

Abstract
Background
An imbalance between excitation and inhibition in the developing central nervous system may result in a pathophysiological outcome. We investigated he mechanistic roles of endocrine activity and gamma-aminobutyric acid type A receptor (GABAAR)-mediated excitation in electroencephalographic seizures caused by the GABAAR-selective anesthetic propofol in neonatal rats.
Methods
Postnatal day 4–6 Sprague Dawley rats underwent a minor surgical procedure to implant electrodes to measure electroencephalographic activity for 1 h before and 1 h after intraperitoneal administration of propofol (40 mg kg−1). Various treatments were administered 15 min before administration of propofol.
Results
Episodes of electroencephalographic seizures and persistent low-amplitude spikes occurred during propofol anesthesia. Multifold increases in serum levels of corticosterone (t(10) = −5.062; P= 0.0005) and aldosterone (t(10) = −5.069; P= 0.0005) were detected 1 h after propofol administration in animals that underwent experimental manipulations identical to those used to study electroencephalographic activity. Pretreatment with bumetanide, the Na+–K+–2Cl– co-transporter inhibitor, which diminishes GABAAR-mediated excitation, eliminated both seizure and spike electroencephalographic activities caused by propofol. Mineralocorticoid and glucocorticoid receptor antagonists, RU 28318 and RU486, depressed electroencephalographic seizures but did not affect the spike electroencephalographic effects of propofol. Etomidate, at a dose sufficient to induce loss of righting reflex, was weak at increasing serum corticosteroid levels and eliciting electroencephalographic seizures. Etomidate given to corticosterone-pretreated rat pups further increased the total duration of electroencephalographic seizures caused by administration of exogenous corticosterone (t(21) = −2.512, P = 0.0203).
Conclusions
Propofol increases systemic corticosteroid levels in neonatal rats, which along with GABAAR-mediated excitation appear to be required for propofol-induced neonatal electroencephalographic seizures. Enhancement of GABAAR activity alone may not be sufficient to elicit neonatal electroencephalographic seizures.
Introduction
The exact mechanisms how neonatal exposure to general anesthetics may affect brain development are unclear. Animal studies indicate that anesthetics are especially harmful if administered at an early postnatal age. In rodents this window of brain vulnerability spans approximately the first 2 postnatal weeks.1 These first 2 weeks of life in rodents are characterized structurally by extensive postnatal neurogenesis and synaptogenesis and functionally by fundamental differences in cellular physiology. One unique property of the brain during this early life period is its increased excitability, which plays an important role regulating signaling pathways that control many developmental processes, including neurogenesis and synaptogenesis.2–4 This normal developmental increased excitability is supported not only by the highest number of excitatory cortical and hippocampal synapses, but also by excitatory effects of the main and otherwise inhibitory neurotransmitter, γ-aminobutyric acid (GABA).5 In immature hippocampal and cortical neurons the intracellular concentration of Cl−, which is the main charge carrier through the GABA type A receptor (GABAAR) channels, is increased due to the relatively high expression of the Na+-K+-2Cl− (NKCC1) Cl− importer and the relatively low expression of the K+-2Cl− (KCC2) Cl− exporter. Consequently, the resulting transmembrane gradient for Cl− supports outward depolarizing Cl− currents upon activation of GABAAR channels. An abnormal increase in GABAAR-mediated excitation is associated with developmental abnormalities.6 In agreement with this we found that bumetanide, that reduces GABAAR-mediated excitation by inhibiting NKCC1 activity,5–8 alleviated developmental side effects of sevoflurane and isoflurane in neonatal rats, including electroencephalographic (EEG) seizures.9–11 Recently, Lim et al. reported that bumetanide prevented post-sevoflurane hyperexcitatory behavior in postnatal day (P) 5 rats.12 Koyama et al. found that bumetanide diminished excitatory and increased sedative effects of the GABAAR-selective anesthetic midazolam in P7, but not in P28 rats.13
We have observed that exposure of neonatal rats to sevoflurane and isoflurane (unpublished observations) was accompanied by a prominent increase in serum levels of the mineralocorticoid hormone, aldosterone and that exogenous aldosterone, administered at high doses, further enhanced EEG seizures caused by sevoflurane.10 Aldosterone together with the glucocorticoids: corticosterone (in rodents) or cortisol (in humans), are corticosteroid hormones produced in the adrenal cortex. The 2 hormones share similar synthetic pathways and the same mineralocorticoid receptors (MR) mediating their actions. Corticosterone acts in the brain either through the high-affinity MRs or the low-affinity glucocorticoid receptors (GR) initiating slow gene transcription-mediated and rapid non-genomic effects. Because of the higher concentration of corticosterone than aldosterone, the majority of MRs in the brain are occupied by corticosterone.14,15 Corticosterone produces a number of proexcitatory effects, such as an increase of presynaptic glutamate release, inhibition of glutamate uptake, induction of expression of the N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors.16,17 Here, using the GABAAR-selective general anesthetic propofol, we tested the hypothesis that both, GABAAR-mediated excitation and increase in corticosteroid levels are involved in mediation of neonatal EEG seizures caused by general anesthetics, whose mechanisms of action include enhancement of GABAAR activity.
Materials and Methods
Animals
All experimental procedures were approved by the University of Florida Institutional Animal Care and Use Committee. Sprague Dawley rats were studied. To control for litter variability we used several pups from each litter for different treatment conditions. At the beginning of each experiment the pups were determined to be well nourished as judged by their stomachs being full of milk (detectable through the transparent abdominal wall). Multiple sets of animals were used in a majority of the experiments.
