

Abstract
Background
Obesity leads to metabolic heart disease (MHD) that is associated with a pathologic increase in myocardial fatty acid (FA) uptake and impairment of mitochondrial function. The mechanism of mitochondrial dysfunction in MHD, which results in oxidant production and decreased energetics, is poorly understood but may be related to excess FAs. Determining the effects of cardiac FA excess on mitochondria can be hindered by the systemic sequelae of obesity. Mice with cardiomyocyte-specific overexpression of the fatty acid transport protein FATP1 have increased cardiomyocyte FA uptake and develop MHD in the absence of systemic lipotoxicity, obesity or diabetes. We utilized this model to assess 1) the effect of cardiomyocyte lipid accumulation on mitochondrial structure and energetic function and 2) the role of lipid-driven transcriptional regulation, signaling, toxic metabolite accumulation, and mitochondrial oxidative stress in lipid-induced MHD.
Methods
Cardiac lipid species, lipid-dependent signaling, and mitochondrial structure / function were examined from FATP1 mice. Cardiac structure and function were assessed in mice overexpressing both FATP1 and mitochondrial-targeted catalase.
Results
FATP1 hearts exhibited a net increase (+12%) in diacylglycerol, with increases in several very long-chain diacylglycerol species (+160-212%, p<0.001) and no change in ceramide, sphingomyelin, or acylcarnitine content. This was associated with an increase in phosphorylation of PKCα and PKCδ, and a decrease in phosphorylation of AKT and expression of CREB, PGC1α, PPARα and the mitochondrial fusion genes MFN1, MFN2 and OPA1. FATP1 overexpression also led to marked decreases in mitochondrial size (-49%, p<0.01), complex II-driven respiration (-28.6%, p<0.05), activity of isolated complex II (-62%, p=0.05), and expression of complex II subunit B (SDHB) (-60% and -31%, p<0.01) in the absence of change in ATP synthesis. Hydrogen peroxide production was not increased in FATP1 mitochondria, and cardiac hypertrophy and diastolic dysfunction were not attenuated by overexpression of catalase in mitochondria in FATP1 mice.
Conclusions
Excessive delivery of FAs to the cardiac myocyte in the absence of systemic disorders leads to activation of lipid-driven signaling and remodeling of mitochondrial structure and function.
Keywords: Mitochondria, Metabolic Heart Disease, Lipid Excess, Obesity
1. Introduction
Metabolic heart disease (MHD) in patients with obesity and diabetes is characterized by cardiac hypertrophy and diastolic dysfunction. In MHD, increased cardiomyocyte uptake of circulating fatty acids (FAs) [1] through cell surface transporters such as FATP1 [2] leads to lipid accumulation in the myocardium [3]. Cardiomyocyte lipid accumulation correlates with diastolic dysfunction [1], impaired mitochondrial energetics [4], and increased oxidative stress [5]. The underlying mechanisms of MHD, including the direct effects of cardiomyocyte FAs on mitochondria, are not well described. It has been hypothesized that increased cardiomyocyte FA uptake could cause mitochondrial dysfunction [5-7]. Possible mechanisms of mitochondrial dysfunction in MHD include respiratory chain uncoupling from ATP synthesis [8], increased reactive oxygen species (ROS) production [6], altered biogenesis [9,10], and/or impaired quality control [11]. However, elucidating the specific role of cardiomyocyte lipid excess in the pathogenesis of MHD can be confounded by systemic metabolic changes that occur with obesity (inflammation, insulin resistance, circulating factors) [12,13]. Thus, this study sought to test the effect of increased cardiomyocyte FA accumulation on mitochondrial structure, function, and oxidant production in the absence of systemic metabolic perturbations.
The hypothesis of this study was that excess cardiomyocyte FA uptake causes mitochondrial dysfunction due to lipid-mediated inhibition of mitochondrial biogenesis and/or energetic function. To address this hypothesis, mice with cardiomyocyte-specific overexpression of the fatty acid transport protein (FATP1) were used as a model of increased cardiomyocyte FA content in the absence of alterations in systemic metabolism. FATP1 mice exhibit increased cardiac FA uptake (4-fold) and utilization (2-fold) and decreased glucose utilization (-50%) [14]. Their cardiac phenotype has been previously described [14]; they develop cardiac hypertrophy and diastolic dysfunction, and thus provide a model of MHD due to cardiac lipid overload. The goals of this study were to 1) determine the effects of FATP1-driven lipid excess on cardiomyocyte mitochondrial structure, function, and oxidant production, and 2) elucidate the mechanisms responsible for MHD in FATP1 hearts by examining toxic lipid accumulation, lipid-driven transcriptional regulation, and oxidative stress on mitochondrial structure and energetic function.
2. Materials and Methods
2.1 Experimental Animals
Cardiomyocyte-specific FATP1 [14] and littermate control mice 5-8 weeks of age were fed normal chow diet and used for all animal studies. Mice with systemic overexpression of catalase in mitochondria (mCAT) [15] were crossed with FATP1 mice and echocardiographic studies were performed at 2-4 months of age. The Institutional Animal Care and Use Committee at Boston University approved this study.
2.2 Lipidomic Analysis
Plasma and hearts from FATP1 and control mice were harvested and flash frozen. Hearts were homogenized in PBS and protein concentration quantified by BCA protein assay. 50μl of the homogenate or plasma was mixed 1:1 with methanol for protein precipitation. The supernatant was spiked with internal standards and was adjusted to 1 ml with 1:1 methanol/water solution for sphingomyelin (SM), ceramide (CER), diacylglycerol (DG) and acylcarnitine (AC). The samples were dried down and reconstituted in the 1:1 methanol/water solution. Samples were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) as previously described [16] at the Metabolomics Facility of the Diabetic Cardiovascular Disease Center at Washington University.
2.3 Immunoblotting
Whole-heart protein lysates made from freeze-clamped hearts using lysis buffer (Hepes pH 7.4 (20mM), β-Glycerol phosphate (50 mM), EGTA (2mM), DTT (1mM), NaF (10mM), NaVO4 (1mM), Triton-X 100 (1%), Glycerol (10%,) and 1 protease inhibitor complete mini tablet-EDTA free/20 ml (Roche)) were subjected to SDS-PAGE with protein content quantified using the Bradford method (Biorad). Following transfer, nitrocellulose membranes were probed with primary antibodies to MFN1 (Neuromab), MFN2 (Sigma-Aldrich AV42420), OPA1 (BD Transduction 612606), phospho-PKCpan (Cell Signaling 9371), phospho-PKC α (Cell Signaling 06-822), total PKC α (EMD 05-154), phospho-PKC δ (Cell Signaling 9376), total PKC δ (Santa Cruz 937), phospho-PKC ε (Santa Cruz sc12355), total PKC ε (EMD 06-991), SDHB (Abcam ab110413), CREB (Pierce MA1-083), GAPDH (Abcam ab9485), and visualized using horseradish peroxidase secondary antibodies with SuperSignal West chemiluinescence substrate and an enhanced chemiluminescence Fujifilm LAS-4000 imager or LI-COR secondary antibodies and the LICOR Odyssey IR imager. Densitometry was quantified using Fujifilm Multigauge or Odyssey software.
