

Abstract
Protein kinase C (PKC) targets cardiac troponin I (cTnI) S43/45 for phosphorylation in addition to other residues. During heart failure, cTnI S43/45 phosphorylation is elevated, and yet there is ongoing debate about its functional role due, in part, to the emergence of complex phenotypes in animal models. The individual functional influences of phosphorylated S43 and S45 also are not yet known. The present study utilizes viral gene transfer of cTnI with phosphomimetic S43D and/or S45D substitutions to evaluate their individual and combined influences on function in intact adult cardiac myocytes. Partial replacement (≤40%) with either cTnIS43D or cTnIS45D reduced the amplitude of contraction, and cTnIS45D slowed contraction and relaxation rates, while there were no significant changes in function with cTnIS43/45D. More extensive replacement (≥70%) with cTnIS43D, cTnIS45D, and cTnIS43/45D each reduced the amplitude of contraction. Additional experiments also showed cTnIS45D reduced myofilament Ca2+ sensitivity of tension. At the same time, shortening rates returned toward control values with cTnIS45D and the later stages of relaxation also became accelerated in myocytes expressing cTnIS43D and/or S45D. Further studies demonstrated this behavior coincided with adaptive changes in myofilament protein phosphorylation. Taken together, the results observed in myocytes expressing cTnIS43D and/or S45D suggest these 2 residues reduce function via independent mechanism(s). The changes in function associated with the onset of adaptive myofilament signaling suggest the sarcomere is capable of fine tuning PKC-mediated cTnIS43/45 phosphorylation and contractile performance. This modulatory behavior also provides insight into divergent phenotypes reported in animal models with cTnI S43/45 phosphomimetic substitutions.
Keywords: Myocyte, Myofilament, Troponin, Protein Kinase C, Phosphorylation
1. INTRODUCTION
The thin filament protein, cardiac troponin I (cTnI) is a phosphorylation target for multiple signaling pathways which modulate myofilament function and cardiac pump performance [1,2]. Several kinases phosphorylate cTnI S23/24 [3,4], and this phosphorylation accelerates Ca2+ dissociation from troponin (Tn), and reduces the myofilament Ca2+ sensitivity [5-7]. This reduced Ca2+ sensitivity translates into accelerated cardiac relaxation to improve or preserve diastolic function [5,7,8], and reduced phosphorylation at this site is linked to diastolic dysfunction during end-stage heart failure [9,10]. In contrast, enhanced phosphorylation of a second cluster of residues at Ser43/45 (S43/45) in cTnI is associated with cardiac dysfunction [11-14]. Protein kinase C (PKC) phosphorylates cTnI S43/45 [15,16], and elevated PKC isoform expression and activity are reported to accompany cardiac dysfunction and end-stage human heart failure [17-23]. PKC-mediated phosphorylation of cTnI and/or phosphomimetic D or E substitutions at S43/45 reduce myofilament Ca2+ sensitivity of tension and actomyosin ATPase activity [15,16,24], and sometimes reduce peak tension, ATPase activity, and peak pressure [15,24-26]. Cardiac TnI with phosphomimetic S43/45 substitutions also reduces maximum sliding speed during in vitro motility assays [24], and maximum unloaded shortening velocity in permeabilized myocytes [26]. Moreover, non-phosphorylatable cTnIS43/45A attenuates the PKC activation response [16,27-29]. Based on this in vitro work, reduced pressure development and accelerated relaxation rate could be expected in animal models expressing phosphomimetic cTnIS43/45.
However, this predicted in vivo functional phenotype is not consistently observed in genetic animal models expressing phosphomimetic S43/45 substitutions. Instead, animal models with differing phenotypes emerged, and the functional role of this cluster in myocardium remains controversial. For example, cTnI containing phosphomimetic D substitutions at the PKC-targeted S23/24, S43/45, and Thr144 (T144) sites fully replaced endogenous cTnI in cTnIAllP mice [30]. Myofilament Ca2+ sensitivity, maximum tension and actomyosin ATPase activity are decreased in these hearts [25,30], and yet only the rate of pressure development is modestly reduced in intact hearts. In a parallel group of mice expressing cTnIS23/24D (cTnIDD), the decreased myofilament Ca2+ sensitivity was accompanied by the anticipated acceleration of cardiac relaxation [30]. A similarly modest cardiac phenotype develops after partial replacement with cTnI S23A/S24/43/45D [31].
A much different cardiac phenotype develops in cTnIPKC-P mice expressing cTnI with S43/45E and T144E substitutions [32]. Endogenous cTnI is replaced by <10% with cTnIPKC-P, which produced significant reductions in maximum myofilament ATPase activity, papillary muscle peak tension, and the amplitude and rates of pressure development and relaxation in isolated, perfused hearts, although the Ca2+ sensitivity of tension remained similar to cTnI [32]. The only response shared by the cTnIAllP, cTnIS23A/S24/S43/S45D, and cTnIPKC-P models is the slowed rate of cardiac pressure development [30-32]. A key difference between these models is the presence or absence of S23/24 substitutions, and yet the phenotype observed in the cTnIDD mouse [30] cannot explain the divergent organ-level function in these animal models. Thus, the explanation(s) for the unexpected phenotypes in animal models and divergence across models are not yet understood. An intact cellular approach has the benefit of providing contractile measurements in the presence of established architecture and signaling pathways within the cell. Thus, the present group of experiments utilizes intact myocytes to serve as a bridge between earlier in vitro and in vivo work.
In addition to understanding the modulatory functional role played by S43/45 phosphorylation, it remains unclear whether S43 or S45 in cTnI independently modulate contractile function. Both S23/24 in cTnI must be phosphorylated to reduce myofilament Ca2+ sensitivity and accelerate relaxation [7,33], yet the individual roles of S43 and S45 are not understood. The enhanced phosphorylation of the individual cTnI S43 and S45 sites observed in failing hearts [11] suggests each residue may play a clinically relevant role. The present group of studies now tests whether cTnI S43 and S45 play an individual or combined role to modulate contractile function.
In the present study, viral-based gene transfer is utilized to replace myofilament cTnI with cTnI phosphomimetic substitutions at one or both of these residues. The results demonstrate cTnI S43 and S45 independently, but not additively, serve as modulatory brakes by slowing and/or reducing contraction. The current studies also show adaptive responses contribute to the cellular phenotype, which suggests there is dynamic modulation of contractile function and could contribute to the divergent phenotypes observed in earlier animal models.
2. METHODS
2.1 Gene Transfer and Cardiac Myocyte Isolation
2.1.1 Mutagenesis and Adenoviral Constructs
For cTnIS43 and/or cTnIS45, site-directed mutagenesis was used to substitute S with D in rat cTnI [29,34]. The cTnI phosphomimetics generated include cTnIS43D, cTnIS45D, and cTnIS43/45D with and without FLAG [29]. The primers used to produce the phosphomimetics were: (substitutions are underlined) 5’-atgccaagaaaaagtctaagatcgatgcctccagaaaacttcagttgaag-3’ (sense) and 5’-cttcaactgaagttttctggaggcatcgatcctagactttttcttggcat-3’ (anti-sense) for cTnIS43D, and 5’-gatctccgccgacagaaagcttcagttgaag-3’ (sense) and 5’-cttcaactgaagctttctgtcggcggagatc-3’ (anti-sense) for cTnIS45D. Primers for cTnIS43/45D were 5’-gccaagaaaaagtctaagatcgatgccgacagaaaacttcag-3’ (sense) and 5’-ctgaagttttctgtcggcatcgatcttagactttttcttggc-3’ (antisense). Recombinant adenoviruses were prepared using the pDC315 shuttle vector and pBHGLoxΔE1,3Cre (Microbix Biosystems Inc, Mississauga,ON) in HEK293 cells [29].