Electroencephalogram recording
EEG recordings were performed in a thermostated chamber (+37 °C) with a continuous supply of oxygen (1.5 l/min). To study the effects of propofol on cortical electrical activity in rat pups ranging from P4 to P6 and from P17 to P18, animals were instrumented for EEG recording as described previously.9–11 In brief, during a 12–15 min minor surgical procedure performed under isoflurane anesthesia (1.6–2.0%), 4 electrodes were implanted bilaterally in occipital and frontal regions of the rat pup’s scull with the left frontal electrode serving as the reference electrode. Continuous EEG recordings were performed using an EEG/electromyogram system (Pinnacle Technology, Lawrence, KS). Acquisition of the EEG was performed using Sirenia software (Pinnacle Technology). Sampling interval per signal was 200 μs (5 kHz). Sirenia Score (Pinnacle Technology), Clampfit 9.2 (Axon Instruments, Union City, CA) and MiniAnalysis (Synaptosoft Inc. Fort Lee, NJ) programs were used for EEG data analysis. Data were filtered offline using a bandpass Bessel (8-pole) 0.04–56-Hz filter.9 Root mean square (RMS) of the EEG was computed for 2.56 s segments. We have previously verified that no obvious differences in EEG activity were detected when EEG electrode implantation was done either immediately before or 1 to 2 days before the start of EEG recording.10 Therefore, in this study the EEG recordings started after recovering from anesthesia for electrode implantation and lasted for 1 hour before and for 1 hour after intraperitoneal injection of 40 mg/kg propofol. At this dose of propofol the rat pups lost their righting reflex in 84.6± 19.59 s (n=4) and did not respond to handling 1 hour after administration of the anesthetic. All animals were without cyanosis and breathing regularly. Our protocol with propofol is a shorter version of the anesthesia protocol originally described by Briner et al. in neonatal rats consisting of 6 injections of propofol (40 mg/kg intraperitoneally for induction for the first 60 min and then 20 mg/kg/h intraperitoneally every h for 6 h in total).18 Neither Briner et al. nor other authors using a single injection of propofol at 75 mg/kg intraperitoneally detected significant changes in blood gases or glucose in neonatal rats.18,19
In order to study the roles of GABAAR-mediated excitation and corticosteroid receptors in the effects of propofol, separate groups of P4-P6 rats received: 1) the NKCC1 inhibitor, bumetanide (1.82 mg/kg, intraperitoneally); 2) the MR antagonist RU28318 (10 mg/kg, intraperitoneally); and 3) the GR antagonist RU486 (10 mg/kg, intraperitoneally), 15 min before intraperitoneal injection of propofol, whereas control animals received either equal volumes of saline or dimethyl sulfoxide vehicle. In order to obtain additional evidence of involvement of corticosteroids in generation of the excitatory EEG patterns in neonatal rats, other experimental groups received corticosterone only (0.2 mg/kg, intraperitoneally), etomidate only (8 mg/kg, intraperitoneally) and corticosterone (0.2 mg/kg, intraperitoneally) 15 min before administration of etomidate (8 mg/kg, intraperitoneally). This dose of corticosterone (0.2 mg/kg) is in the range of glucocorticoid doses administered to children in the early postnatal period to alleviate respiratory distress syndrome and to modulate the inflammatory response associated with cardiopulmonary bypass.20,21 The EEG effect of etomidate was tested because it inhibits the adrenal synthesis of corticosteroids, but enhances GABAAR activity. It took 58.4± 8.68 s (n=4) to observe loss of righting reflex by the P4–P6 rats injected with etomidate (8 mg/kg, intraperitoneally). To determine a dose for etomidate that would give similar levels of anesthesia, we used a study in adult rats comparing propofol and etomidate requirements to achieve similar levels of burst suppression.22 This study determined that the dose for etomidate was on average 1-quarter or slightly less than the corresponding propofol dose. Based on this information, a ratio of 1:5 for etomidate and propofol was chosen. It is important to stress that etomidate was used in this study as a drug that enhances GABAAR activity and interferes with synthesis of corticosteroids. It was not a goal of this study to compare the effects of propofol and etomidate at equipotent doses.
EEG patterns characterized by an amplitude at least 3 times higher than baseline and rhythmic (>2 Hz) activity that lasted for at least 3 s and abruptly reverted to baseline were defined as seizure-like EEG patterns. In most cases these patterns start as high-frequency low-amplitude activity that developed to increased amplitude and decreased frequency and then abruptly reverted to baseline activity.9–11 Continuous isolated spikes that appeared in EEGs recorded in anesthetized rat pups were not considered EEG seizures and were analyzed separately. The investigators analyzing the EEGs were blinded to the experimental conditions and all EEGs were reviewed by 3 independent reviewers. About 5% of animals exhibited episode(s) of seizure-like EEG patterns before the start of anesthesia and were not included in the data analysis. The sample sizes in this study were based on previous experience with the same experimental techniques.9–11
Measurement of serum corticosterone and aldosterone
In order to assess serum levels of corticosterone and aldosterone in conditions identical to those used to study the EEG effects of propofol and etomidate, blood samples were collected 1 h after administration of propofol or etomidate to a separate group of animals that underwent surgery for electrode implantation and 1 h separation from the dams before anesthetic administration. Serum corticosterone and aldosterone were measured using commercial ELISA kits (Cayman Chemical Company, Ann Arbor, USA) following the manufacturer’s instructions.
Drugs
Propofol and etomidate were purchased from APP Pharmaceuticals, LLC (Schaumburg, IL) and Hospira, Inc. (Lake Forest, IL), respectively. Intralipid, corticosterone and RU486 were acquired from Sigma-Aldrich (St. Louis, MO). RU28318 was purchased from R&D Systems, Inc. (Minneapolis, MN). Bumetanide (Ben Venue Laboratories, Inc., Bedford, OH) was purchased from Bedford Laboratories™ (Bedford, OH).
Statistical Analysis
SigmaPlot 12.5 software (Systat Software, Inc., Point Richmond, CA) was used for statistical analyses. Single comparisons were tested using the t test. All comparisons were run as two-tailed tests. The normality assumption test was performed using the Shapiro–Wilk test with the P value set at 0.05. Data for properties of EEG seizures in the corticosterone only and corticosterone plus etomidate groups were log transformed before analysis. The numerical values are expressed as mean ± S.E. A P ≤ 0.05 was considered significant.