2.4 PCR Array and RT-PCR
Frozen hearts were ground under liquid nitrogen and total RNA was extracted with the mirVana miRNA Isolation Kit (Applied Biosystems). Total RNA was treated with DNAse before cDNA synthesis with the High Capacity RNA-to-cDNA Kit (Applied Biosystems). RT2 Profiler PCR arrays were performed per manufacturer instructions (SABiosciences) using the catalogued Mitochondria (PAMM-087Z), Mitochondrial Metabolism (PAMM-008Z) and PPAR Targets (PAMM-149Z) array configurations. Quanitative PCR was performed with TaqMan Universal PCR Master Mix and TaqMan primers (Applied Biosystems) specific for mouse PPARα (Mm00440939_m1), PGC1α (Mm01208835_m1), SDHB (Mm00458272_m1), MFN1 (Mm01289369_m1), MFN2 (Mm00500120_m1), OPA1 (Mm00453873_m1), CREB1 (Mm00501604_m1), HSL (Mm00495359 m1), ATGL (Mm00503040 m1), DGAT1 (Mm00515643 m1), DGAT2 (Mm00499536 m1), and GAPDH (4352339E) using the Applied Biosystems Step One Plus Real Time PCR Systems. Relative mRNA expression was normalized to GAPDH expression using the ΔΔCT method.
2.5 Electron Micrographs
Hearts were harvested, minced (4×4×1 mm), and placed in 2.5% glutaraldehyde/2.0% paraformaldehyde in 0.1 M cacodylate buffer fixative solution overnight. Specimens were then post-fixed in 1.0% osmium tetroxide in 0.15M cacodylate buffer, dehydrated using a graded acetone series, and infiltrated and embedded with an epoxy resin at 65° overnight. Specimens were thick sectioned (200nm) to search for areas of longitudinal fiber orientation, thin sectioned (75nm) using a diamond knife onto a copper grid, stained with 4% aqueous uranyl acetate and lead citrate, and visualized using a CCD camera at 4,000× magnification. Image analysis was performed blinded to genotype with free-hand tracing of mitochondria to estimate size using ImageJ and grid counting to estimate mitochondrial number per visual field.
2.6 Mitochondrial Isolation
Cardiac mitochondria were isolated as previously described [17]. In brief, hearts were harvested, washed and minced in ice-cold relaxation buffer (KCl 100mM, EGTA 5mM, HEPES 5mM pH 7.0 with KOH). Heart pieces were then homogenized in 2ml of HES buffer (HEPES 5mM, EDTA 1mM, Sucrose 250mM pH 7.4) in a Teflon motor-drive homogenizer. The homogenized solution was centrifuged at 500×g for 10 minutes at 4°C. The pellet was discarded and the supernatant was centrifuged at 9000×g for 15 minutes at 4°C. The mitochondrial pellet was resuspended in 50-100μl of HES buffer. Protein was quantified using bicinchoninic acid (BCA, Thermo Scientific) assay.
2.7 Mitochondrial Oxygen Consumption Rate
Oxygen consumption rates were monitored using a Seahorse XF24 oxygen flux analyzer as previously described [17]. Isolated mitochondria were loaded in a 24-well Seahorse plate on ice (5-10μg per well) and 440μl of ice-cold mitochondrial assay solution (MAS: 70mM sucrose, 220mM mannitol, 5mM KH2PO4, 5mM MgCl2, 2mM HEPES, 1mM EGTA, 0.2%BSA fatty acid-free, pH 7.4) plus 10× substrates (complex I: 50mM pyruvate and 50mM malate; complex II: 50mM succinate and 20μm rotenone in MAS) were added on top. The 4 sequential injection ports of the Seahorse cartridge contained the following: Port A- 50μl of 10× substrate and 2.5mM ADP, Port B- 55μl of 20μM oligomycin, Port C- 60μl of 40mM FCCP, Port D- 65μl of 40μM antimycin A. State III was determined after port A injection, state IV after port B, and uncoupled after port C. Antimycin A was used as a control because it blocks the electron transport chain to minimize mitochondrial oxygen consumption. The results are reported as ρmol oxygen/min/μg protein.
2.8 Mitochondrial ATP Synthesis Rate
ATP synthesis rates in isolated heart mitochondria were determined using the luciferin/luciferase-based ATP Bioluminescence Assay Kit CLS II (Roche) with minor modifications [18]. In short, 10 μg of heart mitochondria were suspended in 75μl buffer A (125mM KCl, 10mM Hepes, 5mM MgCl2 and 2mM K2HPO4, pH7.44) to determine complex I-(pyruvate/ malate, 5mM final) or complex II- (succinate, 5mM final) driven ATP synthesis. Following standard practice, succinate-driven ATP generation was measured in the presence of complex I inhibitor rotenone (0.5μM) to avoid the reverse electron transfer effect [19]. Measurements were repeated in the presence of oligomycin, an inhibitor of respiratory complex V to determine the rates of non-mitochondrial ATP production. The background of the assay was determined using mitochondria in the absence of substrates. The measurements for all samples were started simultaneously by adding 75 μl of luciferin/luciferase buffer containing 1mM ADP (0.5mM final). The initial slope of the increase in ATP-supported luciferase chemiluminescence was used to determine the rate of ATP production after subtraction of the background and non-mitochondrial values. Using an ATP standard provided in the kit, the slopes were converted to nmoles/min/mg protein.
2.9 Complex II Activity Assay
Complex II enzyme activity of isolated mitochondria was measured using microplate assay kit (Abcam/Mitosciences ab109908/MS241), as previously described [20]. In this assay kit, complex II was immunocaptured within the wells of the microplate. The production of ubiquinol by complex II was coupled to the reduction of the dye DCPIP (2,6-diclorophenolindophenol) and a decrease in its absorbance at 600 nm was measured spectrophotometrically. The assay was performed in the presence of succinate as a substrate. Enzymatic activity was normalized to mitochondrial protein concentration.
2.10 Mitochondrial ROS Production Rate
Mitochondrial H2O2 production was measured using the Amplex Ultra Red-Horseradish peroxidase method (Invitrogen) as described previously [21] with minor modifications. This assay is based on the H2O2-dependent oxidation of nonfluorescent Amplex Ultra Red (50μM) to fluorescent resorufin red by horseradish peroxidase (2 units/ml). In short, 10μg mitochondria were suspended in 50μl reaction buffer (125mM KCl, 10mM HEPES, 5mM MgCl2, 2mM K2HPO4, pH 7.44) to determine complex I- (pyruvate / malate, 5mM) or complex II- (succinate, 5mM) driven H2O2 production with and without inhibitors (rotenone 2μM, antimycin A, 0.5μM). Mitochondrial H2O2 production was measured after the addition of 50μl of reaction buffer containing horseradish peroxidase and Amplex Ultra Red. Fluorescence was followed at an excitation wavelength of 545nm and an emission wavelength of 590nm for 20 minutes. The slope of the increase in fluorescence was converted to the rate of H2O2 production with a standard curve. All of the assays were performed at 25°C, and results reported as ρmoles/min/mg protein.
