

Abstract
Diabetic hearts are subject to more extensive ischemia/reperfusion (ISC/REP) damage. This study examined the efficiency of citric acid cycle (CAC) flux and the transfer of cytosolic reducing equivalents into the mitochondria for oxidative support of cardiac work following ISC/REP in hearts of c57bl/6 (NORM) and type 2 diabetic, db/db mouse hearts. Flux through the CAC and malate-aspartate shuttle (MA) were monitored via dynamic 13C NMR of isolated hearts perfused with 13C palmitate + glucose. MA flux was lower in db/db than NORM. Oxoglutarate malate carrier (OMC) was elevated in the db/db heart, suggesting a compensatory response to low NADHc. Baseline CAC flux per unit work (rate-pressure-product, RPP) was similar between NORM and db/db, but ISC/REP reduced the efficiency of CAC flux/RPP by 20% in db/db. ISC/REP also increased UCP3 transcription, indicating potential for greater uncoupling. Therefore, ISC/REP induces inefficient carbon utilization through the CAC in hearts of diabetic mice due to the combined inefficiencies in NADHc transfer per OMC content and increased uncoupling via UCP3. Ischemia and reperfusion exacerbated pre-existing mitochondrial defects and metabolic limitations in the cytosol of diabetic hearts. These limitations and defects render diabetic hearts more susceptible to inefficient carbon fuel utilization for oxidative energy metabolism.
Keywords: db/db mouse, malate-aspartate shuttle, oxoglutarate-malate carrier, uncoupling protein, mitochondria, reducing equivalents
1.0 Introduction
Patients with type II diabetes mellitus (DM2) are at an increased risk for cardiovascular disease. In fact, cardiovascular complications are the leading cause of diabetes related morbidity and mortality [1]. While some controversy exists as to the whether diabetic hearts are more susceptible to injury, most in vivo animals studies suggest that the diabetic myocardium is more sensitive to dysfunction following ischemic injury [2]. Both the diabetic heart and the metabolically related cardiac phenotype of the PPARα over-expressing mouse heart (MHC-PPARα), show exacerbated post-ischemic dysfunction [3]. Yet, the distinctions in post-ischemic metabolic recovery of diabetic versus normal myocardium are at best, only superficially understood, and mechanisms regulating the production and oxidation of reducing equivalents for mitochondrial energy production have not been directly studied in the intact, post-ischemic diabetic hearts. This study probed the efficiency of carbon flux in the production of NADH by citric acid cycle (CAC) flux in the support of contractile recovery of intact, post-ischemic diabetic hearts. The protocols enabled a focus on mechanisms linking CAC flux to energy dissipation and the transfer and oxidation of cytosolic NADH into mitochondria.
Characterized by a decrease in glucose oxidation and an increase in fatty acid oxidation (FAO), DM2 is related to not only an increase in delivery of long chain fatty acids (LCFA) into the myocardium but also decreased insulin signaling and activation of peroxisome proliferator-activated receptor-α signaling [4–6]. Changes in myocardial fuel metabolism, a decrease in glucose oxidation and an increase in fatty acid oxidation, drive diabetic cardiomyopathy [4]. The DM2 heart is characterized by substrate inflexibility due to insulin responsiveness; reduced glucose uptake and oxidation with increased long chain fatty acid (LCFA) delivery and oxidation [4, 7]. As glucose availability becomes limited, the diabetic myocardium relies nearly completely on fatty acid oxidation (FAO) for energy production [3, 8].
Cardiac inefficiency, the ratio of cardiac work to myocardial O2 consumption (MVO2), is considered to be an underlying cause of cardiac dysfunction in both type I and type II diabetic myocardium due to consequences of impaired mitochondrial function [7, 9]. While previous work has shown both high insulin and high glucose enhance the pressure-volume area – MVO2 relationship during post-ischemic recovery in the db/db mouse heart, the relationship between reducing equivalent production from carbon-based fuels and cardiac function in the postischemic db/db heart remains unclear [10, 11]. To date, only oxygen consumption rates and substrate oxidation rates have been measured in diabetic hearts with studies of respiratory function in isolated mitochondria. This study provides the first detailed analysis of the efficiency of carbon flux through the CAC for oxidative, mitochondrial NADH production and the coordination of CAC flux with the transfer of cytosolic NADH following ischemia/reperfusion in hearts of diabetic animals.