Results
Propofol caused hyperexcitatory electroencephalographic activity and increased serum levels of corticosteroids
Analysis of the EEG activity of P4–P6 rats after administration of propofol revealed patterns of EEG seizures in 7 of 10 studied animals (Fig. 1A–C). These were similar to those previously seen during anesthesia with sevoflurane or isoflurane.10,11 The duration of individual propofol-caused episodes of EEG seizures ranged from 4 to 61 s. In addition to seizure-like EEG activity, the post-propofol administration period was characterized by the presence of continuous isolated spikes in EEGs (Fig. 1A,B,D). The frequency of the propofol-caused EEG spikes ranged from 0.068 Hz to 1.45 Hz (Fig. 1D). Propofol caused neither EEG seizures nor spikes in P17–P18 rats (n=4).
Figure 1.
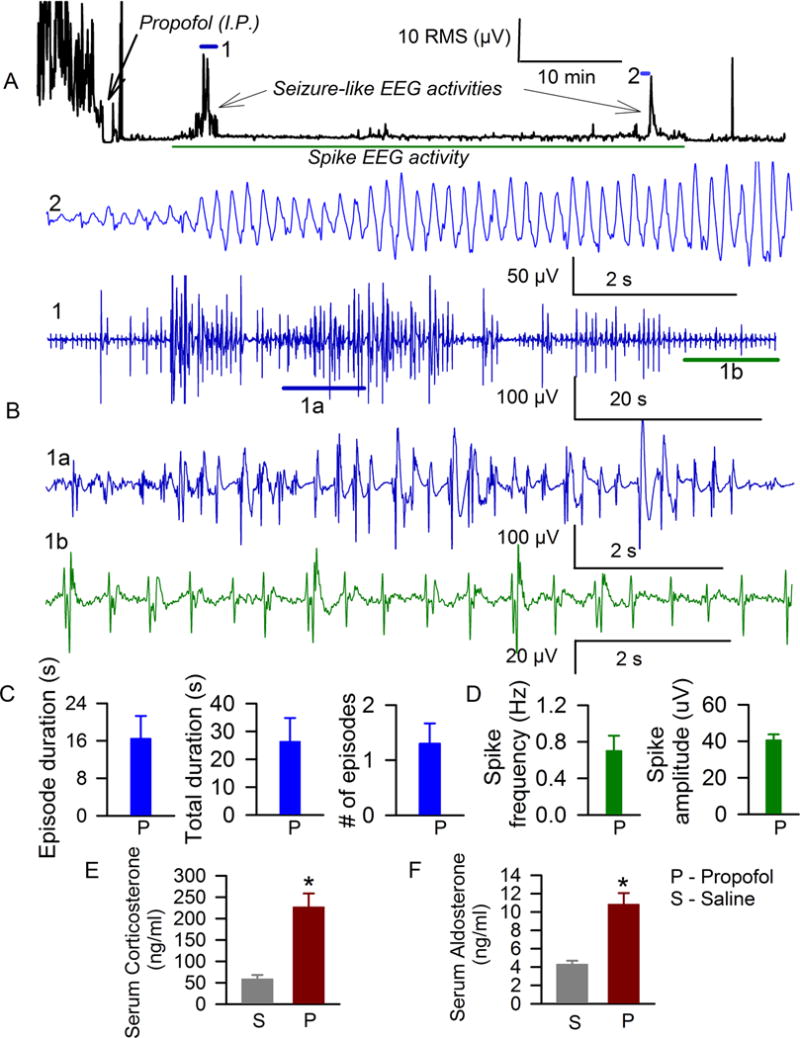
Anesthesia of P4–P6 rats with propofol caused episodes of electroencephalographic seizures and continuous isolated spikes in electroencephalograms. A: Root mean square (RMS) of the electroencephalogram in a P4 rat before and after administration of propofol. Horizontal lines mark occurrence of episodes of electroencephalographic seizures (1 and 2) and spike electroencephalographic activity, which are shown at expanded time scale in B. B: Examples of episode of electroencephalographic seizures in a P4 rat after administration of propofol. Horizontal lines (1a and 1b) indicate segments of electroencephalogram shown below at an expanded time scale. 1a – electroencephalographic seizures; 1b – electroencephalographic spike activity. C: Histograms showing variables of electroencephalographic seizures in P4–P6 rats anesthetized with propofol (P, n=10). D: Histograms showing frequencies and amplitudes of electroencephalogram spike activities in P4–P6 rats anesthetized with propofol. Histograms showing serum levels of corticosterone (F) and aldosterone (E) in P4–P6 rats that received saline (n=6) or propofol (n=6). *, P= 0.0005 (E) and *, P=0.0002 (F) vs. serum levels of corticosterone and aldosterone in P4–P6 rats injected with saline.
In order to test whether anesthesia of neonatal rats with propofol is associated with a change in systemic levels of corticosteroids we measured the serum levels of corticosterone and aldosterone in separate groups of P4–P6 rats that underwent experimental manipulations identical to those used to study EEG activity. Propofol or equal volumes of vehicle were administered 1 hour after completion of surgery for implantation of EEG electrodes. Serum levels of corticosterone in the blood samples collected 1 hr after administration of propofol revealed a significant increase in levels of corticosterone in animals anesthetized with propofol (t(10) = −5.062; P= 0.0005, Fig. 1E). The serum levels of aldosterone in rats anesthetized with propofol were also higher than in rats that received vehicle (t(10) = −5.069; P= 0.0002, Fig. 1F).
Inhibition of NKCC1 importer or corticosteroid receptors diminished electroencephalographic seizures caused by propofol
In order to test whether developmental GABAAR-mediated excitation plays a role in propofol-caused hyperexcitatory EEG patterns, a group of P4–P6 rats received bumetanide 15 min before administration of propofol to block NKCC1 Cl− transporter. Control rats received an equal volume of saline prior to propofol. EEG activity during anesthesia with propofol in animals pretreated with saline was not different from EEG activity recorded in animals anesthetized with propofol that did not receive any pretreatments. Therefore, data from saline-pretreated and nonpretreated animals were combined. No EEG seizures or spike activity were detected during anesthesia with propofol in P4–P6 rats pretreated with bumetanide (Fig. 2A–D). A representative EEG, recorded from a bumetanide-pretreated P6 rat after administration of propofol, is shown in Fig. 2C.
Figure 2.