2.11 Echocardiographic Measurements
LV dimensions and systolic function were measured in non-anesthetized mice, and diastolic function was assessed by transmitral and tissue Doppler echocardiography in anesthetized mice as we have previously described [22].
2.12 Statistical Analysis
Results are presented as means ± SEM. Unpaired two-tailed t-tests were used for most experiments to determine statistical differences between two means and a P value < 0.05 was considered significant. Statistical analysis of mitochondrial size utilized a non-parametric Mann-Whitney test. Lipidomic data was analyzed using Welch's t-test.
3. Results
3.1 Long-chain diacylglycerol species are increased in FATP1 hearts
FATP1 functions to increase the intracellular accumulation of circulating long-chain FAs, and its overexpression in the cardiomyocyte leads to cardiac hypertrophy and diastolic dysfunction [14] (Supplemental Figure 1). These long-chain FAs can then be metabolized or stored. Several lipid metabolites such as ceramide (CER), acylcarnitine (AC), and diacylglycerol (DG) can function as signaling molecules and contribute to lipotoxicity [7], but the effect of FATP1 overexpression on these lipid classes is not known. Therefore, we determined the effect of FATP1 overexpression on steady state levels of these lipid species in the heart and plasma using LC-MS/MS. In the heart, there was a modest increase in the level of dihydrosphingmyelin (DHSM) 24:0 in FATP1 mice (Figure 1), but there were no differences in other sphingomyelin (SM), AC, or CER species. In contrast, there was dramatic remodeling of DG content. Very long chain DGs (18:2-22:6, 18:1-22:6, and 18:0-20:4) were increased (+160%, +212%, and +56.8%, respectively), while DG 18:2-18:2 and DG 16:0-18:2 were modestly decreased (-53.8% and -32.5%, respectively) – leading to a net increase of +12% in the DG pool. This was associated with a decrease in expression of genes of DG metabolism (HSL, ATGL, DGAT1, and DGAT2). No differences were detected in the plasma levels of CER or SM in FATP1 mice (Supplemental Figure 2), consistent with the lack of systemic effects of cardiac-specific FATP1 overexpression. Thus, increased FA uptake into cardiomyocytes due to overexpression of FATP1 is associated with remodeling of DG content and composition.
Figure 1. FATP1 hearts exhibit no change in acylcarnitine, sphingomyelin, or ceramide content and remodeling of diacylglycerol composition with increased total diacylglycerol content.
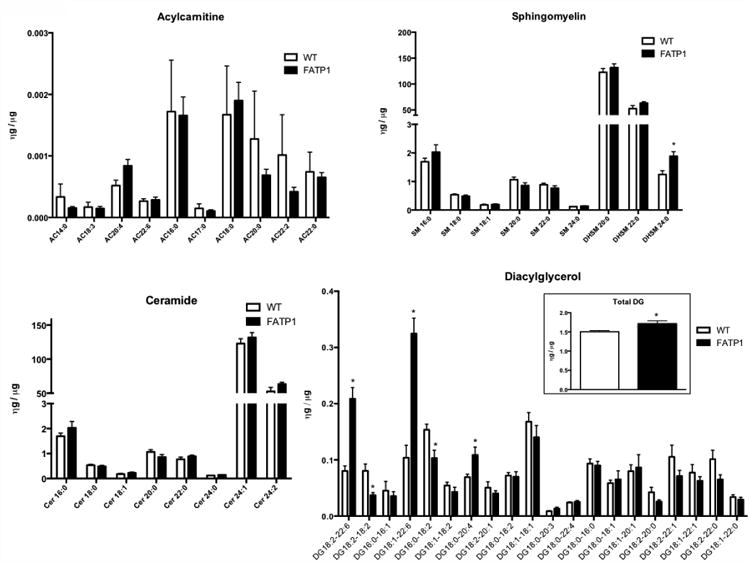
Cardiac acylcarnitine, ceramide, sphingomyelin, and diacylglycerol (DG) content by chain length measured by LC-MS in nanograms relative to heart mass in micrograms. (n = 4-5. * = p<0.001 vs wild-type, error bars are mean ± standard error).
3.2 Increased PKC phosphorylation, decreased AKT phosphorylation, and decreased expression of CREB and PGC1α in FATP1 hearts
A canonical DG-mediated signaling pathway involves activation of protein kinase C (PKC). Preliminary experiments showed that overall PKC activity, as assessed by phosphorylation of a common motif in PKC isoforms (α, β, δ, ε, η and θ) was increased (+381%) in FATP1 hearts (Supplemental Figure 3). Specifically, phosphorylation of the classical isoform PKC α and the novel isoform PKC δ was increased (+31% and +245%, respectively), but phosphorylation of another novel isoform, PKC ε was unchanged (Figure 2A). We next examined potential consequences of increased PKC activity. AKT activation (by phosphorylation) was decreased (-25%) (Figure 2B), and expression of the transcription factor cAMP-responsive element binding protein (CREB) was decreased at both the transcript (-50%) and protein (-50%) level (Figure 2C). CREB is a direct transcriptional activator of the mitochondrial master regulator PGC1 α [23], and in association with its decrease, PGC1α transcript was also decreased (-46%). Thus, in FATP1 hearts, increased PKC activity is associated with inhibition of AKT, down-regulation of CREB, and decreased expression of PGC1α.
Figure 2. FATP1 hearts have increased PKC activation, decreased AKT activation, and decreased expression of CREB and mitochondrial regulator genes.
