

Abstract
The cardiac conduction system coordinates electrical activation through a series of interconnected structures, including the atrioventricular node (AVN), the central connection point that delays impulse propagation to optimize cardiac performance. Although recent studies have uncovered important molecular details of AVN formation, relatively little is known about the transcriptional mechanisms that regulate AV delay, the primary function of the mature AVN. We identify here MyoR as a novel transcription factor expressed in Cx30.2+ cells of the AVN. We show that MyoR specifically inhibits a Cx30.2 enhancer required for AVN-specific gene expression. Furthermore, we demonstrate that MyoR interacts directly with Gata4 to mediate transcriptional repression. Our studies reveal that MyoR contains two nonequivalent repression domains. While the MyoR C-terminal repression domain inhibits transcription in a context-dependent manner, the N-terminal repression domain can function in a heterologous context to convert the Hand2 activator into a repressor. In addition, we show that genetic deletion of MyoR in mice increases Cx30.2 expression by 50% and prolongs AV delay by 13%. Taken together, we conclude that MyoR modulates a Gata4-dependent regulatory circuit that establishes proper AV delay, and these findings may have wider implications for the variability of cardiac rhythm observed in the general population.
INTRODUCTION
The cardiac conduction system is responsible for coordinating electrical activity in the vertebrate heart (1, 2). Each heartbeat is initiated in the sinoatrial node (SAN) and propagated through the atrioventricular node (AVN), His bundle, bundle branches, and Purkinje fibers. Specifically, the AVN connects the atria with the ventricles and delays impulse propagation to optimize cardiac performance (3). Environmental or genetic factors that perturb atrioventricular (AV) delay can impact a variety of common cardiovascular diseases, such as congestive heart failure, atrial fibrillation, and AV block (4). Therefore, detailed understanding of the transcriptional mechanisms controlling formation and function of the AVN will clarify how normal rhythm is established and pathological arrhythmias arise.
Human and mouse genetic studies have demonstrated that the transcription factors Tbx5 and Nkx2.5 function during development to specify lower AV nodal cells and promote their morphogenesis (5–10). Interestingly, while Tbx5 and Nkx2.5 are expressed broadly within the developing heart to activate transcription, the inhibitory T-box transcription factors Tbx2 and Tbx3 are confined to AV node progenitor cells and repress working cardiomyocyte-specific genes (i.e., Nppa and Cx43) by antagonizing Nkx2.5 (11–15). In addition, well-characterized signaling pathways influence these transcriptional mechanisms to refine AVN morphogenesis. While Notch signaling patterns early AVN development (16), the BMP pathway regulates subsequent AVN morphogenesis through Smad-dependent activation of Tbx2 (17–19). In zebrafish, Foxn4 has also been implicated in AVC patterning via direct activation of Tbx2b (20), but it remains uncertain whether Foxn4 functions similarly in higher vertebrates. Although these studies have begun to characterize pathways that impact early AVN progenitor specification, the transcriptional mechanisms that regulate AV delay, the primary function of the mature AVN, remain to be completely elucidated.
In order to gain insight into the mechanisms that regulate AVN-specific function, we focused on transcriptional regulation of Cx30.2/Gjd3, a gap junction protein required for normal AV delay in mice (21, 22). Cx30.2 expression is controlled by a distal upstream enhancer that directs expression to the AVN, and analysis of this element revealed that Gata4 activates Cx30.2 expression to establish normal AV delay (23). Given that Gata4 is broadly expressed within the heart and has numerous functions that are critical for normal cardiac development (24), how Gata4 regulates transcription in a tissue-specific manner remains incompletely understood. Tissue-enriched cofactors provide one potential mechanism for dictating such cell-type specificity. For example, the FOG1 and FOG2 proteins are cofactors that regulate GATA-dependent gene expression in specific tissues (25–29). Given that Gata4 plays a central and pervasive role in cardiac gene expression programs (30), we reasoned that additional Gata4-interacting cofactors must exist to regulate tissue-specific gene expression and hypothesized that an AVN-specific Gata4 cofactor could thus modulate its transcriptional activity. In the present study, we used the Cx30.2 enhancer element as a tool to identify novel transcriptional cofactors of Gata4-dependent gene expression in the AVN.
Transcription factors belonging to the beta helix-loop-helix (bHLH) family are prominent regulators of lineage commitment and tissue-specific gene expression (31). For example, the proneural bHLH transcription factors Mash1 and NeuroD turn on genes required for neuronal differentiation (32, 33). In skeletal muscle, a hierarchy of bHLH factors, including MyoD, myogenin, Myf5, and MRF4, orchestrate muscle differentiation (34). During heart formation, the bHLH proteins Hand1 and Hand2 are critical for morphogenesis of the left and right ventricle, respectively, while Twist1 has been implicated in outflow tract development (35, 36). Interestingly, Hand2, but not Hand1, has been shown to function as a Gata4 coactivator of heart-specific gene expression (37). Whether there exist additional cardiac tissue-enriched bHLH transcription factors that modulate Gata4 transcriptional activity, however, remains to be clarified.
MyoR (also called musculin in mice and ABF-1 in humans) is an inhibitory bHLH transcription factor that was identified based on its skeletal muscle enrichment (38–40), and combined deletion of MyoR and Tcf21/capsulin impairs formation of specific facial muscles necessary for mastication (41). Here, we show that MyoR is also expressed in the AVN of developing mice and represses Gata4-dependent activation of the Cx30.2 enhancer. We demonstrate that MyoR and Gata4 interact directly with one another, and we define their minimal interaction domains. Interestingly, MyoR binds to Gata4 via a protein domain that also binds to Tbx5, but each transcription factor utilizes a unique binding surface to interact with Gata4. We also show that MyoR contains two discrete repression domains that are required to inhibit transcriptional activity. In particular, the N-terminal MyoR repression domain can function in a heterologous context to convert Hand2 from an activator into a repressor. Consistent with our in vitro experiments, we find that genetic ablation of MyoR increases Cx30.2 expression and delays AV conduction. These results implicate the MyoR repressor in a Gata4-dependent transcriptional circuit that establishes normal cardiac conduction.
MATERIALS AND METHODS
Flow cytometry and microarray analysis.
All Cx30.2-lacZ mice used in the present study contain a 10.8-kb fragment of genomic DNA that encompasses the endogenous Cx30.2 enhancer fused to a minimal HSP promoter and lacZ coding sequence as described previously (23). Male Cx30.2-lacZ mice were crossed to female ICR mice to generate Cx30.2-lacZ hemizygous day 12.5 embryos, and transgene positive animals were identified by PCR genotyping by using lacZ primers. Embryonic hearts were dissected from individual embryos and placed into ice-cold phosphate-buffered saline (PBS), minced extensively, and digested in 0.25% trypsin (Invitrogen) for 30 min at 37°C with gentle trituration every 10 min. Digested cells were strained and resuspended in a minimal volume of Dulbecco modified Eagle medium (DMEM; Invitrogen) and incubated with an equal volume of 2 mM fluorescein digalactoside (Invitrogen) for 1 min at 37°C. This cell suspension was diluted 10-fold with ice-cold DMEM and placed on ice for 1 h prior to fluorescence-activated cell sorting (FACS) analysis. FACS was performed at the UT Southwestern Flow Cytometry Core Facility, and Cx30.2-β-Gal+ and Cx30.2-β-Gal− cells were sorted directly into TRIzol (Invitrogen) for RNA purification. Total RNA was submitted to the UT Southwestern Medical Center Microarray Core Facility, and microarray analysis was performed in triplicate for each sample using an Illumina Mouse-6 v1.1 BeadChip Array. Cx30.2-EGFP mice were generated in a similar fashion to the Cx30.2-lacZ transgenic line (23), except that an enhanced green fluorescent protein (EGFP) cassette replaced the lacZ reporter gene. Cx30.2-EGFP mice were processed like Cx30.2-lacZ mice, except that the AVC region was dissected prior to trypsin digestion and fluorescent labeling was not necessary. Reverse transcription-PCR (RT-PCR) analysis was performed with cDNA created by the oligo(dT) method (Invitrogen) and the indicated coding sequence primers using GoTaq polymerase (Promega, Madison, WI) in a MasterCycler EP gradient thermal cycler (Eppendorf, Westbury, NY) for 30 to 35 cycles.
Tissue microdissection and qPCR analysis.
For AVC dissection of embryonic day 10.5 (E10.5) hearts, the atria and ventricles were carefully removed. The remaining tissue, comprising the AVC, was trimmed and frozen in liquid nitrogen prior to RNA isolation and cDNA synthesis. Real-time PCR was performed as previously described (68) using gene-specific probes (ABI, Foster City, CA). PCRs were performed in an ABI Prism 7000 sequence detection system and analyzed using the ΔΔCT method using GAPDH for normalization according to the manufacturer's instructions. Each quantitative PCR (qPCR) was performed in triplicate and averaged before final values were calculated.
Section immunostaining.
E16.5 Cx30.2-EGFP mouse hearts were dissected in cold PBS, fixed in 4% paraformaldehyde for 30 min at 4°C, equilibrated in 30% sucrose, embedded in blocks containing tissue freezing medium (Electron Microscopy Sciences, Hatfield, PA), and snap-frozen prior to sectioning with a cryostat. Consecutive 8-μm sections were obtained, and the position of the AVN was identified based on anatomical landmarks from a survey using hematoxylin and eosin staining and Hcn4 immunostaining. Consecutive sections through the AVN were stained with GFP (Life Technologies, Grand Island, NY), Gata4 (Santa Cruz Biotechnology, Dallas, TX), MyoR (Santa Cruz Biotechnology), and Hcn4 (Alomone Labs, Jerusalem, Israel) primary antibodies, followed by detection with Alexa Fluor-labeled secondary antibodies (Life Technologies) according to the manufacturer's suggested protocol.
DNA constructs.
The Cx30.2-Luc reporter construct was generated by subcloning a 4.1-kb Cx30.2 enhancer fragment (−4.1 to 0.0 relative to the translation start site) using KpnI-XhoI into pGL2 E1b LUC, which contains a minimal TATA box, followed by the luciferase coding sequence. The Gata4, MyoR, and Tbx5 GST and His fusion constructs were generated by PCR amplification of their coding regions using primers containing BamHI and EcoRI linkers. Each coding sequence amplicon was digested and cloned directly into pGEX 6P1 (GE Healthcare Life Sciences, Pittsburgh, PA) or PRSET A (Life Technologies). Gal4-MyoR fusion eukaryotic expression constructs were created by PCR amplification of the appropriate fragment using primers containing EcoRI and SalI linkers. Digested MyoR amplicons were directly cloned into the pM vector (Clontech, Mountain View, CA). For chimeric MyoR-Hand2 constructs, PCR was performed on the N terminus of MyoR using reverse primers that contained an overlapping segment of the Hand2 sequence, and this amplicon was used as a megaprimer to amplify the C-terminal portion of Hand2; the Hand2-MyoR construct was created similarly. The outside primers for each chimeric construct contained EcoRI and SalI linkers that were used to clone directly into the pM vector.
