

Abstract
Promoters often contain multiple binding sites for a single factor. The yeast HO gene contains nine highly conserved binding sites for the SCB (Swi4/6-dependent cell cycle box) binding factor (SBF) complex (composed of Swi4 and Swi6) in the 700-bp upstream regulatory sequence 2 (URS2) promoter region. Here, we show that the distal and proximal SBF sites in URS2 function differently. Chromatin immunoprecipitation (ChIP) experiments show that SBF binds preferentially to the left side of URS2 (URS2-L), despite equivalent binding to the left-half and right-half SBF sites in vitro. SBF binding at URS2-L sites depends on prior chromatin remodeling events at the upstream URS1 region. These signals from URS1 influence chromatin changes at URS2 but only at sites within a defined distance. SBF bound at URS2-L, however, is unable to activate transcription but instead facilitates SBF binding to sites in the right half (URS2-R), which are required for transcriptional activation. Factor binding at HO, therefore, follows a temporal cascade, with SBF bound at URS2-L serving to relay a signal from URS1 to the SBF sites in URS2-R that ultimately activate gene expression. Taken together, we describe a novel property of a transcription factor that can have two distinct roles in gene activation, depending on its location within a promoter.
INTRODUCTION
Transcription factor binding to a gene promoter is an essential step in gene activation, making it a critical target of gene regulation. One mechanism of regulating transcription factor binding is by controlling binding site accessibility through alterations of chromatin (1). At some promoters, nucleosomes can occlude transcription factor binding sites, and nucleosomes must be remodeled or evicted by transcriptional coactivators to permit protein binding (2–5). Three transcriptional coactivators promote chromatin changes at many promoters in yeast to facilitate gene activation: SWI/SNF, SAGA, and Mediator. These three transcriptional coactivators are conserved from yeast to humans and are required for activation of many eukaryotic genes (6), including the yeast HO gene (7).
The HO gene encodes an endonuclease that initiates yeast mating type switching by cleavage at the mating type locus followed by gene conversion. HO regulation is extremely complex, and many transcriptional regulators were first identified in studies of HO regulation, including SWI/SNF and the Gcn5 histone acetyltransferase. The HO promoter is unusually long, with regulatory elements extending ∼1,900 bp upstream of the transcription start site. HO expression is regulated by numerous DNA binding factor binding sites in this long upstream regulatory sequence, which can be subdivided into two regions, upstream regulatory sequence 1 (URS1) and URS2 (8). HO is expressed only in mother cells following mitotic division; this asymmetric mother-specific expression is achieved via daughter cell accumulation of the Ash1 transcription factor, which binds URS1 and recruits the Rpd3(L) histone deacetylase complex (9–11). HO activation is sequential, with ordered recruitment of transcription factors and coactivators first to URS1 and then to URS2 (12, 13). Concomitant with this ordered recruitment, waves of nucleosome eviction follow transcriptional coactivator binding along the HO promoter (14). Coactivator recruitment to URS1 and URS2 is mediated by two cell cycle-regulated transcription factors, Swi5 and SCB binding factor (SBF; composed of Swi4 and Swi6), respectively. HO activation starts with Swi5 binding to sites within nucleosome-depleted regions at −1800 and −1300 and the recruitment of the SWI/SNF, SAGA, and Mediator coactivators. SBF binding to URS2, however, is occluded by nucleosomes, and eviction of these nucleosomes depends upon Swi5-dependent events at URS1. The SBF binding allows for coactivator recruitment to URS2, which ultimately activates HO expression. Thus, SBF binding at URS2 appears to be the penultimate activator of HO expression.
SBF is a heterodimeric protein complex composed of two subunits, Swi4 and Swi6 (15). The SBF complex recognizes the SCB (Swi4/6-dependent cell cycle box) CRCGAAA consensus sequence and activates transcription at numerous promoters to control the G1/S transition, or START. SBF activity is negatively regulated by two inhibitory factors, Stb1 and Whi5 (16–19). Stb1 and Whi5 stably associate with SBF and recruit the Rpd3(L) histone deacetylase (20–22). Stb1 and Whi5 are hyperphosphorylated by cyclin-dependent kinases (CDKs) during late G1, causing them to dissociate from the SBF-bound promoters, allowing activation of transcription.
HO URS2 contains nine sites that possess the SCB CRCGAAA consensus sequence required for SBF binding and gene activation. DNA fragments containing SBF sites from either the left, middle, or right thirds of URS2 display equivalent SBF binding in vitro by electrophoretic mobility shift assays (EMSAs) (11). However, chromatin immunoprecipitation (ChIP) assays show that strong SBF binding in vivo occurs primarily on the left side of URS2 (11). These results suggest that either SBF binding is influenced by proximity to upstream remodeling events occurring at URS1 (the left side of URS2 is a “good neighborhood” for binding) or that the left-most sites share a currently unknown sequence feature that make them preferred binding sites in vivo (these SBF sites have a different “address” than those on the right side). Surprisingly, despite the gradient of SBF binding from left to right, SBF sites throughout URS2 display a high degree of conservation (23). The driving force for this study was the enigma of nine binding sites for a single factor conserved through a 700-bp promoter region, contrasted with strong binding at only a subset of these binding sites.
Our results show that SBF binding at HO URS2 is significantly more complicated than previously modeled. ChIPs examining SBF binding at various HO promoter mutants indicate that SBF enrichment is dependent on proximity to URS1, supporting a “neighborhood” model of SBF binding. Further supporting this model, HO mutants containing only the left half (ΔR-URS2) or right half (ΔL-URS2) of URS2 are fully capable of activating HO expression. SBF enrichment, however, does not always correlate with HO expression. Unexpectedly, mutation of URS2-R SBF sites resulted in loss of HO expression despite near WT levels of SBF binding at the remaining sites at URS2-L. Furthermore, SBF binding at URS2-R was absolutely dependent on functional URS2-L SBF sites. These results suggest a cascade model whereby SBF loading onto URS2-L allows for limited, but essential, SBF binding at URS2-R. As the right-half SBF sites are essential for HO expression, the cascade SBF binding model at URS2 reconciles the paradoxical relationship between SBF binding and site conservation at HO.