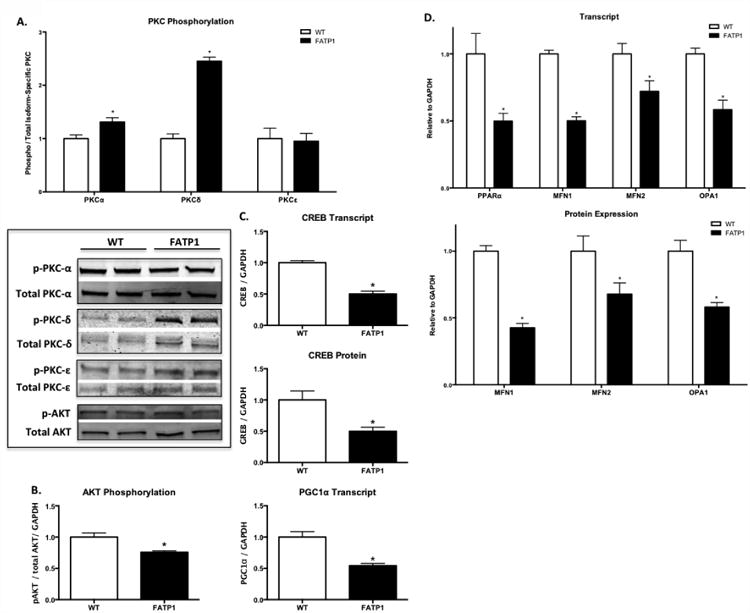
A. Expression of phosphorylated PKC (pan PKC isoform antibody; α, β, δ, ε, η, θ) normalized to GAPDH and phospho-PKCα, phospho-PKCδ, and phospho-PKCε normalized to total PKCα, PKCδ, and PKCε, respectively. B. Phosphorylated AKT relative to total AKT expression normalized to GAPDH. C. CREB transcript and protein expression and PGC1α transcript normalized to GAPDH. D. Transcript level of MFN1, MFN2, and OPA1 normalized to GAPDH. C. Protein expression of MFN1, MFN2, and OPA1 immunoblot relative to GAPDH. Each replicate represents one heart. (n = 7-9. * = p<0.05 vs wild-type, error bars are mean ± standard error).
3.3 FATP1 hearts have decreased PPARα and mitochondrial fusion gene expression
PGC1α is known to regulate PPARα expression [24], which was decreased by 50% in FATP1 hearts (Figure 2D). The PPARα/PGC1α axis induces transcription of fatty acid oxidation genes and upregulates mitochondrial biogenesis and oxidative capacity [25-27]. Likewise, the transcript levels for several PPAR-dependent FA oxidation enzymes were decreased (Supplemental Table 1). In addition to regulation of mitochondrial metabolism, PGC1α mediates the transcription of genes governing mitochondrial fusion (MFN1 and MFN2) [28] to regulate mitochondrial size. FATP1 hearts exhibited decreased transcript levels of all three mitochondrial fusion genes, mitofusin 1 (MFN1, -50%), mitofusin 2 (MFN2, -28%), and optic atrophy 1 (OPA1, -42%), that was associated with decreased protein expression (-58%, -33%, and -50%, respectively, Figure 2D). There was no change in the transcript for the fission regulator DRP1 (data not shown). These alterations in the transcriptional profile of FATP1 hearts suggest a decrease in mitochondrial fusion.
3.4 Increased lipid accumulation and altered mitochondrial morphology in FATP1 hearts
Electron micrographs (EMs) were examined to determine the effect of FATP1 overexpression on subcellular lipid accumulation and mitochondrial morphology. FATP1 hearts showed an 8-fold increase in lipid droplet count (Figure 3A), in agreement with previously published studies showing increased lipid uptake with FATP1 overexpression [14]. Analysis of mitochondrial morphology showed two distinct populations of mitochondria. Mitochondrial size (median mitochondrial area) in the predominant population was decreased by 52% (Figure 3B). FATP1 hearts also had a second distinct population (approximately 1.6% of total mitochondria) of vacuolated mitochondria (Figure 3C) with aberrant cristae structure. Despite the changes in size, mitochondrial volume density was unchanged in FATP1 hearts as assessed by EM (Figure 3D) and by protein expression of the mitochondrial mass marker VDAC1 (Figure 3E).
Figure 3. FATP1 hearts exhibit lipid accumulation, decreased mitochondrial size, debris accumulation, and unchanged mitochondrial volume density and mass.
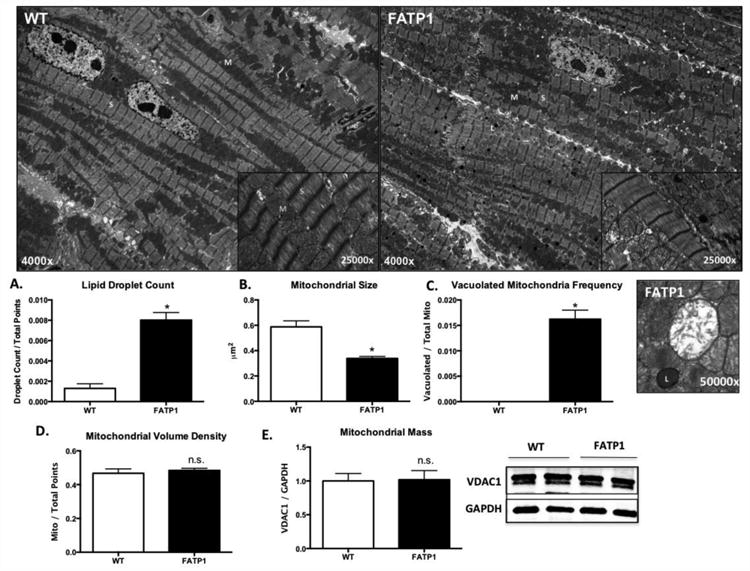
Representative electron micrograph with arrows to mitochondria (M), sarcomere (S), lipid droplet (L). A. Lipid droplet count quantified as lipid droplets divided by total grid intersections counted after elimination of blank space and nucleus. B. Mitochondrial size (median area) quantified by freehand tracing in four standard quadrants per image. C. Vacuolated mitochondrial count quantified by grid intersections containing vacuolated mitochondria relative to total counted mitochondria grid points and representative image. D. Mitochondria volume density as quantified on electron micrograph relative to total counted grid points. E. Mitochondrial mass quantified by immunoblot density of the outer mitochondrial membrane marker VDAC1 relative to the loading control GAPDH. Each replicate was compiled from the mean of three EM images from one heart (n = 4, * = p<0.05 vs wild-type, error bars are mean ± standard error).
3.5 Complex II-driven oxygen consumption is decreased in FATP1 cardiac mitochondria with preserved ATP production
Coincident with the changes in mitochondrial morphology and PGC1α expression, respiration was decreased in mitochondria isolated from FATP1 hearts. Complex I-driven (pyruvate + malate) ADP-stimulated (state III) oxygen consumption was unchanged (Figure 4A). However, complex II-driven (succinate + rotenone) state III respiration was decreased by 33% in FATP1 mitochondria, and this decrease persisted after FCCP-induced uncoupling (-26%). Interestingly, there was a 57% decrease in complex-II driven oligomycin-inhibited respiration (state IV), leading to a 54% increase in the respiratory control ratio (state III/state IV; data not shown) indicative of a decrease in proton leak and increased mitochondrial efficiency.
Figure 4. Mitochondria from FATP1 hearts exhibit decreased complex II oxygen consumption rate, unchanged ATP synthesis rate, and decreased SDHB transcript and protein and decreased complex II activity.