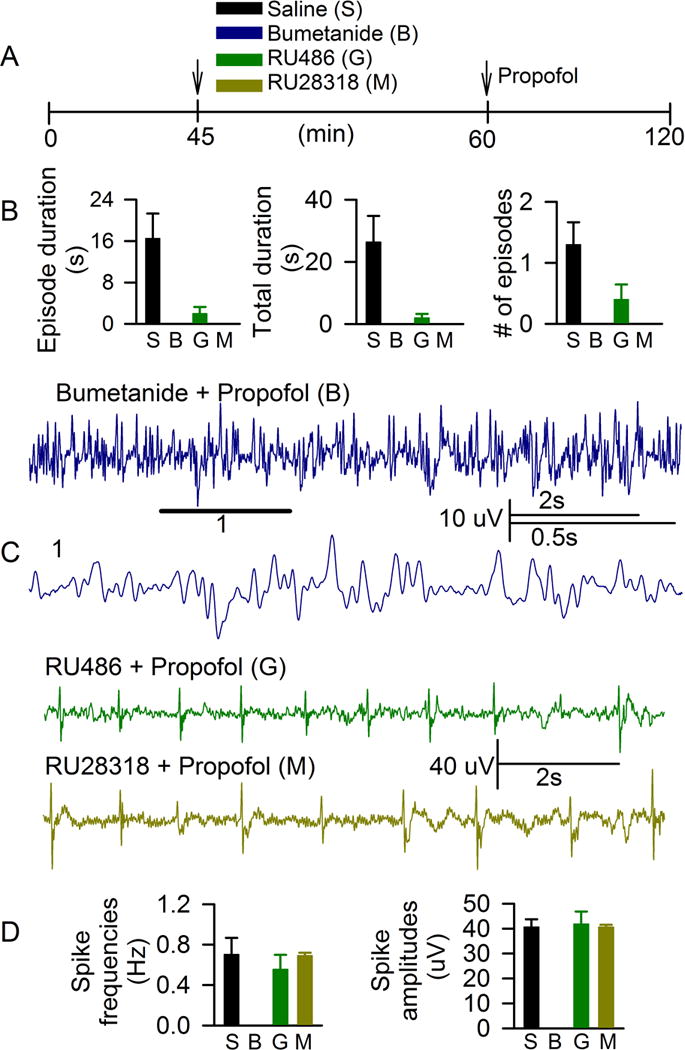
The NKCC1 Cl− transporter inhibitor, bumetanide, depressed both electroencephalographic seizures and spikes, while the corticosteroid receptor antagonists, RU28318 and RU486, depressed seizure electroencephalogram patterns only caused by propofol in P4–P6 rats. A: Illustration of the experimental protocol. Arrows indicate time points of treatment administrations. B: Histograms showing variables of electroencephalographic seizures in rats anesthetized with propofol that received saline as pretreatment (S, n=10), bumetanide (B, no seizure-like activity, n=4), RU486 (G, n=5) and RU28318 (M, no seizure-like activity, n=3). C: Example of electroencephalogram spike activity in P4 rats, anesthetized with propofol that received as RU28318 pretreatment, RU486 or bumetanide. Electroencephalogram spike activity was not detected in rats pretreated with bumetanide before injection of propofol (Bumetanide + Propofol). Horizontal line marks a fragment of electroencephalogram shown at expanded time scale below. D: Histograms showing frequencies and amplitudes of electroencephalogram spike activities in rats anesthetized with propofol, that received saline as pretreatment (S), bumetanide (B, no spike activity), RU486 (G) and RU28318 (M).
To assess whether the propofol-caused increase in corticosteroid levels contributes to hyperexcitatory EEG events detected during anesthesia with propofol separate groups of animals were pretreated before administration of propofol with either RU28318, a MR antagonist, or RU486, a GR antagonist. Among 5 animals pretreated with RU486 before administration of propofol only 1 animal had a single episode of EEG seizures lasting for 4 s (Fig. 2B). No seizure-like EEG activity was detected in animals anesthetized with propofol after pretreatment with RU28318 (Fig. 2B). Neither RU486 nor RU28318 depressed EEG spike activity (Fig. 2C,D). The vehicle for RU486 and RU28318, dimethyl sulfoxide, administered at equal volumes, did not have any obvious effects on EEG activity.
Etomidate, at a dose sufficient to induce loss of righting reflex in neonatal rats, was weak at eliciting EEG seizures, but markedly enhanced EEG seizures caused by exogenous corticosterone.
Our findings suggest complementary roles of GABAAR-mediated excitation and increases in corticosteroid levels in EEG seizures caused by propofol in neonatal rats. In order to obtain further evidence for or against this possibility, we tested the effects of etomidate, another anesthetic that enhances GABAAR activity, but disrupts the adrenal synthesis of corticosteroids. We found that serum levels of corticosterone in blood samples, collected 1 hr after administration of etomidate in P4–P6 rats that underwent experimental manipulations identical to those used to study EEG activity were higher than in those treated with vehicle (t(10) = −3.337; P = 0.0075, Fig. 3A,B), but lower than in those treated with propofol (t(10) = −3.289; P = 0.0082). Similarly, serum levels of aldosterone were elevated in animals injected with etomidate when compared to the saline-treated rats (t(10) = −5.529; P = 0.0003, Fig. 3A,C), but lower than in those treated with propofol (t(10) = −2.730; P = 0.0212). The latter data passed the Shapiro-Wilk test for normality (P = 0.558) and the equality of variance test (F-Test; P = 0.169).
Figure 3.