MATERIALS AND METHODS
All yeast strains used in this study are listed in Table 1 and are isogenic in the W303 background (24). Standard genetic methods were used for strain construction (25, 26). In general, promoter manipulations in the study were constructed using the delitto perfetto method (27) with transformation of the appropriate PCR products or linearized plasmids. Oligonucleotides used in strain construction are available upon request. Plasmids used for strain construction are listed in Table 2, and details on plasmid construction are available upon request. Plasmid pZC09, provided by Zaily Connell and Tim Formosa, was used to add a V5 epitope tag to the C terminus of SWI4 using a PCR strategy (28).
TABLE 1.
Strain list
| Figurea | Strain | Description |
|---|---|---|
| 1 | DY150 | MATa ade2 can1 his3 leu2 trp1 ura3 |
| DY15855 | MATa HO(LX4 sbf)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY16483 | MATa HO(LX5 sbf)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY16942 | MATa SWI4-V5::His3MX ade2 can1 his3 leu2 trp1 ura3 | |
| DY17503 | MATa SWI4-V5::His3MX HO(LX4 sbf)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY17505 | MATa SWI4-V5::His3MX HO(LX5 sbf)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY150 | MATa ade2 can1 his3 leu2 trp1 ura3 | |
| DY16251 | MATa HO(−948 to −629 deleted; replaced by 329 bp from CDC39)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| 3 | DY150 | MATa ade2 can1 his3 leu2 trp1 ura3 |
| DY17307 | MATa HO(URS2-R[−619 to −125 deleted])::KanMX ade2 can1 his3 leu2 lys2 trp1 ura3 | |
| DY17309 | MATa HO(URS2-L[−948 to −610, −287 to −125 deleted])::KanMX ade2 can1 his3 leu2 lys2 trp1 ura3 | |
| DY6758 | MATa swi6::TRP1 ade2 can1 his3 leu2 trp1 ura3 | |
| DY17311 | MATa swi6::TRP1 HO(URS2-R[−619 to −125 deleted])::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY17315 | MATa swi6::TRP1 HO(URS2-L[−948 to −610, −287 to −125 deleted])::KanMX ade2 can1 his3 leu2 lys2 trp1 ura3 | |
| 4 | DY14674 | MATa HO(MCS at −1082)::KanMX ade2 can1 his3 leu2 trp1 ura3 |
| DY15151 | MATa HO(900 bp from CDC39 at −1082)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY15152 | MATa HO(700 bp from CDC39 at −1082)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY15153 | MATa HO(500 bp from CDC39 at −1082)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY15154 | MATa HO(300 bp from CDC39 at −1082)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY15643 | MATa HO(221 bp from CDC39 at −1082)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY16942 | MATa SWI4-V5::His3MX ade2 can1 his3 leu2 trp1 ura3 | |
| DY17901 | MATa SWI4-V5::His3MX HO(700 bp from CDC39 at −1082)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY17903 | MATa SWI4-V5::His3MX HO(300 bp from CDC39 at −1082)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY17905 | MATa SWI4-V5::His3MX HO(170 bp from CDC39 at −1082)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| 5 | DY150 | MATa ade2 can1 his3 leu2 trp1 ura3 |
| DY16679 | MATa HO(−948 to −170 inverted)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY15112 | MATa HO(−928 to −233 deleted; replaced by −530 to −229, −927 to −532)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY16680 | MATa HO(−948 to −629 deleted; replaced by −610 to −291)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY16942 | MATa SWI4-V5::His3MX ade2 can1 his3 leu2 trp1 ura3 | |
| DY17645 | MATa SWI4-V5::His3MX HO(−948 to −170 inverted)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY17648 | MATa SWI4-V5::His3MX HO(−928 to −233 deleted; replaced by −530 to −229, −927 to −532)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| 6 | DY150 | MATa ade2 can1 his3 leu2 trp1 ura3 |
| DY 17857 | MATa HO(−610 to −170 deleted; replaced by 440 bp from CDC39)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY17652 | MATa HO(RX4 sbf)::KanMX SWI4-V5::His3MX ade2 can1 his3 leu2 trp1 ura3 | |
| DY17653 | MATa HO(RX5 sbf)::KanMX SWI4-V5::His3MX ade2 can1 his3 leu2 trp1 ura3 | |
| 7 | DY16942 | MATa SWI4-V5::His3MX ade2 can1 his3 leu2 trp1 ura3 |
| DY17760 | MATa SWI4-V5::His3MX HO(RX4 sbf)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY17505 | MATa SWI4-V5::His3MX HO(LX5 sbf)::KanMX ade2 can1 his3 leu2 trp1 ura3 | |
| DY17461 | MATa GALp::CDC20::ADE2 SWI4-V5::His3MX ade2 can1 his3 leu2 trp1 ura3 |
Figure in which the indicated strains were used.
TABLE 2.
Plasmid list
| Plasmid | Description |
|---|---|
| M1679 | HO (nt −1902 to −929), in BSKS+ |
| M2775 | CDC39, in pRS316 |
| M2817 | HO (nt −3553 to +2584), in pUC18 |
| M5488 | HO (nt −1930 to −155), with MfeI/BsrG1/MluI/NheI/NotI polylinker at −1052, in pRS306 |
| M5536 | HO (nt −1948 to +2584), with −929 to −233 deleted and replaced by −530 to −229, −927 to −532), in pBR322 |
| M5543 | HO (nt −1930 to −155), with 900 bp from CDC39 inserted at −1052, in pRS306 |
| M5544 | HO (nt −1930 to −155), with 700 bp from CDC39 inserted at −1052, in pRS306 |
| M5545 | HO (nt −1930 to −155), with 500 bp from CDC39 inserted at −1052, in pRS306 |
| M5546 | HO (nt −1930 to −155), with 300 bp from CDC39 inserted at −1052, in pRS306 |
| M5588 | HO (nt −1930 to −155), with 221 bp from CDC39 inserted at −1052, in pRS306 |
| M5543 | HO (nt −1930 to −155), with 900 bp from CDC39 inserted at −1052, in pRS306 |
| M5715 | HO (nt −592 to −126), with four mutant Sbf sites, in pUC57-AMP |
| M5717 | HO (nt −3553 to +2584), with first four Sbf sites mutated, in pUC18 |
| M5718 | HO (nt −3553 to +2584), with first five Sbf sites mutated, in pUC18 |
| pCH341 | HO (nt −1930 to −155), with NotI site at −1052, in pRS306 (provided by Pam Geyer) |
| pCORE-UH | Kluvermyces lactis URA3 and HphMX4, in pFA6 (see reference 27) |
| pZC09 | C-terminal V5 tagging vector with TEV site, Gly linker, and His3MX marker (provided by Zaily Connell and Tim Formosa) |
Cell cycle synchronization was performed by galactose withdrawal and readdition with a GALp::CDC20 strain grown at 25°C in yeast extract-peptone (YP) medium containing 2% galactose and 2% raffinose (13). A high degree of synchrony was demonstrated by examination of budding indices and analysis of cycle-regulated mRNAs (data not shown). In all other experiments, cells were grown at 30°C in yeast extract-peptone-adenine-dextrose (YPAD) medium (26).
