog no. QT01161286), and GGATTTGAATCACGTTTGTGTCAT (Fwd) and ACACCTGCTAATTTTACTGGCAA (Rev) for Hprt. Primers for pre-mRNAs were TGCCTAGTTGCTTGTCTCTG (Fwd) and CTCCTGTGTGTCGCTGTC (Rev) for Irf1, GCTTTGCTGGAACATCTCCT (Fwd) and ACTCTGGTCCCCAATGACAG (Rev) for Mx2, and CTGCAGGCCACCAACTACAA (Fwd) and GGACAACCACAAACATCATCAG (Rev) for IκBa. miScript primer assays (Qiagen) were used for the miRNAs miR-872 (catalog no. MS00012719) and let-7c (catalog no. MS00005852) and for the snRNA Rnu6-2 (catalog no. MS00033740).
Antibodies and reagents.
Antibodies against tyrosine-phosphorylated STAT1 (catalog no. 9171; Cell Signaling), STAT1 (BD Biosciences [catalog no. 610185] for Western blotting and Santa Cruz [catalog no. sc-346] for ChIP), tyrosine-phosphorylated STAT2 (catalog no. 07-224; Upstate), STAT2 (catalog no. 07-140; Upstate), tyrosine-phosphorylated STAT3 (catalog no. 9131; Cell Signaling), STAT3 (catalog no. 9139; Cell Signaling), IRF1 (catalog no. sc-13041; Santa Cruz), pan-extracellular signal-regulated kinase (pan-ERK) (catalog no. 610123; BD Biosciences), alpha-tubulin (catalog no. T9026; Sigma-Aldrich), Dicer (catalog no. 30226; Santa Cruz), and p53 (catalog no. MONX10194; Monosan) were used for Western blotting. For immunofluorescence, an anti-STAT1 antibody (catalog no. 610185; BD Biosciences) was used. For chromatin immunoprecipitation (ChIP), antibodies against STAT1 (2 μg/sample; catalog no. sc-346X; Santa Cruz), RNA polymerase II (RNAPII) (5 μg/sample; catalog no. sc-899; Santa Cruz), NF-κB (4 μg/sample; catalog no. sc-372; Santa Cruz), and an IgG control (preimmune serum [17]; 2 μl/sample) were used. For stimulation of cells, 5 ng/ml mouse IFN-γ (eBioscience) and 250 U/ml mouse IFN-β (PBL Interferon Source) were used. For pulse-chase stimulation of immortalized MEFs, cells were stimulated with IFN-γ for 5 min (“pulse”), washed twice with medium to remove IFN, and incubated with conditioned medium for the indicated time (“chase”). For stimulation of cells, 5 μg/ml lipoteichoic acid (LTA) (InvivoGen) was used. Flavopiridol was obtained through the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, and was used at a final concentration of 0.5 μM 15 min prior to IFN treatment. 5,6-Dichlorobenzimidazole 1-β-d-ribofuranosyl benzimidazole (DRB; 100 μM; Sigma-Aldrich) was added to the cells 15 min prior to IFN treatment. Additionally, the following inhibitors were used and were added to the cells simultaneously with IFN: actinomycin D (ActD; 1 μg/ml; Sigma-Aldrich), sodium pervanadate (Na3VO4; 100 μM; Sigma-Aldrich), and cycloheximide (CHX; 20 μg/ml; Sigma-Aldrich). When combined with LTA, ActD and CHX were added 60 min after LTA addition.
Western blotting and immunoprecipitation.
Procedures for Western blotting and immunoprecipitation (IP) have been described previously (17). In brief, for the preparation of whole-cell extracts, cells were washed with ice-cold phosphate-buffered saline (PBS) and were lysed in a buffer containing 10 mM Tris-HCl (pH 7.5), 50 mM NaCl, 30 mM NaPPi, 50 mM NaF, 2 mM EDTA, 1% Triton X-100, and 1× protease inhibitor (Roche). Extracts were cleared by centrifugation at 13,200 rpm and 4°C. For input control, 50 μl of the extract was mixed with 25 μl of SDS buffer and was boiled for 10 min. For immunoprecipitation of STAT1 or STAT2, 250 μl of whole-cell extract was mixed with 4 μg of an anti-STAT1 antibody (catalog no. sc-346; Santa Cruz) or anti-STAT2 (catalog no. 07-140; Upstate) antibody, respectively, and incubated on a rotating wheel at 4°C overnight. On the next day, 50 μl of protein G Dynabeads (Invitrogen; blocked with 0.05% bovine serum albumin [BSA]) was added to the IP samples, which were then incubated on a rotating wheel for 2 h. After washing, elution was carried out with 20 μl of SDS buffer following Western blot analysis using an anti-STAT1 or anti-STAT2 antibody. Detection was carried out using fluorescence-labeled secondary antibodies IRDye800 and IRDye700 (Li-Cor) for infrared detection (Odyssey infrared imaging system; Li-Cor).
RNA interference (RNAi)-mediated silencing.
A total of 5 × 105 STAT1 WT MEFs were seeded in 6-cm plates in DMEM supplemented with 10% FCS without antibiotics. The medium was replaced 24 h later with 5 ml fresh medium, and cells were transfected with a mixture of 100 pmol Dicer1 ON-TARGETplus SMARTpool small interfering RNA (siRNA) (L-040892-00-0005; Dharmacon) diluted in 500 μl Opti-MEM I and 20 μl Lipofectamine RNAiMAX reagent diluted in 500 μl Opti-MEM I (both from Invitrogen). After 48 h, cells were washed and were incubated for an additional 16 h in s medium without siRNAs, followed by pulse-chase stimulation with IFN-β and whole-cell extract preparation or miRNA isolation.
ChIP.
ChIP was carried out as described previously (5) with the following modifications. For sonication, a Bioruptor sonicator (Diagenode) was used to yield 300- to 500-bp DNA fragments. For immunoprecipitation, protein G Dynabeads (Invitrogen) were used. The amount of immunoprecipitated DNA was quantified by qPCR using Kapa Sybr Fast qPCR universal mix (Peqlab). All qPCRs were run on a Mastercycler ep realplex 2 system (Eppendorf). The values for immunoprecipitated DNA were normalized to that for input DNA (percentage of input). The antibodies used for ChIP are specified under “Antibodies and reagents” above. The primers used for ChIP were Irf1 (GAS) Fwd (GGAGCACAGCTGCCTTGTACTT) and Rev (CCCACTCGGCCTCATCATT), Irf1 (Start) Fwd (TCCCGCTAAGTGTTTAGATTTC) and Rev (TTCGGTTCGGCTTAGACTG), Irf1 (exon 4) Fwd (TGCCTAGTTGCTTGTCTCTG) and Rev (CTCCTGTGTGTCGCTGTC), Mx2 (ISRE) Fwd (ACCCAGCCAAGGCCCCCTTA) and Rev (GCAGCTGCCAGGGCTCAGAC), and IκBa (prx) Fwd (AAGAAGGGTTCTTGCAGAGGGCT) and Rev (TCGTCCTCCACTGAGAAGCCTAAA).
Immunofluorescence microscopy.
Cells were fixed with 1.8% formaldehyde and were incubated overnight at 4°C with an anti-STAT1 antibody (catalog no. 610185; BD Biosciences). After washing, the samples were incubated with Alexa Fluor 488-conjugated anti-mouse secondary antibodies (Molecular Probes) for 30 min, washed, and mounted with fluorescence mounting medium (Dako). Analysis was carried out using a Zeiss Axioplan 2 microscope.
RESULTS AND DISCUSSION
Y701 dephosphorylation of IFN-activated STAT1 is dependent on ongoing transcription.