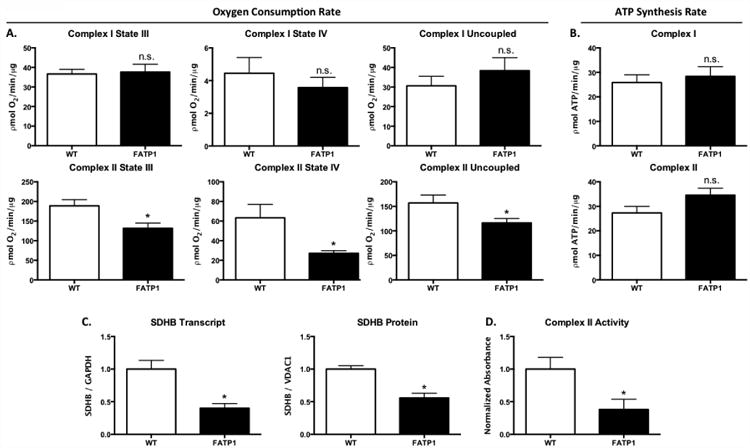
Oxygen consumption rate (OCR) in picomoles of oxygen per minute per microgram of isolated mitochondria and ATP synthesis rate as picomoles of ATP per minute per microgram of isolated mitochondria. A. Complex I substrate = 5mM pyruvate and 5mM malate. B. Complex II substrate = 5mM succinate and 2μM rotenone. C. Transcript level and protein expression of succinate dehydrogenase subunit B (SDHB) normalized to GAPDH and VDAC1, respectively. D. Complex II activity measured as mOD per minute per microgram of protein. (n = 6-8 for OCR and n = 4 for ATP synthesis. n = 5 for qRT-PCR and IB, n = 3-4 for complex activity. * = p<0.05 vs wild-type. N.S. = not significant. Error bars are mean ± standard error).
Decreased complex II-driven oxygen consumption may be due to impaired ATP production and/or increased efficiency of ATP production per mole of oxygen consumed. Measurement of the maximal rate of ATP synthesis from isolated mitochondria showed that FATP1 overexpression did not affect the rate of maximal ATP synthesis with complex I or complex II substrates (Figure 4B). Thus, FATP1 overexpression leads to a decrease in complex II-driven respiration, but preserved capacity to generate ATP suggestive of an increase in ATP synthesis efficiency.
3.6 FATP1 hearts have decreased mitochondrial complex II transcript, protein, and activity
To delineate the mechanism of decreased maximal complex II mitochondrial respiration in FATP1 hearts, complex II expression and activity were measured. Complex II subunit B (SDHB) transcript was decreased by 60% and protein expression was decreased by 31% (Figure 4C). Likewise, complex II activity as measured by the production of ubiquinol by the isolated complex was decreased by 62% (Figure 4D). Thus, FATP1 overexpression causes downregulation of complex II expression resulting in decreased complex II-driven maximal respiration.
3.7 ROS production is unchanged from FATP1 cardiac mitochondria and lack of protective effect of catalase overexpression in FATP1 mice
Cardiac mitochondrial ROS production is increased in models of obesity in which FA oxidation is increased [5]. In mice with diet-induced obesity, we found that the rate of cardiac mitochondrial H2O2 production is increased in association with mitochondrial dysfunction [29]. Contrary to expectations, FATP1 mitochondria showed no change in H2O2 production when supplied with complex I substrate, and there was a trend towards decreased ROS production with complex II substrate (Figure 5A) - suggesting that increased FA uptake and decreased mitochondrial oxygen consumption in FATP1 hearts is not associated with mitochondrial oxidant overproduction.
Figure 5. Mitochondria from FATP1 hearts exhibit unchanged complex II H2O2 production, and FATP1×mCAT mice show no change in cardiac hypertrophy or diastolic dysfunction.
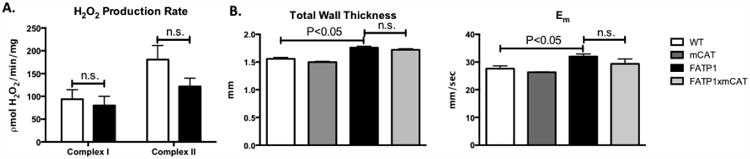
A. H2O2 production rate measured as picomoles of H2O2 per minute per milligram of isolated mitochondria. B. Total wall thickness and myocardial relaxation velocity (Em) from FATP × mCAT hearts by echocardiography. (n = 4 for H2O2 production. n = 3-5 for echocardiography. n.s. = not significant. Error bars are mean ± standard error).
Measurement of isolated mitochondrial ROS production may not fully describe potential sources or subcellular compartments of FA-mediated cardiomyocyte ROS. To further determine whether mitochondrial oxidative damage and/or signaling contribute to the development of FATP1-mediated cardiac dysfunction, mice overexpressing the antioxidant catalase in the mitochondrial matrix (mCAT) were crossed with FATP1 mice, and cardiac structure and function were assessed by echocardiography. Consistent with the lack of increased oxidant production in FATP1 mitochondria, mCAT had no effect on cardiac hypertrophy (total wall thickness) or diastolic function in FATP1 mice (Em) (Figure 5B). These findings suggest that structural remodeling and diastolic dysfunction in FATP1 hearts are not mediated by excessive mitochondrial oxidant production.
4. Discussion
An unresolved question is the role of FA excess, per se, in cardiac mitochondrial dysfunction in MHD [5]. The FATP1 mouse is a unique model of FA-induced cardiomyopathy in which only the cardiomyocyte is exposed to excess FA, thereby allowing investigation of the effect of sustained cardiomyocyte lipid accumulation on mitochondrial structure and function in the absence of confounding systemic factors. This study provides several new findings. First, in FATP1 hearts the intracellular lipid pool is altered with a net increase in DG levels. Second, increased DG levels are associated with activation of PKC, inhibition of CREB and AKT, and decreased expression of the PGC1α/PPARα axis. Third, in FATP1 hearts mitochondrial size is decreased in association with down-regulation of MFN1, MFN2, and OPA1. Fourth, in FATP1 hearts mitochondrial respiration is decreased but maximal ATP production is preserved. Lastly, in FATP1 hearts mitochondrial ROS is not increased, and mitochondrial catalase does not ameliorate the cardiac phenotype.
Structural mitochondrial remodeling
In the FATP1 hearts mitochondrial size is decreased. Lipid excess in other organs has been shown to cause mitochondrial fragmentation [30,31]. A decrease in mitochondrial size may reflect a decrease in fusion due to decreased expression of MFN1, MFN2, and OPA1, all of which were markedly reduced at the transcript and protein level in FATP1 hearts. The transcription of MFN1 and MFN2 in the heart is regulated in part by PGC1α [28,32], which was decreased in FATP1 hearts. Additionally, OPA1 transcription is promoted by AKT [33,34], which was inhibited in FATP1 hearts. Taken together, these findings suggest that sustained cardiomyocyte FA excess leads to down-regulation of transcriptional regulators of mitochondrial fusion. The functional consequences of decreased mitochondrial fusion remain to be determined.