We hypothesized that the contributions of cytosolic NADH to mitochondrial oxidative energy production is limited, due to impaired ability for glucose uptake in the diabetic heart, rendering oxidative metabolism increasingly reliant on NADH production via the CAC. Published studies suggest the increase in MVO2 is attributable to the increase in UCP3 [12, 13], but no direct measurements of the actual NADH producing pathways have been performed beyond the indirect substrate preference or substrate oxidation rates. The current studied explores how the contributions of both cytosolic and mitochondrial NADH to oxidative energy metabolism in the mitochondria are linked to the demand for oxidative carbon flux through the CAC in postischemic, diabetic hearts.
Recently, changes UCP3 were shown to contribute to cardiac inefficiency through mitochondrial uncoupling in the post-ischemic, reperfused state [14]. Previous work in normal hearts has demonstrated the sensitivity of NMR detection of 13C enrichment rates of intramyocellular glutamate to the coupled processes of transfer of reducing equivalents produced in the cytosol into the mitochondria via exchange of malate for α-ketoglutarate within the malate-aspartate shuttle and flux through the citric acid cycle [15–20]. Here, we exploit the 13C NMR detection of these intracellular events in the intact myocardium to investigate changes in CAC flux as an oxidative source of NADH and its balance with transfer of reducing equivalents from cytosolic NADH into mitochondria through the oxaloacetate-malate carrier (OMC) [15–20]. The findings suggest that despite adaptive responses in mitochondrial OMC expression, the availability of cytosolic NADH remains limiting, and together with elevated expression of uncoupling proteins, exacerbate the metabolic inefficiencies of postischemic contractile dysfunction in the diabetic heart.
2.0 Material and Methods
2.1 Animal model
Male db/db, BKS.Cg- +Leprdb/+Leprdb/OlaHsd obtained from Harlan, were studied at 12 weeks of age. The background strain, c57bl/6, were obtained from Harlan and studied at 12 weeks of age [21, 22]. At this age, db/db mice were consistently and severely hyperglycemic and exhibit altered substrate metabolism and cardiac efficiency [13]. Blood glucose from both db/db and c57bl/6 was monitored. Mice had free access to food and water while being housed under controlled temperature and lighting. All experimental procedures were approved by the University of Illinois at Chicago Animal Care and Use Committee.
Isolated heart protocols
12-week old animals were heparinized (50 U/10 g, i.p.) and anesthetized with ketamine (80 mg/kg, i.p.) plus xylazine (12 mg/kg, i.p.). Hearts were excised and retrogradely perfused (80 cm H2O) with modified Krebs-Henseleit buffer (118.5 mM NaCl, 4.7 mM KCl, 1.5 mM CaCl2, 1.2 mM MgSO4 and 1.2 mM KH2PO4) equilibrated with 95% O2/5% CO2, at 37°C, and containing 0.4 mM 12C palmitate/ fatty acid free albumin complex (3:1 molar ratio) and 10 mM glucose. Perfusion media contained 0.4 mM palmitate for both c57bl/6 and db/db. The use of a physiologically normal palmitate concentration allowed for direct comparison of normal and diabetic hearts for elucidating fundamental metabolic mechanisms within the cardiomyocyte in the absence of confounding variables introduced by high exogenous lipids and glucose [7, 23–26]. Previous findings indicate that in the functioning, whole heart, elevated fat content influences lipid storage in the cardiomyocyte but cannot override the metabolic demand of mechanical work and does not result in elevated LCFA oxidation [27]. Protocols were designed to use similar substrates for isolated heart perfusions, as per previously published studies examining oxygen consumption and mitochondrial uncoupling in the diabetic, postischemic heart. As thusly indicated hearts were perfused with LCFA plus glucose and no lactate [7, 23–26]
Hearts undergoing ISC/REP were subjected to 8 min no flow ischemia followed by 10 minutes of reperfusion. After 10 minutes of reperfusion, hearts were switched to 13C-enriched media ([4,6,8,10,12,14,16,-13C7] palmitate) for 30 minutes at baseline workload for mice on a regular chow diet (db/db, N=9; c57bl/6, N =6) and undergoing 8 minutes no flow ischemia/reperfusion (ISC/REP) (db/db, N=8; c57bl/6, N =6). [4,6,8,10,12,14,16,-13C7] palmitate was used for ease of analysis for the C4 carbon of glutamate in the 13C-NMR spectra due to overlapping resonances from the C4 position of glutamate and C2 position of acyl intermediates. Sequential 13C-NMR spectra were collected and hearts were frozen in liquid N2 cooled tongs for biochemical analysis [28, 29]. Mouse hearts are more susceptible to ISC/REP than other species, and small changes in time of global ischemia have resulted in large reductions in recovery contractile recovery [6]. For the purposes of this study, we subjected hearts to 8 minutes of ischemia, which produced a 50% reduction in the recovery of RPP throughout reperfusion in the db/db model.