2.1.2 Adult Myocyte Isolation and Culture
Animal handling protocols and procedures were performed in accordance with institutional guidelines and were approved by the University Committee on the Use and Care of Animals at the University of Michigan. The isolation of Ca2+-tolerant myocytes from adult Sprague Dawley rats was performed as previously described in detail [29]. Briefly, myocytes were plated on laminin coated coverslips in DMEM containing 5% fetal bovine serum (FBS) and antibiotics (P/S; penicillin 50 U/ml; streptomycin 50μg/ml) for 2 hrs followed by incubation with a multiplicity of infection (MOI) of 50-500 recombinant adenoviral constructs (e.g. cTnI, cTnIS43D, cTnIS45D, cTnIS43/45D) diluted in serum-free DMEM plus P/S. Gene transfer of cTnI phosphomimetics was carried out using 200 MOI, unless otherwise indicated in the text. After 1 hr, M199 media supplemented with P/S was added back to each well. Media was changed the next day, and then every other day for quiescent myocytes cultured in 6 well plates. Media was replaced every 12 hrs in electrically paced cells (0.2 Hz). Electrically paced cells were used for studies on cellular shortening and measurement of Ca2+ transients and quiescent cells were used for all other studies.
2.2 Western Detection of Protein and Phosphorylation Levels and Stoichiometry
2.2.1 Western Analysis
Replacement of endogenous cTnI with FLAG-tagged cTnI or phosphomimetic substitutions, maintenance of myofilament stoichiometry, and changes in myofilament phosphorylation were monitored by protein separation using SDS-PAGE followed by Western blot detection of total TnI (FLAG and non-tagged cTnI) on PVDF membranes. Expression and replacement of endogenous cTnI with exogenous constructs and cTnI replacement in intact versus permeabilized myocytes were determined by immunoblot analysis, as described earlier [29]. To evaluate thin filament stoichiometry, blots were re-probed for tropomyosin (Tm; 1:1000; Sigma-Aldrich Co, St Louis,MO) or troponin T (TnT; 1:500; Fitzgerald-II, Acton,MA), which were detected using horse-radish peroxidase conjugated secondary antibody (Ab; 1:2000; Cell Signaling Tech, Beverly, MA) in samples collected 2 and 4 days post-gene transfer. In a subset of experiments, the polyclonal primary antibodies (Ab) listed below and HRP-conjugated secondary Abs (1:2000, Cell Signaling Tech) were used to detect phosphorylation. Primary Abs for these studies included phosphorylated cTnI at S23/24 (pS23/24; 1:1000, Cell Signaling Tech) and T144 (pT144; 1:500; Abcam, Cambridge,MA), phosphorylated phospholamban (PLB) Ser16 (pS16, 1:1000; EMD-Millipore, Billerica,MA) and Thr17 (pT17, 1:5000; Badrilla Ltd, Leeds, UK), and cardiac myosin binding protein C (cMyBP-C) Ser273 (pS273, 1:2500), Ser282 (pS282, 1:2500), and Ser302 (pS302, 1:5000), which were kind gifts from S. Sadayappan [35]. The following primary Abs also were used to detect total cMyBP-C (1:1000; Santa Cruz Biotech, Inc, Santa Cruz,CA), total PLB (1:1000; EMD-Millipore), SERCA2 (1:1000; Santa Cruz Biotech, Inc), protein phosphatase 2A (PP2A; 1:500; Millipore) and methylated PP2A (1:500; EMD-Millipore). Each primary Ab was detected with the appropriate HRP-conjugated secondary Ab, as described for cTnI. The ratio of phosphorylated to total expression of the individual protein was then normalized to the non-treated control values, which was set to 1.0 for quantitative analysis of phosphorylated proteins by immunoblots. The ratios calculated for stoichiometry and SR Ca2+ handling proteins were similarly normalized to the non-treated control values (set to 1.0).
For studies on phosphatase inhibition, myocytes were incubated in media ± calyculin A (CalA; 10 nM; EMD-Millipore) for 10 min at 37°C, and immediately collected into ice-cold sample buffer, and stored at −80°C until proteins were separated by SDS-PAGE and analyzed on immunoblots. The cTnI pS23/24 and cMyBP-C pS282 for these studies is expressed as the percent change from basal values. The influence of cellular acidosis was evaluated by comparing cTnI pS23/24 in myocytes incubated in pH 7.4 versus 6.8 media for 30 min at 37°C. Samples were collected into ice-cold sample buffer, proteins separated by SDS-PAGE, and cTnI pS23/24 detected as described above. As with the phosphatase studies, results are expressed as the percent change from basal values.
2.3 Sarcomere Incorporation
Intact and permeabilized myocytes were evaluated by immunoblot analysis for cTnI [29] as indirect support for sarcomere incorporation. In addition, cardiac myocyte localization of non- and FLAG-tagged cTnI within cardiac myocytes was analyzed by indirect immunohistochemistry (IHC) with dual primary antibodies [29]. Immunostained myocytes were imaged on a Fluoview 500 laser scanning confocal microscope (Olympus, Center Valley, PA). Projection images were de-convoluted with Autoquant X (Media Cybernetics, Rockville, MD), and non-treated cardiac myocytes served as controls.
2.4 Myocyte Contractile Function and Ca2+ Transients
2.4.1 Intact Myocyte Function
Contractile function was evaluated using sarcomere shortening and re-lengthening measurements in intact myocytes 2 and 4 days post-gene transfer. Paced myocytes were mounted in a 37°C cell chamber and perfused with M199 media [29]. Signal-averaged traces collected with a CCD camera (Ionoptix LLC, Milton,MA) were analyzed for resting sarcomere length, peak shortening amplitude (% baseline), shortening and re-lengthening rates, time to peak (TTP), and times to 25%, 50%, and 75% re-lengthening (TTR25%, TTR50%, TTR75%, respectively), as described previously [36,37]. Ca2+-transient and sarcomere shortening traces also were recorded in Fura-2AM-loaded myocytes. Signal-averaged Ca2+ traces were analyzed for resting and peak Ca2+ amplitude ratios, rates of Ca2+ rise and decay, and times to 25%, 50%, and 75% decay (TTD25%, TTD50%, and TTD75%, respectively) in addition to contractile function [38].
2.4.2 Isometric tension measurements
Calcium-dependent isometric tension was measured in permeabilized non-treated controls and myocytes expressing cTnI and cTnIS45D after 4 days post-gene transfer. Individual myocytes were mounted to a piezoelectric motor (Model 315C, Aurora Scientific, Aurora,ON) and a force transducer (Model 403A, Aurora Scientific) in high relaxing (HR) buffer composed of pCa (−log [Ca2+]) 9.0, 10 mM EGTA, 20 mM Imidazole, 1 mM free Mg2+, 4 mM free ATP, 14.5 mM creatine phosphate and sufficient KCl for an ionic strength of 180 mM at 15°C. Ion concentrations for each pCa were calculated using MATLAB [39]. Myocytes were permeabilized in HR with 0.1% Triton X-100, sarcomere length (SL) was set to 2.0 or 2.3 μm, and active tension measured over a pCa (−log [Ca2+]) range of 9.0 to 4.5 using the slack test approach [29]. Tension-pCa curves were fit to the Marquardt-Levenburg non-linear, least squares algorithm for the Hill equation, where P is the fractional tension, K is the midpoint or –log [Ca2+] producing 50% peak tension (pCa50) and nH is the Hill coefficient in the equation:
2.5 Data analysis and statistics
Quantitative results are expressed as mean ± SEM. Multiple groups were compared using a one-way analysis of variance (ANOVA) and post-hoc Newman-Keuls tests, and a 2-way ANOVA and post-hoc Tukey's test was used to compare contractile function in myocytes expressing cTnIFLAG or cTnIS43/45DFLAG at 2 and 4 days. An unpaired Student's t-test was used to compare 2 groups. The temporal influence at multiple MOIs was compared by two-way ANOVA and post-hoc Bonferonni tests. Statistical significance was set at p<0.05 for all comparisons.
3. RESULTS
3.1 Myofilament Expression and Sarcomere Incorporation of cTnIS43/45 Phosphomimetics
Two days after gene transfer FLAG-tagged wild-type cTnI, cTnIS43D, cTnIS45D and cTnIS43/45D replaced 35-45% of endogenous cTnI, and this increased to 65-85% replacement by day 4 (Fig. 1A,B), which agrees with earlier work [29,34,38]. Total cTnI protein expression levels and the TnI/Tm (Tm; Fig. 1C) and TnI/TnT (not shown) stoichiometry were not different from the ratios in non-treated control myocytes. Gene transfer of 500 MOI cTnIFLAG and cTnIS43/45DFLAG confirmed this stoichiometry is maintained in myocytes (Table S1). The comparable expression of cTnI constructs in intact and detergent- permeabilized myocytes is consistent with their myofilament incorporation (Fig. 1D). Sarcomere localization was confirmed by immunohistochemistry, which showed an overlapping, striated labeling pattern for FLAG and TnI in myocytes expressing each FLAG-tagged cTnI construct (Fig. 1E).