While downregulation of PGC1α expression is typically associated with a decrease in mitochondrial biogenesis, overall mitochondrial mass was unaffected in the FATP1 heart -suggesting a decrease in mitochondrial turnover. Lipid excess in the pancreatic beta cell decreases mitochondrial turnover by impairing autophagy [31]. Additionally, MFN2, which is decreased in the FATP1 heart, may have a role in regulating autophagy [35]. It is possible that the appearance of a population of large vacuolated mitochondria in FATP1 hearts reflects a block in autophagic flux and potentially mitophagy mediated by lipid excess.
Functional mitochondrial remodeling
Structural remodeling was associated with decreased complex II activity as reflected by a decrease in maximal state III oxygen consumption with complex II substrates, and was associated with a decrease in the expression of complex II subunit B that is mediated, at least in part, by a decrease in transcription. It is likely that decreased complex II expression is mediated by the observed decrease in PGC1α, which is known to regulate SDHB expression via NRF1 and NRF2 [36].
Surprisingly, decreased complex II state III oxygen consumption was not associated with a decrease in maximal complex II-mediated ATP production. This dissociation between respiration and ATP production may reflect a decrease in mitochondrial proton leak, as suggested by the concomitant decrease in oligomycin-inhibited state IV respiration. A larger decrease in proton leak, when compared to state III respiration, would increase mitochondrial coupling efficiency and thereby help to maintain ATP production. The observed increase in coupling efficiency in FATP1 mitochondria differs from observations in diabetic mice (db/db and ob/ob) in which there is decreased coupling efficiency [8]. However, in diabetic mice uncoupling is thought to be related to increased mitochondrial ROS, which are not increased in FATP1 mitochondria (discussed below). The mitochondrial isolation we used selectively enriches sub-sarcolemmal mitochondria, making it possible that impaired function of inter-fibrillar mitochondria was missed. However, this is unlikely as sub-sarcolemmal mitochondria appear to be more affected in MHD, while inter-fibrillar populations are relatively spared [37].
Remodeling of the lipid pool and DG-mediated signaling
The major consequence of FATP1 overexpression is increased transport of long chain FAs with no change in steady state triglyceride (TG), cholesterol ester or cardiolipin levels [14]. Therefore, quantitative and/or qualitative changes in the intracellular lipid pool may play a central role in mediating mitochondrial remodeling. FATP1 overexpression had no effect on myocardial content of potentially toxic CER or AC species. However, there was a substantial remodeling of the DG pool with 2-3 fold increases in some very long chain DG (18:2-22:6, 18:1-22:6, and 18:0-20:4) resulting in a 12% increase in net DG content. The myocardial lipid profile in FATP1 mice appears to be less toxic than that of the acyl-CoA synthetase (ACS) overexpressing mouse [38] in which accumulation of DG, TG and CER is associated with a more severe cardiac phenotype with systolic dysfunction and increased mortality.
DG is a major regulator of PKC, which was activated in FATP1 hearts and associated with decreases in CREB expression, AKT phosphorylation and PGC1α expression. Activation of PKC can induce mitochondrial fragmentation and dysfunction [39,40], possibly via inhibition of AKT [41] and/or CREB [42,43]. CREB is a direct transcriptional activator of PGC1 α [23], and AKT regulates mitochondrial biogenesis via activation of the CREB-PGC1 α axis [44]. Thus, increased DG provides a plausible mechanism by which PGC1α is down-regulated in FATP1 hearts.
The coordinated down-regulation of PPARα and PGC1α in FATP1 hearts that we observed is somewhat surprising because PPARα promotes lipid utilization [45], which is increased in the FATP1 heart [14]. In contrast to the FATP1 mouse, in diabetic ob/ob and db/db mice the changes in cardiac PPARα and PGC1α are discordant, with increased PPARα and decreased PGC1α expression. This is associated with decreased mitochondrial function (as in the FATP1 heart) but also with increased ROS production, increased uncoupling, and impaired cardiac function [37,46]. Likewise, cardiomyocyte overexpression of PPARα has pathologic consequences [47], and mimics the diabetic phenotype [48,49]. Since in the FATP1 heart ATP production was preserved, it appears that the coordinated decrease in PPARα and PGC1α in the FATP1 heart leads to mitochondrial remodeling that is of benefit in the setting of nutrient excess.
Role of mitochondrial ROS
Mitochondrial ROS are increased in other models of MHD [5,29] and overexpression of antioxidants can ameliorate pathologic cardiac remodeling in diabetic mice [50-53] and protect mitochondrial structure and function [54,55]. Surprisingly, mitochondrial H2O2 production was not increased in FATP1 hearts. Furthermore, transgenic overexpression of mitochondrial-targeted catalase, which attenuates cardiac oxidative stress in other models [15,56], did not exert a beneficial effect on the cardiac phenotype in FATP1 mice. Thus, ROS do not appear to play a role in mediating the cardiac phenotype in FATP1 mice.
Conclusion
Overexpression of FATP1 in the heart leads to DG accumulation, altered intracellular signaling, coordinated down-regulation of PPARα/PGC1α, and both structural and functional mitochondrial remodeling (Figure 6). There are several noteworthy divergences from other models of MHD related to diabetes or altered lipid metabolism. In particular, mitochondrial remodeling in response to cardiac myocyte-specific FA excess, per se, leads to compensated function with sustained ability to generate ATP in the absence of pathologic ROS production. These findings suggest that systemic factors beyond those related to increased FA levels play an important role in mediating the impaired mitochondrial function in MHD.
Figure 6. Model of effect of FATP1 overexpression on mitochondrial structure and function.
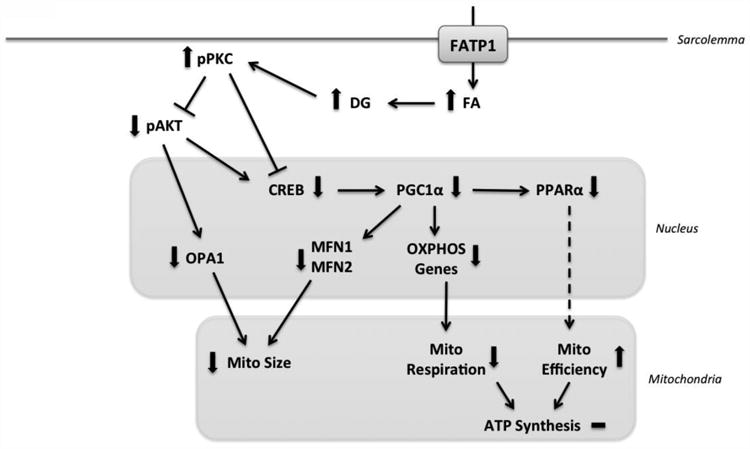
FATP1 overexpression in the cardiomyocyte leads to fatty acid (FA) excess, which causes diacylglycerol (DG) accumulation and protein kinase C activation (pPKC), which inhibits CREB and AKT and leads to decreased expression of PGC1α, MFN1, MFN2 and OPA1 to decrease mitochondrial size and respiration.