A water-filled latex balloon was fitted into the left ventricle and set to a diastolic pressure of 5 mmHg. Left ventricular developed pressure (LVDP) and heart rate (HR) were continuously recorded with a pressure transducer and digital recording system (Powerlab, AD Instruments, Colorado Springs, CO). Rate-pressure product (RPP) was calculated as the product of heart rate and developed pressure. Temperature was maintained at 37°C.
2.2 NMR spectroscopy and tissue chemistry
Using previously established methods, dynamic 13C-spectra from intact, beating hearts were collected as previously reported [30–33]. Sequential, proton-decoupled 13C NMR spectra were acquired (2 min each) with natural 13C abundance correction using previously reported NMR methods (Fig. 1) [30, 31]. Magnetic field homogeneity was optimized by shimming to a proton line width of 10–20 Hz.
Figure 1.

Representative spectra from a perfused db/db heart after 8 min ischemia followed by 10 minutes reperfusion. A. Dynamic-mode 13C spectra from isolated db/db heart perfused with 0.4 mM [4,6,8,10,12,14,16 – 13C7] palmitate + 10 mM unlabeled glucose. Spectra display progressive 13C enrichment of glutamate over 30 minutes. The signals from the 4-, 3-, and 2-carbons of glutamate produced by oxidation of 13C palmitate, at 34, 28, and 56 ppm respectively (C4, C3, and C2). C. 13C NMR spectrum of extract from db/db heart perfused with 13C palmitate + 10 mM unlabeled glucose. The resonance signals from the 4- and 3-carbons of glutamate at 34 and 28 ppm respectively (C4 and C3) are produced by oxidation of 13C palmitate and display multiplets from J13C-13C coupling due to glutamate isotopomer formation. Note the absence of 13C enriched glutamine at 32 ppm the spectrum, which displays enrichment at only the 1.1% natural abundance level that indicates no significant conversion of 13C glutamate to glutamine in the heart.
Tissue metabolites were extracted from frozen heart tissue using 7% perchloric acid and neutralized with KOH. Tissue extracts were analyzed by enzymatic assay either spectrophotometrically or fluorometrically for metabolite content (aspartate, α-ketoglutarate, citrate) using previously described methods [34–36]. Glutamate concentration was determined with glutamate dehydrogenase and diaphorase (Roche L-Glutamic acid colorimetric kit). In vitro high-resolution 1H and 13C NMR spectra of tissue extracts reconstituted in 0.5 mL of D2O were collected with a 5 mm 13C probe (Fig. 1) (Bruker Instruments, Billerica, MA). Analysis of 13C spectra was performed to determine fractional enrichment of [2-13C] acetyl CoA [37, 38].