The level of STAT1 Y701 phosphorylation induced by IFN-β, a type I IFN, decreased with the duration of IFN-β treatment in murine bone marrow-derived macrophages (BMDMs) (Fig. 1A). The diminishing amounts of activated STAT1 were paralleled by the profile of STAT1 recruitment to the GAS element in the Irf1 promoter (Fig. 1B) and to the ISRE in the Mx2 promoter (Fig. 1C). The promoter occupancy of STAT1 correlated with the transcription rate, which was assessed by Irf1 and Mx2 precursor mRNA (pre-mRNA) measurements (Fig. 1D and E). The profile of Irf1 pre-mRNA was similar to that of Irf1 mature mRNA (Fig. 1D and F), whereas the level of Mx2 mature mRNA was still increasing when Mx2 pre-mRNA had already reached a plateau (Fig. 1E and G). The low stability of Irf1 mRNA (5, 18) and the high stability of Mx2 mRNA (18) explain why Irf1 but not Mx2 mRNA levels correlate with the pre-mRNA amounts and hence with the rate of transcription. Taking these findings together, we conclude that the duration of IFN-β responses was restricted at the level of both STAT1 activation and STAT1-dependent transcription.
FIG 1.

Tyrosine dephosphorylation of IFN-activated STAT1 is dependent on ongoing transcription. (A) Kinetics of STAT1 Y701 dephosphorylation after type I IFN stimulation. BMDMs were stimulated with IFN-β for the indicated times, and cell extracts were analyzed by Western blotting using antibodies to Y701-phosphorylated STAT1 (pY-STAT1), STAT1, and tubulin. (B and C) Occupancy of STAT1 at the Irf1 (B) and Mx2 (C) promoters. BMDMs were stimulated with IFN-β for the indicated times, and STAT1 recruitment was assessed by ChIP for the corresponding GAS (B) and ISRE (C) sites. Signals were normalized to input DNA. Error bars represent standard deviations. (D to G) Kinetics of IFN-β-induced transcription. BMDMs were stimulated with IFN-β; total RNA was isolated; and the levels of Irf1 and Mx2 pre-mRNA (D and E) and mature mRNA (F and G) were analyzed by qRT-PCR. Error bars indicate standard deviations for biological replicates (n = 3). (H) Actinomycin D (ActD), but not cycloheximide (CHX), inhibits transcription. BMDMs were stimulated with IFN-β in the presence or absence (w/o) of ActD or CHX for the indicated times. Total RNA was isolated, and the levels of Irf1 pre-mRNA were analyzed as for panels D to G. (I) Tyrosine dephosphorylation of STAT1 and STAT2 requires ongoing transcription but not translation. BMDMs were stimulated with IFN-β in the presence or absence of ActD or CHX for the indicated times. Cell extracts were analyzed by Western blotting using antibodies to tyrosine-phosphorylated STAT1 (pY-STAT1), tyrosine-phosphorylated STAT2 (pY-STAT2), total STAT1, total STAT2, IRF1, and tubulin. Note that a lack of IRF1 induction in cells treated with ActD or CHX confirmed a block in transcription or translation, respectively. (J) IFN-β-induced STAT1 Y701 phosphorylation is prolonged upon transcription inhibition. BMDMs were stimulated with IFN-β in the presence or absence of ActD. Cell extracts were analyzed by Western blotting as for panel A. (K) Ongoing transcription is required for tyrosine dephosphorylation of STAT3. BMDMs were treated as for panel I, and cell extracts were analyzed by Western blotting using antibodies to tyrosine-phosphorylated STAT3 (pY-STAT3), total STAT3, and tubulin (as a loading control). (L) Tyrosine dephosphorylation of IFN-γ-activated STAT1 requires both ongoing transcription and translation. BMDMs were stimulated with IFN-γ for the indicated times, and tyrosine phosphorylation of STAT1 was analyzed as described for panel I. (M) Translation is required for Y701 dephosphorylation of IFN-γ-activated STAT1, and JAK2 inhibition abolishes this requirement. BMDMs were stimulated with IFN-γ in the presence or absence of CHX or the JAK2 inhibitor P6, or both, for the indicated times. Cell extracts were analyzed by Western blotting as for panel I. (N) JAK2 inhibition by P6 substitutes for SOCS1 function. WT and Socs1−/− BMDMs were stimulated with IFN-γ for the indicated times, and P6 was added 30 min after IFN-γ stimulation. Cell extracts were analyzed by Western blotting as for panel I. (O) The JAK2 inhibitor P6 fails to block IFN-β-induced STAT1 and STAT2 tyrosine phosphorylation. BMDMs were stimulated with IFN-β or IFN-γ and were simultaneously treated with P6 at the indicated concentrations. Cell extracts were analyzed by Western blotting as for panel I, with additional antibodies to tyrosine-phosphorylated STAT2 (pY-STAT2), total STAT2, and pan-ERK (as a loading control).
To test whether ongoing transcription and de novo protein synthesis were required for the downregulation of STAT1 activity, BMDMs were treated with the transcription inhibitor actinomycin D (ActD) or the translation inhibitor cycloheximide (CHX), added simultaneously with IFN-β. ActD but not CHX inhibited transcription, as confirmed by Irf1 pre-mRNA measurements (Fig. 1H). Both Y701 dephosphorylation of STAT1 and Y689 dephosphorylation of STAT2 were impaired when transcription was blocked, but they were not affected when translation was inhibited (Fig. 1I). As expected, the induction of the IFN target IRF1, which is strongly increased at 180 min of IFN-β treatment, was prevented by both inhibitors (Fig. 1I). The impaired tyrosine dephosphorylation in ActD-treated cells was observed also after 360 min of IFN-β stimulation, although some dephosphorylation was detectable at this time point (Fig. 1J). Inhibition of transcription but not translation also resulted in a decreased rate of STAT3 Y705 dephosphorylation in IFN-β-treated BMDMs (Fig. 1K). Thus, tyrosine dephosphorylation of STAT1, STAT2, and STAT3, the major IFN-β-activated STATs, proceeds only if transcription is permitted, whereas de novo protein synthesis is not required. These data reveal that ongoing transcription is required for STAT1, STAT2, and STAT3 inactivation after IFN-β stimulation, and they suggest that this mode of regulation is conserved among STATs.
To address the effects of transcription and protein synthesis on STAT1 inactivation in the type II IFN response, Y701 dephosphorylation was examined in BMDMs stimulated with IFN-γ. ActD prevented efficient Y701 dephosphorylation of IFN-γ-activated STAT1 (Fig. 1L), resembling the pattern for IFN-β signaling. However, in contrast to the IFN-β pathway, in the IFN-γ pathway STAT1 Y701 dephosphorylation was also inhibited by CHX (Fig. 1L). IFN-γ signaling is negatively regulated by suppressor of cytokine signaling 1 (SOCS1), a STAT1 target induced by IFNs (19). To test whether the impaired Y701 dephosphorylation in IFN-γ responses under conditions of blocked translation resulted from the lack of SOCS1 induction, we interrupted IFN-γ signaling using the JAK2 inhibitor pyridone 6 (P6) (the 50% inhibitory concentration [IC50] is 1 nM for JAK2) (20) 30 min after IFN-γ treatment. Upon P6 treatment, Y701 dephosphorylation was no longer affected by blocking of translation (Fig. 1M). Further, P6 was capable of substituting for the missing SOCS1, as revealed by complete Y701 dephosphorylation in SOCS1-deficient cells in the presence of P6 (Fig. 1N). Similar analysis of responses to type I IFNs was precluded by the poor ability of P6 to inhibit IFN-β-induced STAT1 and STAT2 tyrosine phosphorylation (Fig. 1O). These data show that the major reason for impaired Y701 dephosphorylation upon translation blockade in the IFN-γ response was unrestricted activity of IFN-γ signaling due to the lack of SOCS1 synthesis. Since transcription inhibition also prevents SOCS1 induction, the role of ongoing transcription in Y701 dephosphorylation cannot be assessed in IFN-γ responses.