FIGURE 1. Protein expression, thin filament stoichiometry, and sarcomere incorporation of cTnI phosphomimetics 2 and 4 days after gene transfer.

A. Representative immunoblots show protein expression of cTnI and cTnI phosphomimetics (cTnIS43D, cTnIS45D, cTnIS43/45D) with and without FLAG tags. The FLAG- and non-tagged cTnI (upper panel), Tm expression (middle panel), and a silver (Ag)-stained gel band (lower panel) are shown to indicate protein loading in each lane. B. Quantitative analysis of the percent replacement with individual FLAG-tagged constructs 2 (left) and 4 (right) days after gene transfer. The number of individual hearts analyzed is indicated by the n value in this panel and panel C. Results in both panels were compared with a 1-way ANOVA (p>0.05) for statistical comparisons. C. Quantitative analysis of thin filament stoichiometry based on the cTnI/Tm expression ratio in myocytes expressing FLAG-tagged cTnI substitutions. Ratios were normalized to time-matched controls for each phosphomimetic cTnI (see Methods) at 2 (left) and 4 (right) days post-gene transfer. Comparable results were obtained for non-tagged constructs (not shown). D. Representative Western blot showing comparable replacement of endogenous cTnI with each construct in intact (I) and permeabilized (P) myocytes. E. Representative confocal projection images for control, cTnIFLAG- and cTnIS43/45DFLAG-expressing cardiac myocytes immunostained for TnI and FITC (left; bar = 10 μm), FLAG and Texas Red (middle), plus the overlay (right). Myocytes expressing cTnIS43DFLAG and cTnIS45DFLAG produced comparable IHC patterns (not shown).
3.2 Influence of cTnIS43/45 Phosphomimetics on Myofilament Function
Phosphomimetic cTnI expression produced time-dependent changes in contractile function. Two days after gene transfer, partial replacement with cTnIS43D or cTnIS45D independently reduced peak shortening amplitude and slowed the shortening rate compared to myocytes expressing cTnI or cTnIFLAG (Fig. 2A). Expression of cTnIS45D also decreased re-lengthening rates, while other measures of shortening and re-lengthening remained unchanged from control values (Fig. 2A). In contrast, cTnIS43/45D slowed shortening rate without changing shortening amplitude even though myofilament replacement was comparable to cTnIS43D and cTnIS45D (Figs. 1,2A). These responses also were not observed after gene transfer of cTnI or cTnIFLAG compared to non-treated controls (Fig. 2A). The results demonstrate cTnIS43 and S45 independently modulate function and thus, differ from the upstream S23/24 tandem cluster, which requires phosphorylation of both residues for a functional response [33]. The limited influence of cTnIS43/45D suggests S43 and S45 may work via separate mechanisms, with the actions of one residue counteracting the other in the combined construct.
FIGURE 2. Contractile function in cardiac myocytes 2 (A) and 4 (B, C) days after cTnI gene transfer.

A. Quantitative analysis of basal contractile function in cTnIS43D-, cTnIS45D-, TnIS43/45D-, cTnI+FLAG-expressing myocytes, and time-matched controls 2 days post-gene transfer. Resting sarcomere length, peak shortening amplitude (%basal), shortening rate and time to peak (TTP), re-lengthing rate and time to 50% re-lengthening (TTR50%) are shown. The legend for each panel includes sample n. Values for cTnI and cTnIFLAG were pooled based on initial studies showing no statistical differences between these groups (p>0.05). B. Quantitative analysis of sarcomere shortening and re-lengthening in intact myocytes 4 days after gene transfer. Myocytes were treated with 200 MOI of recombinant cTnIS43D, cTnIS45D, and cTnIS43/45D adenovirus and compared to non-treated controls (*p<0.05) and myocytes expressing cTnIFLAG (^p<0.05) by 1-way ANOVA and post-hoc analyses in panels A, B and D. C. A quantitative comparison of functional measurements made in myocytes 4 days post-gene transfer of 50-500 MOI cTnIFLAG or cTnIS43/45DFLAG. Statistical comparisons were performed using a 2-way ANOVA and post-hoc Tukey's test (*p<0.05 vs. cTnIFLAG; ^p<0.05 vs. 50 MOI cTnIS43/45DFLAG). D. Analysis of pCa50 measured in permeabilized myocytes at a SL of 2.0 and 2.3 µm compared to non-treated controls (*p<0.05, 1-way ANOVA). Myocytes expressing cTnIS45D were evaluated based on their significant impact on shortening amplitude. Maximum tension (P0) at each SL was not different (p>0.05) among myocyte groups (Po kN/m2; SL 2.0 μm: Con = 6.07±0.81; cTnI = 6.51±0.82; cTnIS45D = 7.81±1.33; SL 2.3 μm: Con = 11.17±1.15; cTnI = 8.96±0.71; cTnIS45D = 9.17±1.16).
By 4 days post-gene transfer, higher cTnIS43/45D expression levels also decreased shortening amplitude and more extensive myofilament cTnIS43D and cTnIS45D expression continued to reduce peak shortening amplitude (Fig. 2B). Shortening rates also slowed significantly in myocytes expressing cTnIS43D, while both shortening and re-lengthening rates unexpectedly reversed course and increased relative to the day 2 values in myocytes expressing cTnIS45D (Fig. 2A,B; p<0.05 2-way ANOVA, Tukey tests). The acceleration of TTP and/or TTR50% in myocytes expressing cTnIS45D is consistent with these findings (Fig. 2B, Fig. S1), and may prevent further slowing of shortening and re-lengthening rates with cTnIS43D expression. These responses are specific for the phosphomimetic cTnI constructs, as similar responses were not observed after gene transfer of cTnIFLAG (Fig. 2B, Fig. S1). The accelerated TTP and TTR50% could result from adaptive response(s) which restore shortening and/or re-lengthening rates. Contractile function in myocytes treated with 50-500 MOI cTnIS43/45DFLAG validated this idea (Fig. 2C). An accelerated TTR50% accompanied the dose-dependent contractile dysfunction observed with 200 MOI cTnIS43/45DFLAG at 4 days, but not cTnIFLAG or at earlier time points (Fig. 2C, Table S2). In contrast, TTR50% remained unchanged and the TTP slowed with 500 MOI cTnIS43/45DFLAG expression (Fig. 2C), and coincided with further reductions in the amplitude and rates of shortening and re-lengthening. Thus, the progressive increases in expression observed over 4 days and the functional comparison to time-matched, cTnI-expressing controls indicates each response is specific for the individual substitution.
The diminished shortening rates observed in cTnIS43D- and cTnIS45D-expressing myocytes are consistent with slowed pressure development in animal models expressing cTnI with phosphomimetic S43/45 substitutions,30,32 and could result from diminished myofilament Ca2+ sensitivity (pCa50). To test this idea, isometric tension was measured in permeabilized myocytes over a range of Ca2+ concentrations in control, cTnI, and cTnIS45D-expressing myocytes after 4 days (Figs. 2D, S2). While Ca2+-activated tension was not different among myocytes at a sarcomere length (SL) of 2.0 μm (Fig. 2D), cTnIS45D significantly reduced the pCa50 compared to controls at a SL of 2.3 μm (Fig. 2D), indicating a length-dependent influence of cTnIS45D on myofilament Ca2+ sensitivity. Previously, cTnIS43/45E exchange into permeabilized myocardium also decreased the pCa50 and peak tension at 2.3 μm [24]. Maximum isometric tension was not different among myocyte groups at either SL in our experiments (Fig. 2D legend). Variable influences on maximum tension and/or pressure also are evident in earlier studies [24, 25,30-32], which could stem from differences in the phosphomimetic residue selected, preparation analyzed and/or the relative amount of cTnI replacement in myofilaments.