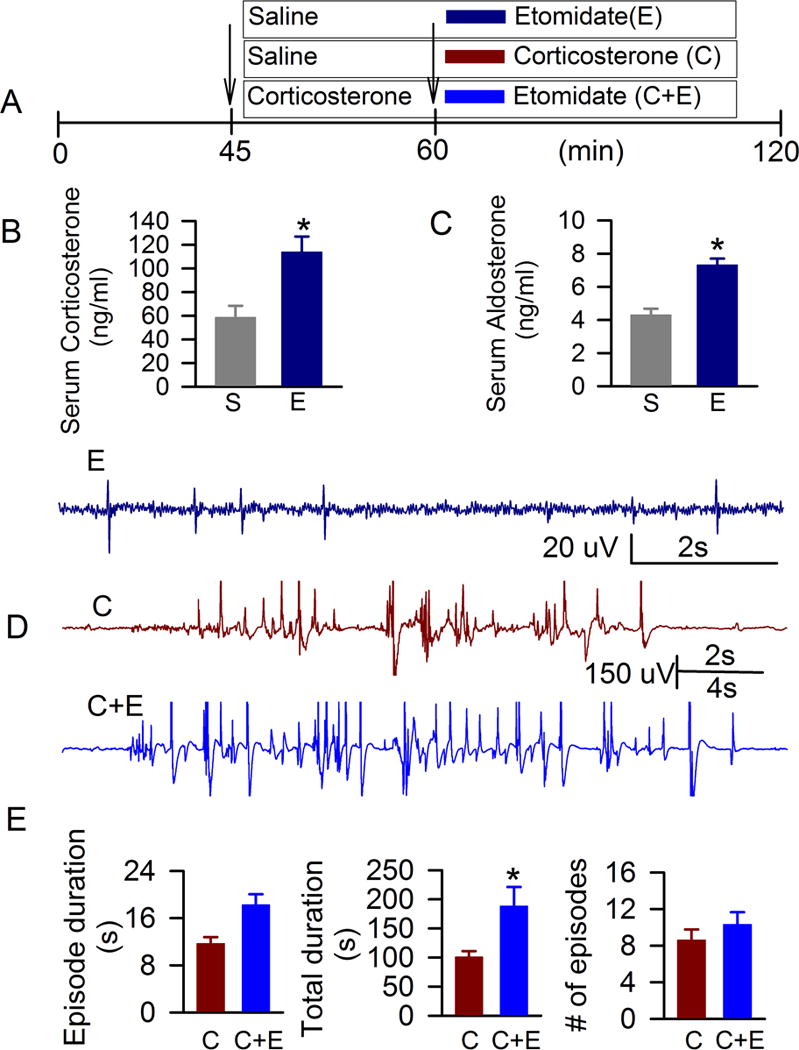
Etomidate caused a moderate increase in serum levels of corticosteroids and weak electroencephalographic seizures, but further enhanced electroencephalographic seizures in the presence of exogenous corticosterone. A: Illustrations of the experimental protocols. Arrows indicate time points of treatment administration. B and C: Histograms showing serum levels of corticosterone and aldosterone in P4–P6 rats that received saline (n=6) or etomidate (n=6). *, P = 0.0075 and #, P = 0.0003 vs. serum levels of corticosterone and aldosterone, respectively, in P4–P6 rats injected with saline. D: Examples of spike electroencephalogram pattern after administration of etomidate (E) in a P5 rat, electroencephalographic seizures in a P4 rat after administration of corticosterone (C) and in a P5 rat pretreated with corticosterone before anesthesia with etomidate (C+E). E: Histograms showing properties of electroencephalographic seizures (mean ± S.E) during a 60 min period in P4–P6 rats after administration of corticosterone (C, n=10) and after administration of etomidate to rats that received corticosterone 15 min earlier (C+E, n=13). *, P = 0.0203 vs corticosterone only.
Etomidate caused EEG seizures in only 2 of 8 P4–P6 rats (Fig. 3A,D,E). The EEGs of animals anesthetized with etomidate exhibited only irregular and abbreviated spikes with frequencies from 0.01 Hz to 1.09 Hz (Fig. 3D). Exogenous corticosterone alone, administered to P4–P6 rats, caused EEG seizures (Fig. 3A,D,E), but not spike-like EEG activity. Etomidate, administered to the corticosterone pretreated P4–P6 rats, further increased the total duration of EEG seizures (t(21) = −2.512, P = 0.0203; data were log transformed). The data passed the Shapiro-Wilk test for normality (P = 0.648) and the equality of variance test (F-Test; P = 0.139). The duration of a single episode and the number of episodes of EEG seizures were not significantly increased (Fig. 3A,D,E).
Discussion
The results of this study provide experimental evidence that propofol administered to neonatal rats causes abnormal excitation and adrenocortical hyperactivation as evident from episodes of EEG seizures, persistent spikes in EEGs and increase in serum levels of corticosteroids, respectively. The EEG seizures caused by propofol were depressed by both MR and GR antagonists and by an inhibitor of the NKCC1 Cl− importer. These findings together with the weak effects of etomidate by itself on serum levels of corticosteroids and EEG seizures, but enhancement by etomidate of EEG seizures caused by exogenous corticosterone, suggest that both, GABAAR-mediated excitation and an increase in corticosteroid levels are required for the proexcitatory effects of propofol in neonatal rats. These findings, especially those with etomidate, suggest that enhancement of GABAAR activity alone may not be sufficient to elicit neonatal EEG seizures.
At the neuronal level, enhancement of GABAAR activity is likely the central mechanism whereby propofol and etomidate induce anesthesia. At a molecular level, such a mechanism is suggested by the results of studies in genetically modified animals, in which the mutation in the same single amino acid of the β3 subunit of GABAARs [β3(N265M)] reduced the ability of propofol and etomidate to cause loss of righting reflex and eliminated their ability to prevent the response to a painful stimulus.23,24 Since the electrochemical gradients for the charge carriers through the GABAAR channels, but not the exact mechanisms of agonist-receptor interaction, determine whether enhancement of GABAAR activity results in excitatory or inhibitory signaling at a given membrane potential, factors, beyond anesthetic-enhanced GABAAR activity, are needed to explain the different excitatory EEG effects of propofol and etomidate in neonatal rats.
Our results support the possibility that a propofol-induced increase in secretion of corticosteroids in neonatal rats is at least 1 of the factors required for the proexcitatory effects of propofol. Corticosterone and aldosterone produce their effects not only via delayed genomic, but also rapid nongenomic pathways,16,17,25 providing a mechanistic basis for their involvement in the acute EEG effects of propofol. Corticosterone binds to MRs with ~10 fold higher affinity than to GRs and is present in brain in higher concentrations than aldosterone.14,15 Therefore, brain MRs primarily mediate the effects of corticosterone while GRs become increasingly involved in mediation of the effects of corticosterone at higher concentrations of the hormone. The depression of propofol-caused EEG seizures in neonatal rats by both MR and GR antagonists implicates an involvement of both, MRs and GRs in mediation of the effects of propofol in neonatal rats. Corticosterone may cause excitation and contribute to the propofol-caused EEG seizures in neonatal rats by increasing presynaptic glutamate release, inhibiting glutamate uptake, inducing expression of NMDA and AMPA receptors.16,17 The finding that the corticosteroid receptor antagonists depress EEG seizures, but unlike bumetanide, do not prevent spike EEG activity implicates multiple mechanisms in mediating the excitatory EEG effects of propofol in neonatal rats.