ActD blocks RNAPII progression by intercalating into DNA (21). Carboxy-terminal domain (CTD) phosphorylation of RNAPII at S2 is increased under conditions of ActD treatment due to the release of CDK9 from negative inhibition (21). Thus, ActD blocks transcription and, in parallel, locks S2-phosphorylated (i.e., elongation-competent) RNAPII at the promoter. To test whether transcription inhibition without a concomitant increase in RNAPII S2 phosphorylation causes defects in Y701 dephosphorylation similar to those with ActD, we employed flavopiridol, a specific inhibitor of the Mediator-associated kinase CDK8 and of the transcription elongation-promoting CTD S2 kinase CDK9 (22, 23). Like ActD treatment, flavopiridol treatment resulted in impaired tyrosine dephosphorylation of both STAT1 and STAT2 in IFN-β-stimulated cells (Fig. 2A).
FIG 2.

ActD and flavopiridol inhibit STAT1 dephosphorylation independently of p53- and miRNA-mediated mechanisms. (A) Transcription inhibition by flavopiridol (FP) impairs STAT1 and STAT2 tyrosine dephosphorylation. BMDMs were either left untreated or pretreated with FP for 15 min and were then stimulated with IFN-β for the indicated times. Cell extracts were analyzed by Western blotting using antibodies to tyrosine-phosphorylated STAT1 (pY-STAT1), tyrosine-phosphorylated STAT2 (pY-STAT2), total STAT1, and total STAT2. (B) ActD and FP do not cause IFN-independent activation of STAT1 and STAT2. BMDMs either were stimulated with IFN-β for the indicated time or remained unstimulated in the presence or absence of ActD or FP. Treatment with ActD or FP started 150 min prior to IFN-β addition. Cell extracts were analyzed by Western blotting as described for panel A. (C and D) ActD and FP inhibit STAT1 dephosphorylation in p53−/− BMDMs. (C) WT and p53-deficient BMDMs were either left untreated or treated with IFN-β or ActD for 30 min. HEK293 (HEK) cells were used as a positive control, because they express large amounts of p53. Cell extracts were analyzed by Western blotting for pY701-STAT1, STAT1, and p53. Note that human p53 (in HEK293 cells) has a high molecular weight and exhibits slower migration than mouse p53 (in BMDMs). (D) p53-deficient BMDMs were treated with IFN-β in the presence or absence of ActD or FP for the indicated times. Cell extracts were analyzed by Western blotting as described for panel A. (E, F, and G) miRNAs do not contribute to Y701 dephosphorylation of STAT1. (E) STAT1 WT MEFs were incubated with siRNA directed against Dicer or with a control siRNA (CTRL). Cell extracts were analyzed by Western blotting using antibodies to Dicer and tubulin. (F) miRNA was isolated from cells treated with Dicer siRNA (siDicer) or control siRNA (siCTRL), and the levels of miR-872 and let-7c were quantified using qRT-PCR. (G) Cells were stimulated with IFN-β for the indicated times, and pY701 dephosphorylation of STAT1 was analyzed by Western blotting.
ActD induces genotoxic stress and activates p53 (24, 25). Importantly, ActD has been reported to cause STAT1 activation in tumor cells in a p53-dependent way, independently of IFN treatment (26). To examine whether these mechanisms could also operate in BMDMs, we employed several strategies. First, we treated BMDMs for 180 min with ActD or flavopiridol without IFN-β stimulation and observed no activation of STAT1, in contrast to IFN-β-stimulated cells (Fig. 2B). Second, we tested the effects of ActD and flavopiridol in BMDMs derived from p53−/− mice (Fig. 2C). Activation of STAT1 by IFN-β was similar in WT and p53−/− cells (Fig. 2C). Importantly, Y701 dephosphorylation proceeded normally in p53−/− cells, but as in WT cells, it was inhibited by ActD and flavopiridol treatment (Fig. 2D). Together, these experiments ruled out a role for a p53-dependent genotoxic-stress-induced pathway in STAT1 activation in ActD-treated BMDMs. Another explanation for prolonged STAT1 activation upon transcription blockade might be missing production of inducible or constitutively expressed miRNAs, which negatively target the type I IFN pathway. However, type I IFNs are poor inducers of miRNAs in murine macrophages (27), and constitutively expressed miRNAs exhibit an average half-life of 5 days (28) so that their levels remain largely unchanged during the 3 to 6 h of ActD treatment in our experiments. Thus, involvement of miRNAs in the negative feedback inhibition of STAT1 is not likely. Yet a cancer-associated polymorphism in the human IFNAR1 gene has been linked to miRNA miR-1231 binding, although no specific function for this miRNA in negative regulation of type I IFN signaling has been reported (29). To broadly investigate miRNA function in STAT1 Y701 dephosphorylation, we employed siRNA to silence the single Dicer gene, Dicer1, in mouse fibroblasts (Fig. 2E). The Dicer knockdown resulted in a reduction of approximately 40% in the levels of miR-872 and let-7 (Fig. 2F). The reported half-lives of miR-872 and let-7 in mouse fibroblasts are approximately 24 and 36 h, respectively (28), in good agreement with the reductions in their levels after 62 h of treatment with Dicer siRNA (siDicer) (Fig. 2F). Importantly, STAT1 Y701 dephosphorylation proceeded normally in Dicer-depleted cells, although the levels of Y701-phosphorylated STAT1 were slightly lower in Dicer-silenced cells than in controls (Fig. 2G). The experiment confirmed that lack of miRNA transcription is not involved in the inhibition of Y701 dephosphorylation by ActD.
In conclusion, transcription, but not de novo protein synthesis, is required for efficient Y701 inactivation in responses to IFN-β, whereas synthesis of a negative regulator plays a major role in STAT1 inactivation in IFN-γ signaling. Nonetheless, a fundamental contribution of transcription to Y701 inactivation in IFN-γ responses cannot be ruled out.
Transcription inhibition does not impede Y701 dephosphorylation of nucleoplasmic STAT1.
The requirement for ongoing transcription in the tyrosine dephosphorylation of IFN-β-stimulated STAT1, STAT2, and STAT3 raised the question of whether STAT binding to the promoter was needed for functional exposure to a tyrosine phosphatase. To examine the role of DNA binding, we employed cells lacking Irf9, the DNA binding subunit of the ISGF3 complex. Irf9−/− BMDMs treated with IFN-β showed impaired tyrosine dephosphorylation of both STAT1 and STAT2 (Fig. 3A). Importantly, nuclear accumulation of STAT1 was not compromised in the absence of Irf9 (Fig. 3B). IFN-β-mediated upregulation of Irf1 transcription, which is driven by the binding of STAT1 homodimers to the Irf1 GAS site, was similar in WT and Irf9−/− cells, providing evidence for the correct functioning of STAT1 homodimers in Irf9−/− cells (Fig. 3C). The dephosphorylation profile in Irf9−/− BMDMs displayed atypical kinetics, though: Y701 phosphorylation levels decreased after 180 min of IFN-β treatment but, unexpectedly, increased again after 360 min (Fig. 3A). Increasing tyrosine phosphorylation could also be observed for STAT2 in the same cells (Fig. 3A). STAT1 and STAT2 protein levels are subject to type I IFN-mediated upregulation (30). Basal constitutive type I IFN signaling is required for the maintenance of steady-state STAT1 and STAT2 protein levels. The amounts of STAT1 and STAT2 are reduced in cells with deficiencies in the type I IFN pathway (30). Consistently, Irf9−/− BMDMs displayed lower levels of STAT1 and STAT2 than WT cells (Fig. 3A). However, the induction of STAT1 and STAT2 after 360 min of IFN-β treatment was much more pronounced in Irf9−/− BMDMs than in WT cells (Fig. 3A). We asked whether the profound increase in STAT1 and STAT2 levels could explain the prolonged tyrosine phosphorylation of these proteins in Irf9−/− cells. Since translation inhibition does not impair STAT1 and STAT2 dephosphorylation in WT cells (Fig. 1I), we decided to prevent the IFN-β-mediated induction of STAT1 and STAT2 protein levels by CHX treatment. Under these conditions, the defective tyrosine dephosphorylation of STAT1 and STAT2 in Irf9−/− cells was no longer observed (Fig. 3D). To address the integrity of STAT1-STAT2 heterodimers in the absence of Irf9, we first examined these complexes in WT BMDMs. Coimmunoprecipitation experiments revealed that STAT1-STAT2 heterodimers exist as stable complexes regardless of IFN stimulation (Fig. 3E). Coimmunoprecipitation of STAT2 using anti-STAT1 antibodies confirmed the formation of STAT1-STAT2 heterodimers in Irf9-deficient cells also (Fig. 3F). We were unsuccessful in coimmunoprecipitating STAT1 using anti-STAT2 antibodies from Irf9−/− cell extracts, presumably because of the combination of poor affinity of anti-STAT2 antibodies and low STAT1 and STAT2 expression in these cells. Together, the results show that STAT1-STAT2 heterodimers lacking Irf9 and incapable of binding to ISRE sites (14) are tyrosine dephosphorylated similarly to functional ISGF3 complexes.