Transient transfections.
Cotransfection experiments were carried out in COS7 cells with Fugene (Promega) according to the manufacturer's suggested protocol. Briefly, 100 ng of reporter plasmid was cotransfected with 30 to 100 ng of Gata4, MyoR, Hand1, LexA-VP16, or Gal4 fusion expression constructs. All experiments were carried out in triplicate at least three times. Reporter expression was normalized by cotransfection of a cytomegalovirus (CMV)-lacZ plasmid, and β-galactosidase activity was used to calculate relative luciferase units. The luciferase activity was determined at 48 to 72 h using a kit from Promega according to the manufacturer's recommendations. Expression of transfected fusion proteins was verified by Western blot analysis of cellular extracts.
Western blotting, coimmunoprecipitation, and ChIP assays.
Flag- and Myc-tagged expression plasmids were transfected into COS7 cells, and whole-cell lysate was prepared 72 h posttransfection. For Western blotting, whole-cell lysates were separated by SDS-PAGE and subjected to immunoblotting with a Myc antibody. Alternatively, lysates were immunoprecipitated with Flag-agarose (Sigma, St. Louis, MO) and washed, followed by SDS-PAGE and Western blotting. Experiments were performed on immunoprecipitated lysates alongside parallel input blots with Myc and Flag antibodies. For chromatin immunoprecipitation (ChIP) experiments, COS7 cells were transfected with the indicated eukaryotic expression constructs and enhancer templates. The cells were cross-linked, harvested, and sheared according to a published protocol (42). Sheared chromatin was immunoprecipitated with a Flag antibody (Sigma, Milwaukee, WI) that had been coupled to protein G-Dynabeads (Invitrogen). The immunoprecipitates were washed extensively, and associated genomic DNA was eluted and used as the template in qPCRs using Cx30.2 enhancer primers. For in vivo ChIP assays, pooled AVC and non-AVC tissue were isolated from E16.5 Cx30.2-lacZ embryos, snap-frozen, and pulverized prior to cross-linking. Preparation of sheared chromatin and subsequent immunoprecipitation were carried out essentially as described above except that a MyoR antibody (Santa Cruz Biotechnology) was used. All qPCRs were performed at least in triplicate, and the resulting average occupancy values were normalized to input and expressed as the fold enrichment relative to control. Sequential ChIP experiments were conducted essentially as previously described (43) using a Gata4 antibody (Santa Cruz Biotechnology) that was biotinylated with a commercially available kit (Thermo Scientific, Grand Island, NY) according to the manufacturer's suggested protocol. Sequentially immunoprecipitated fragments were interrogated by qPCR, and enhancer occupancy was calculated relative to mock immunoprecipitation controls and compared to the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) coding sequence.
GST pulldown assays.
Protein-protein interaction experiments were carried out using standard methods. Briefly, the indicated proteins were in vitro translated using a coupled transcription-translation kit (Promega) in a 50-μl reaction mixture containing [35S]methionine and other reagents recommended by the manufacturer. Beads containing normalized amounts of Gata4 or MyoR glutathione S-transferase (GST) fusion proteins, expressed and purified as described previously (44), were equilibrated in buffer A (50 mM NaCl, 20 mM Tris-HCl [pH 8.0], 0.05% NP-40, 0.25% bovine serum albumin [BSA], 1 mM phenylmethylsulfonyl fluoride, and 1 mM dithiothreitol) for 30 min at 4°C. Subsequently, 5 μl of in vitro-translated protein was added, and the reaction mixtures were incubated at 4°C for 2 h. For competition assays, highly purified recombinant His-MyoR was added during the incubation period. The beads were then washed three times with buffer A and once with buffer A without BSA, and bound 35S-labeled proteins were visualized on a 10 to 15% SDS-PAGE gel, stained and destained, submerged in fluorographic solution (Amplify; GE Healthcare Life Sciences), dried, and exposed overnight at −80°C.
Mouse breeding.
All animal procedures were approved by the Institutional Animal Care and Use Committee at UT Southwestern Medical Center. Cx30.2-lacZ transgenic and MyoR-null mice have been previously described (23, 38). Cx30.2-lacZ and Cx30.2-EGFP male mice were crossed with ICR female mice, and the vaginal plug was considered to be 0.5 days postcoitus (dpc). Similarly, for experiments involving embryonic heart microdissection, wild-type C57BL/6 males and females were crossed, and the vaginal plug was considered to be 0.5 dpc. To generate MyoR+/+; Cx30.2-lacZ/+ and MyoR−/−; Cx30.2-lacZ/+ animals, Cx30.2-lacZ/+ mice were first crossed with MyoR−/− mice to obtain Cx30.2-lacZ/+; MyoR+/− mice, which were intercrossed to generate the indicated strains. MyoR+/+ and MyoR−/− animals for qPCR and EKG analysis were generated by intercrossing MyoR+/− mice.
β-Galactosidase assays.
β-Galactosidase assays were performed as described previously (23). Briefly, embryos were removed at E16.5 and placed into ice-cold PBS, and AVCs were microdissected and flash-frozen in liquid nitrogen. Embryonic AVCs were subsequently lysed in 100 μl of passive lysis buffer (Promega) and homogenized by repeated passage through a pipette tip. Protein lysates were incubated on ice for 30 min with frequent agitation and were centrifuged to remove debris. Lysate (20 μl) was incubated with LacZ assay buffer (10 mM MgCl2, 50 mM β-mercaptoethanol, 67 mM Na3PO4 and 0.88 mg/ml ONPG [o-nitrophenyl-β-d-galactopyranoside]) for 1 h at 37°C prior to the addition of 1 ml of stop solution (1 M Na2CO3) and the A420 measurement. Reactions were performed in triplicate, and samples of the indicated genotypes were averaged together to generate the final results. A sample of each lysate (10 μl) was used for the BCA assay (Pierce, Rockford, IL) to determine protein concentrations and normalize the β-galactosidase activity of each sample.
EKG analysis.
Electrocardiograms (EKGs) were performed as previously described (23) on adult mice at 4 to 6 weeks of age anesthetized with 50 μg/kg of pentobarbital delivered by intraperitoneal injection. Mice were laid down on a heating pad to maintain core body temperature, and limb leads were placed subcutaneously. Lead II EKGs were recorded at a sampling rate of 4,000 Hz (ADInstruments, Colorado Springs, CO) at rest. EKG intervals were measured by using the LabChart software package (ADInstruments) by an investigator who was blinded to the genotypes of the mice. Comparisons in all experiments were made between MyoR−/− and wild-type age-matched littermates.
Statistics.
All data are expressed as means ± the standard errors of the mean. P values were determined by using the Student two-tailed t test, and statistical significance was considered for P <0.05.
RESULTS
Identification of MyoR as an AVN-enriched transcript.
We previously generated a transgenic Cx30.2-lacZ mouse line marking cells of the developing AVN (23). In order to identify genes enriched in the developing AVN, we performed microarray analysis on Cx30.2-lacZ+ cells at E12.5 (Fig. 1A). As expected, Cx30.2 transcripts were enriched 70-fold in isolated Cx30.2-lacZ+ cells, and several ion channel subunits known to be expressed in the AVN (e.g., Kcne1 and Cacna2d2) emerged at the top of our gene list (45), thereby confirming our cell isolation strategy (Fig. 1B). Moreover, Tbx2, Tbx3, and Tbx5, which are known transcriptional regulators of AVN specification and/or morphogenesis (2), were enriched in Cx30.2-lacZ+ cells. Unexpectedly, our microarray analysis also identified enrichment of MyoR, a transcriptional repressor known to regulate facial muscle formation (41), in Cx30.2-lacZ+ cells.
FIG 1.
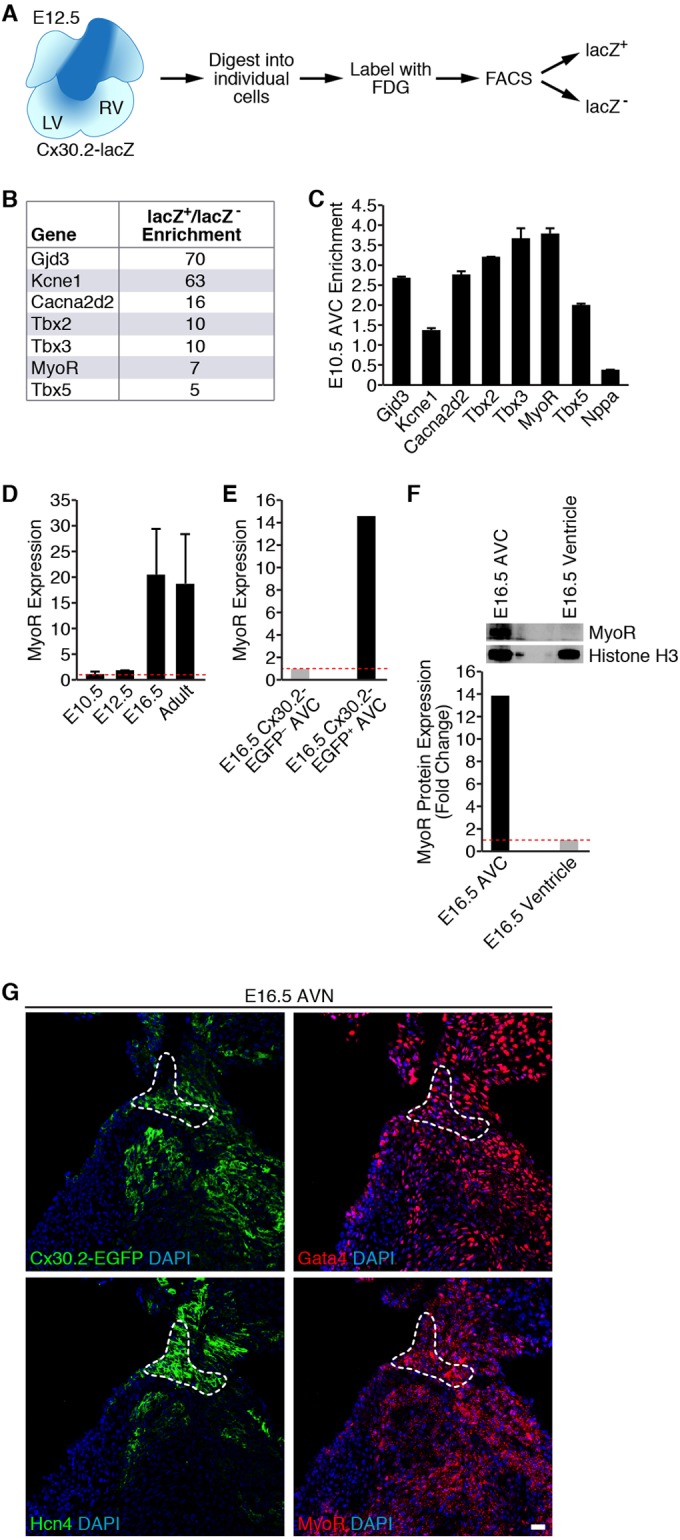
Transcriptional profiling of Cx30.2-lacZ+ cells identifies MyoR as an AVC-enriched transcript. (A) Strategy used for purifying E12.5 Cx30.2-lacZ+ AVC cells by flow cytometry. (B) Partial list of genes found to be enriched in microarray analysis. Fold enrichment refers to transcript levels in Cx30.2-lacZ+ cells relative to lacZ-null cells. Note that MyoR transcripts were 7-fold more abundant in Cx30.2-lacZ+ cells within the E12.5 heart. (C) qRT-PCR analysis of microdissected E10.5 AVC tissue demonstrating enrichment of MyoR. Fold enrichment refers to transcript levels in dissected AVC tissue versus whole heart. (D) Developmental analysis of MyoR expression by qRT-PCR analysis. MyoR expression in the whole heart at a particular time point relative to E10.5 is shown. MyoR transcripts were found to peak during late gestation (E16.5). (E) qRT-PCR analysis of MyoR expression level in Cx30.2-EGFP+ AVC cells isolated at E16.5. MyoR expression is shown relative to transcript levels in EGFP− AVC cells. (F) Western blot analysis of AVC and ventricular nuclear extracts dissected from E16.5 embryos (top) with the indicated antibodies. Band quantitation and normalization demonstrated ∼14-fold enrichment of MyoR in the E16.5 AVC relative to ventricular tissue. (G) Immunostaining of consecutive sections through an E16.5 Cx30.2-EGFP heart for GFP, Gata4, Hcn4, and MyoR, demonstrating localization of Cx30.2, Gata4, and MyoR to the Hcn4+ AVN (dashed outline). All sections were counterstained with DAPI to stain cell nuclei. Scale bar, 20 μm. LV, left ventricle; RV, right ventricle; FDG, fluorescein digalactopyranoside; AVC, atrioventricular canal; AVN, atrioventricular node.