This series of experiments demonstrates that the GABA-ergic anesthetic propofol causes an increase in serum levels of corticosterone and aldosterone in P4–P6 rats, an age period in rats when adrenocortical hormone secretion is less responsive to stress while the brain is more vulnerable to the effects of corticosteroids. A large body of evidence indicates that increased glucocorticoid levels resulting from pathophysiological conditions, therapeutic interventions or stress can have profound effects on cognitive and emotional functions that may become apparent only later in life.26–31 The delays between anesthetic exposures and functional abnormalities reported in animal studies32,33 support a potential role of the corticoids in the adverse developmental effects of general anesthetics. Despite the fact that etomidate disrupts synthesis of corticosteroids, we observed an increase in serum corticosteroid levels in animals treated with etomidate, though it was notably smaller than that in rats treated with propofol. It is plausible that changes in corticosteroid levels caused by etomidate may result from 2 opposite effects of the anesthetic – a) inhibition of the adrenal synthesis of corticosteroids,34 and b) stimulation of signal transduction pathways resulting in corticosteroid secretion, an effect similar to that caused by propofol.
Collectively, our data demonstrate that a propofol-caused increase in serum levels of corticosteroids, along with developmental GABAAR-mediated excitation, are required for the acute effects of propofol to induce EEG seizures in neonatal rats.
The KCC2 Cl− exporter expression in human neocortex begins to increase at 40 weeks after conception and reaches its adult level at the end of the first year after birth,8 supporting the applicability of anesthetic-enhanced neuronal activity to human neonates. Propofol may further decrease the KCC2/NKCC1 ratio by elevating systemic levels of aldosterone, which increases NKCC1 protein expression via a rapid mechanism that does not involve changes in mRNA expression.35 Importantly, prematurely born low birth weight neonates have elevated systemic levels of aldosterone even without exposure to general anesthetics.36 Clinical reports that support the presence of excessive GABAAR-mediated neuronal excitability are that bumetanide reduced seizures in a 6-week-old baby with intractable multifocal seizures37 and alleviated symptoms in 3–11 year old children with autism or Asperger syndrome,38 disease states that are characterized by abnormal excitation/inhibition ratio in the brain. Finally, 2 clinical studies in the United States and Europe are under way to evaluate the antiepileptic properties of bumetanide in neonatal seizures.39
Acknowledgments
We would like to thank Bruno Panzarini (Undergraduate Student, University of Florida) for technical assistance and Dr. Terrie Vasilopoulos (Assistant Professor, University of Florida) for help with statistical analysis.
Funding: Supported by grant No. R01 GM93036-01A1 from the National Institute of Health/National Institute of General Medical Sciences (to A.E.M.), and the Jerome H. Modell, M.D., F.A.H.A. Endowed Professorship (to N.G.). The funding agencies were not involved in the design and conduct of the study, data analysis, and production of the manuscript.
Footnotes
The authors declare no conflicts of interest.
Reprints will not be available from the authors.
DISCLOSURES:
Name: Jesse Willis, BS
Contribution: Jesse Willis has seen the original study data, helped conduct the study, analyze and interpret the data, and critically review the manuscript.
Attestation: Jesse Willis approved the final manuscript. Jesse Willis attests to the integrity of the original data and the analysis reported in this manuscript.
Name: Wanting Zhu, BS
Contribution: Wanting Zhu has seen the original study data, helped conduct the study, analyze and interpret the data, and critically review the manuscript.
Attestation: Wanting Zhu approved the final manuscript.
Name: Julio Perez-Downes, BS
Contribution: Julio Perez-Downes has seen the original study data, helped conduct the study, analyze and interpret the data, and critically review the manuscript.
Attestation: Julio Perez-Downes approved the final manuscript.
Name: Sijie Tan, PhD
Contribution: Sijie Tan has seen the original study data, helped conduct the study, analyze and interpret the data, and critically review the manuscript.
Attestation: Sijie Tan approved the final manuscript.
Name: Changqing Xu, MD, PhD
Contribution: Changqing Xu has seen the original study data, helped conduct the study, analyze and interpret the data, and critically review the manuscript.
Attestation: Changqing Xu approved the final manuscript.
Name: Christoph Seubert, MD, PhD
Contribution: Christoph Seubert has seen the original study data, helped design the study, analyze and interpret the data, and write the manuscript.
Attestation: Christoph Seubert approved the final manuscript. Christoph Seubert attests to the integrity of the original data and the analysis reported in this manuscript.
Name: Nikolaus Gravenstein, MD
Contribution: Nikolaus Gravenstein has seen the original study data, helped design the study, analyze and interpret the data, and write the manuscript.
Attestation: Nikolaus Gravenstein approved the final manuscript.
Name: Anatoly Martynyuk, PhD, DSc
Contribution: Anatoly E. Martynyuk has seen the original study data, was involved in the conception of the study idea, design, data analysis and interpretation, and wrote the paper. Anatoly Martynyuk is the archival author.
Attestation: Anatoly Martynyuk approved the final manuscript. Anatoly Martynyuk attests to the integrity of the original data and the analysis reported in this manuscript.
This manuscript was handled by: Gregory J. Crosby, MD
Contributor Information
Jesse Willis, Department of Anesthesiology, University of Florida College of Medicine, Gainesville, Florida.
Wanting Zhu, Department of Anesthesiology, University of Florida College of Medicine, Gainesville, Florida.
Julio Perez-Downes, Lake Erie College of Osteopathic Medicine, Bradenton, Florida.
Sijie Tan, Department of Anesthesiology, University of Florida College of Medicine, Gainesville, Florida.