ChIPs were performed as described previously (13, 29) by using mouse monoclonal antibody to the V5 epitope (SV5-Pk1; Abcam) or anti-histone H3 (07-690; Upstate) and antibody-coated magnetic beads (rabbit and pan mouse IgG beads; Life Technologies). Samples prepared for ChIPs were cross-linked in 1% formaldehyde overnight on ice. ChIP assays were analyzed by real-time qPCR as described previously (30). ChIP qPCR primers are available upon request. For all logarithmically grown strains, each ChIP sample was first normalized to the ChIP signal at the CLN1 promoter and then to its respective input DNA sample. In the cell synchrony experiment, the ChIP values were normalized only to input DNA, and the ChIP signal at the URS2-L was compared to that at URS2-R. Error bars in ChIP assays reflect the standard deviation from two biological samples or two independent IPs.
MNase mapping of nucleosome positions was preformed as described previously (31). Mononucleosomes were prepared as described for ChIPs, with the exceptions that cells were formaldehyde cross-linked for only 5 min at room temperature and chromatin was only lightly sheared prior to micrococcal nuclease digestion.
RNA was isolated from logarithmically growing cells, and HO mRNA levels were measured by reverse transcription-quantitative PCR (RT-qPCR) as described previously (29). RNA expression was normalized to RPR1 expression and graphed relative to wild-type (WT) expression. Error bars reflect the standard deviations from two biological samples. RT-qPCR primers are available upon request.
RESULTS
URS2-R SBF sites cannot compensate for left-half site mutants.
Binding of the SBF factor to URS2 is a required step in HO expression. We have previously shown that SBF binds predominantly to URS2-L (11) despite the presence of multiple additional SBF sites within URS2-R. Furthermore, we have demonstrated via an in vitro EMSA that these sites bind SBF equally well (11). To further investigate the role of SBF binding preferentially to URS2-L, we mutated the first four (LX4 sbf) (Fig. 1A) or five (LX5 sbf) SBF sites in URS2 to determine the effect of these mutations on HO expression. HO expression in these SBF site mutant strains was greatly reduced compared to that of the WT, nearly as much as the decrease observed in a strain lacking the Swi5 activator that initiates events at the HO promoter (data not shown). This result demonstrates that the first several SBF sites of URS2 are required for HO expression. The downstream SBF sites cannot compensate for HO activation when the left-half binding sites are mutated, despite the fact that all of the SBF sites show equal evolutionary conservation (23). ChIP experiments were next performed to measure SBF binding to WT, LX4 sbf, and LX5 sbf promoters, using strains with a V5 epitope tag at the C terminus of the Swi4 subunit of SBF. Consistent with our previous SBF ChIP data (11), mutation of the first four or five SBF sites of URS2 largely eliminates SBF binding to URS2 (Fig. 1B).
FIG 1.

Downstream SBF sites cannot compensate for left-half site mutants. Diagrams on the left represent the URS2 portion of the HO promoter, and numbers within the rectangles indicate SBF site locations. Mutations in an SBF site are indicated by an X. CDC39 replacement is indicated by a gray block. (A) HO mRNA levels were measured for strains with the wild-type, LX4 sbf, and LX5 sbf versions of the HO promoter. (B) Binding of the Swi4-V5 subunit of SBF to the wild-type, LX4 sbf, and LX5 sbf versions of the HO promoter was determined by ChIP followed by qPCR. HO SBF ChIP enrichment was normalized to that of CLN1 and graphed relative to wild-type enrichment. Location of ChIP primers is indicated on the diagram. (C) HO mRNA levels were measured for strains with the wild-type HO promoter and CDC39 replacing URS2L.
It was possible that inhibitory sequences present in URS2-L somehow prevented utilization of the downstream SBF sites. To address this question, we replaced the left half of the URS2 sequence with a fragment of equivalent size from the CDC39 open reading frame (ORF). Consistent with expression data from our SBF site mutants, HO expression was again greatly diminished. This result reinforces the critical importance of the first several SBF sites in HO activation and furthermore demonstrates that upstream inhibitory sequences were not responsible for the failure of SBF to activate transcription from the right half of URS2. The slightly elevated HO expression observed with the CDC39 replacement relative to the SBF site mutants is likely due to the dual role of SBF as both an activator (32) and a repressor of transcription (11, 16, 17).
Two models: neighborhood versus address.
We considered two possible models to explain the selective binding of SBF to URS2-L: a neighborhood model and an address model. The neighborhood model predicts that SBF binding is dependent on changes in chromatin, and additionally that chromatin changes that occur first at URS1 (14) facilitate chromatin changes at URS2 but only at sites in close proximity; this results in chromatin changes occurring at URS2-L in preference to the sites at URS2-R. In contrast, the address model predicts that sequences surrounding the SBF sites play an important role in determining SBF binding in vivo that could not be revealed by the in vitro EMSA; for example, there could be stimulatory sequence elements in URS2-L or inhibitory elements in URS2-R. To differentiate between these two models for SBF binding at HO URS2, we constructed multiple HO promoter rearrangements, insertions, and deletions (Fig. 2). Nucleosome dynamics are known to play a critical role in HO activation (14), so we wanted to consider nucleosome positions when making changes to the promoter. We therefore mapped nucleosome positions along the HO promoter using primers spaced every 30 bp along the promoter to amplify DNA prepared from mononucleosomes isolated after MNase digestion (Fig. 2) (31). We then used this nucleosome positioning data as a guide when making deletions, insertions, or rearrangements to the promoter.
FIG 2.