2.3 Kinetic Analysis of Isotope Enrichment and Oxidative Rates
Kinetic analysis of isotopic enrichment rates provided quantitative measures of flux through the CAC, rates of LCFA oxidation, and the rate of cytosolic NADH transfer through malate/α-ketoglutarate exchange between the cytosol and mitochondria [15–17, 19, 20]. Briefly, the oxidation of 13C enriched LCFA to form [2-13C] acetyl CoA results in enrichment of the first span of the CAC to form [4-13C] α-ketoglutarate within the mitochondria. Competition for [4-13C] α-ketoglutarate, as a substrate for either oxidation via α-ketoglutarate dehydrogenase or efflux via OMC from the mitochondria in exchange for cytosolic malate, determines the extent and rate of the production of [2 or 3-13C] succinate or alternatively, interconversion of [4-13C] α-ketoglutarate into [4-13C] glutamate in the cytosol. Subsequent oxidation of [2 or 3-13C] succinate in the mitochondria through the second span of the CAC results in labeling of oxaloacetate at either the 2- or 3-carbon positions, in equal probability. Condensation of [2 or 3-13C] oxaloacetate with newly-formed acetyl CoA recycles the 13C label to the 2- and 3-positions of citrate to re-enter the first span of the CAC and produce α-ketoglutarate that is enriched with 13C at the 2- and 3-carbon sites. This relabeled α-ketoglutarate is also available for either further oxidation within the mitochondria or exchange to the cytosol through OMC. The rates of appearance of the isotope at each of the 4-, and 2- or 3-carbon positions of glutamate can then be detected, because glutamate is in sufficient abundance in the cell for NMR detection from the intact heart. Since the transamination of 13C-enrichedα-ketoglutarate to form 13C-enriched glutamate, which is 90% cytosolic, is approximately 50-fold faster than the process of exchange for malate across the mitochondrial membrane via OMC, the enrichment of glutamate is also dependent on the net forward flux of intermediates through the malate-aspartate shuttle [15, 16, 37]. Thus, the rates of enrichment of glutamate are specific to flux through the CAC and the rate of exchange via OMC, which represents net forward flux through the malate-aspartate shuttle.
With sequential collection of 13C NMR spectra from hearts supplied 13C-enriched LCFA, both the rates of CAC flux and net forward flux through the malate-aspartate shuttle can quantified following direct detection of the end point 13C enrichment levels of glutamate and quantification of the key CAC intermediates that determine the concentration history of influx and efflux through each metabolite pool of the CAC and glutamate [37, 39]. Note also that the absence of appreciable conversion of glutamate to glutamine under well-oxygenated conditions in the myocardium does not result in transfer of 13C to glutamine (Figure 1) that further simplifies the rate analysis of isotope flux through each compartment pool.
Metabolic flux measurements were thusly assessed using previously detailed methods. Measurements are fully quantitative that, unlike other approaches, provide total flux with accounting for both labeled and unlabeled intermediates, and is not limited to only measures of substrate use rates or substrate competition [15, 27, 36, 39, 40]. Data were analyzed using well-established methods that have been previously described in detail under varied experimental conditions in the heart [15, 16, 18, 31, 36, 37, 39].
2.4 Protein expression
Protein expression was measured by Western blot in whole heart tissue. 30 mg of frozen tissue was homogenized in 1X lysis buffer (20mM Tris-HCL, 150mM NaCl, 1mM EGTA, 1mM EDTA, 2.5mM sodium pyrophosphate, 1mM β-glycerophosphate, 1mM Na3VO4, 1μg/ml leupeptin, 1mM PMSF). Tissue lysates were resolved on SDS-PAGE and transferred to PVDF membranes (db/db, N=3; c57b3l/6, N=3; ISC/REP db/db, N=3; c57bl/6, N=3). Membranes were probed by SLC25A11 (Abcam), UCP3 (Abcam), UCP2 (R&D), and PPARα (Abcam) primary antibodies [41]. Western band intensity was analyzed by NIH Image J software and normalized by calsequestrin (Thermal Scientific) as the loading control.
2.5 RNA extraction and quantitative
RT-PCR Quantitative RT-PCR was performed for OMC, UCP2, UCP3, PPARα. Total RNA was extracted from frozen heart tissue by using an RNeasy Lipid Tissue kit (Qiagen), according to the manufacturer’s instructions. RNA quantity was determined at 260 nm (NanoDrop 1000 Spectrometer, Thermal Scientific). Single-stranded cDNA was synthesized from the prepared RNA by using High Capacity cDNA Reverse Transcription kit (Applied biosystems), and gene products were determined by quantitative RT-PCR by using Fast SYBR Green Master Mix (Applied biosystems) with an ABI ViiA7 instrument. The following cycle profile was used: 1 cycle at 95°C for 20 sec, 40 cycles of 95°C for 1 sec, 60°C for 20 sec (db/db, N=5; c57bl/6, N=5; ISC/REP db/db, N=5; c57bl/6, N=5). The mRNA levels were determined by a comparative CT method and normalized by Cyclophilin A [42]. Primers for PPARα have been previously described [41, 42]. Primers for SLC25A11 (OMC), UCP2, and UCP3 were purchased from and specially designed by IDT. Primer sequences are as follows:
| SLC25A11: | 5-CCTTCACCACTCAACTGCAT-3 5-CCTAAGTCTGTCAAGTTCCTGT-3 |
| UCP2: | 5-GCAAGACGAGACAGAGGAAC-3 5-TTAGAGAAGCTTGACCTTGGAG-3 |
| UCP3: | 5-GTCACCATCTCAGCACAGTT-3 5-ATGCCTACAGAACCATCGC-3 |
2.6 Statistical Analysis
Student’s t-test or ANOVA, where appropriate. When ANOVA was used, intergroup statistics were analyzed using one-way ANOVA analysis with the Tukey post-test. Statistical significance was established at 5% probability (P < 0.05). All reported values are reported as averages ± SEM.