Changqing Xu, Department of Anesthesiology, University of Florida College of Medicine, Gainesville, Florida.
Christoph Seubert, Department of Anesthesiology, University of Florida College of Medicine, Gainesville, Florida.
Nikolaus Gravenstein, Department of Anesthesiology and the McKnight Brain Institute, University of Florida College of Medicine, Gainesville, Florida.
Anatoly Martynyuk, Department of Anesthesiology and the McKnight Brain Institute, University of Florida College of Medicine, Gainesville, Florida.
References
- 1.Sanders RD, Hassell J, Davidson AJ, Robertson NJ, Ma D. Impact of anaesthetics and surgery on neurodevelopment: an update. Br J Anaesth. 2013;110(Suppl 1):153–72. doi: 10.1093/bja/aet054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rakic P, Bourgeois JP, Eckenhoff MF, Zecevic N, Goldman-Rakic PS. Concurrent overproduction of synapses in diverse regions of the primate cerebral cortex. Science. 1986;232:232–5. doi: 10.1126/science.3952506. [DOI] [PubMed] [Google Scholar]
- 3.Harris KM, Jensen FE, Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci. 1992;12:2685–705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanchez RM, Jensen FE. Maturational aspects of epilepsy mechanisms and consequences for the immature brain. Epilepsia. 2001;42:577–85. doi: 10.1046/j.1528-1157.2001.12000.x. [DOI] [PubMed] [Google Scholar]
- 5.Glykys J1, Dzhala VI, Kuchibhotla KV, Feng G, Kuner T, Augustine G, Bacskai BJ, Staley KJ. Differences in cortical versus subcortical GABAergic signaling: a candidate mechanism of electroclinical uncoupling of neonatal seizures. Neuron. 2009;63:657–72. doi: 10.1016/j.neuron.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tyzio R1, Nardou R, Ferrari DC, Tsintsadze T, Shahrokhi A, Eftekhari S, Khalilov I, Tsintsadze V, Brouchoud C, Chazal G, Lemonnier E, Lozovaya N, Burnashev N, Ben-Ari Y. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science. 2014;343:675–9. doi: 10.1126/science.1247190. [DOI] [PubMed] [Google Scholar]
- 7.Khazipov R, Khalilov I, Tyzio R, Morozova E, Ben-Ari Y, Holmes GL. Developmental changes in GABAergic actions and seizure susceptibility in the rat hippocampus. Eur J Neurosci. 2004;19:590–600. doi: 10.1111/j.0953-816x.2003.03152.x. [DOI] [PubMed] [Google Scholar]
- 8.Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, Delpire E, Jensen FE, Staley KJ. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–13. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- 9.Edwards DA, Shah HP, Cao W, Gravenstein N, Seubert CN, Martynyuk AE. Bumetanide alleviates epileptogenic and neurotoxic effects of sevoflurane in neonatal rat brain. Anesthesiology. 2010;112:567–75. doi: 10.1097/ALN.0b013e3181cf9138. [DOI] [PubMed] [Google Scholar]
- 10.Cao W, Pavlinec C, Gravenstein N, Seubert CN, Martynyuk AE. Roles of aldosterone and oxytocin in abnormalities caused by sevoflurane anesthesia in neonatal rats. Anesthesiology. 2012;117:791–800. doi: 10.1097/ALN.0b013e318266c62d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seubert CN, Zhu W, Pavlinec C, Gravenstein N, Martynyuk AE. Developmental Effects of Neonatal Isoflurane and Sevoflurane Exposure in Rats. Anesthesiology. 2013;119:358–64. doi: 10.1097/ALN.0b013e318291c04e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim BG, Shen FY, Kim YB, Kim WB, Kim YS, Han HC, Lee MK, Kong MH, Kim YI. Possible role of GABAergic depolarization in neocortical neurons in generating hyperexcitatory behaviors during emergence from sevoflurane anesthesia in the rat. ASN Neuro. 2014 Mar 6; doi: 10.1042/AN20140004. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koyama Y, Andoh T, Kamiya Y, Morita S, Miyazaki T, Uchimoto K, Mihara T, Goto T. Bumetanide, an inhibitor of cation-chloride cotransporter isoform 1, inhibits γ-aminobutyric acidergic excitatory actions and enhances sedative actions of midazolam in neonatal rats. Anesthesiology. 2013;119:1096–108. doi: 10.1097/ALN.0b013e31829e4b05. [DOI] [PubMed] [Google Scholar]
- 14.De Kloet ER. Brain corticosterone receptor balance and homeostatic control. Front Neuroendocrinol. 1991;12:95–164. [Google Scholar]
- 15.Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117:2505–11. doi: 10.1210/endo-117-6-2505. [DOI] [PubMed] [Google Scholar]
- 16.Karst H, Berger S, Turiault M, Tronche F, Schutz G, Joels M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci USA. 2005;102:19204–07. doi: 10.1073/pnas.0507572102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Timmermans W, Xiong H, Hoogenraad CC, Krugers HJ. Stress and excitatory synapses: from health to disease. Neuroscience. 2013;248:626–36. doi: 10.1016/j.neuroscience.2013.05.043. [DOI] [PubMed] [Google Scholar]
- 18.Briner A, Nikonenko I, De Roo M, Dayer A, Muller D, Vutskits L. Developmental Stage-dependent persistent impact of propofol anesthesia on dendritic spines in the rat medial prefrontal cortex. Anesthesiology. 2011;115:282–93. doi: 10.1097/ALN.0b013e318221fbbd. [DOI] [PubMed] [Google Scholar]
- 19.Yu D, Jiang Y, Gao J, Liu B, Chen P. Repeated exposure to propofol potentiates neuroapoptosis and long-term behavioral deficits in neonatal rats. Neurosci Lett. 2013;534:41–6. doi: 10.1016/j.neulet.2012.12.033. [DOI] [PubMed] [Google Scholar]