Neighborhood and address models. Top, diagrams of the HO promoter, with rectangles indicating URS1 and URS2. Swi5 site locations in URS1 are indicated, and numbers indicate SBF sites in URS2. Dashed lines present in panels B and C indicate deletions of either the right-half or the left-half URS2 sequence. The right-hand rectangle in panel D represents a URS2 sequence inversion. Reordered URS2 rectangles in panel E represent a URS2 rearrangement in which the right half of URS2 was moved in front of what was originally the left half of URS2. Rectangle between URS1 and URS2 in panel F indicates the location of insertions tested in this paper. Right, predicted HO expression for all tested constructs according to the neighborhood and address models, as described in the text. Lower, H3 nucleosome enrichment over the HO promoter. Enrichment peaks indicate the midpoints of nucleosome positions, and determined nucleosome locations are represented below by circles with two diagonal lines. Nine nucleosomes can be observed along the HO promoter, centered at the following positions: bp −1628, −1478, −1208, −1058, −908, −698, −548, −358, and −198 with respect to the ATG.
Two constructs were of particular importance in distinguishing between the neighborhood and address models: ΔL-URS2 and URS1-insert-URS2 (Fig. 2C and F). Under the neighborhood model, we expect deletion of URS2-L (ΔL-URS2) to now allow equivalent chromatin changes to occur but now at URS2-R, making it a viable target for SBF binding and subsequent HO activation. In contrast, the address model predicts that the left half of URS2 is inherently more active than the right half, predicting that this ΔL-URS2 deletion should exhibit sharply reduced HO expression. The neighborhood model also predicts that insertions between URS1 and URS2 (URS1-insert-URS2) should move URS2 too far from URS1 to be effective, resulting in a decrease in HO expression that is proportional to the size of the insertion. In the address model, the proximity of URS2 to URS1 is not relevant, so insertions should not affect HO expression.
Each half of URS2 is capable of supporting HO expression.
To distinguish between the neighborhood and address models, we first examined the two URS2 deletion halves (ΔR-URS2 and ΔL-URS2) for HO expression. In agreement with the neighborhood model, both ΔR-URS2 and ΔL-URS2 expressed HO at WT or higher levels (Fig. 3A). Both URS2 halves are therefore capable of supporting full HO expression. Previous work showed that expression of HO in the complete absence of URS2 is no longer dependent on SBF (32), so it was important to test the necessity of SBF in the HO expression observed in our constructs. We therefore tested the effect of deleting the Swi6 subunit of SBF on the activity of the same promoter constructs. The swi6 mutation markedly reduced expression of the ΔR-URS2 and ΔL-URS2 promoters, demonstrating that these promoters are still largely dependent upon SBF and that the shorter URS2 does not completely bypass the normal requirements for activation seen at native HO (Fig. 3B). Interestingly, the ΔR-URS2 and ΔL-URS2 promoter mutants displayed higher levels of HO expression than the native promoter in a swi6 strain, suggesting that full-length URS2 is required to prevent inappropriate gene activation by Swi5 bound at URS1.
FIG 3.

Left- and right-half deletions of HO URS2 are capable of HO expression. Diagrams on the left represent the URS2 portion of the HO promoter, and numbers within the rectangles indicate SBF site locations. Dashed lines indicate deletions of either the right-half or the left-half URS2 sequence. (A) HO mRNA levels were measured for SWI6 strains with the wild-type, ΔR-URS2, and ΔL-URS2 versions of the HO promoter. (B) HO mRNA levels were measured for swi6 strains with the wild-type, ΔR-URS2, and ΔL-URS2 versions of the HO promoter.
Insertions between URS1 and URS2 reduce HO expression and SBF binding.
To directly investigate whether the proximity of URS1 to URS2 affects HO expression, we inserted “neutral” sequences of various size between them. We initially chose exonic sequences from metazoan genes for this purpose, but to our surprise these inserts contained sequences capable of promoter activation independent of other HO activators (data not shown). However, exonic sequences from yeast ORFs did not produce this inappropriate activation, so these were used instead. For the studies shown here, we inserted fragments from the CDC39 gene, ranging in size from 170 bp to 900 bp (Fig. 4), although we observed similar results with inserts derived from a different gene (data not shown).
FIG 4.

Increased distance between URS1 and URS2 reduces HO expression and SBF binding. (A) The diagram of the HO promoter has rectangles indicating URS1 and URS2, lines indicating Swi5 site locations in URS1, numbers indicating SBF sites in URS2, and a rectangle between URS1 and URS2 indicating the insertions. HO mRNA levels were measured for strains with HO promoters with insertions of the indicated size. (B) The diagram is as described for panel A, with the location of ChIP primers indicated. Binding of the Swi4-V5 subunit of SBF to the indicated versions of the HO promoter was determined by ChIP followed by qPCR. Location of ChIP primers is indicated on the diagram.
In agreement with the neighborhood model, HO expression decreased in all but one of our CDC39 insertions, and HO expression levels inversely correlated with the size of the insertion (Fig. 4A). Introducing an Swi4-V5 tag into a set of the same strains produced comparable results (data not shown) and allowed analysis of SBF occupancy by ChIP in parallel with the HO analysis. We found that SBF binding also inversely correlated with the size of CDC39 insertion (Fig. 4B). These results suggest that the proximity of URS1 is an important feature in the activation of URS2 by binding of SBF, consistent with the idea that remodeling of the chromatin structure initiated at URS1 has a limited range and allows SBF to access sites only within a favorable window.
URS2 rearrangements support the neighborhood model of URS1 interaction with URS2.
The deletion constructs tested in Fig. 3 altered the spatial context of URS1 with respect to the TATA and potentially removed URS2 sequences required for the normal regulation of HO activation. To investigate HO expression and SBF binding to the two URS2 halves in a full-length URS2 setting, two manipulations of full-length URS2 were constructed: URS2-flip and URS2R→L (Fig. 2D and E). Both the URS2-flip and URS2R→L constructs place the right-half of URS2 immediately adjacent to URS1, either by inversion of the URS2 sequence or by rearrangement of the two URS2 halves. RNA analysis of the URS2-flip and URS2R→L constructs revealed HO expression comparable to that of the WT (Fig. 5A). Additionally, the URS2-flip result indicated that the orientation of the SBF binding sequence CACGAAA does not affect HO activation. This result is somewhat surprising given that all of the SBF binding sites in the HO promoter are present in the same orientation (33). Expression of the URS2R→L promoter demonstrated that the positional arrangement of the two URS2 halves has little effect on HO activation.