3.0 Results
3.1 Hemodynamics
Values for physiological function for each group are displayed in the Table. Under pre-ischemic baseline conditions, mechanical performance, as assessed by rate-pressure-product (RPP) was similar between normal hearts, represented by c57bl/6 and the diabetic, db/db hearts. Consistent with previous studies showing increased susceptibility of the diabetic heart to ischemia/reperfusion damage, the db/db hearts displayed significantly lower recovery of RPP upon reperfusion than c57bl/6 [16, 43].
3.2 Malate/Aspartate Shuttle Activity
Using net forward flux through the oxoglutarate malate carrier protein (OMC) to asses M/A shuttle activity, we show that M/A shuttle activity is significantly lower in the db/db compared to c57bl/6 at baseline and following ISC/REP (Fig. 2). However, expression of OMC protein was significantly higher in the db/db than the c57bl/6 heart at baseline and following ISC/REP (Fig. 3). Messenger transcript levels for OMC were also significantly higher in the db/db heart than the c57bl/6 heart at baseline and following ISC/REP. Thus, upregulation of OMC in the db/db heart is an effect of the diabetic phenotype, rather than result of ISC/REP. This is the first indication of an altered malate-aspartate shuttle component in the diabetic heart, which in light of reduced flux through OMC, appears compensatory
Figure 2.

Malate/Aspartate Shuttle Activity. C57bl/6: N = 6; c57bl/6 + ISC/REP: N = 6; db/db: N = 6; db/db + ISC/REP: N = 7. *P < 0.05 vs. c57bl/6; † P < 0.05 vs. c57bl6 ISC/REP.
Figure 3.
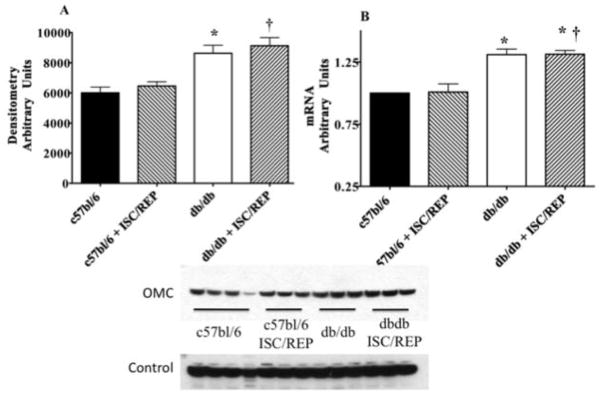
Myocardial Oxoglutarate Malate Carrier (OMC). A. Myocardial OMC protein content. B. Myocardial OMC mRNA content. C. Western blot from perfused c57bl/6 and db/db tissue with and without ISC/REP. Calsequestrin was used for control protein. *P < 0.05 vs. c57bl/6; † P < 0.05 vs. c57bl6 ISC/REP.
3.3 Oxidative Efficiency of Cardiac Function
Under baseline pre-ischemic conditions, CAC flux per unit work (CAC/RPP) is similar for the diabetic heart compared to the c57bl/6 (Fig. 4). The normal c57bl/6 heart is resistant ISC/REP and CAC/unit work remains similar. However, CAC/unit work significantly increases as a result of ISC/REP in the diabetic heart.
Figure 4.

Metabolic Efficiency of Cardiac Function: Citric acid cycle (CAC) flux normalized to rate-pressure product (Vcac/RPP) *P < 0.05, vs. db/db; † P < 0.05 vs. c57bl6 ISC/REP.