FIG 3.

Inhibition of transcription is permissive for Y701 dephosphorylation of nucleoplasmic STAT1. (A) STAT1 and STAT2 tyrosine dephosphorylation in the absence of Irf9. WT and Irf9−/− BMDMs were stimulated with IFN-β for the indicated times. Levels of tyrosine-phosphorylated STAT1 and STAT2, total STAT1 and STAT2, and tubulin (used as a loading control) were assessed by Western blotting. Note that total STAT1 and STAT2 levels strongly increased with time of IFN-β treatment in Irf9−/− cells, in contrast to levels in WT cells (compare the 0-min with the 360-min time points). (B) IFN-β-activated STAT1 accumulates in the nucleus in the absence of IRF9. Irf9−/− BMDMs were treated with IFN-β for the indicated times, and immunofluorescence analysis was performed using STAT1-specific antibodies. (C) IFN-β-induced transcription of Irf1 proceeds in the absence of Irf9. WT and Irf9−/− BMDMs were treated with IFN-β, and total RNA was isolated and was analyzed by qRT-PCR. Error bars indicate standard deviations for 3 biological replicates. (D) Ongoing tyrosine dephosphorylation of STAT1 and STAT2 in Irf9−/− BMDMs with a block in protein synthesis. Irf9−/− BMDMs were treated as described in the legend to panel A in the presence or absence of CHX and were analyzed by Western blotting. (E) Amounts of STAT1 and STAT2 heterodimers remain stable regardless of IFN-β treatment. BMDMs were stimulated with IFN-β for the indicated times, and immunoprecipitation (IP) was carried out using antibodies against STAT1 (top) or STAT2 (bottom), or beads only as a control (CTRL). Immunoprecipitated complexes were analyzed by Western blotting using antibodies to STAT1 and STAT2. (IN, input control [20%]). (F) STAT1 and STAT2 form complexes in Irf9-deficient cells. STAT1 was immunoprecipitated in Irf9−/− BMDMs as described in the legend to panel E. (G) Y701 dephosphorylation of WT and DNA binding-deficient STAT1 after IFN-γ stimulation. Immortalized MEFs expressing either WT STAT1 or a DNA binding-deficient STAT1 mutant (K336A) were stimulated with IFN-γ for the indicated times and were analyzed by Western blotting as described for panel A. (H) Y701 dephosphorylation of the DNA binding-deficient STAT1 mutant. MEFs expressing STAT1 K336A were stimulated with IFN-γ for 5 min (“pulse”), followed by a chase for the indicated times in the presence or absence of the JAK2 inhibitor P6. (I) Y701 dephosphorylation of MEFs expressing WT or K336A STAT1 after stimulation with IFN-β or IFN-γ. MEFs expressing WT or K336A STAT1 were pulsed with IFN-β or IFN-γ for 5 min, followed by a chase for the indicated times. STAT1 tyrosine phosphorylation was analyzed by Western blotting as described for panel A.
These data address STAT1 tyrosine dephosphorylation in the pool of STAT1-STAT2 heterodimers but not in the pool of STAT1 homodimers, since homodimers are still functional and capable of DNA binding in the absence of Irf9: IFN-β induces STAT1 and STAT2 levels and increases the STAT1 homodimer-driven transcription of Irf1 in Irf9−/− cells (Fig. 3A and C). To directly address the role of DNA binding in Y701 dephosphorylation of STAT1 homodimers, we employed fibroblasts expressing solely a STAT1 K336A mutant, which is incapable of chromatin recruitment and induction of IFN-γ target genes, despite nuclear accumulation (13). IFN-γ treatment of STAT1 K336A-expressing cells revealed impaired Y701 dephosphorylation (Fig. 3G). This was expected, since the K336A mutant does not induce transcription and consequently does not induce SOCS1 expression to turn off IFN-γ signaling in these cells. In order to show that the K336A mutant was capable of targeting by STAT1 Y701 phosphatase, we used the JAK2 inhibitor P6. To circumvent the slow kinetics of STAT1 dephosphorylation in fibroblasts, we employed a pulse-chase protocol for IFN stimulation: 5 min of IFN treatment followed by washout and incubation in the absence of IFNs. Inhibition of IFN-γ signaling by treatment with the JAK2 inhibitor P6 30 min after IFN-γ stimulation resulted in Y701 dephosphorylation of the K336A mutant (Fig. 3H), demonstrating that dephosphorylation can proceed in the absence of DNA binding. Upon IFN-β treatment, Y701 dephosphorylation of the K336A mutant progressed similarly to that for WT STAT1 (Fig. 3I), suggesting that transcription, which is required for Y701 dephosphorylation (Fig. 1I) by the K336A-containing ISGF3 complex, proceeds normally. This is in agreement with previous studies showing that ISGF3 containing the K336A mutation is transcriptionally active (13). In sum, these results provide evidence that, in the DNA-unbound state, dephosphorylation of both STAT1 homodimers and ISGF3 is an unregulated process. The dependence of STAT1 and STAT2 dephosphorylation on ongoing transcription, as shown for IFN-β signaling (Fig. 1I), but not on DNA binding implies that STAT inactivation constitutes a regulated step only if the transcription factor has been recruited to the promoter.
Transcription inhibition prolongs STAT1 occupancy at target promoters.
The regulation of Y701 dephosphorylation of DNA-bound STAT1 by a mechanism that requires ongoing transcription suggests either that transcription directly facilitates the activity of a tyrosine phosphatase toward DNA-bound STAT1 or that the transcription machinery feeds back to decrease STAT1 promoter occupancy. In the latter scenario, less promoter-bound STAT1 would result in larger amounts of free (not DNA-bound) STAT1, which is readily dephosphorylated in an unregulated fashion.
To address the control of STAT1 promoter occupancy by the transcription process, we first examined whether transcription blockade and the accompanying persistent Y701 phosphorylation result in prolonged association of STAT1 with target promoters. IFN-β-induced STAT1 promoter occupancy decreased with the duration of IFN-β treatment (Fig. 4A and B). The decrease of STAT1 occupancy at the Irf1 promoter was more pronounced than that of ISGF3 at the Mx2 promoter (Fig. 4A and B). Importantly, the decrease in STAT1 promoter occupancy was almost blocked when transcription was inhibited by ActD treatment (Fig. 4A and B). Thus, the reduced STAT1 tyrosine dephosphorylation in ActD-treated cells correlated with a slower decrease in STAT1 promoter occupancy. IFN-β-induced recruitment of RNA polymerase II (RNAPII) to the transcription start site (TSS) was not impaired by ActD treatment (Fig. 4C and D). As expected, the progression of RNAPII into the gene body was inhibited by ActD treatment, as revealed by RNAPII occupancy in exon 4 of Irf1 (Fig. 4E).