Although the microarray results suggested AVN enrichment of MyoR expression, we wanted to validate these results using an alternative approach. Therefore, we manually dissected the AV canal (AVC) region of E10.5 hearts to isolate tissue containing AV nodal cells. Compared to the whole heart, gene expression analysis of E10.5 AVC tissue demonstrated enrichment of AVC-specific markers (Cacna2d2, Tbx2, and Tbx3) and depletion of the chamber-specific marker Nppa (15), suggesting that microdissection was successful (Fig. 1C). Importantly, MyoR expression was ∼3.5-fold enriched in E10.5 AVC, which compares favorably with the established regulator of AVN formation, Tbx3. Taken together, these results demonstrate that MyoR is expressed in the developing heart with particular enrichment in Cx30.2+ AV nodal cells.
To determine whether MyoR expression is developmentally regulated, we performed qPCR analysis on RNA prepared from whole hearts isolated from staged embryos and adults. Interestingly, MyoR expression increases during embryogenesis and peaks during late gestation with an ∼20-fold increase at E16.5 compared to E10.5 (Fig. 1D). Although MyoR expression reaches a maximum at E16.5, it is worth noting that Gata4 and Tbx5 are ∼70- and ∼10-fold more abundant than MyoR, respectively, at the same developmental time point (data not shown). Given that MyoR expression peaks at E16.5, we microdissected the AVC of E16.5 Cx30.2-EGFP and isolated Cx30.2-EGFP+ cells by flow cytometry for gene expression analysis. Using this approach, we found that MyoR was ∼14-fold enriched in Cx30.2-EGFP+ AV nodal cells compared to Cx30.2-EGFP− AVC cells at E16.5, confirming that MyoR is enriched in Cx30.2-EGFP+ cells just prior to birth (Fig. 1E).
To determine whether MyoR protein has a similar expression pattern, we dissected E16.5 AVC and the remaining non-AVC (atrial and ventricular) tissue for Western blot analysis. Consistent with our qRT-PCR analysis, MyoR protein was expressed in the E16.5 AVC ∼14-fold more than non-AVC tissue (Fig. 1F). We also performed immunostaining of Cx30.2-EGFP E16.5 heart sections to assess the in situ localization of MyoR protein in relation to Cx30.2, Gata4, and the AVN marker Hcn4. This experiment revealed that Cx30.2, Gata4, and MyoR are coexpressed in the Hcn4+ AVN (Fig. 1G). Interestingly, we found that MyoR localizes to both the nucleus and cytoplasm, raising the intriguing possibility that nucleocytoplasmic shuttling regulates MyoR transcriptional activity. Collectively, our gene and protein expression data demonstrate that MyoR is a novel AVN-enriched transcription factor and suggest that it may function within the AVN during late gestation.
MyoR inhibits Gata4-dependent Cx30.2 activation.
Given that MyoR is expressed in Cx30.2+ AV nodal cells, we hypothesized that it regulates Cx30.2 gene expression. Since Hand2, another bHLH family member, directly interacts with Gata4 to regulate cardiac gene transcription (37), we wondered whether MyoR similarly targets Gata4 to modulate Cx30.2 gene expression. In order to test this possibility, we performed transient-transfection analysis using a Cx30.2-Luc reporter construct (Fig. 2A) and various combinations of Gata4, MyoR, and Hand1 (Fig. 2B). As expected, Gata4 activated Cx30.2 reporter expression ∼3.5-fold (Fig. 2B), while the addition of MyoR almost completely abolished Gata4-dependent gene activation. In contrast, Hand1, which does not interact with Gata4 (37), does not repress Gata4-dependent activation of the Cx30.2 reporter, suggesting that repression is specific to MyoR rather than a general property of bHLH factors in this system. Thus, these results demonstrate that MyoR inhibits Gata4-dependent transcription of a Cx30.2 reporter gene, which is consistent with previous experimental evidence showing that MyoR is a transcriptional repressor (38, 40).
FIG 2.
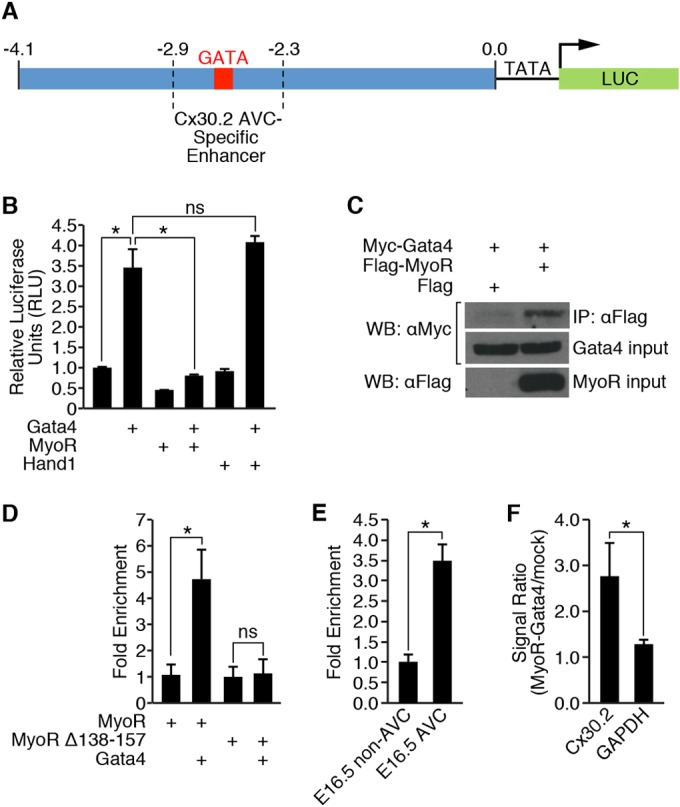
MyoR inhibits Gata4-dependent activation of the Cx30.2 enhancer. (A) Schematic depiction of the Cx30.2-Luc reporter construct used in panel B that contains the Cx30.2 enhancer fused to a minimal TATA box and luciferase coding sequence. The 4.1-kb enhancer fragment encompasses the −2.9/−2.3 region that contains the minimal AVC-specific Cx30.2 enhancer (23). (B) Transient-transfection analysis in COS cells. A total of 30 ng of Cx30.2-Luc reporter was transfected, along with 100 ng of Gata4 and/or 100 ng of bHLH transcription factor (MyoR or Hand1). A 10-ng portion of CMV-lacZ was also cotransfected as an internal control. MyoR repressed Gata4-dependent transcriptional activity driven by Cx30.2-Luc, while Hand1, another bHLH transcription factor expressed in the heart, did not. (C) Coimmunoprecipitation analysis between MyoR and Gata4. Myc-Gata4 and either Flag vector alone or Flag-MyoR were cotransfected into COS7 cells. Whole-cell lysates were precipitated with a Flag antibody and probed with a Myc antibody (top panel). A 10% input was also run separately and probed with either a Myc antibody (middle panel) or a Flag antibody (lower panel). Myc-Gata4 was only detected after Flag immunoprecipitation in the presence of Flag-MyoR but not Flag alone. (D) A ChIP experiment was performed in COS7 cells transfected with a Cx30.2 enhancer template and the indicated proteins. MyoR associated with the Cx30.2 enhancer in a Gata4-dependent manner, but the association with the enhancer was strongly diminished in the presence of a MyoR mutant (MyoRΔ138-157) that does not bind to Gata4. (E) MyoR ChIP from dissected E16.5 AVC and non-AVC (atrial and ventricular) tissue showed that MyoR preferentially associates with the Cx30.2 enhancer in AVC tissue compared to non-AVC tissue. (F) Sequential MyoR and Gata4 ChIP experiment from E16.5 heart tissue demonstrated that both MyoR and Gata4 simultaneously associate with the Cx30.2 enhancer in vivo. *, P < 0.05; ns, not significant.
Since MyoR was able to repress Gata4-dependent transcriptional activity, we also wondered whether MyoR and Gata4 could interact with one another. Therefore, we performed coimmunoprecipitation analysis using a Flag antibody on cells transfected with Myc-Gata4, along with empty vector or Flag-MyoR (Fig. 2C). This experiment showed that Gata4 was immunoprecipitated with a Flag antibody in a MyoR-dependent fashion, demonstrating that these two proteins do interact. Since MyoR interacts with Gata4 to repress Cx30.2 transcriptional activation, we also wanted to evaluate whether MyoR could associate with Gata4 on the Cx30.2 enhancer. Therefore, we performed ChIP assays on cells overexpressing MyoR alone or in combination with Gata4. Although MyoR weakly associated with the Cx30.2 enhancer in the absence of Gata4, we observed augmentation of MyoR binding (∼4.5-fold) in the presence of Gata4 (Fig. 2D). This result demonstrates that MyoR associates with the Cx30.2 enhancer and that this interaction is largely Gata4 dependent. Taken together, our results reveal that MyoR interacts with Gata4 on the Cx30.2 enhancer to repress Cx30.2 gene activation and suggest that MyoR mediates its transcriptional effects by interacting directly with Gata4.