While UCP2 transcription is elevated in the db/db heart, there is no difference in protein content (Fig. 5A). This finding is consistent with others showing that the regulation of UCP2 levels occur post-translationally [44]. As a consequence of ISC/REP, message levels for UCP2 were significantly higher in the diabetic heart compared to db/db at baseline and c75bl/6 following ISC/REP. (Fig. 5B). In contrast to UCP2 protein expression, UCP3 protein expression is significantly higher in the diabetic heart, compared to the normal heart (Fig. 5C). While the duration of the current experimental protocol was likely too brief to accommodate changes in protein levels, a prolonged reperfusion period might result in a higher UCP3 protein expression as a result of translation from increased UCP3 mRNA expression (Fig. 5D). Consistent with the diabetic phenotype, PPARα protein expression is significantly elevated in hearts of db/db mice versus the control group (Fig 6A). PCR transcript analysis from normal and db/db hearts, revealed significantly elevated PPARα messaging in the db/db heart post ischemia/reperfusion compared to the normal c57bl/6 heart (Fig. 6B).
Figure 5.

UCP2 and UCP3 Content and Expression. A. UCP2 protein content. B. UCP2 mRNA content. C. UCP3 protein content. D. UCP3 mRNA content. E. Western blot for UCP2 from perfused c57bl/6 and db/db tissue with and without ISC/REP. F. Western blot for UCP3 from perfused c57bl/6 and db/db tissue with and without ISC/REP. Calsequestrin was used for control protein. *P < 0.05, vs. c57bl/6; † P < 0.05 vs. c57bl6 ISC/REP; ‡ P < 0.05 vs. db/db.
Figure 6.

PPARα Content and Expression. A. PPARα protein content. B. PPARα mRNA content. C. Western blot for PPARα from perfused c57bl/6 and db/db tissue with and without ISC/REP. Calsequestrin was used for control protein. *P < 0.05, vs. c57bl/6; † P < 0.05, vs. c57bl6 ISC/REP.
Although circulating fats are variable and trend high, regulatory factors involved in insulin response and mechanical work find and determine fatty acid oxidation to ATP generation and not concentration of exogenous fatty acid which has a greater affect on TAG and lipid dynamics.
4.0 Discussion
The current study is the first demonstration of inefficient citric acid cycle (CAC) flux in the production of mitochondrial NADH in concert with limited cytosolic NADH production and transfer for oxidative support of mechanical function in post-ischemic, diabetic hearts. The model used in this study of DM2, the leptin receptor deficient db/db mouse, is known to display increased oxygen consumption without a proportionate increase in cardiac work following administration of long chain fatty acids [43]. This inefficient oxygen use has been attributed, at least in part, to mitochondrial uncoupling and elevated levels of uncoupling protein 3 (UCP3) [2, 45]. Uncoupling proteins, in particular UCP2 and UCP3, are a class of anion carrier proteins providing an alternate reentry route for H+ into the mitochondria, which is not coupled to ATP synthesis [46]. More recent work suggests that changes in regulation and expression of the mitochondrial anion carrier protein, UCP3, contribute to cardiac inefficiency through mitochondrial uncoupling in the post-ischemic, reperfused state [25].
At baseline pre-ischemic conditions, CAC flux per unit work (CAC/RPP) is similar for the diabetic heart compared to the c57bl/6 (Fig. 4B). This finding is consistent with a report by Harmancey et al, wherein insulin resistance improved coupling between glycolytic rates and glucose oxidation, with improvements in metabolic efficiency of carbohydrate utilization [24]. However, the expression of UCP3 in hearts of animals with high sucrose diet-induced insulin resistance is directionally different from UCP3 expression models, accounting for different levels of uncoupling among models that appear dependent on circulating free fatty acids levels [24, 25]. In this current study, CAC/unit work remains unchanged between non-ischemic and post ISC/REP conditions in normal hearts, but ISC/REP results in a significant increase in CAC/unit work in the diabetic heart. Post ISC/REP, the efficiency of carbon flux though the CAC and oxidative metabolism in supporting the mechanical performance of the diabetic heart is significantly compromised.