3.3 Adaptations Mediated via Myofilament Phosphorylation
The influence of cTnI phosphomimetics on myofilament Ca2+ sensitivity is consistent with the reduced amplitude and rate of shortening, but the accelerated TTP and TTR50% values observed with cTnIS43D and cTnIS45D on day 4 are more likely due to adaptive changes, such as alterations in Ca2+ handling. Previously, a similar mismatch was associated with changes in Ca2+ handling [40], and thus, shortening and Ca2+ transients in Fura-2AM-loaded myocytes were measured 2 and 4 days after gene transfer to test this possibility (Fig. 3A,B). On day 2, contractile function was similar to non-Fura-loaded myocytes (results not shown), and the Ca2+ transient amplitude and/or rates were similar across myocyte groups (Fig. 3A). The reduced shortening amplitude previously observed at 4 days also developed in Fura-2AM-loaded myocytes (results not shown), and peak Ca2+ and the rates of Ca2+ release and decay were not different from controls (Fig. 3B). However, an accelerated TTD50% accompanied the faster TTR50% observed at day 4 in cTnIS43D and cTnIS45D-expressing myocytes (Fig. 3B). Resting Ca2+ levels also were slightly elevated in myocytes expressing cTnI or cTnIS43/45D (Fig. 3B). Taken together, these results indicate the cTnI phosphomimetics directly influence function at 2 days, and may include adaptive TTD50% changes by 4 days post-gene transfer. The day 4 response is not associated with detectable changes in myosin heavy chain isoform expression (Fig. S3).
FIGURE 3. Quantitative analysis of Ca2+ handling in electrically paced, Fura-2AM-loaded adult myocytes 2 (A) and 4 (B) days after gene transfer.

The basal Ca2+ ratio, peak Ca2+ transient amplitude (% baseline), rates of Ca2+ release and re-uptake, and time to 50% decay (TTD50%) were evaluated for each Ca2+ transient. Statistical differences between cTnI phosphomimetics and controls (*p<0.05; ANOVA) and/or cTnI-expressing myocytes (^p<0.05) are indicated in each panel. Sample n is shown in each legend.
Targets likely to contribute to this adaptation in Ca2+ decay include enhanced PLB phosphorylation and/or up-regulated SERCA2A expression in the sarcoplasmic reticulum (SR). Increased PLB phosphorylation can develop at the PKA targeted S16 site or Ca2+/Calmodulin-dependent protein kinase II (CaMKII)-targeted T17 site [41]. Immunoblot analysis indicated PLB/SERCA2A, pS16-PLB/PLB and pT17-PLB/PLB in cTnI phosphomimetic myocytes were not different from controls (Fig. 4A,B).
FIGURE 4. Representative immunoblots and quantitative analysis of proteins contributing to Ca2+ uptake into the sarcoplasmic reticulum (SR).

A. Representative immunoblots for SERCA2A expression (upper left), pS16-PLB (middle left), and pT17-PLB (upper right) relative to total PLB expression (lower panels). B. Quantitative analysis of pS16-PLB (left) and pT17-PLB (middle) relative to total PLB expression, and PLB/SERCA2A ratio (right). The ratios in each group were normalized to the appropriate control values and compared by ANOVA (p>0.05). Sample n is shown in the title for each panel.
Alterations in myofilament phosphorylation also could contribute to the return of shortening and re-lengthening rates toward control values and/or the faster TTP interval observed in cTnI phosphomimetic-expressing myocytes at day 4 (Fig. 2B). In radiolabeled myocytes, a significant increase in cTnI phosphorylation was detected by day 4 (Fig. S4A,B). Representative myofilament proteins were further analyzed on immunoblots using phospho-specific antibodies. In these experiments, there were no changes in cTnI pS23/24 on day 2 (Fig. S4C), but elevated cTnI pS23/24 develops by day 4 (Fig. 5A,B). In contrast, pT144 did not change across the myocyte groups on day 4 (Fig. 5A,B). The reduced myofilament Ca2+ sensitivity and accelerated dP/dtmin in intact hearts expressing cTnIS23/24D8 indicates the adaptive increase in pS23/24 observed here could attenuate the slowed re-lengthening observed with cTnI phosphomimetics (Fig. 2). Phosphorylation of S23/24 and T144 were not significantly different from controls in cTnIS43/45D-expressing myocytes (Fig. 5A,B). Further analysis of myocytes expressing FLAG-tagged cTnI indicated pS23/24 was evenly distributed between FLAG and the remaining cTnI (Fig. 5 C,D). Quantitative analysis of similar blots also indicated the elevated pS23/24 is stochastically distributed among myocyte groups on day 2 (Fig. 5D).
FIGURE 5. Analysis of myofilament protein phosphorylation in myocytes expressing wild type and phosphomimetic cTnI.

A. Representative pS23/24 (upper left) and pT144 (upper right) relative to total cTnI expression (lower panels) detected by immunoblot 4 days after gene transfer. B. Quantitative analysis of pS23/24- and pT144-cTnI detected in cTnI immunoblots. The normalized ratio of phosphorylated to total cTnI in each group was compared by ANOVA in both panels (*p<0.05 vs control). The legend shown to the right of this panel applies to panels B, D and F, with sample n indicated in each panel. C. Representative immunoblots showing pS23/24 for myocytes expressing FLAG-tagged cTnI constructs 2 and 4 days after gene transfer. D. Quantitative analysis of the %pS23/24 present as FLAG (% pFLAG) compared to % cTnIFLAG (%FLAG) expression relative to total cTnI 2 days after gene transfer (p>0.05 by ANOVA). E. Representative immunoblots showing cMyBP-C pS273 (upper left), pS282 (upper middle), pS302 (upper right) and total cMyBP-C protein expression (lower panels) in adult myocytes 4 days after gene transfer. F. Quantitative analysis of the phospho-/total cMyBP-C ratios for pS273-, pS282-, and pS302-cMyBP-C. Normalized phosphorylation ratios for these cMyBP-C residues were calculated as described in panel B.
To determine whether adaptive responses also develop in thick filament proteins, cMyBP-C phosphorylation levels of the S273 (pS273), S282 (pS282), and S302 (pS302) residues within the cardiac-specific m-motif were evaluated using site-specific phospho-antibodies (Fig. 5E,F) [35]. A trend toward increased pS273 levels in cMyBP-C was detected on day 4 but was highly variable and not statistically significant compared to controls (Fig. 5E,F). Interestingly, pS282 was significantly enhanced in myocytes expressing the phosphomimetic cTnI substitutions compared to controls at the same time point, while pS302 levels were similar across myocyte groups expressing different cTnI constructs (Fig. 5E,F). Cross-bridge cycling rates increase in response to cMyBP-C phosphorylation [42], and the elevated phosphorylation detected here may help to restore shortening and/or re-lengthening rates in myocytes expressing phosphomimetic cTnI, although proof will require additional studies. The elevated cTnI S43/45 phosphorylation levels reported with chronic dysfunction [11-13], and phosphorylation of other myofilament residues in our experiments suggests prolonged cTnI modification leads to adaptive sarcomere phosphorylation to modulate cross-bridge cycling and/or fine tune contractile performance.
The enhanced phosphorylation at some residues (cTnI pS23/24; cMyBP-C pS282), and variability in the phosphorylation of other residues (cMyBP-C pS273; Figs. 4B, 5) raised questions about the mechanism(s) responsible for adaptation. The possibility phosphatase activity contributed to adaptation was examined by comparing cTnI pS23/24 and cMyBP-C pS282 in the presence versus absence of the phosphatase inhibitor, CalA. The enhanced pS23/24 and pS282 produced by CalA resulted in a similar level of overall phosphorylation across myocyte groups (Fig. 6A-D), and resulted in much smaller phosphorylation changes in cTnIS43D- or cTnIS45D- versus cTnI-expressing myocytes (Fig. 6B,D).
FIGURE 6. Representative immunoblot (A, C) and quantitative (B, D) analyses of cTnI pS23/24 (A, B) and cMyBP-C pS282 (C,D) in day 4 adult myocytes treated with the phosphatase inhibitor, calyculin A (CalA, 10 nM).