While these experiments demonstrate that MyoR can associate with the Cx30.2 enhancer, they do not address whether this interaction takes place in the developing heart. Therefore, we dissected E16.5 AVC and non-AVC tissue to perform ChIP experiments using a MyoR antibody. Consistent with our in vitro data, MyoR associated more strongly with the Cx30.2 enhancer in the AVC relative to the atria and ventricles (Fig. 2E). Furthermore, sequential ChIP experiments with MyoR and Gata4 antibodies demonstrated that both MyoR and Gata4 simultaneously occupy the Cx30.2 enhancer in vivo (Fig. 2F). Based on these results, we conclude that MyoR and Gata4 cooccupy the Cx30.2 enhancer in AVC tissue, and these findings are consistent with both MyoR enrichment in the AVN and its proposed role in AVN gene expression.
MyoR and Gata4 interact directly.
Previous work has demonstrated that Hand2, but not Hand1, functions as a Gata4 coactivator due to its unique ability to interact directly with Gata4 (37). Given the tight link between Gata4 interaction and the functional impact of Hand2, we speculated that MyoR inhibits Gata4 via direct protein-protein interaction. To test this notion, we carried out GST pulldown assays using bead-bound GST-Gata4 protein and the 35S-labeled MyoR C-terminal deletions shown in Fig. 3A. This experiment demonstrated that full-length MyoR interacts directly with Gata4 (Fig. 3B, lane 1) in the absence of additional cellular cofactors. Furthermore, while C-terminal deletion of MyoR to amino acid residue 157 (construct 1-157) did not affect its interaction with Gata4 (Fig. 3B, lane 2), deletion of an additional 19 residues (construct 1-138) abolished this interaction (Fig. 3B, lane 3), suggesting that amino acids 138 to 157 of MyoR are necessary to bind Gata4; these residues correspond to the second helix (H2) of the MyoR bHLH domain. Notably, none of the other C-terminal deletion mutant MyoR proteins bound directly to GST-Gata4 (Fig. 3B, lanes 4 to 6). To confirm these results, we created an internal deletion mutant of MyoR (1-201Δ138-157) that lacked the H2 segment. Although full-length MyoR was able to bind directly to Gata4, the internal deletion mutant was unable to do so (Fig. 3B, lanes 7 to 8). To further substantiate this finding, we performed ChIP assays in cells overexpressing a MyoR mutant devoid of its Gata4-interaction domain (Δ138-157) alone or in combination with Gata4. Although full-length and mutant MyoR both bound weakly to the Cx30.2 enhancer, the interaction of MyoRΔ138-157 with DNA was not augmented in the presence of Gata4 (Fig. 2D). Altogether, these results demonstrate that MyoR interacts directly with Gata4 via helix 2 of its bHLH domain and support the notion that this interaction is required for association of MyoR with the Cx30.2 enhancer.
FIG 3.
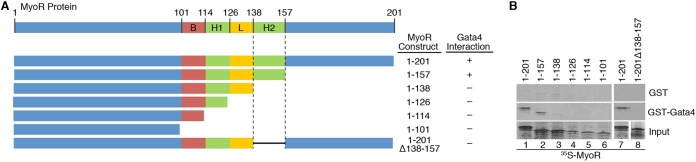
Gata4 interacts directly with the H2 domain of MyoR. (A) Schematic diagram of the MyoR deletion proteins used in GST pulldown experiments with summarized Gata4 interaction data. (B) In vitro-translated 35S-labeled MyoR proteins were incubated with either GST alone or GST-Gata4 to test for direct interaction. Input proteins are shown in the bottom panel. Full-length MyoR and a C-terminal deletion to residue 157 interact directly with Gata4 (lanes 1 and 2). MyoR C-terminal deletion beyond amino acid 157 or internal deletion of residues 138 to 157 abrogates interaction with Gata4 (lanes 3 to 8), demonstrating that helix 2 (H2) of the bHLH domain mediates direct contact with Gata4. B, basic domain; H1, helix 1 domain; L, loop domain; H2, helix 2 domain.
In addition to identifying the Gata4-binding domain of MyoR, we wanted to map the reciprocal MyoR interaction surface on Gata4. Thus, we performed GST pulldown assays with bead-bound GST-MyoR and the 35S-labeled Gata4 deletion constructs depicted in Fig. 4A. Again, we confirmed that GST-MyoR interacts directly with full-length Gata4 (Fig. 4B, lane 1). Deletion of the C-terminal 68 residues of Gata4 (1–332) was dispensable for binding to MyoR (Fig. 4B, lane 2), but removal of the next 38 residues or more (constructs 1-294, 1-260, and 1-177) severely diminished this interaction (Fig. 4B, lanes 3 to 5). Additional Gata4 deletion proteins that retained residues 294 to 332 (177-332, 177-332Δ265-294, 1-332Δ265-294, and 1-332Δ178-261) were still able to bind MyoR (Fig. 4B, lanes 6 to 9), suggesting that the Gata4 protein domain just C-terminal to the second zinc finger is necessary for this interaction. To further confirm these findings, we generated a Gata4 internal deletion mutant protein lacking the presumptive MyoR interaction domain (1-440Δ294-332) and performed a GST pulldown assay with full-length MyoR. As expected, this experiment showed that a mutant Gata4 lacking residues 294 to 332 failed to interact with MyoR (Fig. 4B, lanes 11 and 12). Taken together, these studies demonstrate that MyoR interacts directly with Gata4 via amino acid residues 294 to 332, which comprise a domain with unknown structure lying adjacent to its C-terminal zinc finger.
FIG 4.

MyoR interacts directly with Gata4 adjacent to the CZF and separate from the Tbx5 binding surface. (A) Schematic diagram of the Gata4 deletion proteins used in GST pulldown experiments with summarized MyoR interaction data. (B) In vitro-translated 35S-labeled Gata4 proteins were incubated with either GST alone (upper panel) or GST-MyoR (middle panel) to test for direct interaction. Input proteins are shown in the lower panel. Full-length Gata4 and deletions that preserve residues 294 to 332 interact directly with MyoR (lanes 1 and 2 and lanes 6 to 9). C-terminal deletion of Gata4 beyond amino acid 332 or internal deletion of residues 294 to 332 eliminates interaction with MyoR (lanes 3 to 5 and 12), showing that the region adjacent to the C-terminal zinc finger (CZF) is responsible for interacting with MyoR. In addition, the Gata4 G295S mutant retains the ability to interact with MyoR (lane 10), suggesting that MyoR and Tbx5 contact Gata4 via distinct binding surfaces. (C) GST pulldown competition assay with 35S-labeled Gata4 interacting with GST, GST-Tbx5, or GST-MyoR in the presence or absence of unlabeled His-tagged MyoR recombinant protein (lanes 1 to 5). Although unlabeled MyoR competes efficiently with GST-MyoR for Gata4 interaction (lanes 4 and 5), it is unable to prevent binding between GST-Tbx5 and Gata4 (lanes 2 and 3), providing additional evidence that the binding of Gata4 to MyoR or Tbx5 is not mutually exclusive. TAD, transactivation domain; NZF, N-terminal zinc finger; CZF, C-terminal zinc finger.
MyoR and Tbx5 bind to unique sites on Gata4.
Although the MyoR interaction site on Gata4 (positions 294 to 332) does not belong to any known protein domain family, this region has been shown previously to interact with Tbx5 (46). A specific GATA4 point mutation (G296S) segregates with autosomal dominant atrial septal defects (ASDs) and disrupts its interaction with Tbx5 (46). This observation provided strong genetic and biochemical evidence for transcriptional synergy between Gata4 and Tbx5 during cardiac morphogenesis. Interestingly, we have demonstrated that Gata4 and Tbx5 coordinately activate the Cx30.2 enhancer (23), suggesting that MyoR may inhibit Gata4-dependent transcriptional activation by perturbing the interaction between Gata4 and Tbx5. Given that Tbx5 interacts with Gata4 via a critical protein-protein interaction domain centered on the orthologous mouse amino acid residue 295, we wanted to test whether MyoR inhibits Cx30.2 transcription by competing with Tbx5 for interaction with Gata4. Since the G295S mutation abrogates the ability of Gata4 to interact with Tbx5 (46), we hypothesized that this mutation alters a common Gata4 binding surface utilized by Tbx5 and MyoR. Surprisingly, the Gata4 G295S mutant protein retained the ability to bind MyoR (Fig. 4B, lane 10), suggesting that Tbx5 and MyoR interact with distinct binding surfaces on Gata4. To confirm this result, we took the complementary approach of performing a GST pull-down competition assay. We first added 35S-labeled full-length Gata4 to bead-bound GST, GST-Tbx5, or GST-MyoR, confirming that Gata4 interacts directly with Tbx5 and MyoR (Fig. 4C, lanes 1, 2, and 4). To test whether MyoR and Tbx5 compete for a common Gata4 binding site, we added unlabeled recombinant MyoR to each binding reaction. Although the addition of unlabeled MyoR displaced 35S-labeled Gata4 (35S-Gata4) from bead-bound GST-MyoR (Fig. 4C, lanes 4 and 5), it was unable to displace 35S-Gata4 from GST-Tbx5 (Fig. 4C, lanes 2 and 3), supporting the notion that Tbx5 and MyoR bind to distinct surfaces of Gata4. Taken together, our results strongly suggest that direct competition with Tbx5 is not a major mechanism by which MyoR represses Gata4-dependent transcriptional activity.
MyoR contains two distinct repression domains.
Although previous studies have established that MyoR is a transcriptional repressor (38, 40), the mechanism by which MyoR inhibits transcription remains unknown. Given that MyoR does not appear to inhibit Cx30.2 expression by competing with Tbx5 for binding to Gata4, we sought to test whether MyoR contains a discrete repression domain. To address this question, we used a trans-repression assay (Fig. 5A) using a synthetic promoter bearing adjacent LexA and GAL4 binding sites driving luciferase (L8G5-Luc) expression. As expected, transfection of LexA-VP16 resulted in robust activation of the L8G5-Luc reporter (Fig. 5B). When Gal4-MyoR was cotransfected with LexA-VP16, we observed potent transcriptional repression, suggesting that MyoR possesses a trans-acting repression domain. In order to map the putative MyoR repression domain, we generated several Gal4 MyoR fusion proteins (Fig. 5B). Deletion of the N-terminal 75 residues (Gal4-MyoR 75-201) had a negligible effect on the ability of Gal4-MyoR to inhibit transcriptional activation by LexA-VP16. Similarly, deletion of the C-terminal 44 residues (Gal4-MyoR 1-157) did not significantly impair the ability of Gal4-MyoR to repress LexA-VP16, suggesting that residues 75 to 157 contain the MyoR repression domain(s). Consistent with these findings, Gal4-MyoR 75-157 retained the ability to repress LexA-VP16-dependent activation. Further deletion of residues 138 to 157 (Gal4-MyoR 75-138) or 75 to 100 (Gal4-MyoR 101-157) decreased, but did not completely abolish, the ability of Gal4-MyoR to repress transcriptional activation by LexA-VP16. These results indicate that MyoR contains two separate trans-repression domains. To confirm and extend these findings, we generated a Gal4-MyoR construct lacking both putative repression domains (75 to 100 and 138 to 157) and assessed its ability to repress LexA-VP16-dependent transcriptional activation; this MyoR mutant protein was unable to repress activation by LexA-VP16. Taken together, these results demonstrate that MyoR contains two discrete repression domains comprised of residues 75 to 100 and 138 to 157 that are both required for transcriptional inhibition.