FIG 5.

Proximity to URS1 is the major determinant of SBF enrichment. (A) The diagrams of the different HO promoter constructs have rectangles representing URS2, and the numbers indicate SBF sites. HO mRNA levels were measured for strains with the indicated versions of the HO promoter. (B) The diagrams are as described for panel A, with the location of ChIP primers indicated. Binding of the Swi4-V5 subunit of SBF to the indicated versions of the HO promoter was determined by ChIP followed by qPCR.
ChIP assays measuring SBF binding are quite informative concerning the two models. Consistent with the neighborhood model, we observe stronger SBF binding to sites closest to URS1, regardless of their original position (Fig. 5B). In contrast, the address model predicts that more SBF will bind to those sequences that are present at the left half of URS2 in the native promoter, which was not seen. This result demonstrates that SBF binding at HO is critically regulated by proximity to the nucleosome remodeling events at URS1.
To completely rule out a requirement for sequences present in URS2-L for HO expression, one additional full-length URS2 construct was generated, URS2RR (Fig. 5A). The URS2RR construct replaces the left half of URS2 with an equivalent piece of sequence from the right half of URS2. This construct essentially duplicates the right half of URS2 at the expense of the left half of URS2, while maintaining the normal size of the entire URS2 region. HO expression from the URS2RR construct was at levels higher than the WT (Fig. 5A), indicating that the left half of URS2 is indeed dispensable for HO activation.
Conserved right-half SBF sites are required for normal expression of HO.
Our results indicate that proximity to URS1 is key for the nucleosome eviction that provides SBF access to its binding sites (14). However, this fails to explain why the additional SBF sites in URS2-R are present and are evolutionarily conserved (23).
To investigate this question, we replaced the URS2-R with an equivalent piece of “neutral” CDC39 sequence. Surprisingly, HO expression was greatly reduced in this construct (Fig. 6). This result suggests that SBF sites present in URS2-R are still required for their activating role despite preferential enrichment of SBF to URS2-L. As it was possible that the replacement of the right half of CDC39 removed sequences involved in the normal regulation of HO expression, we next made point mutations in the SBF sites in the right half of an otherwise native URS2, creating the RX4 sbf and RX5 sbf constructs (Fig. 6). These mutants demonstrated an even greater reduction in HO expression levels. These results, taken together with earlier results of SBF site mutations in URS2-L (Fig. 1), indicate that functional SBF sites are required in both halves of URS2 for HO promoter activation.
FIG 6.
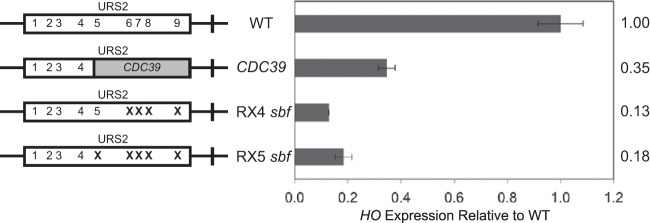
Right-half SBF sites are required for normal expression of HO. Diagrams on the left represent the URS2 portion of the HO promoter, and numbers within the rectangles indicate SBF site locations. Mutations in an SBF site are indicated by an X. CDC39 replacement is indicated by a gray block. HO mRNA levels were measured for strains with the indicated versions of the HO promoter.
Left-half SBF sites are required for SBF binding at the right half of URS2.
We have previously shown that SBF binds predominately to URS2-L (11). In this study, we have demonstrated that functional SBF sites are required in both halves of URS2 for effective promoter activation. To reconcile these two results, as well as better explain the mutual dependence of SBF sites in both halves of URS2 for HO expression, we examined SBF binding to URS2 mutants lacking either left-half or right-half SBF sites (Fig. 7A). In strains lacking the right-half SBF sites at URS2 (R4X sbf), ChIP assays showed that SBF binding to URS2-L was largely unaffected by the downstream site mutations. This result indicates that the preferential loading of SBF onto URS2-L is not dependent on sites present in URS2-R. However, as shown above, this promoter is defective for HO expression, indicating that SBF binding to URS2-L is not sufficient for promoter activation. SBF ChIP to the URS2 mutant lacking left-half SBF sites (LX5 sbf), however, revealed a large defect in SBF binding to URS2-R. This result demonstrates that SBF binding to URS2-R is dependent on upstream SBF binding to URS2-L. Taken together, these results suggest that SBF is initially loaded onto URS2-L due to proximity to URS1 and that this incipient binding is required for subsequent, albeit diminished or transient, downstream SBF binding and ultimate activation of HO.
FIG 7.

Left-half SBF sites are required for SBF binding to the right half of URS2. (A) Diagrams on the left represent the URS2 portion of the HO promoter, and numbers within the rectangles indicate SBF site locations, with the location of ChIP primers indicated. Mutations in an SBF site are indicated by red X's. Binding of the Swi4-V5 subunit of SBF to the indicated versions of the HO promoter was determined by ChIP followed by qPCR. (B) Cells containing the GALp::CDC20 allele were synchronized by galactose withdrawal and readdition. The 0-min time point represents the G2/M arrest, before release. Cells were harvested at the indicated time points, and samples were processed for both RNA and chromatin. HO expression was measured by RT-qPCR, and binding of the Swi4-V5 subunit of SBF was determined by ChIP followed by qPCR. The ChIP values for URS2-L enrichment, URS2-R enrichment, and HO expression make use of the left y axis. The URS2-L/URS2-R ratio is plotted against the right y axis, and this represents the ratio of URS2-L enrichment divided by URS2-R enrichment. Error bars reflect the standard deviations from two independent ChIP experiments from the same set of synchronized cells.
Early SBF enrichment occurs primarily at the left half of URS2.
The explanation proposed above predicts differences in the timing of SBF binding to sites in URS2. To test this, we measured SBF binding by ChIP using cells synchronized by GALp::CDC20 arrest and release, which typically results in peak HO expression at 40 min after release from G2/M (14). Samples were taken for ChIP every 5 min from the synchronized cells, and enrichment at URS2-L relative to that at URS2-R was determined. SBF binding at URS2-L and URS2-R had similar kinetic profiles; rising, peaking, and declining at approximately the same times (Fig. 7B). The two profiles are not identical, however, with much more rapid binding seen at the left half of URS2 than at the right half. This can be seen most clearly in the purple curve showing the ratio of URS2-L/URS2-R binding (Fig. 7B). This result supports a model of SBF loading onto URS2-L as a prerequisite event to URS2-R SBF binding and HO expression.