Supplementary Material
Highlights.
We examine if cardiomyocyte lipid excess leads to mitochondrial dysfunction.
Lipid excess causes mitochondrial remodeling in FATP1 transgenic mice.
FATP1 hearts have decreased mitochondrial size and respiration, unchanged ATP.
ROS is not increased in FATP1 mitochondria.
Acknowledgments
This work was supported by NIH K08 HL109158 (EJM), NIH R01 HL064750 (WSC), NIH N01 HV28178 (WSC), NHMRC (Australia) CJ Martin Fellowship APP1037603 (AS), RACP Marjorie Hooper Fellowship (AS), Boston University Evans Center Fellowship (ML), NIH P30 DK020579 (Washington University Diabetes Research Center), NIH P20 HL113444 (JES)
Abbreviations
- FATP1
Fatty acid transport protein 1
- FA
Fatty acids
- MHD
Metabolic heart disease
- ROS
Reactive oxygen species
- PPARα
Peroxisome proliferator-activated receptor α
- PGC1α
PPARγ coactivator-1 α
- DG
Diacylglycerol
- CREB
Cyclic AMP responsive element binding protein
- PKC
Protein kinase C
- ETC
Electron transport chain
- ATP
Adenosine triphosphate
- ADP
Adenosine diphosphate
- SM
Sphingomyelin
- CER
Ceramide
- AC
Acyl carnitine
- EM
Electron micrographs
- SDHB
Succinate dehydrogenase subunit B
- mCAT
Mitochondrial catalase
- ACS
Acyl-CoA synthetase
Footnotes
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Leichman JG, Aguilar D, King TM, Vlada A, Reyes M, Taegtmeyer H. Association of plasma free fatty acids and left ventricular diastolic function in patients with clinically severe obesity. Am J Clin Nutr. 2006;84:336–41. doi: 10.1093/ajcn/84.1.336. [DOI] [PubMed] [Google Scholar]
- 2.García-Rúa V, Otero MF, Lear PV, Rodríguez-Penas D, Feijóo-Bandín S, Noguera-Moreno T, et al. Increased expression of fatty-acid and calcium metabolism genes in failing human heart. PLoS ONE. 2012;7:e37505. doi: 10.1371/journal.pone.0037505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, et al. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. Faseb J. 2004;18:1692–700. doi: 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- 4.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–51. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 5.Boudina S, Abel ED. Diabetic Cardiomyopathy Revisited. Circulation. 2007;115:3213–23. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 6.Wende AR, Abel ED. Lipotoxicity in the heart. Biochimica Et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2010;1801:311–9. doi: 10.1016/j.bbalip.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldberg IJ, Trent CM, Schulze PC. Lipid Metabolism and Toxicity in the Heart. Cell Metabolism. 2012;15:805–12. doi: 10.1016/j.cmet.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boudina S, Abel ED. Mitochondrial uncoupling: a key contributor to reduced cardiac efficiency in diabetes. Physiology (Bethesda) 2006;21:250–8. doi: 10.1152/physiol.00008.2006. [DOI] [PubMed] [Google Scholar]
- 9.Ren J, Pulakat L, Whaley-Connell A, Sowers JR. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J Mol Med. 2010;88:993–1001. doi: 10.1007/s00109-010-0663-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finck BN, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1(PGC-1) regulatory cascade in cardiac physiology and disease. Circulation. 2007;115:2540–8. doi: 10.1161/CIRCULATIONAHA.107.670588. [DOI] [PubMed] [Google Scholar]
- 11.Chen L, Knowlton AA. Mitochondrial dynamics in heart failure. Congest Heart Fail. 2011;17:257–61. doi: 10.1111/j.1751-7133.2011.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ge F, Hu C, Hyodo E, Arai K, Zhou S, Lobdell H, et al. Cardiomyocyte triglyceride accumulation and reduced ventricular function in mice with obesity reflect increased long chain Fatty Acid uptake and de novo Fatty Acid synthesis. J Obes. 2012;2012:205648. doi: 10.1155/2012/205648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan MC, Arany Z. The many roles of PGC-1 α in muscle - recent developments. Metab Clin Exp. 2014;63:441–51. doi: 10.1016/j.metabol.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiu HC, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, et al. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. 2005;96:225–33. doi: 10.1161/01.RES.0000154079.20681.B9. [DOI] [PubMed] [Google Scholar]
- 15.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–11. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 16.Fan M, Sidhu R, Fujiwara H, Tortelli B, Zhang J, Davidson C, et al. Identification of Niemann-Pick C1 disease biomarkers through sphingolipid profiling. J Lipid Res. 2013;54:2800–14. doi: 10.1194/jlr.M040618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liesa M, Luptak I, Qin F, Hyde BB, Sahin E, Siwik DA, et al. Mitochondrial Transporter ATP Binding Cassette Mitochondrial Erythroid Is a Novel Gene Required for Cardiac Recovery After Ischemia/Reperfusion. Circulation. 2011;124:806–13. doi: 10.1161/CIRCULATIONAHA.110.003418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mansouri A, Muller FL, Liu Y, Ng R, Faulkner J, Hamilton M, et al. Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind-limb skeletal muscle during aging. Mech Ageing Dev. 2006;127:298–306. doi: 10.1016/j.mad.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 19.Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–65. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li FCH, Yen JC, Chan SHH, Chang AYW. Bioenergetics failure and oxidative stress in brain stem mediates cardiovascular collapse associated with fatal methamphetamine intoxication. PLoS ONE. 2012;7:e30589. doi: 10.1371/journal.pone.0030589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muller FL, Liu Y, Abdul-Ghani MA, Lustgarten MS, Bhattacharya A, Jang YC, et al. High rates of superoxide production in skeletal-muscle mitochondria respiring on both complex I- and complex II-linked substrates. Biochem J. 2008;409:491–9. doi: 10.1042/BJ20071162. [DOI] [PubMed] [Google Scholar]
- 22.Qin F, Siwik DA, Luptak I, Hou X, Wang L, Higuchi A, et al. The polyphenols resveratrol and S17834 prevent the structural and functional sequelae of diet-induced metabolic heart disease in mice. Circulation. 2012;125:1757–64. S1–6. doi: 10.1161/CIRCULATIONAHA.111.067801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–83. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- 24.Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Molecular and Cellular Biology. 2000;20:1868–76. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huss JM, Torra IP, Staels B, Giguère V, Kelly DP. Estrogen-related receptor alphadirects peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Molecular and Cellular Biology. 2004;24:9079–91. doi: 10.1128/MCB.24.20.9079-9091.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huss JM, Kopp RP, Kelly DP. Peroxisome proliferator-activated receptor coactivator-1 alpha (PGC-1 alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1 alpha. J Biol Chem. 2002;277:40265–74. doi: 10.1074/jbc.M206324200. [DOI] [PubMed] [Google Scholar]