A. Representative immunoblot comparing cTnI pS23/24 in the absence and presence of CalA. The black vertical line in this panel and panel C indicates separation by additional lanes on the same immunoblot. B. Quantitative analysis of the normalized ratio of cTnI pS23/24 relative to total cTnI. The change in phosphorylation observed in the presence versus absence of CalA is expressed as the Δ% change (right panel). In this panel and panel D, quantitative results were compared by ANOVA (*p<0.05 vs. control), and sample n is provided in each legend. C. Representative immunoblot showing cMyBP-C pS282 in the absence and presence of CalA (day 4). D. Quantitative analysis of cMyBP-C pS282 after CalA (left), and the Δ% change in phosphorylation produced by CalA (right panel). The pS282 results are expressed as a normalized fraction of total cMyBP-C.
The possibility adaptive signaling at cTnI pS23/24 could also result from alterations in the cellular environment was tested by incubating myocytes in acidic media equivalent to the level observed with myocardial ischemia (pH 6.8) [43-45]. Interestingly, acidosis increased cTnI pS23/24 to comparable levels as CalA, such that overall phosphorylation was no longer different across myocyte groups (Fig. 7A,B). These results also are consistent with earlier organ level studies [43]. Acidosis-induced shifts in pS23/24 also were greater for cTnI than phosphomimetic cTnIs when results from cTnI-expressing myocytes were pooled and compared to cTnI phosphomimetics (Fig. 7C), which suggests dynamic sarcomere modulation is not unique to expression of cTnI substitutions.
FIGURE 7. Representative immunoblot (A) and quantitative analyses (B,C) of cTnI pS23/24 in response to acidosis (pH 6.8).
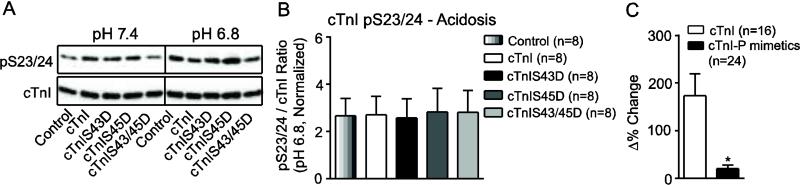
A. Representative immunoblot comparing cTnI pS23/24 in the presence and absence of acidosis treatment in myocytes 4 days post-gene transfer. The vertical black line indicates additional lanes separated samples on the same blot. B. Quantitative analysis of the normalized cTnI pS23/24 / total cTnI ratio. Results were compared by 1-way ANOVA (p>0.05). C. Analysis of the Δ% change in cTnI pS23/24 when results from control and cTnI-expressing myocytes were pooled and compared to the pooled set of cTnI-phosphomimetic-expressing myocytes (cTnIS43D, cTnIS45D, cTnIS43/45D) using a Student's unpaired t-test (*p<0.05).
Alterations in protein phosphatase 2A (PP2A) were evaluated to determine whether this phosphatase contributes to the adaptive response produced by cTnI phosphomimetics in a final group of experiments. Protein phosphatase 1 (PP1) and 2A (PP2A) de-phosphorylate myofilament proteins [46-48] and influence myofilaments in intact hearts [48,49]. Total PP2A expression normalized to a Sypro-stained blot band was comparable across the myocyte groups (Fig. 8A). In contrast, less PP2A methylation was detected for myocytes expressing cTnI phosphomimetics compared to cTnI (Fig. 8B). This shift towards lower PP2A methylation in phosphomimetic- versus cTnI-expressing myocytes is consistent with the adaptive myofilament phosphorylation observed with cTnI S43D or S45D.
FIGURE 8. Protein phosphatase 2A (PP2A) expression (A,B) and a working model of adaptive phosphorylation (C).

A. Representative immunoblot showing methylated (upper), total PP2A (middle) and a Sypro-stained blot band (lower panel) to indicate protein load). B. Quantitative analysis of the normalized ratio of methylated PP2A relative to total PP2A. Values obtained in myocytes expressing phosphomimetic cTnI were compared to cTnI-expressing myocytes using a 1-way ANOVA and post-hoc tests (*p<0.05 vs. cTnI). C. Working model of the kinase and phosphatase activity balance in the sarcomere under basal conditions (model 1) and the changes to this balance during adaptive myofilament protein phosphorylation (models 2 and 3) produced by phosphomimetic cTnI and acidosis. An expanded portion of the sarcomere is included for each model to demonstrate the relative myofilament distribution and/or activity (active vs. inactive) of kinases and phosphatases. The legend explaining inactive and active kinases and phosphatases is shown below model 1. Results from the present study provide evidence to support model 2 (reduced phosphatase activity), but kinase activity also may contribute to this adaptive response (model 3).
4. DISCUSSION
The current studies show cTnIS43D and cTnIS45D independently act as a brake on cellular contractile function (Figs. 2-4), and these functional changes developed after partial myofilament replacement with the individual phosphomimetics (Fig. 1). Comparable reductions in function require higher expression levels of cTnIS43/45D (Figs. 1,2). These findings, together with earlier studies, suggest PKC-mediated phosphorylation of cTnI S43 or S45 contributes to cardiac dysfunction under pathophysiological conditions [11-13,50]. Based on the present studies, cTnI S43 and S45 phosphorylation produce similar changes in contractile function, but appear to utilize separate mechanisms. As a result, one residue appears to block the influence of the other on peak shortening when both residues are modified on <50% cTnI. In addition, this work demonstrates that cellular adaptation(s) contributed to a partial return of shortening or re-lengthening rates toward a basal state after more extensive replacement with cTnIS43D or cTnIS45D (Figs. 2B,5). These adaptations included enhanced thick (cMyBP-CS282) and thin (cTnIS23/24) filament phosphorylation (Fig. 5). The environmental perturbation of acidosis similarly enhanced cTnI pS23/24 (Fig. 7). This shared response suggests signaling pathways fine-tune myofilament function in response to internal and external stressors. Differences in pS23/24 levels were removed by phosphatase inhibition (Fig. 6), and there was reduced PP2A methylation associated with cTnI phosphomimetic expression (Fig. 8). Additional protein targets, other phosphatases and/or kinase activity also may contribute to this response, but require further investigation in the future. Regardless of the trigger, this signaling behavior appears to play a role in restoring contractile function back towards the original steady-state. An absence of this signaling response could accelerate the onset of end-stage heart failure when PKC activity [17-20,22] and cTnI S43/S45 phosphorylation [11-14,31,50] are elevated during periods of cardiac dysfunction.
4.1 Mechanistic Insight into the cTnI S43 and S45-mediated Functional Responses
The S43 and S45 residues are positioned on different faces of the H1(I) α helix of cTnI [51], which could result in each residue operating via separate mechanisms. The cTnI pS23/24-induced acceleration of Ca2+ dissociation from TnC6, 7 translates into an accelerated relaxation rate [5,8], and cTnI S43D and S45D may act similarly, given that S23/24D, S43/45E, and S45D (Figs. 2D, S2) each produce rightward shifts in myofilament Ca2+ sensitivity [5,8,24]. Thin filament incorporation of cTnIS43/45D or cTnIS43/45E also accelerates Ca2+ dissociation from cTnC [52]. Disrupted interactions between cTnI S45 and E18 in the N-helix of cTnC are predicted to contribute to this response [53,54]. A similar or separate unknown interaction could be involved in the cTnIS43 mechanism. The current results also indicate the transduction mechanism for cTnIS43 and S45 may involve more than interactions with TnC to alter the Ca2+ dissociation rate. Some TnC mutations also reduce myofilament Ca2+ sensitivity, but differentially affect Ca2+ association versus dissociation in TnC [55]. Additional conformational changes in the C- and N-lobes of cTnC observed with cTnIS43/45, but not cTnI S23/24 phosphomimetics, are consistent with a differential influence on Ca2+ on and off rates [30,54].