FIG 5.

MyoR requires two nonequivalent repression domains to inhibit transcription. (A) Diagram depiction of the trans-repression assay system used for these experiments. The system utilizes a synthetic reporter construct containing eight LexA binding sites adjacent to five Gal4 binding sites. The LexA DNA-binding domain is fused with the HSV VP16 transactivation domain to mediate potent transcriptional activation. Candidate repressors are fused to the Gal4 DNA-binding domain and assessed for inhibitory activity in transient-transfection experiments. (B) A total of 100 ng of Lex8-Gal5-Luc was cotransfected with 100 ng of LexA-VP16 and 200 ng of the indicated Gal4-MyoR deletion construct. Also, 10 ng of CMV-lacZ was transfected for normalization purposes. The data are represented as the fold activation compared to LexA-VP16 alone (100%). N-terminal deletion to amino acid 75 or C-terminal deletion to residue 157 does not impair the ability of Gal4-MyoR to repress transcription by LexA-VP16. C-terminal deletion to residue 138 or N-terminal deletion to residue 100 partially impairs Gal4-MyoR repression. However, deletion of both putative repression domains (75-100 and 138-157) completely eliminates the ability of Gal4-MyoR to inhibit LexA-VP16-dependent transcriptional activation. (C) Assessment of repression activity by various Hand2-MyoR chimeric Gal4 constructs. As expected, Gal4-MyoR represses and Gal4-Hand2 activates LexA-VP16 mediated transcriptional activity. A Gal4 fusion protein containing MyoR 1-100, which includes the N-terminal repression domain (75-100), and the C terminus of Hand2, potently repressed transcriptional activation mediated by LexA-VP16. In contrast, a Gal4 fusion protein containing the Hand2 N terminus fused with the MyoR C terminus, including the C-terminal repression domain, was unable to inhibit LexA-VP16 activation of reporter gene expression. (D) Protein alignment between amino acids 75 to 100 of MyoR and the corresponding region of Hand2 with conserved residues shaded black and similar residues shaded gray. Based on the alignment, MyoR contains a short peptide sequence (KKPLP) that is not present in Hand2.
The N terminus of MyoR converts Hand2 into a repressor.
The bHLH family comprises a diverse set of transcription factors that can activate and/or repress transcription (31). Hand1 and Hand2 are expressed in the developing heart and play crucial roles during ventricular morphogenesis (35). Although Hand2 binds Gata4 to cooperatively activate transcription on several cardiac promoters (37), Hand1 does not interact with Gata4 and has no effect on Gata4-dependent transcription (Fig. 2B). Since MyoR and Hand2 are bHLH family members with opposing effects on Gata4-dependent transcription, we could directly test additional characteristics of the MyoR repression domains. Thus, we generated chimeric MyoR-Hand2 proteins to assess (i) the ability of both MyoR repression domains to function in a heterologous context and (ii) the functional equivalence of the two domains. Using the L8G5-Luc reporter, Gal4-MyoR potently inhibited transcription mediated by LexA-VP16, while Gal4-Hand2 augmented LexA-VP16 transcriptional activity (Fig. 5C), a finding consistent with the notion that Hand2 functions as a transcriptional activator (37). However, when the N terminus of MyoR (domain 1-100) was fused to the bHLH domain and C terminus of Hand2 (domain 97-214), this Gal4 fusion protein repressed LexA-VP16-dependent activation. Conversely, when the N terminus of Hand2 (domain 1-97) was fused to the bHLH domain and C terminus of MyoR (domain 101-201), this Gal4 fusion protein augmented transcriptional activity mediated by LexA-VP16. These experiments suggest that the ability of MyoR and Hand2 to function as a repressor and activator, respectively, is determined by unique sequences contained within the N terminus of each protein. Given that residues 75 to 100 constitute the minimal repression domain within the MyoR N terminus, we searched for amino acids in this region that diverge from Hand2. Sequence alignment of this region between MyoR and Hand2 revealed several unique residues, including a Proline-rich motif (KKPLP) that is only present in MyoR (Fig. 5D). Collectively, these studies demonstrate that the MyoR N-terminal repression domain (domain 75-100), but not the C-terminal repression domain (domain 138-157), can function in a heterologous context and is sufficient to convert Hand2 into a repressor.
MyoR regulates Cx30.2 expression in vivo.
Given that MyoR is enriched in the AVN (Fig. 1) and directly inhibits Gata4-dependent Cx30.2 expression in cell culture (Fig. 2B), we wished to evaluate whether MyoR regulates Cx30.2 expression in vivo. To address this question, we bred Cx30.2-lacZ transgenic mice with MyoR−/− animals (41) to obtain MyoR+/−; Cx30.2-lacZ/+ double-transgenic mice. Then, double-transgenic mice were bred with MyoR+/− animals to generate MyoR+/+; Cx30.2-lacZ/+ and MyoR−/−; Cx30.2-lacZ/+ littermate mice to compare lacZ expression in a wild-type versus MyoR-null background. When we performed whole-mount X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) staining on E16.5 hearts from these mice, we did not detect any obvious changes in the pattern of Cx30.2-lacZ expression (data not shown), demonstrating that MyoR does not alter the spatial distribution of Cx30.2-lacZ transcripts. However, this experiment did not address whether MyoR influences the absolute levels of Cx30.2-lacZ expression, since whole-mount X-Gal staining is qualitative when performed under saturating conditions. Therefore, we conducted colorimetric determination of β-galactosidase activity in the AVC region under linear assay conditions to obtain a more quantitative assessment of lacZ expression (23). Using this approach, we found that the AVC of MyoR−/−; Cx30.2-lacZ/+ mice contained ∼2-fold more X-Gal activity compared to the AVC of MyoR+/+; Cx30.2-lacZ/+ mice at E16.5 (Fig. 6A), an observation consistent with the notion that MyoR represses Cx30.2 enhancer activity in vivo. Although this experiment suggests that MyoR functions as an AVN-specific transcriptional repressor in vivo, it does not address whether MyoR directly inhibits endogenous Cx30.2 gene expression. Thus, we compared Cx30.2 expression levels in the AVC of MyoR-null mice compared to wild-type littermates (Fig. 6B). This experiment demonstrated that Cx30.2 expression was elevated in MyoR-null mice, although the results were not as dramatic as our findings with the Cx30.2-lacZ transgenic line. Taken together, these studies demonstrate that MyoR functions in vivo to repress Cx30.2 enhancer activity and endogenous Cx30.2 expression.
FIG 6.
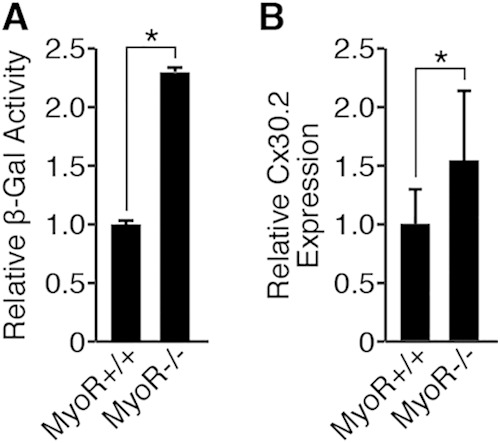
MyoR represses Cx30.2 expression in vivo. (A) The AVC region of MyoR+/+; Cx30.2-lacZ/+ (n = 3) and MyoR−/−; Cx30.2-lacZ/+ (n = 2) littermates was dissected at E16.5, and lysates were assessed for β-galactosidase activity as a quantitative measure of Cx30.2-lacZ expression. Enzyme activity is shown relative to MyoR+/+; Cx30.2-lacZ/+ AVC levels. (B) Endogenous Cx30.2 expression was measured by qRT-PCR in the E16.5 AVC of MyoR+/+ (n = 10) and MyoR−/− (n = 10) animals. Cx30.2 gene expression is shown relative to the AVC of MyoR+/+ mice. *, P < 0.05.
MyoR modulates cardiac electrical activity.
We previously demonstrated that Gata4 heterozygous mice have shortened PR intervals as a result of reduced Cx30.2 expression, suggesting that a Gata4-dependent regulatory circuit regulates AV conduction delay (23). Given our results implicating MyoR in Gata4-dependent regulation of Cx30.2, we wished to determine whether MyoR also impacts AV conduction delay. Since MyoR represses Gata4, we hypothesized that MyoR-null mice would have a prolonged PR interval due to increased Cx30.2 expression; this would contrast with the shortened PR interval observed in mice with reduced Cx30.2 expression due to Gata4 haploinsufficiency (23) or genetic ablation (21). To test this hypothesis, we interbred MyoR+/− mice to generate MyoR+/+ and MyoR−/− mice for EKG analysis (Fig. 7A). From these experiments, we observed an increase in the PR interval of MyoR-null mice compared to wild-type littermates (39.6 ± 1.1 ms versus 44.6 ± 2.0 ms; P < 0.05) (Fig. 7B), thus indicating prolonged AV conduction. Importantly, none of the other EKG parameters, including heart rate (HR), QRS interval, and QT interval, differed significantly between the two groups of mice. Interestingly, one MyoR-null mouse exhibited more severe AV conduction defects, such as Mobitz I and Mobitz II second degree AVB (data not shown), which were never observed in wild-type littermate mice. Collectively, these studies demonstrate that MyoR modulates AV conduction delay.
FIG 7.
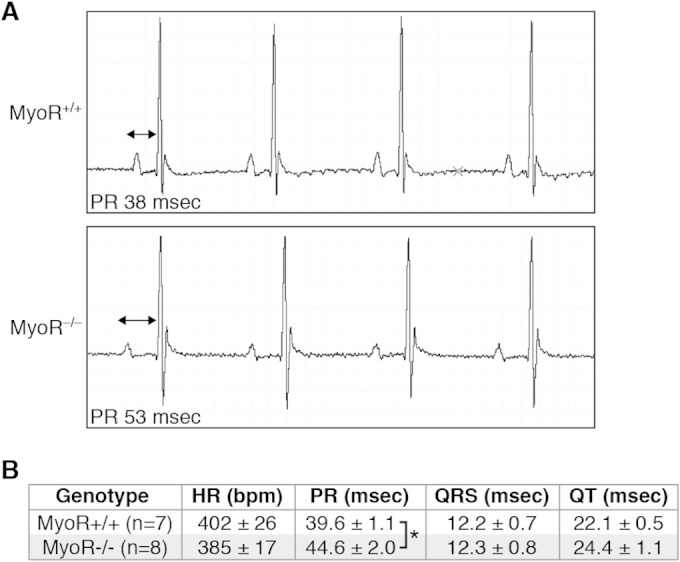
MyoR modulates AV delay. (A) EKG analysis was performed on 4-week-old MyoR+/+ and MyoR−/− mice, and representative tracings from heart rate-matched littermates are shown. Note the mild prolongation in the PR interval (double-headed arrow) of MyoR−/− mice compared to MyoR+/+ mice. (B) Summary of EKG analysis on MyoR+/+ (n = 7) and MyoR−/− (n = 8) mice. The PR interval is prolonged in MyoR−/− mice compared to MyoR+/+ mice, while the HR, QRS interval, and QT interval are similar between groups. *, P < 0.05.