DISCUSSION
The HO gene is under complex regulation and has an unusually large promoter with two distinct regulatory regions, URS1 and URS2. Two DNA-binding factors, Swi5 and SBF, bind sequentially to URS1 and URS2, respectively, and successively recruit coactivators to both promoter regions (12, 14, 33–35). SBF is the ultimate activator of HO expression, and its ability to bind to URS2 is the major determinant of promoter activation. Here, we show that signals from the upstream URS1 region influence chromatin changes at URS2 but only at sites within a limited distance. We also show that there is a spatiotemporal cascade of SBF binding at URS2, with SBF bound at URS2-L relaying a signal from URS1 to the SBF sites in URS2-R, facilitating SBF binding here and ultimately activating transcription (Fig. 8).
FIG 8.
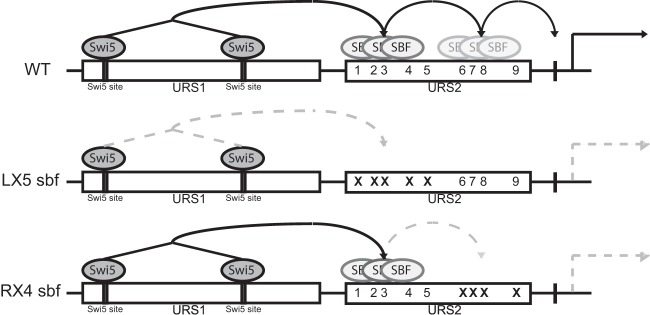
Model of mutual dependency of SBF binding. Diagrams of the HO promoter with rectangles indicating URS1 and URS2. Swi5 site locations in URS1 are indicated, and numbers indicate SBF sites in URS2. Swi5 and SBF transcription factors are represented by dark- and light-gray ovals, respectively. Signal propagation along the HO promoter is indicated by arrows. Generation of HO RNA is specified by the solid arrow, while the absence of production of HO expression is represented by dashed arrows. Top (wild type), predicted signal propagation in the native promoter. Swi5-dependent coactivator recruitment to URS1 allows for SBF loading onto the left half of URS2 via an unknown mechanism. Left-half SBF loading allows for downstream right-half URS2 SBF binding and ultimate activation of HO. Middle, loss of URS2-L SBF sites blocks URS2-L SBF loading and prevents downstream SBF signal propagation. Bottom, loss of URS2-R sites allows for URS2-L SBF loading but prevents the URS2-R SBF binding that is required for HO activation.
Our previous studies of HO revealed the puzzling conclusion that the nine SBF-binding sites in URS2 appeared to bind SBF equivalently in vitro and are all equally conserved, but only those adjacent to URS1 were highly occupied. Here, we used a variety of promoter manipulations to show that the major determinant of SBF site binding is proximity to URS1 and not some unrecognized feature of the distal sites (Fig. 4 and 5). Rather than having specific sequence elements, our results are consistent with events at URS1 stimulating nucleosome eviction, making the adjacent sites in URS2 a better “neighborhood” for efficient SBF binding. Proximal sites that displayed weak SBF binding were equally able to take advantage of URS1 to activate HO expression when placed in proximity to it, and SBF binding was lost for all of URS2 when it was placed farther away with DNA insertions.
These results explain the differential occupancy of SBF sites in URS2 but not the conservation of all sites, and our results with point mutations confirm that the SBF sites in URS2-R that are poorly occupied are nonetheless required for HO activation. This contradiction can be resolved by proposing that SBF binding at URS2-L is dependent on URS1 but independent of URS2-R and that binding to URS2-R is dependent on binding to URS2-L. Time course experiments strongly support this model, revealing pronounced enrichment of SBF during early promoter activation at URS2-L relative to the amount at URS2-R (Fig. 7B, URS2-L/URS2-R ratio). Taken together, these results suggest that URS1 creates a limited chromatin neighborhood conducive for SBF binding in URS2-L, and this is a prerequisite for SBF binding to URS2-R. This proximal binding is required for promoter activation but has lower occupancy and therefore produces lower signal in ChIP experiments than binding at URS2-L.
The molecular mechanism by which SBF binding to URS2-L stimulates binding to URS2-R remains an important area for future study. During the cell cycle, nucleosomes are evicted from the left half of URS1 before they are evicted from the right half (14). We speculate that sequential chromatin dynamics allow for SBF to be loaded onto left-half URS2 sites, and SBF at these left-half sites serves to recruit coactivators necessary to derepress the remainder of URS2 that allows the limited, but required, SBF binding at proximal sites that ultimately activates HO expression.
We note that SBF appears to have a limited window of promoter activation in other contexts as well, as most sites for genes activated by SBF are within a few hundred bases of the transcriptional start site (36). This is consistent with classic studies showing that yeast activators work at relatively short distances from core promoters (37, 38). It seems likely that SBF at the right-half sites recruits RNA Pol II for gene activation. We suggest that the left-half sites are too far from the TATA to activate transcription and instead function as recipients of the signal from URS1.
Regulation of the HO gene exhibits several unique features. One is that HO is expressed only in mother cells (39). The second distinctive feature is that two distinct DNA binding factors, Swi5 and SBF, are required for HO activation (7). The HO promoter has evolved such that both factors are necessary for activation; either one, alone, is inadequate. Swi5 and SBF are both cell cycle-regulated factors, but they function as activators at different times within the cell cycle. Swi5 activates in early M phase and SBF activates in late G1. Importantly, Swi5 acts before cell separation, while SBF acts after cells are separated. We suggest that this requirement of HO activators that function both before and after cell separation, along with the Ash1 repressive factor expressed in M phase that localizes primarily to daughter cells, is required to build a mother-specific promoter (7, 9–11).