Slowed TnC Ca2+ binding also could result in the slowed shortening rates observed with cTnI phosphomimetics (Figs. 2,3), reduced loaded shortening velocity in cTnIAllP myocardium [25], and decreased rate of pressure development in both cTnIAllP and cTnIPKC-P mouse hearts [30,32]. While differential influences on Tn-Ca2+ association and dissociation remain a possible mechanism, the rate limiting step in binding is usually attributed to Ca2+ diffusion, and cTnIS43/45D increased rather than decreased thin filament Ca2+ association [52]. Burkart and colleagues [24] proposed decreased crossbridge detachment rates as an alternative to explain the slower sliding speeds observed with cTnIS43/45E in motility assays. This mechanism would produce slowed re-lengthing, but not necessarily explain other aspects of the functional response (Figs 2,3). Instead, the influence of S43 or S45 may extend beyond direct interactions between cTnI-cTnC, which is in keeping with the stabilized thin filament inactive state observed in biochemical studies with cTnIS45E [56].
4.2 Myocyte Studies Serve as a Bridge for Integration of Earlier Work on cTnIS43/45
Unlike studies on cTnI pS23/24, in vitro myofilament responses do not necessarily predict the in vivo cardiac phenotype produced by cTnIS43/45 phosphomimetics. At first glance, this divergence is suprising given that cTnIS23/24 and cTnIS43/45 phosphomimetics each increase cTnC Ca2+ dissociation rate and reduce myofilament Ca2+ sensitivity of tension [6,7,24,52]. However, a multitude of cardiac phenotypes are observed in genetic animal models designed to test the influence of PKC-targeted cTnI phosphorylation on cardiac performance. There is currently no animal model designed to examine the functional response to S43/45 phosphomimetics alone. Instead, animal models utilize cTnI containing changes in this cluster plus PKC-targeted T144 phosphomimetic and/or a S23/24 substitution [30-32]. Complete myofilament replacement with cTnI phosphomimetics at PKC-targeted cTnI S23/24, S43/45 and T144 in cTnIAllP mice produced only a modest decrease cardiac pressure development rate under basal conditions [30]. In contrast, significant reductions in the rate and amplitude of pressure development, ejection fraction and/or relaxation rate are reported with substantially less cTnI S43/45E and T144E replacement in the cTnIPKC-P or cTnIS23A/S24/43/45D mouse models [31,32]. Neither the differences in myofilament Ca2+ sensitivity between these models nor the substituted residues can fully account for the divergent phenotypes observed in these models. Overall, the distinct phenotypes are difficult to reconcile with the in vitro influence of the cTnI phosphomimetics on Tn or thin filament behavior.
The focus on temporal responses to cTnIS43/45 phosphomimetics in the present study indicates adaptive signaling played a significant role in modifying cellular function, and probably factors into functional differences between these genetic animal models. The heightened cTnI pS23/24 is consistent with the restoration of re-lengthening rate observed on day 4 (Figs. 2,5) [5,8]. In addition, accelerated cross-bridge cycling produced by augmented cMyBP-C phosphorylation is consistent with partial restoration of shortening and/or re-lengthening rate (Figs. 2,5) [42,57]. Thus, adaptive signaling could contribute to the lack of change in developed pressure and relaxation rates observed in cTnIAllP and cTnIS23A/S24/43/45D mouse models, along with additional modifications to other residues and/or proteins [30,31]. The substantial systolic and diastolic impairment observed in isolated, perfused hearts with far less cTnIPKC-P replacement32 than the cTnIAllP mouse [30] is consistent with the direct influence of S43/45 phosphorylation on function. Adaptive signaling may be absent in this model due to the low levels of cTnI phosphomimetic expression, and/or may depend on a neurohormonal or temporal component. While compensatory signaling was not detected in cTnIAllP and cTnIPKC-P mice [30,32], these changes may be difficult to detect in intact hearts due to a host of factors known to influence post-translational modifications [58]. In contrast, the cTnIS23A/S24/43/45D mouse model developed alterations in Ca2+ cycling protein expression and phosphorylation [31]. A remaining question is whether the S23A/S24D substitution produces a different complement of adaptive signaling behavior in this mouse compared to results obtained in the present study (Figs. 3-5). However, the elevated cTnI pS23/24 observed with acidosis and cTnIS43/45 phosphomimetics indicates adaptive signaling is a shared response (Figs. 5,7). This behavior is predicted to be dynamic and could differ between the animal models. Most importantly, our results indicate a thorough exploration of dynamic temporal-, load- , and substitution-dependent adaptations initiated by and within the sarcomere is needed for understanding the earlier animal models.
4.3 The sarcomere as a dynamic signaling scaffold
The development of adaptive phosphorylation indicates the sarcomere acts as dynamic signaling scaffold, in addition to serving as the basic functional unit responsible for cardiac pump performance. Many signaling pathways are known to target myofilament proteins, but the organization of signaling clusters within the sarcomere is not well defined. This idea of a dynamic sarcomere scaffold is now integrated with PKC-mediated cTnI S43 and/or S45 phosphorylation into an initial set of models (Fig. 8C). In model 1, kinase and phosphatase activity are predicted to be evenly matched under physiological conditions (Fig. 8C). Transient environmental and/or neurohormonal signals may briefly tilt the balance toward kinase and/or phosphatase activity near myofilament target proteins, such as cTnI. When activated PKC phosphorylates cTnI S43 or S45 under these conditions, our results indicate they act as a modulatory brake on systolic and diastolic cardiac function. This prediction is consistent with the proposal that this cluster plays a role in beat-to-beat regulation of the heart [24], by returning cardiac pump performance to a basal steady state level. In response to longer duration exposure to environmental stressors and/or sustained sarcomeric protein modifications, decreased phosphatase activity then initiates an adaptive response within the sarcomere (model 2). This response is based on the evidence for reduced phosphatase activity observed with cTnI S43/45 phosphomimetic expression 4 days after gene transfer (Figs. 6,8A,B). The present results do not rule out the possibility that increased kinase activity contributes to this adaptive behavior (Fig. 8C, model 3), but specific kinases remain to be identified. There is a growing recognition destabilization of the balance between kinase and phosphatase activity may contribute to cardiac dysfunction [59,60], and this dynamic modulation within the sarcomere could play an important role during compensated cardiac dysfunction. A loss of adaptive behavior also could contribute to decompensated dysfunction and/or heart failure. Detailed insights into the spatio-temporal organization and balance between sarcomeric kinase and phosphatase activity are now needed to understand the in vivo contribution of adaptive signaling under physiological and pathophysiological conditions.
Supplementary Material
Highlights.
Phosphomimetic cTnIS43D and cTnIS45D individually modulate myocyte function.
The cTnI S43 and S45 phosphomimetics reduce systolic and diastolic function.
Adaptive behavior is initiated in myocytes expressing cTnIS43D and cTnIS45D
Changes in phosphatase activity contribute to this adaptive behavior.
The sarcomere acts as a dynamic scaffold in myocytes.