FIG 4.

Transcription inhibition by ActD prolongs STAT1 but not NF-κB occupancy at target promoters. (A and B) Transcription inhibition prolongs STAT1 occupancy at the Irf1 and Mx2 promoters. BMDMs were stimulated with IFN-β in the presence or absence of ActD for the indicated times. STAT1 occupancy at the corresponding GAS (A) and ISRE (B) sites was assessed by ChIP. Signals were normalized to input DNA. Error bars represent standard deviations (n = 3). (C and D) ActD treatment does not alter RNAPII recruitment to the Irf1 and Mx2 promoters. BMDMs were stimulated with IFN-β in the presence or absence of ActD for the indicated times. (C and D) The binding of RNAPII to the Irf1 (C) and Mx2 (D) promoters was analyzed by ChIP. Signals were normalized to input DNA. Error bars represent standard deviations (n = 3). (E) ActD treatment inhibits the progression of RNAPII into the gene body. BMDMs were stimulated with IFN-β in the presence or absence of ActD for the indicated times. The recruitment of RNAPII to Irf1 exon 4 was assessed by ChIP. Signals were normalized to input DNA. Error bars represent standard deviations (n = 3). (F and G) Transcription inhibition does not prolong NF-κB occupancy at the Iκba promoter. (F) BMDMs were stimulated with LTA for 60 min, followed by ActD treatment for an additional 60 min. RNA was isolated, and Iκba pre-RNA was analyzed by qRT-PCR. Error bars indicate standard deviations for 3 biological replicates. (G) BMDMs were stimulated as described in the legend to panel F, and ActD or cycloheximide (CHX) was added 60 min after LTA stimulation. The binding of NF-κB to the Iκba promoter was analyzed by ChIP. Signals were normalized to input DNA. Error bars represent standard deviations (n = 3).
To test whether blockade of transcription had more general effects on the dynamics of chromatin association of transcription factors, we examined NF-κB occupancy at the Iκba promoter in lipoteichoic acid (LTA)-stimulated BMDMs treated with ActD. LTA causes rapid Toll-like receptor 2 (TLR2)-dependent NF-κB activation but not type I IFN production in BMDMs, so that an undesired interaction with STAT1 was excluded (31). Cells were stimulated with LTA for 60 min, followed by ActD treatment for an additional 60 min. Under these conditions, Iκba transcription was completely abolished (Fig. 4F). Importantly, such a regime allowed accumulation of the NF-κB inhibitor IκB prior to the transcription blockade so that aberrant inactivation of NF-κB was avoided, as confirmed by inhibiting translation (Fig. 4G, CHX control). The occupancy of NF-κB at the Iκba promoter displayed transient kinetics, with a peak after 60 min of LTA treatment (Fig. 4G). A similar occupancy profile was observed upon ActD treatment, indicating that transcription inhibition did not impair NF-κB dissociation from the promoter (Fig. 4G). The data suggest that the effects of ActD on STAT1 promoter binding are specific.
Like ActD treatment, flavopiridol treatment resulted in impaired tyrosine dephosphorylation of both STAT1 and STAT2 in IFN-β-stimulated cells (Fig. 2A). Under these conditions, the transcription of Irf1 and Mx2 in IFN-β-stimulated cells was prevented, as confirmed by measurements of Irf1 and Mx2 pre-mRNAs (Fig. 5A and B). Flavopiridol enhanced STAT1 occupancy at both the Irf1 and Mx2 genes throughout IFN-β treatment (Fig. 5C and D). In contrast to ActD treatment (Fig. 4A and B), STAT1 occupancy was consistently higher in flavopiridol-treated cells at 60 min of IFN-β stimulation, but a decrease in STAT1 occupancy was not completely abolished (Fig. 5C and D). Importantly, STAT1 occupancy at 360 min of IFN-β treatment in the presence of flavopiridol was as high as that at 60 min without flavopiridol (Fig. 5C and D). Thus, the increased levels of Y701-phosphorylated STAT1 in flavopiridol-treated cells correlated with higher STAT1 promoter occupancy. The occupancy of RNAPII at the TSSs of both the Irf1 and Mx2 genes was not affected by flavopiridol (Fig. 5E and F).
FIG 5.

Transcription inhibition by flavopiridol and DRB prolongs STAT1 promoter occupancy. (A and B) Flavopiridol (FP) inhibits IFN-β-induced transcription. BMDMs that had been left untreated (w/o) or pretreated with FP for 15 min were stimulated with IFN-β for the indicated time. Total RNA was isolated, and the levels of Irf1 (A) and Mx2 (B) pre-mRNAs were analyzed by qPCR. Error bars indicate standard deviations (n = 3). (C and D) Flavopiridol increases and prolongs STAT1 occupancy at the Irf1 and Mx2 promoters. BMDMs were either left untreated or pretreated with FP for 15 min; then they were stimulated with IFN-β for the indicated times. STAT1 occupancy at Irf1-GAS (C) and Mx2-ISRE (D) sites was analyzed by ChIP. Error bars represent standard deviations (n = 3). Note that STAT1 promoter occupancy at 360 min of IFN-β treatment in the presence of FP was as high as that at 60 min without FP. (E and F) IFN-β-induced RNAPII recruitment to the Irf1 and Mx2 promoters is not altered in the presence of FP. BMDMs either remained untreated or were pretreated with FP for 15 min; then they were stimulated with IFN-β for the indicated time. RNAPII occupancy at the Irf1 (E) and Mx2 (F) promoters was analyzed by ChIP. Signals were normalized to input DNA. Error bars indicate standard deviations (n = 3). (G) Transcription inhibition by DRB impairs STAT1 and STAT2 tyrosine dephosphorylation. BMDMs were either left untreated or pretreated with DRB for 15 min; then they were stimulated with IFN-β for the indicated times. Cell extracts were analyzed by Western blotting using antibodies to tyrosine-phosphorylated STAT1 (pY-STAT1), tyrosine-phosphorylated STAT2 (pY-STAT2), total STAT1, and total STAT2. (H and I) DRB increases and prolongs STAT1 occupancy at the Irf1 and Mx2 promoters. BMDMs were either left untreated or pretreated with DRB for 15 min; then they were stimulated with IFN-β for the indicated times. STAT1 occupancy at Irf1-GAS (H) and Mx2-ISRE (I) sites was analyzed by ChIP. Error bars represent standard deviations (n = 3). (J and K) DRB treatment does not alter RNAPII recruitment to target promoters. BMDMs either remained untreated or were pretreated for 15 min with DRB; then they were stimulated with IFN-β for the indicated time. RNAPII occupancy at the Irf1 (J) and Mx2 (K) promoters was analyzed by ChIP as described for panels E and F. Signals were normalized to input DNA. Error bars indicate standard deviations (n = 3).
DRB (5,6-dichloro-1-β-ribo-furanosyl benzimidazole) is a transcription inhibitor (21) that reversibly targets CDK9 and, in contrast to flavopiridol, also inhibits CDK7, albeit with 3-fold-lower efficiency (32). Thus, DRB targets transcription elongation and, to a lesser extent, transcription initiation. Consistently, DRB treatment of BMDMs prevented IFN-β-induced expression of Irf1 (Fig. 5G). Like ActD and flavopiridol treatments, DRB treatment inhibited STAT1 Y701 dephosphorylation (Fig. 5G) and extended IFN-β-induced STAT1 association with the Irf1 and Mx2 promoters (Fig. 5H and I) but did not block RNAPII recruitment (Fig. 5J and K). We consistently observed that DRB enhanced STAT1 occupancy at 60 min of IFN-β stimulation. With this regard, DRB resembled flavopiridol, which is also a kinase inhibitor, rather than the DNA-intercalating ActD.