Deleting the URS2 region completely results in a promoter that is activated by Swi5 in anaphase but is independent of SBF (32, 40). However, this promoter is expressed in daughter cells (10), and thus removing the dependence on both Swi5 and SBF eliminates the mother cell specificity. The promoter constructs with either the left half or the right half of URS2 deleted were still active (Fig. 3). However, there was significant residual expression of these promoters in an swi6 mutant lacking SBF. It is likely that this residual promoter activity is due to Swi5. Although Swi5 activates a downstream promoter element at HO, Swi5 functions as a traditional activator at other genes, where the Swi5 binding site is closer to the TATA element (29). In the native HO promoter, the closest Swi5 binding site is 1,200 bp from the TATA, but in these URS2-L and URS-R deletion constructs, Swi5 is 500 bp closer. The HO promoter shows strong evolutionary conservation, including the conserved SBF sites scattered throughout URS2. We suggest that evolution has selected a promoter with the URS2 region of sufficient size so that Swi5 is too far away from the TATA site to activate transcription.
To our knowledge, this is the first study to indicate that a single transcription factor can have two distinct roles in gene activation, depending on its location within a promoter. Significantly, it is the poorly occupied SBF sites in URS2-R that ultimately activate transcription, and thus factor enrichment does not always correlate with functional importance. Additionally, it is likely that the less occupied URS2-R sites would have been dismissed as inconsequential or background by current techniques such as ChIP-Seq. As such, these data sets should be used carefully, as such experiments may not always detect the relevant transcription factor binding sites functionally required for gene activation. Furthermore, many regulatory regions in higher eukaryotes contain multiple binding sites for a single transcription factor, and studies have suggested that this leads to cooperative DNA binding by the factor (41, 42). However, cooperative interactions are not required for gene activation (43). It seems possible that, like at HO, promoters in higher eukaryotes also contain multiple binding sites for which a single transcription factor can serve distinct roles in activation.
ACKNOWLEDGMENTS
We thank Tim Formosa, Emily Parnell, and Dean Tantin for comments on the manuscript and members of the Stillman lab for helpful advice throughout the course of this project. We thank Pam Geyer for plasmid pCH341 with a NotI site between HO URS1 and URS2, Francesca Storic for plasmid pCORE-UH, Zaily Connell and Tim Formosa for plasmid pZC09 for V5 epitope tagging, and Shinya Takahata for designing many of the primers used.
This work was supported by National Institutes of Health grant GM39067, awarded to D.J.S. R.M.Y. was supported by an NIH training grant (T32 DK007115).
REFERENCES
- 1.Li B, Carey M, Workman JL. 2007. The role of chromatin during transcription. Cell 128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 2.Adkins MW, Howar SR, Tyler JK. 2004. Chromatin disassembly mediated by the histone chaperone Asf1 is essential for transcriptional activation of the yeast PHO5 and PHO8 genes. Mol Cell 14:657–666. doi: 10.1016/j.molcel.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 3.Biddick RK, Law GL, Chin KK, Young ET. 2008. The transcriptional coactivators SAGA, SWI/SNF, and mediator make distinct contributions to activation of glucose-repressed genes. J Biol Chem 283:33101–33109. doi: 10.1074/jbc.M805258200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwabish MA, Struhl K. 2007. The Swi/Snf complex is important for histone eviction during transcriptional activation and RNA polymerase II elongation in vivo. Mol Cell Biol 27:6987–6995. doi: 10.1128/MCB.00717-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verdone L, Wu J, van Riper K, Kacherovsky N, Vogelauer M, Young ET, Grunstein M, Di Mauro E, Caserta M. 2002. Hyperacetylation of chromatin at the ADH2 promoter allows Adr1 to bind in repressed conditions. EMBO J 21:1101–1111. doi: 10.1093/emboj/21.5.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weake VM, Workman JL. 2010. Inducible gene expression: diverse regulatory mechanisms. Nat Rev Genet 11:426–437. doi: 10.1038/nrg2781. [DOI] [PubMed] [Google Scholar]
- 7.Stillman DJ. 2013. Dancing the cell cycle two-step: regulation of yeast G1-cell-cycle genes by chromatin structure. Trends Biochem Sci 38:467–475. doi: 10.1016/j.tibs.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nasmyth K. 1985. At least 1400 base pairs of 5′-flanking DNA is required for the correct expression of the HO gene in yeast. Cell 42:213–223. doi: 10.1016/S0092-8674(85)80117-3. [DOI] [PubMed] [Google Scholar]
- 9.Bobola N, Jansen RP, Shin TH, Nasmyth K. 1996. Asymmetric accumulation of Ash1p in postanaphase nuclei depends on a myosin and restricts yeast mating-type switching to mother cells. Cell 84:699–709. doi: 10.1016/S0092-8674(00)81048-X. [DOI] [PubMed] [Google Scholar]
- 10.Sil A, Herskowitz I. 1996. Identification of an asymmetrically localized determinant, Ash1p, required for lineage-specific transcription of the yeast HO gene. Cell 84:711–722. doi: 10.1016/S0092-8674(00)81049-1. [DOI] [PubMed] [Google Scholar]
- 11.Takahata S, Yu Y, Stillman DJ. 2011. Repressive chromatin affects factor binding at yeast HO (homothallic switching) promoter. J Biol Chem 286:34809–34819. doi: 10.1074/jbc.M111.281626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cosma MP, Tanaka T, Nasmyth K. 1999. Ordered recruitment of transcription and chromatin remodeling factors to a cell cycle- and developmentally regulated promoter. Cell 97:299–311. doi: 10.1016/S0092-8674(00)80740-0. [DOI] [PubMed] [Google Scholar]
- 13.Bhoite LT, Yu Y, Stillman DJ. 2001. The Swi5 activator recruits the Mediator complex to the HO promoter without RNA polymerase II. Genes Dev 15:2457–2469. doi: 10.1101/gad.921601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takahata S, Yu Y, Stillman DJ. 2009. FACT and Asf1 regulate nucleosome dynamics and coactivator binding at the HO promoter. Mol Cell 34:405–415. doi: 10.1016/j.molcel.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andrews BJ, Herskowitz I. 1989. The yeast SWI4 protein contains a motif present in developmental regulators and is part of a complex involved in cell-cycle-dependent transcription. Nature 342:830–833. doi: 10.1038/342830a0. [DOI] [PubMed] [Google Scholar]