- 27.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–56. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin OJ, Lai L, Soundarapandian MM, Leone TC, Zorzano A, Keller MP, et al. A Role for Peroxisome Proliferator-Activated Receptor γ Coactivator-1 in the Control of Mitochondrial Dynamics During Postnatal Cardiac Growth. Circulation Research. 2014;114:626–36. doi: 10.1161/CIRCRESAHA.114.302562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sverdlov AL, Elezaby A, Behring JB, Bachschmid MM, Luptak I, Tu VH, et al. High fat, high sucrose diet causes cardiac mitochondrial dysfunction due in part to oxidative post-translational modification of mitochondrial complex II. Journal of Molecular and Cellular Cardiology. 2014 doi: 10.1016/j.yjmcc.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Molina AJA, Wikstrom JD, Stiles L, Las G, Mohamed H, Elorza A, et al. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes. 2009;58:2303–15. doi: 10.2337/db07-1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Las G, Serada SB, Wikstrom JD, Twig G, Shirihai OS. Fatty acids suppress autophagic turnover in β-cells. J Biol Chem. 2011;286:42534–44. doi: 10.1074/jbc.M111.242412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soriano FX, Liesa M, Bach D, Chan DC, Palacín M, Zorzano A. Evidence for amitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-gamma coactivator-1 alpha, estrogen-related receptor-alpha, and mitofusin 2. Diabetes. 2006;55:1783–91. doi: 10.2337/db05-0509. [DOI] [PubMed] [Google Scholar]
- 33.Parra V, Verdejo HE, Iglewski M, Del Campo A, Troncoso R, Jones D, et al. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFκB-Opa-1 signaling pathway. Diabetes. 2014;63:75–88. doi: 10.2337/db13-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Din S, Mason M, Völkers M, Johnson B, Cottage CT, Wang Z, et al. Pim-1 preserves mitochondrial morphology by inhibiting dynamin-related protein 1 translocation. Proc Natl Acad Sci USa. 2013;110:5969–74. doi: 10.1073/pnas.1213294110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao T, Huang X, Han L, Wang X, Cheng H, Zhao Y, et al. Central role of mitofusin 2 in autophagosome-lysosome fusion in cardiomyocytes. J Biol Chem. 2012;287:23615–25. doi: 10.1074/jbc.M112.379164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen JQ, Cammarata PR, Baines CP, Yager JD. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochimica Et Biophysica Acta. 2009;1793:1540–70. doi: 10.1016/j.bbamcr.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dabkowski ER, Baseler WA, Williamson CL, Powell M, Razunguzwa TT, Frisbee JC, et al. Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterationsin spatially distinct mitochondrial proteomes. AJP: Heart and Circulatory Physiology. 2010;299:H529–40. doi: 10.1152/ajpheart.00267.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, et al. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813–22. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nowak G, Bakajsova D, Samarel AM. Protein kinase C-epsilon activation induces mitochondrial dysfunction and fragmentation in renal proximal tubules. Am J Physiol Renal Physiol. 2011;301:F197–208. doi: 10.1152/ajprenal.00364.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patergnani S, Marchi S, Rimessi A, Bonora M, Giorgi C, Mehta KD, et al. PRKCB/protein kinase C, beta and the mitochondrial axis as key regulators of autophagy. Autophagy. 2013;9:1367–85. doi: 10.4161/auto.25239. [DOI] [PubMed] [Google Scholar]
- 41.Motley ED, Kabir SM, Eguchi K, Hicks AL, Gardner CD, Reynolds CM, et al. Protein kinase C inhibits insulin-induced Akt activation in vascular smooth muscle cells. Cell Mol Biol (Noisy-Le-Grand) 2001;47:1059–62. [PubMed] [Google Scholar]
- 42.Mehta KD. Emerging role of protein kinase C in energy homeostasis: A brief overview. World J Diabetes. 2014;5:385–92. doi: 10.4239/wjd.v5.i3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Drosatos K, Bharadwaj KG, Lymperopoulos A, Ikeda S, Khan R, Hu Y, et al. Cardiomyocyte lipids impair β-adrenergic receptor function via PKC activation. Am J Physiol Endocrinol Metab. 2011;300:E489–99. doi: 10.1152/ajpendo.00569.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, He L, Zeng N, Sahu D, Cadenas E, Shearn C, et al. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) signaling regulates mitochondrial biogenesis and respiration via estrogen-related receptor α (ERRα) J Biol Chem. 2013;288:25007–24. doi: 10.1074/jbc.M113.450353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–38. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- 46.Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112:2686–95. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 47.Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, et al. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci USa. 2003;100:1226–31. doi: 10.1073/pnas.0336724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duncan JG. Peroxisome proliferator activated receptor-alpha (PPARα) and PPARgamma coactivator-1 alpha (PGC-1α) regulation of cardiac metabolism in diabetes. Pediatr Cardiol. 2011;32:323–8. doi: 10.1007/s00246-011-9889-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aubert G, Vega RB, Kelly DP. Perturbations in the gene regulatory pathways controlling mitochondrial energy production in the failing heart. Biochimica Et Biophysica Acta (BBA) - Molecular Cell Research. 2013;1833:840–7. doi: 10.1016/j.bbamcr.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shen X, Zheng S, Metreveli NS, Epstein PN. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes. 2006;55:798–805. doi: 10.2337/diabetes.55.03.06.db05-1039. [DOI] [PubMed] [Google Scholar]
- 51.Cai L, Wang Y, Zhou G, Chen T, Song Y, Li X, et al. Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J Am Coll Cardiol. 2006;48:1688–97. doi: 10.1016/j.jacc.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 52.Ye G, Metreveli NS, Donthi RV, Xia S, Xu M, Carlson EC, et al. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes. 2004;53:1336–43. doi: 10.2337/diabetes.53.5.1336. [DOI] [PubMed] [Google Scholar]
- 53.Liang Q, Carlson EC, Donthi RV, Kralik PM, Shen X, Epstein PN. Overexpression of metallothionein reduces diabetic cardiomyopathy. Diabetes. 2002;51:174–81. doi: 10.2337/diabetes.51.1.174. [DOI] [PubMed] [Google Scholar]
- 54.Dai DF, Rabinovitch P. Mitochondrial oxidative stress mediates induction of autophagy and hypertrophy in angiotensin-II treated mouse hearts. Autophagy. 2011;7:917–8. doi: 10.4161/auto.7.8.15813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dai DF, Hsieh EJ, Liu Y, Chen T, Beyer RP, Chin MT, et al. Mitochondrial proteome remodelling in pressure overload-induced heart failure: the role of mitochondrial oxidative stress. Cardiovascular Research. 2012;93:79–88. doi: 10.1093/cvr/cvr274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–97. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.