We wanted to test the requirement for ongoing transcription in Y701 dephosphorylation without the use of chemical transcription inhibitors. To this end, we made use of BMDMs derived from mice expressing solely the STAT1β isoform (STAT1β/β mice) (15). STAT1 occurs in cells in two isoforms: STAT1α is the full-length isoform, whereas STAT1β is an isoform that lacks the C-terminal 38 amino acids but otherwise is identical with STAT1α. STAT1β homodimers were previously regarded as not transcriptionally active, but a recent study using STAT1β/β mice demonstrated that STAT1β homodimers retain significant transcriptional activity (15). BMDMs from STAT1β/β mice exhibit reduced induction of GAS-driven genes in response to IFN-γ, but not all target genes are affected to the same extent (15). The expression of ISRE-driven genes is not grossly affected in STAT1β/β BMDMs stimulated by IFN-β, indicating that the ISGF3 complex is fully functional in these cells. The Irf1 and Socs1 genes were shown to be significantly less induced in IFN-γ-stimulated STAT1β/β BMDMs than in IFN-γ-stimulated WT BMDMs (15). IFN-β treatment of STAT1β/β BMDMs and analysis of pre-mRNA expression of Irf1 and Socs1 demonstrated that STAT1β homodimers were less transcriptionally active at these genes in type I IFN responses also (Fig. 6A and B). However, similarly to the reported responses to IFN-γ, STAT1β was still significantly active, since the pre-mRNA levels of Irf1 and Socs1 were approximately 50% lower in STAT1β/β BMDMs than in WT cells (Fig. 6A and B). Strikingly, ChIP experiments revealed longer occupancy of STAT1β than of WT STAT1 at the Irf1 and Socs1 promoters (Fig. 6C and D). This finding further supported our notion that processive transcription limits STAT1 promoter occupancy.
FIG 6.
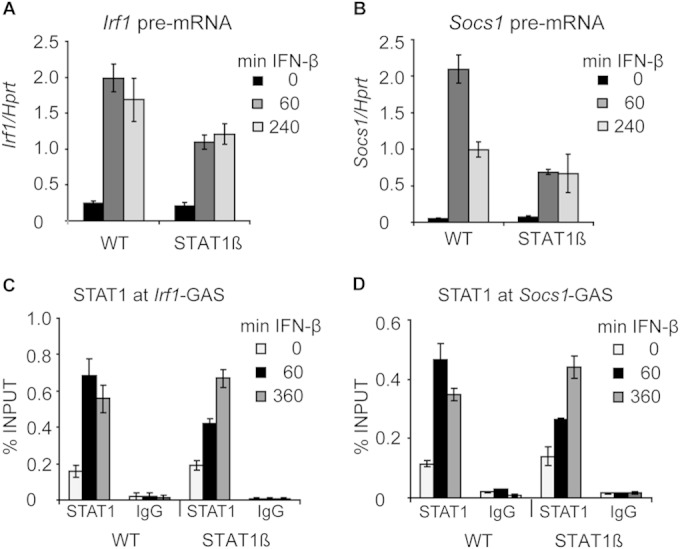
Analysis of IFN-β-induced gene expression and STAT1 promoter occupancy in BMDMs expressing solely the STAT1β isoform. (A and B) IFN-β-induced transcription in WT and STAT1β BMDMs. Cells were stimulated with IFN-β; total RNA was isolated; and the levels of Irf1 (A) and Socs1 (B) pre-mRNAs were analyzed by qRT-PCR. Error bars indicate standard deviations for biological replicates (n = 3). (C and D) STAT1 promoter occupancy in WT and STAT1β BMDMs. BMDMs were stimulated with IFN-β for the indicated times. STAT1 occupancy at the Irf1-GAS (C) and Socs1-GAS (D) sites was analyzed by ChIP. Error bars represent standard deviations (n = 3).
In sum, all three transcription inhibitors, despite different mechanisms of action, prolonged STAT1 promoter occupancy and STAT1 Y701 phosphorylation in response to IFN-β. Similarly, reduction of STAT1 transcriptional activity by genetic means caused longer promoter association of STAT1. Together, these results demonstrate that STAT1 inactivation can proceed only after the establishment of a transcription-competent RNAPII complex.
Processive transcription is required for downregulation of STAT1 promoter occupancy but not for Y701 dephosphorylation.
Prolonged STAT1 accumulation at the target promoters under conditions preventing processive transcription might be the consequence of impaired STAT1 Y701 dephosphorylation. We therefore asked whether a block in Y701 dephosphorylation is sufficient for enhanced promoter accumulation of STAT1. To answer this question, we inhibited Y701 dephosphorylation by treatment of cells with the irreversible tyrosine phosphatase inhibitor pervanadate, which is known to prevent STAT1 Y701 dephosphorylation (33). As expected, pervanadate treatment resulted in inhibition of tyrosine dephosphorylation of STAT1 and STAT2 (Fig. 7A) and persistent nuclear accumulation of STAT1 (Fig. 7B) in IFN-β-stimulated cells. Surprisingly, STAT1 occupancy at Irf1 and Mx2 promoters decreased with time in the presence as well as in the absence of pervanadate (Fig. 7C and D). In agreement with the declining STAT1 occupancy, pervanadate treatment was permissive for IFN-β-induced transcription of IFN-stimulated genes (Fig. 7E). Thus, regardless of the amounts of Y701-phosphorylated STAT1 in the nucleus, the IFN-β-stimulated promoter occupancy of STAT1 decreases with time of processive transcription. Together, these data establish that the regulated step of STAT1 inactivation in macrophages is STAT1 promoter occupancy, not STAT1 Y701 dephosphorylation.
FIG 7.

Processive transcription is required for downregulation of STAT1 promoter occupancy but not for Y701 dephosphorylation. (A) Pervanadate (Na3VO4) inhibits the tyrosine dephosphorylation of STAT1 and STAT2. BMDMs were stimulated with IFN-β in the presence or absence of Na3VO4 for the indicated times. Cell extracts were analyzed by Western blotting using antibodies to Y701-phosphorylated STAT1 (pY-STAT1) and STAT2 (pY-STAT2), total STAT1 and STAT2, and tubulin (used as a loading control). (B) Na3VO4 treatment results in persistent STAT1 nuclear accumulation. BMDMs were stimulated with IFN-β in the presence or absence of Na3VO4 for the indicated times. Cellular localization of STAT1 was assessed by immunofluorescence using STAT1-specific antibodies. (C and D) Inhibition of tyrosine dephosphorylation does not prevent a decrease in STAT1 promoter occupancy. BMDMs were stimulated with IFN-β in the presence or absence of Na3VO4 for the indicated times. STAT1 recruitment was assessed by ChIP for Irf1-GAS (C) and Mx2-ISRE (D) sites. Signals were normalized to input DNA. Error bars represent standard deviations (n = 3). (E) Na3VO4 treatment is permissive for IFN-β-induced transcription. BMDMs were stimulated with IFN-β in the presence or absence of Na3VO4. Total RNA was isolated, and the levels of Irf1, Mx2, Socs1, and Ifit1 mRNAs were quantified using qRT-PCR. Error bars indicate standard deviations (n = 3).