DISCUSSION
Here we show that MyoR is expressed in the developing AVN and represses Gata4-dependent activation of the Cx30.2 enhancer. We demonstrate that MyoR directly interacts with Gata4 via a protein domain that also binds to Tbx5, but each transcription factor utilizes a unique binding surface. We also show that MyoR contains two discrete repression domains, and the N-terminal repression domain can function in a heterologous context to convert Hand2 into a repressor. Consistent with these results, we find that genetic ablation of MyoR increases Cx30.2 expression and delays AV conduction, thus implicating MyoR in a Gata4-dependent transcriptional circuit that establishes normal cardiac rhythm.
Pharyngeal mesoderm links MyoR to heart development.
Previous studies have demonstrated that deletion of MyoR and capsulin results in complete absence of specific muscles of mastication (41). Consistent with this phenotype, MyoR is developmentally expressed in splanchnic pharyngeal mesoderm (SPM), which contains cranial skeletal muscle precursor cells (47–49). Interestingly, the SPM also contains the second heart field (SHF), which contributes cells to the inflow tract (IFT), SAN, atria, right ventricle, outflow tract, and AVN (50). In fact, mounting experimental evidence supports the novel idea that a common subset of precursor cells within the SPM contributes to both the muscles of mastication and SHF-derived structures within the heart (47). Interestingly, MyoR transcripts have been previously detected in the SHF-derived sinus venosus/IFT region of the developing chick heart (51). In the present study, we provide evidence that MyoR functions within the mouse AVN, a structure that is also derived in part from the SHF. Perhaps not unexpectedly, MyoR thus functions within two distinct derivatives of the SPM, the muscles of mastication and the AVN. The implication for such functional duality is that MyoR must be capable of executing unique tissue-specific transcriptional programs depending upon the developmental context.
Individual transcription factors can potentially activate multiple gene expression programs by cooperating with tissue-specific cofactors. Although Gata4 is abundant within the heart, for example, it is not known to be expressed in the developing facial musculature. Therefore, we postulate that Gata4 serves as a heart-specific cofactor that directs MyoR-dependent gene regulation within the heart. Likewise, MyoR may serve as a Gata4 cofactor to distinguish the gene expression signature of the AVN from other regions of the heart. Previous studies have established that cardiomyocyte specification and organogenesis rely upon a densely interconnected regulatory network of cardiac transcription factors (i.e., Gata4, Mef2c, and Tbx5) (52). Given that these transcription factors are widely expressed in the heart, however, it remains unclear how individual transcription factors modulate gene expression programs within specific cardiac cell types. Our findings provide support for a model in which Gata4 serves as a critical node during heart development upon which additional cofactors act to impact lineage-specific gene expression (24). Indeed, Gata4 interacts with several transcription factors (Hand2 and MyoR) and coactivators (Fog1, Fog2, and CBP/p300) that directly influence various aspects of cardiac morphogenesis and gene expression. Based on the results of our experiments, we postulate that MyoR and Hand2 form an antagonistic pair of Gata4 cofactors that modulate cell type-specific gene expression depending upon their relative abundance. Similarly, we speculate that additional antagonistic pairs of transcriptional cofactors may fine-tune Gata4-dependent transcription in various subdomains within the developing heart.
Transcriptional repression and cardiac conduction.
Multiple transcriptional repressors have been show to influence formation and function of the conduction system. For example, Tbx2 and Tbx3 are expressed within the central conduction system to repress working cardiomyocyte-specific genes (11, 12). To accomplish this function, they interact with Nkx2.5 and prevent transcriptional activation of the chamber-specific genes Nppa and Cx43 to reinforce conduction cell identity. In a similar fashion, the SAN-specific repressor Shox2 inhibits Nkx2.5-dependent activation of atrial gene expression (53). The AVC repressors Msx1 and Msx2 repress chamber-specific Cx43 expression (54); similarly, Irx3 is expressed in the AV bundle and represses Cx43 expression (55). Collectively, these examples highlight a common theme by which tissue-specific transcriptional repressors prevent chamber-specific gene expression within the conduction system. In contrast, the transcriptional repressor Id2 functions within AV bundle cells to regulate cell cycle exit rather than to repress chamber-specific gene expression (56). Thus, cell cycle regulation is a second method by which transcriptional repressors influence conduction system formation. Here we demonstrate that the transcriptional repressor MyoR fine-tunes Cx30.2 expression within the AVN. Thus, MyoR-dependent modulation of a gap junction protein defines a third strategy for transcriptional repressors to influence conduction system function. These repressive strategies are unlikely to be mutually exclusive, however, so we cannot exclude the possibility that MyoR also affects cardiac conduction by additional mechanisms. Taken together, we propose that transcriptional repressors can modulate cardiac conduction by three distinct mechanisms: (i) repression of chamber-specific genes, (ii) regulation of cell cycle exit, and (iii) modulation of gap junction protein levels.
Previous studies have established that some bHLH repressors inhibit transcription by interacting with histone deacetylase-containing corepressor complexes (57), such as N-Cor/SMRT, NuRD, and Co-REST, while others function in a dominant-negative fashion (58). We demonstrate here that MyoR interacts with Gata4 and inhibits transcription via two separable, but nonequivalent, repression domains (Fig. 8). Interestingly, the N-terminal repression domain can function in a heterologous context, while the C-terminal repression domain cannot. Moreover, the N terminus of MyoR converts Hand2 into a repressor, while the N terminus of Hand2 converts MyoR into an activator. Thus, these findings support the idea that the ability of MyoR and Hand2 to function as a repressor and activator, respectively, is entirely dictated by the impact of their N terminus on transcription. Consistent with this idea, we identified a proline-rich sequence (KKPLP) within the N-terminal repression domain of MyoR that is absent from Hand2 (Fig. 5D). Given that SH3 domains can bind to proline-rich motifs (59, 60), we searched the mouse genome for histone-modifying proteins that contain an SH3 domain and identified a single protein, PRMT2. However, we were unable to demonstrate that PRMT2 potentiates MyoR-dependent repression (J. P. Harris and N. V. Munshi, unpublished data), and the precise mechanism by which MyoR inhibits transcription remains unknown. Since the C-terminal repression (H2) domain is required when deleted from MyoR but is unable to inhibit transcription when transferred to Hand2, our studies suggest that this domain is necessary for repression only in a specific context. To reconcile these findings, we speculate that the MyoR H2 domain indirectly influences transcription by mediating a structural role that can be fulfilled by the H2 domain of another bHLH family member such as Hand2.
FIG 8.
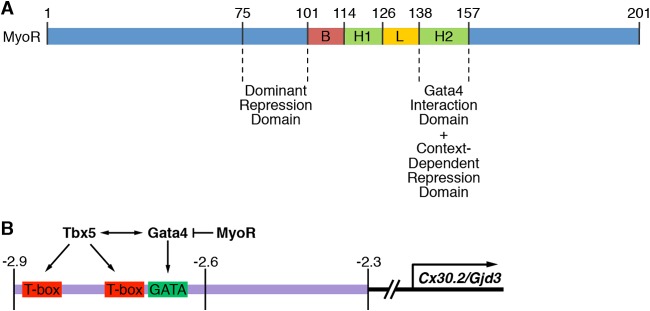
Model for MyoR-dependent regulation of Cx30.2 expression. (A) Summary of MyoR functional domains identified in the present study. Residues 138 to 157 serve as a Gata4 interaction and context-dependent repression domain. The domain encompassing amino acids 75 to 100 functions as a trans-repression domain even in a heterologous context. (B) Tbx5 and Gata4 regulate Cx30.2 expression via a 0.6-kb minimal regulatory element that directs expression to the AVN. Based on the present study, we propose that MyoR functions within AV nodal cells as a Gata4-specific transcriptional repressor to decrease Cx30.2 gene expression and thus modulate AV delay. Given the results of our protein-protein interaction experiments, we conclude that MyoR does not inhibit transcriptional activation by competing with Tbx5 for Gata4 interaction. Instead, we speculate that MyoR possesses a trans-repression domain that recruits as-yet unidentified corepressors to inhibit Gata4- and Tbx5-dependent activation of Cx30.2 expression.
MyoR as a candidate quantitative trait locus (QTL).
Previous twin studies have provided strong evidence that cardiac conduction is a heritable trait (61). Based on the finding that EKG parameters are more correlated in monozygotic versus dizygotic twins, it has been estimated that 34% of the PR interval is heritable. Although AV delay has been correlated with specific mutations in transcription factors, ion channels, and gap junction proteins, it remains unclear whether such mutations account for a significant proportion of PR interval variability in the general population (2). More recently, genomewide association studies have identified numerous single nucleotide polymorphisms (SNPs) that associate with PR interval length in the general population (62, 63). Although the influence of each individual SNP is modest, the overall impact across the entire population is predicted to be significant. In other words, PR interval is a continuous variable that is likely to be shaped by the contribution of multiple QTLs with modest individual effects. From our experimental results, it is clear that MyoR is not required for AVN formation or function per se; indeed, the PR interval of MyoR knockout mice is only ∼13% longer than wild-type mice. Rather, we suggest that MyoR contributes to the PR interval in mice by modulating AV conduction in concert with a collection of additional QTLs such as Tnni3k (64). Since mutations in MyoR would not be predicted to significantly impact fitness, SNPs within the MyoR locus could contribute to PR interval variability in the general human population. Especially given the results of recent large-scale whole-exome sequencing efforts in diverse human populations (65, 66), we anticipate that individuals with homozygous loss-of-function MyoR mutations are likely to exist. Since Cx31.9, the human ortholog of mouse Cx30.2, has not been detected in the human cardiac conduction system (67), we predict that MyoR operates through additional conserved pathways to impact PR interval variability in humans. Although the validation of these hypotheses must await the accumulation of large sequencing data sets, the search for additional QTLs that contribute to PR interval variability in the general population will remain an area of active investigation.