ACKNOWLEDGEMENTS
The technical assistance of Gail Romanchuk is gratefully acknowledged. This work was supported by National Institutes of Health Grant R01-HL-067254, NIGMS T32 GM007315 (SEL), American Heart Association pre-doctoral award 12PRE8830022 (SEL), and NIH R25 HL108842. Our work utilized the Core Facilities (Morphology and Image Analysis, and Cell and Molecular Biology Cores) of the Michigan Diabetes Research and Training Center (supported by National Institutes of Health Grant P60-DK20572).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
None
LITERATURE CITED
- 1.Solaro RJ, Henze M, Kobayashi T. Integration of troponin I phosphorylation with cardiac regulatory networks. Circ Res. 2013;112:355–366. doi: 10.1161/CIRCRESAHA.112.268672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Solaro RJ, Kobayashi T. Protein phosphorylation and signal transduction in cardiac thin filaments. J Biol Chem. 2011;286:9935–9940. doi: 10.1074/jbc.R110.197731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Layland J, Solaro RJ, Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc Res. 2005;66:12–21. doi: 10.1016/j.cardiores.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 4.Westfall MV. Post-translational modification of troponin. In: Jin J-P, editor. Troponin: Regulator of muscle contraction. Nova Science; New York: 2013. pp. 163–203. [Google Scholar]
- 5.Yasuda S, Coutu P, Sadayappan S, Robbins J, Metzger JM. Cardiac transgenic and gene transfer strategies converge to support an important role for troponin I in regulating relaxation in cardiac myocytes. Circ Res. 2007;101:377–386. doi: 10.1161/CIRCRESAHA.106.145557. [DOI] [PubMed] [Google Scholar]
- 6.Robertson SP, Johnson JD, Holroyde MJ, Kranias EG, Potter JD, Solaro RJ. The effect of troponin I phosphorylation on the Ca2+-binding properties of the Ca2+-regulatory site of bovine cardiac troponin. J Biol Chem. 1982;257:260–263. [PubMed] [Google Scholar]
- 7.Zhang R, Zhao J, Mandveno A, Potter JD. Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circ Res. 1995;76:1028–1035. doi: 10.1161/01.res.76.6.1028. [DOI] [PubMed] [Google Scholar]
- 8.Takimoto E, Soergel DG, Janssen PM, Stull LB, Kass DA, Murphy AM. Frequency- and afterload-dependent cardiac modulation in vivo by troponin I with constitutively active protein kinase A phosphorylation sites. Circ Res. 2004;94:496–504. doi: 10.1161/01.RES.0000117307.57798.F5. [DOI] [PubMed] [Google Scholar]
- 9.Christopher B, Pizarro GO, Nicholson B, Yuen S, Hoit BD, Ogut O. Reduced force production during low blood flow to the heart correlates with altered troponin I phosphorylation. J Muscle Res Cell Motil. 2009;30:111–123. doi: 10.1007/s10974-009-9180-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bodor GS, Oakeley AE, Allen PD, Crimmins DL, Ladenson JH, Anderson PA. Troponin I phosphorylation in the normal and failing adult human heart. Circulation. 1997;96:1495–1500. doi: 10.1161/01.cir.96.5.1495. [DOI] [PubMed] [Google Scholar]
- 11.Zhang P, Kirk JA, Ji W, dos Remedios CG, Kass DA, Van Eyk JE, et al. Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart. Circulation. 2012;126:1828–1837. doi: 10.1161/CIRCULATIONAHA.112.096388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong X, Sumandea CA, Chen YC, Garcia-Cazarin ML, Zhang J, Balke CW, et al. Augmented phosphorylation of cardiac troponin I in hypertensive heart failure. J Biol Chem. 2012;287:848–857. doi: 10.1074/jbc.M111.293258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walker LA, Walker JS, Ambler SK, Buttrick PM. Stage-specific changes in myofilament protein phosphorylation following myocardial infarction in mice. J Mol Cell Cardiol. 2010;48:1180–1186. doi: 10.1016/j.yjmcc.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noguchi T, Hunlich M, Camp PC, Begin KJ, El-Zaru M, Patten R, et al. Thin-filament-based modulation of contractile performance in human heart failure. Circulation. 2004;110:982–987. doi: 10.1161/01.CIR.0000139334.43109.F9. [DOI] [PubMed] [Google Scholar]
- 15.Noland TA, Jr., Kuo JF. Protein kinase C phosphorylation of cardiac troponin I and troponin T inhibits Ca2+-stimulated MgATPase activity in reconstituted actomyosin and isolated myofibrils, and decreases actin-myosin interactions. J Mol Cell Cardiol. 1993;25:53–65. doi: 10.1006/jmcc.1993.1007. [DOI] [PubMed] [Google Scholar]
- 16.Noland TA, Jr., Raynor RL, Jideama NM, Guo X, Kazanietz MG, Blumberg PM, et al. Differential regulation of cardiac actomyosin S-1 MgATPase by protein kinase C isozyme-specific phosphorylation of specific sites in cardiac troponin I and its phosphorylation site mutants. Biochemistry. 1996;35:14923–14931. doi: 10.1021/bi9616357. [DOI] [PubMed] [Google Scholar]
- 17.Bowling N, Walsh RA, Song G, Estridge T, Sandusky GE, Fouts RL, et al. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation. 1999;99:384–391. doi: 10.1161/01.cir.99.3.384. [DOI] [PubMed] [Google Scholar]
- 18.Jalili T, Takeishi Y, Song G, Ball NA, Howles G, Walsh RA. PKC translocation without changes in gαq and PLC-beta protein abundance in cardiac hypertrophy and failure. Am J Physiol Heart Circ Physiol. 1999;277:H2298–H2304. doi: 10.1152/ajpheart.1999.277.6.H2298. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Liu X, Sentex E, Takeda N, Dhalla NS. Increased expression of protein kinase C isoforms in heart failure due to myocardial infarction. Am J Physiol Heart Circ Physiol. 2003;284:H2277–H2287. doi: 10.1152/ajpheart.00142.2002. [DOI] [PubMed] [Google Scholar]
- 20.Bayer AL, Heidkamp MC, Patel N, Porter M, Engman S, Samarel AM. Alterations in protein kinase C isoenzyme expression and autophosphorylation during the progression of pressure overload-induced left ventricular hypertrophy. Mol Cell Biochem. 2003;242:145–152. [PubMed] [Google Scholar]
- 21.Johnsen DD, Kacimi R, Anderson BE, Thomas TA, Said S, Gerdes AM. Protein kinase C isozymes in hypertension and hypertrophy: Insight from shhf rat hearts. Mol Cell Biochem. 2005;270:63–69. doi: 10.1007/s11010-005-3781-x. [DOI] [PubMed] [Google Scholar]
- 22.Belin RJ, Sumandea MP, Allen EJ, Schoenfelt K, Wang H, Solaro RJ, et al. Augmented protein kinase C-α-induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circ Res. 2007;101:195–204. doi: 10.1161/CIRCRESAHA.107.148288. [DOI] [PubMed] [Google Scholar]
- 23.Simonis G, Briem SK, Schoen SP, Bock M, Marquetant R, Strasser RH. Protein kinase C in the human heart: Differential regulation of the isoforms in aortic stenosis or dilated cardiomyopathy. Mol Cell Biochem. 2007;305:103–111. doi: 10.1007/s11010-007-9533-3. [DOI] [PubMed] [Google Scholar]
- 24.Burkart EM, Sumandea MP, Kobayashi T, Nili M, Martin AF, Homsher E, et al. Phosphory-lation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J Biol Chem. 2003;278:11265–11272. doi: 10.1074/jbc.M210712200. [DOI] [PubMed] [Google Scholar]
- 25.Hinken AC, Hanft LM, Scruggs SB, Sadayappan S, Robbins J, Solaro RJ, et al. Protein kinase C depresses cardiac myocyte power output and attenuates myofilament responses induced by protein kinase a. J Muscle Res Cell Motil. 2012;33:439–448. doi: 10.1007/s10974-012-9294-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lester JW, Hofmann PA. Role for PKC in the adenosine-induced decrease in shortening velocity of rat ventricular myocytes. Am J Physiol Heart Circ Physiol. 2000;279:H2685–H2693. doi: 10.1152/ajpheart.2000.279.6.H2685. [DOI] [PubMed] [Google Scholar]
- 27.Pi Y, Kemnitz KR, Zhang D, Kranias EG, Walker JW. Phosphorylation of troponin I controls cardiac twitch dynamics: Evidence from phosphorylation site mutants expressed on a troponin I-null background in mice. Circ Res. 2002;90:649–656. doi: 10.1161/01.res.0000014080.82861.5f. [DOI] [PubMed] [Google Scholar]
- 28.Montgomery DE, Wolska BM, Pyle WG, Roman BB, Dowell JC, Buttrick PM, et al. Alpha-adrenergic response and myofilament activity in mouse hearts lacking PKC phosphorylation sites on cardiac TnI. Am J Physiol Heart Circ Physiol. 2002;282:H2397–H2405. doi: 10.1152/ajpheart.00714.2001. [DOI] [PubMed] [Google Scholar]