Our findings reveal that the transcription machinery communicates to the promoter-bound transcriptional complexes the status of transcription: establishment of processive transcription diminishes STAT1 occupancy, whereas a perturbation in transcription maintains high occupancy. STAT1 bound to DNA is protected from efficient Y701 dephosphorylation, while unbound STAT1 is subject to rapid dephosphorylation and recycling to the IFN receptor. By these means, permanent monitoring of the cytokine receptor activity and precise adjustment of the transcriptional output are ensured. Such a sensor function of continuous STAT activation and inactivation has been proposed by mathematical modeling (34, 35). Coupling of processive transcription with a reduction in promoter occupancy could result from a promoter displacement of STAT1 or from prevention of STAT1 recruitment after the onset of transcription. In both scenarios, the most likely regulators are chromatin modifiers and/or chromatin remodelers. Dynamic changes in histone modifications and chromatin remodeling operate in concert to reversibly switch the chromatin conformation between open and closed (36, 37). Transcription factor access can be facilitated through histone modifications such as acetylation, which loosens the nucleosome-DNA contacts, or by nucleosome eviction at the binding site, or by combinations of both. An example of regulation of transcription factor occupancy by signal-dependent chromatin changes is the two-wave recruitment of NF-κB in lipopolysaccharide (LPS)-stimulated macrophages, whereby the second wave of NF-κB appears at the target promoters only after a stimulus-dependent gain in the accessibility of NF-κB binding sites (38). Conversely, lineage-specific transcription factors can, in a signal-independent manner, maintain chromatin in an open configuration, which is characteristic for the particular lineage. In this way, the transcription factor Pu.1 preserves nucleosome-depleted sites at positions of macrophage-specific enhancers (39). The observed negative regulation of STAT1 occupancy after the onset of transcription might result from a gradual change from open to closed chromatin. The recruitment of several chromatin modifiers, including histone deacetylases or the RVB and BRG1 chromatin remodelers, has been associated with the induction of IFN-stimulated genes (40–44). The removal of these enzymes from the target genes might help to reestablish a closed chromatin conformation, thereby reducing STAT1 promoter occupancy. An important player in the repositioning of evicted nucleosomes is the INO80 remodeling complex, which also facilitates the exchange of the transcription-promoting histone variant H2A.Z with the nucleosome-stabilizing H2A (45). Although INO80 is not critical for type I IFN-induced gene expression in HEK293 cells (43), in combination with lineage-specific chromatin regulators, such as Pu.1, INO80 might contribute to restricting STAT1 promoter occupancy in macrophages. We assume that cell type-specific aspects feed in to modulate the duration of STAT1 signaling: our findings demonstrate that despite constitutive IFN presence, STAT1 is efficiently inactivated within several hours in BMDMs, whereas the dephosphorylation kinetics appears slower in MEFs, in which an IFN-free chase is beneficial for the assessment of STAT1 inactivation. Future studies should explore the involvement of general as well as cell type-specific histone marks and nucleosome configuration in the control of STAT1 occupancy. One can also speculate that a second signal (other than IFN) might restructure the chromatin configuration at the STAT1 target genes such that the promoter occupancy of STAT1 is shortened or prolonged. Such mechanisms would provide additional ways of regulating IFN/STAT1 signaling. Notably, pathogens might have developed strategies to interfere with IFN signaling by restricting STAT1 turnover at the promoter: persistent STAT1 chromatin association without concomitant transcription of STAT1 target genes has been observed in Toxoplasma gondii-infected cells (46). The results of our study have broader implications for STAT biology, since inactivation of STAT2 and STAT3, like that of STAT1, is impaired under conditions of blocked transcription.
ACKNOWLEDGMENTS
We thank Marton Janos for critical reading of the manuscript.
This work was supported by Austrian Science Fund (FWF) grants W1220-B09 DP (Molecular Mechanisms of Cell Signaling), P22806-B11, and P27538-B21 to P.K., by Austrian Science Fund grant SFB-F28 to B.S., and by the European Union Seventh Framework Programme Marie Curie Initial Training Networks (FP7-PEOPLE-2012-ITN) for the project INBIONET under grant agreement PITN-GA-2012-316682.
REFERENCES
- 1.Stark GR, Darnell JE Jr. 2012. The JAK-STAT pathway at twenty. Immunity 36:503–514. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Shea JJ, Plenge R. 2012. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 36:542–550. doi: 10.1016/j.immuni.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casanova JL, Holland SM, Notarangelo LD. 2012. Inborn errors of human JAKs and STATs. Immunity 36:515–528. doi: 10.1016/j.immuni.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ungureanu D, Vanhatupa S, Kotaja N, Yang J, Aittomaki S, Janne OA, Palvimo JJ, Silvennoinen O. 2003. PIAS proteins promote SUMO-1 conjugation to STAT1. Blood 102:3311–3313. doi: 10.1182/blood-2002-12-3816. [DOI] [PubMed] [Google Scholar]
- 5.Bancerek J, Poss ZC, Steinparzer I, Sedlyarov V, Pfaffenwimmer T, Mikulic I, Dolken L, Strobl B, Muller M, Taatjes DJ, Kovarik P. 2013. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity 38:250–262. doi: 10.1016/j.immuni.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tenoever BR, Ng SL, Chua MA, McWhirter SM, Garcia-Sastre A, Maniatis T. 2007. Multiple functions of the IKK-related kinase IKKε in interferon-mediated antiviral immunity. Science 315:1274–1278. doi: 10.1126/science.1136567. [DOI] [PubMed] [Google Scholar]
- 7.Donnelly RP, Kotenko SV. 2010. Interferon-lambda: a new addition to an old family. J Interferon Cytokine Res 30:555–564. doi: 10.1089/jir.2010.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, Zhu W, Tremblay M, David M, Shuai K. 2002. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol 22:5662–5668. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krämer OH, Knauer SK, Greiner G, Jandt E, Reichardt S, Guhrs KH, Stauber RH, Bohmer FD, Heinzel T. 2009. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev 23:223–235. doi: 10.1101/gad.479209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antunes F, Marg A, Vinkemeier U. 2011. STAT1 signaling is not regulated by a phosphorylation-acetylation switch. Mol Cell Biol 31:3029–3037. doi: 10.1128/MCB.05300-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyer T, Marg A, Lemke P, Wiesner B, Vinkemeier U. 2003. DNA binding controls inactivation and nuclear accumulation of the transcription factor Stat1. Genes Dev 17:1992–2005. doi: 10.1101/gad.268003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mertens C, Zhong M, Krishnaraj R, Zou W, Chen X, Darnell JE Jr. 2006. Dephosphorylation of phosphotyrosine on STAT1 dimers requires extensive spatial reorientation of the monomers facilitated by the N-terminal domain. Genes Dev 20:3372–3381. doi: 10.1101/gad.1485406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sadzak I, Schiff M, Gattermeier I, Glinitzer R, Sauer I, Saalmuller A, Yang E, Schaljo B, Kovarik P. 2008. Recruitment of Stat1 to chromatin is required for interferon-induced serine phosphorylation of Stat1 transactivation domain. Proc Natl Acad Sci U S A 105:8944–8949. doi: 10.1073/pnas.0801794105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kimura T, Kadokawa Y, Harada H, Matsumoto M, Sato M, Kashiwazaki Y, Tarutani M, Tan RS, Takasugi T, Matsuyama T, Mak TW, Noguchi S, Taniguchi T. 1996. Essential and non-redundant roles of p48 (ISGF3γ) and IRF-1 in both type I and type II interferon responses, as revealed by gene targeting studies. Genes Cells 1:115–124. doi: 10.1046/j.1365-2443.1996.08008.x. [DOI] [PubMed] [Google Scholar]