- 16.Costanzo M, Nishikawa JL, Tang X, Millman JS, Schub O, Breitkreuz K, Dewar D, Rupes I, Andrews B, Tyers M. 2004. CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell 117:899–913. doi: 10.1016/j.cell.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 17.de Bruin RA, McDonald WH, Kalashnikova TI, Yates J III, Wittenberg C. 2004. Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell 117:887–898. doi: 10.1016/j.cell.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 18.Ho Y, Costanzo M, Moore L, Kobayashi R, Andrews BJ. 1999. Regulation of transcription at the Saccharomyces cerevisiae start transition by Stb1, a Swi6-binding protein. Mol Cell Biol 19:5267–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Bruin RA, Kalashnikova TI, Wittenberg C. 2008. Stb1 collaborates with other regulators to modulate the G1-specific transcriptional circuit. Mol Cell Biol 28:6919–6928. doi: 10.1128/MCB.00211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang H, Carey LB, Cai Y, Wijnen H, Futcher B. 2009. Recruitment of Cln3 cyclin to promoters controls cell cycle entry via histone deacetylase and other targets. PLoS Biol 7:e1000189. doi: 10.1371/journal.pbio.1000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang D, Kaluarachchi S, van Dyk D, Friesen H, Sopko R, Ye W, Bastajian N, Moffat J, Sassi H, Costanzo M, Andrews BJ. 2009. Dual regulation by pairs of cyclin-dependent protein kinases and histone deacetylases controls G1 transcription in budding yeast. PLoS Biol 7:e1000188. doi: 10.1371/journal.pbio.1000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takahata S, Yu Y, Stillman DJ. 2009. The E2F functional analogue SBF recruits the Rpd3(L) HDAC, via Whi5 and Stb1, and the FACT chromatin reorganizer, to yeast G1 cyclin promoters. EMBO J 28:3378–3389. doi: 10.1038/emboj.2009.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bai L, Ondracka A, Cross FR. 2011. Multiple sequence-specific factors generate the nucleosome-depleted region on CLN2 promoter. Mol Cell 42:465–476. doi: 10.1016/j.molcel.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas BJ, Rothstein R. 1989. Elevated recombination rates in transcriptionally active DNA. Cell 56:619–630. doi: 10.1016/0092-8674(89)90584-9. [DOI] [PubMed] [Google Scholar]
- 25.Rothstein R. 1991. Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Meth Enzymol 194:281–302. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- 26.Sherman F. 1991. Getting started with yeast. Meth Enzymol 194:3–21. doi: 10.1016/0076-6879(91)94004-V. [DOI] [PubMed] [Google Scholar]
- 27.Storici F, Lewis LK, Resnick MA. 2001. In vivo site-directed mutagenesis using oligonucleotides. Nat Biotechnol 19:773–776. doi: 10.1038/90837. [DOI] [PubMed] [Google Scholar]
- 28.Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953–961. [DOI] [PubMed] [Google Scholar]
- 29.Voth WP, Yu Y, Takahata S, Kretschmann KL, Lieb JD, Parker RL, Milash B, Stillman DJ. 2007. Forkhead proteins control the outcome of transcription factor binding by antiactivation. EMBO J 26:4324–4334. doi: 10.1038/sj.emboj.7601859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eriksson P, Biswas D, Yu Y, Stewart JM, Stillman DJ. 2004. TATA-binding protein mutants that are lethal in the absence of the Nhp6 high-mobility-group protein. Mol Cell Biol 24:6419–6429. doi: 10.1128/MCB.24.14.6419-6429.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sekinger EA, Moqtaderi Z, Struhl K. 2005. Intrinsic histone-DNA interactions and low nucleosome density are important for preferential accessibility of promoter regions in yeast. Mol Cell 18:735–748. doi: 10.1016/j.molcel.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 32.Breeden L, Nasmyth K. 1987. Similarity between cell-cycle genes of budding yeast and fission yeast and the Notch gene of Drosophila. Nature 329:651–654. doi: 10.1038/329651a0. [DOI] [PubMed] [Google Scholar]
- 33.Nasmyth K. 1985. A repetitive DNA sequence that confers cell-cycle START (CDC28)-dependent transcription of the HO gene in yeast. Cell 42:225–235. doi: 10.1016/S0092-8674(85)80118-5. [DOI] [PubMed] [Google Scholar]
- 34.Tebb G, Moll T, Dowser C, Nasmyth K. 1993. SWI5 instability may be necessary but is not sufficient for asymmetric HO expression in yeast. Genes Dev 7:517–528. doi: 10.1101/gad.7.3.517. [DOI] [PubMed] [Google Scholar]
- 35.McBride HJ, Brazas RM, Yu Y, Nasmyth K, Stillman DJ. 1997. Long-range interactions at the HO promoter. Mol Cell Biol 17:2669–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E. 2006. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics 7:113. doi: 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Struhl K. 1984. Genetic properties and chromatin structure of the yeast gal regulatory element: an enhancer-like sequence. Proc Natl Acad Sci U S A 81:7865–7869. doi: 10.1073/pnas.81.24.7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guarente L, Hoar E. 1984. Upstream activation sites of the CYC1 gene of Saccharomyces cerevisiae are active when inverted but not when placed downstream of the “TATA box.” Proc Natl Acad Sci U S A 81:7860–7864. doi: 10.1073/pnas.81.24.7860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nasmyth K. 1983. Molecular analysis of a cell lineage. Nature 302:670–676. doi: 10.1038/302670a0. [DOI] [PubMed] [Google Scholar]
- 40.Nasmyth K, Adolf G, Lydall D, Seddon A. 1990. The identification of a second cell cycle control on the HO promoter in yeast: cell cycle regulation of SWI5 nuclear entry. Cell 62:631–647. doi: 10.1016/0092-8674(90)90110-Z. [DOI] [PubMed] [Google Scholar]
- 41.Giniger E, Ptashne M. 1988. Cooperative DNA binding of the yeast transcriptional activator GAL4. Proc Natl Acad Sci U S A 85:382–386. doi: 10.1073/pnas.85.2.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carey M, Lin YS, Green MR, Ptashne M. 1990. A mechanism for synergistic activation of a mammalian gene by GAL4 derivatives. Nature 345:361–364. doi: 10.1038/345361a0. [DOI] [PubMed] [Google Scholar]
- 43.Xu HE, Kodadek T, Johnston SA. 1995. A single GAL4 dimer can maximally activate transcription under physiological conditions. Proc Natl Acad Sci U S A 92:7677–7680. doi: 10.1073/pnas.92.17.7677. [DOI] [PMC free article] [PubMed] [Google Scholar]