- 29.Lang SE, Robinson DA, Wu HC, Herron TJ, Wahr PA, Westfall MV. Myofilament incorporation and contractile function after gene transfer of cardiac troponin I Ser43/45Ala. Arch Biochem Biophys. 2013;535:49–55. doi: 10.1016/j.abb.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakthivel S, Finley NL, Rosevear PR, Lorenz JN, Gulick J, Kim S, et al. In vivo and in vitro analysis of cardiac troponin I phosphorylation. J Biol Chem. 2005;280:703–714. doi: 10.1074/jbc.M409513200. [DOI] [PubMed] [Google Scholar]
- 31.Bilchick KC, Duncan JG, Ravi R, Takimoto E, Champion HC, Gao WD, et al. Heart failure-associated alterations in troponin I phosphorylation impair ventricular relaxation-afterload and force-frequency responses and systolic function. Am J Physiol Heart Circ Physiol. 2007;292:H318–H325. doi: 10.1152/ajpheart.00283.2006. [DOI] [PubMed] [Google Scholar]
- 32.Kirk JA, MacGowan GA, Evans C, Smith SH, Warren CM, Mamidi R, et al. Left ventricular and myocardial function in mice expressing constitutively pseudophosphorylated cardiac troponin I. Circ Res. 2009;105:1232–1239. doi: 10.1161/CIRCRESAHA.109.205427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang R, Zhao J, Potter JD. Phosphorylation of both serine residues in cardiac troponin I is required to decrease the Ca2+ affinity of cardiac troponin C. J Biol Chem. 1995;270:30773–30780. doi: 10.1074/jbc.270.51.30773. [DOI] [PubMed] [Google Scholar]
- 34.Westfall MV, Borton AR. Role of troponin I phosphorylation in protein kinase C-mediated enhanced contractile performance of rat myocytes. J Biol Chem. 2003;278:33694–33700. doi: 10.1074/jbc.M305404200. [DOI] [PubMed] [Google Scholar]
- 35.Sadayappan S, Gulick J, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, et al. Cardiac myosin binding protein-C phosphorylation in a β-myosin heavy chain background. Circulation. 2009;119:1253–1262. doi: 10.1161/CIRCULATIONAHA.108.798983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Helmes M, Lim CC, Liao R, Bharti A, Cui L, Sawyer DB. Titin determines the Frank-Starling relation in early diastole. J Gen Physiol. 2003;121:97–110. doi: 10.1085/jgp.20028652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coutu P, Metzger JM. Genetic manipulation of calcium handling proteins in cardiac myocytes. I. Experimental studies. Am J Physiol Heart Circ Physiol. 2005;288:H601–H612. doi: 10.1152/ajpheart.00424.2004. [DOI] [PubMed] [Google Scholar]
- 38.Westfall MV, Lee AM, Robinson DA. Differential contribution of troponin I phosphorylation sites to the endothelin-modulated contractile response. J Biol Chem. 2005;280:41324–41331. doi: 10.1074/jbc.M506043200. [DOI] [PubMed] [Google Scholar]
- 39.Reitz FB, Pollack GH. Labview virtual instruments for calcium buffer calculations. Comput Methods Programs Biomed. 2003;70:61–69. doi: 10.1016/s0169-2607(01)00196-1. [DOI] [PubMed] [Google Scholar]
- 40.Haim TE, Dowell C, Diamanti T, Scheuer J, Tardiff JC. Independent FHC-related cardiac troponin T mutations exhibit specific alterations in myocellular contractility and calcium kinetics. J Mol Cell Cardiol. 2007;42:1098–1110. doi: 10.1016/j.yjmcc.2007.03.906. [DOI] [PubMed] [Google Scholar]
- 41.Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res. 2012;110:1646–1660. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL. Acceleration of crossbridge kinetics by protein kinase a phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ Res. 2008;103:974–982. doi: 10.1161/CIRCRESAHA.108.177683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mundina-Weilenmann C, Vittone L, Cingolani HE, Orchard CH. Effects of acidosis on phosphorylation of phospholamban and troponin I in rat cardiac muscle. Am J Physiol Cell Physiol. 1996;270:C107–114. doi: 10.1152/ajpcell.1996.270.1.C107. [DOI] [PubMed] [Google Scholar]
- 44.Allen DG, Orchard CH. Myocardial contractile function during ischemia and hypoxia. Circ Res. 1987;60:153–168. doi: 10.1161/01.res.60.2.153. [DOI] [PubMed] [Google Scholar]
- 45.Nayler WG, Ferrari R, Poole-Wilson PA, Yepez CE. A protective effect of a mild acidosis on hypoxic heart muscle. J Mol Cell Cardiol. 1979;11:1053–1071. doi: 10.1016/0022-2828(79)90394-8. [DOI] [PubMed] [Google Scholar]
- 46.Jideama NM, Crawford BH, Hussain AK, Raynor RL. Dephosphorylation specificities of protein phosphatase for cardiac troponin I, troponin T, and sites within troponin T. Int J Biol Sci. 2006;2:1–9. doi: 10.7150/ijbs.2.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deshmukh PA, Blunt BC, Hofmann PA. Acute modulation of PP2A and troponin I phosphorylation in ventricular myocytes: Studies with a novel PP2A peptide inhibitor. Am J Physiol Heart Circ Physiol. 2007;292:H792–H799. doi: 10.1152/ajpheart.00225.2006. [DOI] [PubMed] [Google Scholar]
- 48.Wijnker PJ, Boknik P, Gergs U, Muller FU, Neumann J, dos Remedios C, et al. Protein phosphatase 2A affects myofilament contractility in non-failing but not in failing human myocardium. J Muscle Res Cell Motil. 2011;32:221–233. doi: 10.1007/s10974-011-9261-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang F, Aiello DL, Pyle WG. Cardiac myofilament regulation by protein phosphatase type 1α and CapZ. Biochem Cell Biol. 2008;86:70–78. doi: 10.1139/o07-150. [DOI] [PubMed] [Google Scholar]
- 50.Ruse CI, Willard B, Jin JP, Haas T, Kinter M, Bond M. Quantitative dynamics of site-specific protein phosphorylation determined using liquid chromatography electrospray ionization mass spectrometry. Anal Chem. 2002;74:1658–1664. doi: 10.1021/ac0157122. [DOI] [PubMed] [Google Scholar]
- 51.Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 52.Liu B, Lopez JJ, Biesiadecki BJ, Davis JP. Protein kinase C phosphomimetics alter thin filament Ca2+ binding properties. PLoS One. 2014;9:e86279. doi: 10.1371/journal.pone.0086279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kobayashi T, Dong WJ, Burkart EM, Cheung HC, Solaro RJ. Effects of protein kinase c dependent phosphorylation and a familial hypertrophic cardiomyopathy-related mutation of cardiac troponin I on structural transition of troponin C and myofilament activation. Biochemistry. 2004;43:5996–6004. doi: 10.1021/bi036073n. [DOI] [PubMed] [Google Scholar]
- 54.Finley NL, Rosevear PR. Introduction of negative charge mimicking protein kinase c phosphorylation of cardiac troponin I. Effects on cardiac troponin C. J Biol Chem. 2004;279:54833–54840. doi: 10.1074/jbc.M408304200. [DOI] [PubMed] [Google Scholar]
- 55.Tikunova SB, Liu B, Swindle N, Little SC, Gomes AV, Swartz DR, et al. Effect of calcium-sensitizing mutations on calcium binding and exchange with troponin C in increasingly complex biochemical systems. Biochemistry. 2010;49:1975–1984. doi: 10.1021/bi901867s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mathur MC, Kobayashi T, Chalovich JM. Negative charges at protein kinase C sites of troponin I stabilize the inactive state of actin. Biophys J. 2008;94:542–549. doi: 10.1529/biophysj.107.113944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang L, Ji X, Barefield D, Sadayappan S, Kawai M. Phosphorylation of cMyBP-C affects contractile mechanisms in a site-specific manner. Biophys J. 2014;106:1112–1122. doi: 10.1016/j.bpj.2014.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walker LA, Medway AM, Walker JS, Cleveland JC, Jr., Buttrick PM. Tissue procurement strategies affect the protein biochemistry of human heart samples. J Muscle Res Cell Motil. 2011;31:309–314. doi: 10.1007/s10974-010-9233-6. [DOI] [PubMed] [Google Scholar]
- 59.Hwang H, Robinson D, Rogers JB, Stevenson TK, Lang SE, Sadayappan S, et al. Agonist activated PKCβII translocation and modulation of cardiac myocyte contractile function. Sci Rep. 2013;3:1971. doi: 10.1038/srep01971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yin X, Cuello F, Mayr U, Hao Z, Hornshaw M, Ehler E, et al. Proteomics analysis of the cardiac myofilament subproteome reveals dynamic alterations in phosphatase subunit distribution. Mol Cell Proteomics. 2010;9:497–509. doi: 10.1074/mcp.M900275-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.