ACKNOWLEDGMENTS
J.P.H. was supported by a Green Fellowship from UT Dallas and UT Southwestern Medical Center. E.N.O. was supported by grants from the National Institutes of Health and the Robert A. Welch Foundation. This study was supported by a K08 Award (HL094699) from the NHLBI, a Career Award for Medical Scientists (no. 1009838) from the Burroughs Wellcome Fund, and a Disease Oriented Clinical Scholars Award from UT Southwestern Medical Center (to N.V.M.).
We thank the UT Southwestern Medical Center Flow Cytometry and Microarray Core Facilities for expert technical assistance, Jose Cabrera for graphical assistance, Ning Liu for critical reading and suggestions for improvement of the manuscript, Antonio Fernandez-Perez for assistance with figures, and members of the Munshi and Olson labs for technical support and scientific discussions.
REFERENCES
- 1.Arnolds DE, Chu A, McNally EM, Nobrega MA, Moskowitz IP. 2011. The emerging genetic landscape underlying cardiac conduction system function. Birth Defects Res A Clin Mol Teratol 91:578–585. doi: 10.1002/bdra.20800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Munshi NV. 2012. Gene regulatory networks in cardiac conduction system development. Circ Res 110:1525–1537. doi: 10.1161/CIRCRESAHA.111.260026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bakker ML, Moorman AF, Christoffels VM. 2010. The atrioventricular node: origin, development, and genetic program. Trends Cardiovasc Med 20:164–171. doi: 10.1016/j.tcm.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Cheng S, Keyes MJ, Larson MG, McCabe EL, Newton-Cheh C, Levy D, Benjamin EJ, Vasan RS, Wang TJ. 2009. Long-term outcomes in individuals with prolonged PR interval or first-degree atrioventricular block. JAMA 301:2571–2577. doi: 10.1001/jama.2009.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basson CT, Bachinsky DR, Lin RC, Levi T, Elkins JA, Soults J, Grayzel D, Kroumpouzou E, Traill TA, Leblanc-Straceski J, Renault B, Kucherlapati R, Seidman JG, Seidman CE. 1997. Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet 15:30–35. doi: 10.1038/ng0197-30. [DOI] [PubMed] [Google Scholar]
- 6.Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG. 1998. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 281:108–111. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- 7.Jay PY, Harris BS, Maguire CT, Buerger A, Wakimoto H, Tanaka M, Kupershmidt S, Roden DM, Schultheiss TM, O'Brien TX, Gourdie RG, Berul CI, Izumo S. 2004. Nkx2-5 mutation causes anatomic hypoplasia of the cardiac conduction system. J Clin Invest 113:1130–1137. doi: 10.1172/JCI200419846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moskowitz IP, Pizard A, Patel VV, Bruneau BG, Kim JB, Kupershmidt S, Roden D, Berul CI, Seidman CE, Seidman JG. 2004. The T-Box transcription factor Tbx5 is required for the patterning and maturation of the murine cardiac conduction system. Development 131:4107–4116. doi: 10.1242/dev.01265. [DOI] [PubMed] [Google Scholar]
- 9.Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE, Seidman JG. 2001. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 106:709–721. doi: 10.1016/S0092-8674(01)00493-7. [DOI] [PubMed] [Google Scholar]
- 10.Pashmforoush M, Lu JT, Chen H, Amand TS, Kondo R, Pradervand S, Evans SM, Clark B, Feramisco JR, Giles W, Ho SY, Benson DW, Silberbach M, Shou W, Chien KR. 2004. Nkx2-5 pathways and congenital heart disease; loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell 117:373–386. doi: 10.1016/S0092-8674(04)00405-2. [DOI] [PubMed] [Google Scholar]
- 11.Hoogaars WM, Tessari A, Moorman AF, de Boer PA, Hagoort J, Soufan AT, Campione M, Christoffels VM. 2004. The transcriptional repressor Tbx3 delineates the developing central conduction system of the heart. Cardiovasc Res 62:489–499. doi: 10.1016/j.cardiores.2004.01.030. [DOI] [PubMed] [Google Scholar]
- 12.Habets PE, Moorman AF, Clout DE, van Roon MA, Lingbeek M, van Lohuizen M, Campione M, Christoffels VM. 2002. Cooperative action of Tbx2 and Nkx2.5 inhibits ANF expression in the atrioventricular canal: implications for cardiac chamber formation. Genes Dev 16:1234–1246. doi: 10.1101/gad.222902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh R, Hoogaars WM, Barnett P, Grieskamp T, Rana MS, Buermans H, Farin HF, Petry M, Heallen T, Martin JF, Moorman AF, ten Hoen PA, Kispert A, Christoffels VM. 2012. Tbx2 and Tbx3 induce atrioventricular myocardial development and endocardial cushion formation. Cell Mol Life Sci 69:1377–1389. doi: 10.1007/s00018-011-0884-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christoffels VM, Hoogaars WM, Tessari A, Clout DE, Moorman AF, Campione M. 2004. T-box transcription factor Tbx2 represses differentiation and formation of the cardiac chambers. Dev Dyn 229:763–770. doi: 10.1002/dvdy.10487. [DOI] [PubMed] [Google Scholar]
- 15.Bakker ML, Boukens BJ, Mommersteeg MT, Brons JF, Wakker V, Moorman AF, Christoffels VM. 2008. Transcription factor Tbx3 is required for the specification of the atrioventricular conduction system. Circ Res 102:1340–1349. doi: 10.1161/CIRCRESAHA.107.169565. [DOI] [PubMed] [Google Scholar]
- 16.Rentschler S, Harris BS, Kuznekoff L, Jain R, Manderfield L, Lu MM, Morley GE, Patel VV, Epstein JA. 2011. Notch signaling regulates murine atrioventricular conduction and the formation of accessory pathways. J Clin Invest 121:525–533. doi: 10.1172/JCI44470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stroud DM, Gaussin V, Burch JB, Yu C, Mishina Y, Schneider MD, Fishman GI, Morley GE. 2007. Abnormal conduction and morphology in the atrioventricular node of mice with atrioventricular canal targeted deletion of Alk3/Bmpr1a receptor. Circulation 116:2535–2543. doi: 10.1161/CIRCULATIONAHA.107.696583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaussin V, Morley GE, Cox L, Zwijsen A, Vance KM, Emile L, Tian Y, Liu J, Hong C, Myers D, Conway SJ, Depre C, Mishina Y, Behringer RR, Hanks MC, Schneider MD, Huylebroeck D, Fishman GI, Burch JB, Vatner SF. 2005. Alk3/Bmpr1a receptor is required for development of the atrioventricular canal into valves and annulus fibrosus. Circ Res 97:219–226. doi: 10.1161/01.RES.0000177862.85474.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh R, Horsthuis T, Farin HF, Grieskamp T, Norden J, Petry M, Wakker V, Moorman AF, Christoffels VM, Kispert A. 2009. Tbx20 interacts with smads to confine tbx2 expression to the atrioventricular canal. Circ Res 105:442–452. doi: 10.1161/CIRCRESAHA.109.196063. [DOI] [PubMed] [Google Scholar]
- 20.Chi NC, Shaw RM, De Val S, Kang G, Jan LY, Black BL, Stainier DY. 2008. Foxn4 directly regulates tbx2b expression and atrioventricular canal formation. Genes Dev 22:734–739. doi: 10.1101/gad.1629408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kreuzberg MM, Schrickel JW, Ghanem A, Kim JS, Degen J, Janssen-Bienhold U, Lewalter T, Tiemann K, Willecke K. 2006. Connexin30.2 containing gap junction channels decelerate impulse propagation through the atrioventricular node. Proc Natl Acad Sci U S A 103:5959–5964. doi: 10.1073/pnas.0508512103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kreuzberg MM, Sohl G, Kim JS, Verselis VK, Willecke K, Bukauskas FF. 2005. Functional properties of mouse connexin30.2 expressed in the conduction system of the heart. Circ Res 96:1169–1177. doi: 10.1161/01.RES.0000169271.33675.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Munshi NV, McAnally J, Bezprozvannaya S, Berry JM, Richardson JA, Hill JA, Olson EN. 2009. Cx30.2 enhancer analysis identifies Gata4 as a novel regulator of atrioventricular delay. Development 136:2665–2674. doi: 10.1242/dev.038562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou P, He A, Pu WT. 2012. Regulation of GATA4 transcriptional activity in cardiovascular development and disease. Curr Top Dev Biol 100:143–169. doi: 10.1016/B978-0-12-387786-4.00005-1. [DOI] [PubMed] [Google Scholar]
- 25.Zhou B, Ma Q, Kong SW, Hu Y, Campbell PH, McGowan FX, Ackerman KG, Wu B, Tevosian SG, Pu WT. 2009. Fog2 is critical for cardiac function and maintenance of coronary vasculature in the adult mouse heart. J Clin Invest 119:1462–1476. doi: 10.1172/JCI38723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crispino JD, Lodish MB, Thurberg BL, Litovsky SH, Collins T, Molkentin JD, Orkin SH. 2001. Proper coronary vascular development and heart morphogenesis depend on interaction of GATA-4 with FOG cofactors. Genes Dev 15:839–844. doi: 10.1101/gad.875201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Svensson EC, Huggins GS, Lin H, Clendenin C, Jiang F, Tufts R, Dardik FB, Leiden JM. 2000. A syndrome of tricuspid atresia in mice with a targeted mutation of the gene encoding Fog-2. Nat Genet 25:353–356. doi: 10.1038/77146. [DOI] [PubMed] [Google Scholar]
- 28.Jacobsen CM, Mannisto S, Porter-Tinge S, Genova E, Parviainen H, Heikinheimo M, Adameyko II, Tevosian SG, Wilson DB. 2005. GATA-4:FOG interactions regulate gastric epithelial development in the mouse. Dev Dyn 234:355–362. doi: 10.1002/dvdy.20552. [DOI] [PubMed] [Google Scholar]
- 29.Beuling E, Bosse T, aan de Kerk DJ, Piaseckyj CM, Fujiwara Y, Katz SG, Orkin SH, Grand RJ, Krasinski SD. 2008. GATA4 mediates gene repression in the mature mouse small intestine through interactions with friend of GATA (FOG) cofactors. Dev Biol 322:179–189. doi: 10.1016/j.ydbio.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stefanovic S, Barnett P, van Duijvenboden K, Weber D, Gessler M, Christoffels VM. 2014. GATA-dependent regulatory switches establish atrioventricular canal specificity during heart development. Nat Commun 5:3680. doi: 10.1038/ncomms4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skinner MK, Rawls A, Wilson-Rawls J, Roalson EH. 2010. Basic helix-loop-helix transcription factor gene family phylogenetics and nomenclature. Differentiation 80:1–8. doi: 10.1016/j.diff.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. 1995. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science 268:836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- 33.Guillemot F, Lo LC, Johnson JE, Auerbach A, Anderson DJ, Joyner AL. 1993. Mammalian achaete-scute homolog 1 is required for the early development of olfactory and autonomic neurons. Cell 75:463–476. doi: 10.1016/0092-8674(93)90381-Y. [DOI] [PubMed] [Google Scholar]