- 20.O’Shea TM, Kothadia JM, Klinepeter KL, Goldstein DJ, Jackson BG, Weaver RG, 3rd, Dillard RG. Randomized placebo-controlled trial of a 42-day tapering course of dexamethasone to reduce the duration of ventilator dependency in very low birth weight infants: outcome of study participants at 1-year adjusted age. Pediatrics. 1999;104:15–21. doi: 10.1542/peds.104.1.15. [DOI] [PubMed] [Google Scholar]
- 21.Graham EM, Atz AM, Butts RJ, Baker NL, Zyblewski SC, Deardorff RL, DeSantis SM, Reeves ST, Bradley SM, Spinale FG. Standardized preoperative corticosteroid treatment in neonates undergoing cardiac surgery: results from a randomized trial. J Thorac Cardiovasc Surg. 2011;142:1523–29. doi: 10.1016/j.jtcvs.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vijn PC1, Sneyd JR. I.v. anaesthesia and EEG burst suppression in rats: bolus injections and closed-loop infusions. Br J Anaesth. 1998;81:415–21. doi: 10.1093/bja/81.3.415. [DOI] [PubMed] [Google Scholar]
- 23.Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, Rudolph U. General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta 3 subunit. FASEB J. 2003;17:250–52. doi: 10.1096/fj.02-0611fje. [DOI] [PubMed] [Google Scholar]
- 24.Zeller A, Arras M, Lazaris A, Jurd R, Rudolph U. Distinct molecular targets for the central respiratory and cardiac actions of the general anesthetics etomidate and propofol. FASEB J. 2005;19:1677–79. doi: 10.1096/fj.04-3443fje. [DOI] [PubMed] [Google Scholar]
- 25.Olijslagers JE, de Kloet ER, Elgersma Y, van Woerden GM, Joëls M, Karst H. Rapid changes in hippocampal CA1 pyramidal cell function via pre- as well as postsynaptic membrane mineralocorticoid receptors. Eur J Neurosci. 2008;27:2542–50. doi: 10.1111/j.1460-9568.2008.06220.x. [DOI] [PubMed] [Google Scholar]
- 26.Frodl T, O’Keane V. How does the brain deal with cumulative stress? A review with focus on developmental stress, HPA axis function and hippocampal structure in humans. Neurobiol Dis. 2013;52:24–37. doi: 10.1016/j.nbd.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 27.Palazidou E. The neurobiology of depression. Br Med Bull. 2012;101:127–45. doi: 10.1093/bmb/lds004. [DOI] [PubMed] [Google Scholar]
- 28.Aisa B, Tordera R, Lasheras B, DelRio J, Ramirez MJ. Cognitive impairment associated to HPA axis hyperactivity after maternal separation in rats. Psychoneuroendocrinology. 2007;32:256–66. doi: 10.1016/j.psyneuen.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 29.Brunson KL. Mechanisms of late onset cognitive decline after early-life stress. J Neurosci. 2005;25:9328–38. doi: 10.1523/JNEUROSCI.2281-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ivy AS, Rex CS, Chen Y, Dubé C, Maras PM, Grigoriadis DE, Gall CM, Lynch G, Baram TZ. Hippocampal dysfunction and cognitive impairments provoked by chronic early-life stress involve excessive activation of CRH receptors. J Neurosci. 2010;30:13005–15. doi: 10.1523/JNEUROSCI.1784-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oomen CA1, Soeters H, Audureau N, Vermunt L, van Hasselt FN, Manders EM, Joëls M, Lucassen PJ, Krugers H. Severe early life stress hampers spatial learning and neurogenesis, but improves hippocampal synaptic plasticity and emotional learning under high-stress conditions in adulthood. J Neurosci. 2010;30:6635–45. doi: 10.1523/JNEUROSCI.0247-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stratmann G, May LD, Sall JW, Alvi RS, Bell JS, Ormerod BK, Rau V, Hilton JF, Dai R, Lee MT, Visrodia KH, Ku B, Zusmer EJ, Guggenheim J, Firouzian A. Effect of hypercarbia and isoflurane on brain cell death and neurocognitive dysfunction in 7-day-old rats. Anesthesiology. 2009;110:849–61. doi: 10.1097/ALN.0b013e31819c7140. [DOI] [PubMed] [Google Scholar]
- 33.Sanders RD, Xu J, Shu Y, Januszewski A, Halder S, Fidalgo A, Sun P, Hossain M, Ma D, Maze M. Dexmedetomidine attenuates isoflurane-induced neurocognitive impairment in neonatal rats. Anesthesiology. 2009;110:1077–85. doi: 10.1097/ALN.0b013e31819daedd. [DOI] [PubMed] [Google Scholar]
- 34.Vanlersberghe C, Camu F. Etomidate and other non-barbiturates. Handb Exp Pharmacol. 2008;182:267–82. doi: 10.1007/978-3-540-74806-9_13. [DOI] [PubMed] [Google Scholar]
- 35.Ding B, Frisina RD, Zhu X, Sakai Y, Sokolowski B, Walton JP. Direct control of Na(+)-K(+)-2Cl(−)-cotransport protein (NKCC1) expression with aldosterone. Am J Physiol Cell Physiol. 2014;306:C66–75. doi: 10.1152/ajpcell.00096.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinerie L, Pussard E, Foix-L’Hélias L, Petit F, Cosson C, Boileau P, Lombès M. Physiological partial aldosterone resistance in human newborns. Pediatr Res. 2009;66:323–8. doi: 10.1203/PDR.0b013e3181b1bbec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kahle KT, Barnett SM, Sassower KC, Staley KJ. Decreased seizure activity in a human neonate treated with bumetanide, an inhibitor of the Na(+)-K(+)-2Cl(−) cotransporter NKCC1. J Child Neurol. 2009;24:572–6. doi: 10.1177/0883073809333526. [DOI] [PubMed] [Google Scholar]
- 38.Lemonnier E, Degrez C, Phelep M, Tyzio R, Josse F, Grandgeorge M, Hadjikhani N, Ben-Ari Y. A randomised controlled trial of bumetanide in the treatment of autism in children. Transl Psychiatry. 2012;2:e202. doi: 10.1038/tp.2012.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pressler RM, Mangum B. Newly emerging therapies for neonatal seizures. Semin Fetal Neonatal Med. 2013;18:216–23. doi: 10.1016/j.siny.2013.04.005. [DOI] [PubMed] [Google Scholar]