- 15.Semper C, Leitner NR, Lassnig C, Parrini M, Mahlakoiv T, Rammerstorfer M, Lorenz K, Rigler D, Muller S, Kolbe T, Vogl C, Rulicke T, Staeheli P, Decker T, Muller M, Strobl B. 2014. STAT1β is not dominant negative and is capable of contributing to gamma interferon-dependent innate immunity. Mol Cell Biol 34:2235–2248. doi: 10.1128/MCB.00295-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A. 1992. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 17.Kovarik P, Stoiber D, Novy M, Decker T. 1998. Stat1 combines signals derived from IFN-γ and LPS receptors during macrophage activation. EMBO J 17:3660–3668. (Erratum, 17:4210.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dolken L, Ruzsics Z, Radle B, Friedel CC, Zimmer R, Mages J, Hoffmann R, Dickinson P, Forster T, Ghazal P, Koszinowski UH. 2008. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA (New York, NY) 14:1959–1972. doi: 10.1261/rna.1136108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, Corbin JE, Cornish AL, Darwiche R, Owczarek CM, Kay TW, Nicola NA, Hertzog PJ, Metcalf D, Hilton DJ. 1999. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell 98:597–608. doi: 10.1016/S0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- 20.Thompson JE, Cubbon RM, Cummings RT, Wicker LS, Frankshun R, Cunningham BR, Cameron PM, Meinke PT, Liverton N, Weng Y, DeMartino JA. 2002. Photochemical preparation of a pyridone containing tetracycle: a Jak protein kinase inhibitor. Bioorg Med Chem Lett 12:1219–1223. doi: 10.1016/S0960-894X(02)00106-3. [DOI] [PubMed] [Google Scholar]
- 21.Bensaude O. 2011. Inhibiting eukaryotic transcription: which compound to choose? How to evaluate its activity? Transcription 2:103–108. doi: 10.4161/trns.2.3.16172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chao SH, Price DH. 2001. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem 276:31793–31799. doi: 10.1074/jbc.M102306200. [DOI] [PubMed] [Google Scholar]
- 23.Rickert P, Corden JL, Lees E. 1999. Cyclin C/CDK8 and cyclin H/CDK7/p36 are biochemically distinct CTD kinases. Oncogene 18:1093–1102. doi: 10.1038/sj.onc.1202399. [DOI] [PubMed] [Google Scholar]
- 24.Nelson WG, Kastan MB. 1994. DNA strand breaks: the DNA template alterations that trigger p53-dependent DNA damage response pathways. Mol Cell Biol 14:1815–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choong ML, Yang H, Lee MA, Lane DP. 2009. Specific activation of the p53 pathway by low dose actinomycin D: a new route to p53 based cyclotherapy. Cell Cycle (Georgetown, Tex) 8:2810–2818. doi: 10.4161/cc.8.17.9503. [DOI] [PubMed] [Google Scholar]
- 26.Youlyouz-Marfak I, Gachard N, Le Clorennec C, Najjar I, Baran-Marszak F, Reminieras L, May E, Bornkamm GW, Fagard R, Feuillard J. 2008. Identification of a novel p53-dependent activation pathway of STAT1 by antitumour genotoxic agents. Cell Death Differ 15:376–385. doi: 10.1038/sj.cdd.4402270. [DOI] [PubMed] [Google Scholar]
- 27.O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. 2007. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A 104:1604–1609. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gantier MP, McCoy CE, Rusinova I, Saulep D, Wang D, Xu D, Irving AT, Behlke MA, Hertzog PJ, Mackay F, Williams BR. 2011. Analysis of microRNA turnover in mammalian cells following Dicer1 ablation. Nucleic Acids Res 39:5692–5703. doi: 10.1093/nar/gkr148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou C, Yu Q, Chen L, Wang J, Zheng S, Zhang J. 2012. A miR-1231 binding site polymorphism in the 3′UTR of IFNAR1 is associated with hepatocellular carcinoma susceptibility. Gene 507:95–98. doi: 10.1016/j.gene.2012.06.073. [DOI] [PubMed] [Google Scholar]
- 30.Gough DJ, Messina NL, Hii L, Gould JA, Sabapathy K, Robertson AP, Trapani JA, Levy DE, Hertzog PJ, Clarke CJ, Johnstone RW. 2010. Functional crosstalk between type I and II interferon through the regulated expression of STAT1. PLoS Biol 8:e1000361. doi: 10.1371/journal.pbio.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN. 2002. TLR4, but not TLR2, mediates IFN-β-induced STAT1α/β-dependent gene expression in macrophages. Nat Immunol 3:392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- 32.Mancebo HS, Lee G, Flygare J, Tomassini J, Luu P, Zhu Y, Peng J, Blau C, Hazuda D, Price D, Flores O. 1997. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev 11:2633–2644. doi: 10.1101/gad.11.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haspel RL, Darnell JE Jr. 1999. A nuclear protein tyrosine phosphatase is required for the inactivation of Stat1. Proc Natl Acad Sci U S A 96:10188–10193. doi: 10.1073/pnas.96.18.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rateitschak K, Karger A, Fitzner B, Lange F, Wolkenhauer O, Jaster R. 2010. Mathematical modelling of interferon-gamma signalling in pancreatic stellate cells reflects and predicts the dynamics of STAT1 pathway activity. Cell Signal 22:97–105. doi: 10.1016/j.cellsig.2009.09.019. [DOI] [PubMed] [Google Scholar]
- 35.Swameye I, Muller TG, Timmer J, Sandra O, Klingmüller U. 2003. Identification of nucleocytoplasmic cycling as a remote sensor in cellular signaling by databased modeling. Proc Natl Acad Sci U S A 100:1028–1033. doi: 10.1073/pnas.0237333100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerhold CB, Gasser SM. 2014. INO80 and SWR complexes: relating structure to function in chromatin remodeling. Trends Cell Biol 24:619–631. doi: 10.1016/j.tcb.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 37.Petty E, Pillus L. 2013. Balancing chromatin remodeling and histone modifications in transcription. Trends Genet 29:621–629. doi: 10.1016/j.tig.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saccani S, Pantano S, Natoli G. 2001. Two waves of nuclear factor κB recruitment to target promoters. J Exp Med 193:1351–1359. doi: 10.1084/jem.193.12.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barozzi I, Simonatto M, Bonifacio S, Yang L, Rohs R, Ghisletti S, Natoli G. 2014. Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol Cell 54:844–857. doi: 10.1016/j.molcel.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zupkovitz G, Tischler J, Posch M, Sadzak I, Ramsauer K, Egger G, Grausenburger R, Schweifer N, Chiocca S, Decker T, Seiser C. 2006. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol Cell Biol 26:7913–7928. doi: 10.1128/MCB.01220-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nusinzon I, Horvath CM. 2003. Interferon-stimulated transcription and innate antiviral immunity require deacetylase activity and histone deacetylase 1. Proc Natl Acad Sci U S A 100:14742–14747. doi: 10.1073/pnas.2433987100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang HM, Paulson M, Holko M, Rice CM, Williams BR, Marié I, Levy DE. 2004. Induction of interferon-stimulated gene expression and antiviral responses require protein deacetylase activity. Proc Natl Acad Sci U S A 101:9578–9583. doi: 10.1073/pnas.0400567101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gnatovskiy L, Mita P, Levy DE. 2013. The human RVB complex is required for efficient transcription of type I interferon-stimulated genes. Mol Cell Biol 33:3817–3825. doi: 10.1128/MCB.01562-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pattenden SG, Klose R, Karaskov E, Bremner R. 2002. Interferon-gamma-induced chromatin remodeling at the CIITA locus is BRG1 dependent. EMBO J 21:1978–1986. doi: 10.1093/emboj/21.8.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yen K, Vinayachandran V, Pugh BF. 2013. SWR-C and INO80 chromatin remodelers recognize nucleosome-free regions near +1 nucleosomes. Cell 154:1246–1256. doi: 10.1016/j.cell.2013.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosowski EE, Nguyen QP, Camejo A, Spooner E, Saeij JP. 2014. Toxoplasma gondii inhibits gamma interferon (IFN-γ)- and IFN-β-induced host cell STAT1 transcriptional activity by increasing the association of STAT1 with DNA. Infect Immun 82:706–719. doi: 10.1128/IAI.01291-13. [DOI] [PMC free article] [PubMed] [Google Scholar]