- 34.Buckingham M, Rigby PW. 2014. Gene regulatory networks and transcriptional mechanisms that control myogenesis. Dev Cell 28:225–238. doi: 10.1016/j.devcel.2013.12.020. [DOI] [PubMed] [Google Scholar]
- 35.Srivastava D. 1999. HAND proteins: molecular mediators of cardiac development and congenital heart disease. Trends Cardiovasc Med 9:11–18. doi: 10.1016/S1050-1738(98)00033-4. [DOI] [PubMed] [Google Scholar]
- 36.Vincentz JW, Barnes RM, Rodgers R, Firulli BA, Conway SJ, Firulli AB. 2008. An absence of Twist1 results in aberrant cardiac neural crest morphogenesis. Dev Biol 320:131–139. doi: 10.1016/j.ydbio.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dai YS, Cserjesi P, Markham BE, Molkentin JD. 2002. The transcription factors GATA4 and dHAND physically interact to synergistically activate cardiac gene expression through a p300-dependent mechanism. J Biol Chem 277:24390–24398. doi: 10.1074/jbc.M202490200. [DOI] [PubMed] [Google Scholar]
- 38.Lu J, Webb R, Richardson JA, Olson EN. 1999. MyoR: a muscle-restricted basic helix-loop-helix transcription factor that antagonizes the actions of MyoD. Proc Natl Acad Sci U S A 96:552–557. doi: 10.1073/pnas.96.2.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robb L, Hartley L, Wang CC, Harvey RP, Begley CG. 1998. Musculin: a murine basic helix-loop-helix transcription factor gene expressed in embryonic skeletal muscle. Mech Dev 76:197–201. doi: 10.1016/S0925-4773(98)00122-1. [DOI] [PubMed] [Google Scholar]
- 40.Massari ME, Rivera RR, Voland JR, Quong MW, Breit TM, van Dongen JJ, de Smit O, Murre C. 1998. Characterization of ABF-1, a novel basic helix-loop-helix transcription factor expressed in activated B lymphocytes. Mol Cell Biol 18:3130–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu JR, Bassel-Duby R, Hawkins A, Chang P, Valdez R, Wu H, Gan L, Shelton JM, Richardson JA, Olson EN. 2002. Control of facial muscle development by MyoR and capsulin. Science 298:2378–2381. doi: 10.1126/science.1078273. [DOI] [PubMed] [Google Scholar]
- 42.Nelson JD, Denisenko O, Bomsztyk K. 2006. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 1:179–185. doi: 10.1038/nprot.2006.27. [DOI] [PubMed] [Google Scholar]
- 43.Medeiros RB, Papenfuss KJ, Hoium B, Coley K, Jadrich J, Goh SK, Elayaperumal A, Herrera JE, Resnik E, Ni HT. 2009. Novel sequential ChIP and simplified basic ChIP protocols for promoter co-occupancy and target gene identification in human embryonic stem cells. BMC Biotechnol 9:59. doi: 10.1186/1472-6750-9-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yie J, Merika M, Munshi N, Chen G, Thanos D. 1999. The role of HMG I(Y) in the assembly and function of the IFN-beta enhanceosome. EMBO J 18:3074–3089. doi: 10.1093/emboj/18.11.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marionneau C, Couette B, Liu J, Li H, Mangoni ME, Nargeot J, Lei M, Escande D, Demolombe S. 2005. Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J Physiol 562:223–234. doi: 10.1113/jphysiol.2004.074047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC, Srivastava D. 2003. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424:443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 47.Tzahor E, Evans SM. 2011. Pharyngeal mesoderm development during embryogenesis: implications for both heart and head myogenesis. Cardiovasc Res 91:196–202. doi: 10.1093/cvr/cvr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tzahor E. 2009. Heart and craniofacial muscle development: a new developmental theme of distinct myogenic fields. Dev Biol 327:273–279. doi: 10.1016/j.ydbio.2008.12.035. [DOI] [PubMed] [Google Scholar]
- 49.Grifone R, Kelly RG. 2007. Heartening news for head muscle development. Trends Genet 23:365–369. doi: 10.1016/j.tig.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 50.Kelly RG. 2012. The second heart field. Curr Top Dev Biol 100:33–65. doi: 10.1016/B978-0-12-387786-4.00002-6. [DOI] [PubMed] [Google Scholar]
- 51.von Scheven G, Bothe I, Ahmed MU, Alvares LE, Dietrich S. 2006. Protein and genomic organisation of vertebrate MyoR and capsulin genes and their expression during avian development. Gene Expr Patterns 6:383–393. doi: 10.1016/j.modgep.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 52.Olson EN. 2006. Gene regulatory networks in the evolution and development of the heart. Science 313:1922–1927. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Espinoza-Lewis RA, Yu L, He F, Liu H, Tang R, Shi J, Sun X, Martin JF, Wang D, Yang J, Chen Y. 2009. Shox2 is essential for the differentiation of cardiac pacemaker cells by repressing Nkx2-5. Dev Biol 327:376–385. doi: 10.1016/j.ydbio.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boogerd KJ, Wong LY, Christoffels VM, Klarenbeek M, Ruijter JM, Moorman AF, Barnett P. 2008. Msx1 and Msx2 are functional interacting partners of T-box factors in the regulation of Connexin43. Cardiovasc Res 78:485–493. doi: 10.1093/cvr/cvn049. [DOI] [PubMed] [Google Scholar]
- 55.Zhang SS, Kim KH, Rosen A, Smyth JW, Sakuma R, Delgado-Olguin P, Davis M, Chi NC, Puviindran V, Gaborit N, Sukonnik T, Wylie JN, Brand-Arzamendi K, Farman GP, Kim J, Rose RA, Marsden PA, Zhu Y, Zhou YQ, Miquerol L, Henkelman RM, Stainier DY, Shaw RM, Hui CC, Bruneau BG, Backx PH. 2011. Iroquois homeobox gene 3 establishes fast conduction in the cardiac His-Purkinje network. Proc Natl Acad Sci U S A 108:13576–13581. doi: 10.1073/pnas.1106911108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moskowitz IP, Kim JB, Moore ML, Wolf CM, Peterson MA, Shendure J, Nobrega MA, Yokota Y, Berul C, Izumo S, Seidman JG, Seidman CE. 2007. A molecular pathway, including Id2, Tbx5, and Nkx2-5, required for cardiac conduction system development. Cell 129:1365–1376. doi: 10.1016/j.cell.2007.04.036. [DOI] [PubMed] [Google Scholar]
- 57.Laherty CD, Yang WM, Sun JM, Davie JR, Seto E, Eisenman RN. 1997. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell 89:349–356. doi: 10.1016/S0092-8674(00)80215-9. [DOI] [PubMed] [Google Scholar]
- 58.Perk J, Iavarone A, Benezra R. 2005. Id family of helix-loop-helix proteins in cancer. Nat Rev Cancer 5:603–614. doi: 10.1038/nrc1673. [DOI] [PubMed] [Google Scholar]
- 59.Yu H, Chen JK, Feng S, Dalgarno DC, Brauer AW, Schreiber SL. 1994. Structural basis for the binding of proline-rich peptides to SH3 domains. Cell 76:933–945. doi: 10.1016/0092-8674(94)90367-0. [DOI] [PubMed] [Google Scholar]
- 60.Andreotti AH, Bunnell SC, Feng S, Berg LJ, Schreiber SL. 1997. Regulatory intramolecular association in a tyrosine kinase of the Tec family. Nature 385:93–97. doi: 10.1038/385093a0. [DOI] [PubMed] [Google Scholar]
- 61.Havlik RJ, Garrison RJ, Fabsitz R, Feinleib M. 1980. Variability of heart rate, P-R, QRS and Q-T durations in twins. J Electrocardiol 13:45–48. doi: 10.1016/S0022-0736(80)80008-2. [DOI] [PubMed] [Google Scholar]
- 62.Pfeufer A, van Noord C, Marciante KD, Arking DE, Larson MG, Smith AV, Tarasov KV, Muller M, Sotoodehnia N, Sinner MF, Verwoert GC, Li M, Kao WH, Kottgen A, Coresh J, Bis JC, Psaty BM, Rice K, Rotter JI, Rivadeneira F, Hofman A, Kors JA, Stricker BH, Uitterlinden AG, van Duijn CM, Beckmann BM, Sauter W, Gieger C, Lubitz SA, Newton-Cheh C, Wang TJ, Magnani JW, Schnabel RB, Chung MK, Barnard J, Smith JD, Van Wagoner DR, Vasan RS, Aspelund T, Eiriksdottir G, Harris TB, Launer LJ, Najjar SS, Lakatta E, Schlessinger D, Uda M, Abecasis GR, Muller-Myhsok B, Ehret GB, Boerwinkle E, Chakravarti A, Soliman EZ, Lunetta KL, Perz S, Wichmann HE, Meitinger T, Levy D, Gudnason V, Ellinor PT, Sanna S, Kaab S, Witteman JC, Alonso A, Benjamin EJ, Heckbert SR. 2010. Genome-wide association study of PR interval. Nat Genet 42:153–159. doi: 10.1038/ng.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Holm H, Gudbjartsson DF, Arnar DO, Thorleifsson G, Thorgeirsson G, Stefansdottir H, Gudjonsson SA, Jonasdottir A, Mathiesen EB, Njolstad I, Nyrnes A, Wilsgaard T, Hald EM, Hveem K, Stoltenberg C, Lochen ML, Kong A, Thorsteinsdottir U, Stefansson K. 2010. Several common variants modulate heart rate, PR interval, and QRS duration. Nat Genet 42:117–122. doi: 10.1038/ng.511. [DOI] [PubMed] [Google Scholar]
- 64.Lodder EM, Scicluna BP, Milano A, Sun AY, Tang H, Remme CA, Moerland PD, Tanck MW, Pitt GS, Marchuk DA, Bezzina CR. 2012. Dissection of a quantitative trait locus for PR interval duration identifies Tnni3k as a novel modulator of cardiac conduction. PLoS Genet 8:e1003113. doi: 10.1371/journal.pgen.1003113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fu W, O'Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, Gabriel S, Rieder MJ, Altshuler D, Shendure J, Nickerson DA, Bamshad MJ, Akey JM. 2013. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, Kang HM, Jordan D, Leal SM, Gabriel S, Rieder MJ, Abecasis G, Altshuler D, Nickerson DA, Boerwinkle E, Sunyaev S, Bustamante CD, Bamshad MJ, Akey JM. 2012. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kreuzberg MM, Liebermann M, Segschneider S, Dobrowolski R, Dobrzynski H, Kaba R, Rowlinson G, Dupont E, Severs NJ, Willecke K. 2009. Human connexin31.9, unlike its orthologous protein connexin30.2 in the mouse, is not detectable in the human cardiac conduction system. J Mol Cell Cardiol 46:553–559. doi: 10.1016/j.yjmcc.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 68.Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, Olson EN. 2008. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev 22:3242–3254. [DOI] [PMC free article] [PubMed] [Google Scholar]