Uncoupling proteins
Mitochondrial energetics in the db/db heart are generally considered to be uncoupled [43, 47] and our findings of flux through intermediary metabolism are consistent with this finding. However, uncoupling need not be the sole source of increased CAC flux. Two things account for the large post-ischemic CAC/RPP in the diabetic heart: 1) inefficient coupling of the CAC to oxidative phosphorylation via the increased levels of UCP3 in the db/db heart and induction of UCP3 post-ISC/REP, and 2) in the absence of normal NADHc production, and thus limited transfer of reducing equivalents via the malate-aspartate shuttle, electron transport activity then relies on a compensatory increase in CAC flux for NADH production.
While UCP2 transcription is elevated in the db/db heart, there is no difference in protein content (Fig. 5A). This finding is consistent with others showing that the regulation of UCP2 levels occur post-translationally [44]. In contrast, UCP3 protein expression is significantly higher in the diabetic heart, compared to the normal heart (Fig. 5C). Higher UCP3 levels would exacerbate oxidative inefficiency of post-ischemic cardiac function in the db/db heart. As a consequence of ISC/REP, message levels for UCP3 increased, suggesting a compensatory mechanism to deal with decrease oxidative stress caused by an increase in mitochondrial membrane potential post ISC/REP (Fig. 5D) [44]. While the duration of the current experimental protocol was probably too brief to accommodate changes in protein levels, during a prolonged reperfusion period the increased UCP3 mRNA might result a higher UCP3 protein expression. Any subsequent increase in UCP3 protein expression would exacerbate existing the inefficiencies of the CAC overtime. Consistent with the diabetic phenotype, PPARα protein expression is significantly higher in hearts of db/db mice versus the control group. PPARα messaging is also significantly elevated in the post ischemic db/db heart compared to the c57bl/6 heart post ISC/REP (Fig. 6). UCP3 is regulated by PPARα as a mechanism for handling excess LCFA in the mitochondria [12, 14, 46, 48].
Other studies using mitochondria isolated from ventricle tissue of DM1, STZ injected rats suggest that UCP3 augments LCFA generation and export from the mitochondria, thus preventing excessive accumulation of LCFA metabolites [49]. In the human study, a positive correlation has been found between serum FFA concentrations and UCP2 and UCP3 protein levels [14]. Interestingly, the regulation of UCP2 and UCP3 levels in the heart occurs through separate, distinct mechanisms. While both UCP2 and UCP3 are sensitive to serum FFA concentrations, evidence shows that PPARα regulates UCP3 levels while a PPARα independent pathway regulates UCP2 levels [13, 14, 50]. The role of uncoupling proteins is generally understood to buffer cellular ROS production by lowering the mitochondrial proton gradient [44]. However, there is no clear agreement of the role of uncoupling proteins in disease states. Human studies in heart failure suggest that increases in UCP3 result in increase in mitochondrial H+ leak, thereby exacerbating cardiac inefficiency [14]. Findings in the leptin deficient, ob/ob mice suggest a role for UCP3 in high fat fed induce mitochondrial uncoupling, but no evidence was shown for UCP regulation of cardiac efficiency. Leptin, which induces PPARα, protects the heart from high-fat diet and regulate intracellular lipid homeostasis in nonadipocytes [50, 51]. Studies in both the leptin receptor deficient db/db and PPARα−/− mice show that UCP3 is sensitive to PPARα regulation [48].
Cytosolic NADH oxidation
In the diabetic, db/db heart, elevated OMC levels appear adaptive for a limited availability of cytosolic NADH to supply the electron transport chain that fuels oxidative phosphorylation in the mitochondria. Nevertheless, the increase in OMC did not offset the apparent deficiency in flux through this reducing equivalent shuttle system in the postischemic diabetic heart. Despite increased OMC, our findings indicate that M/A flux remains low in the diabetic heart, a likely consequence of reduced cytosolic NADH/NAD+. Despite elevated OMC content, fueling the electron transport chain with electrons from cytosolic NADH is limited in the diabetic heart, thereby relying more on mitochondrial NADH generation via CAC. Therefore, the reduced contribution of cytosolic NADH, from glycolysis to oxidative, mitochondrial energy metabolism, forces greater reliance on long chain fatty acid oxidation to supply reducing equivalents for the electron transport chain. This observation represents a newly identified mechanism contributing to the well-known reliance of the diabetic heart on high levels of fatty acid oxidation [7, 52].
In both models of DM1 and DM2, there is a reduction in glucose utilization and glucose uptake [6, 46]. Under conditions of either reduced NADHc availability or an inability to respond to demand due to insulin resistance, mitochondria in diabetic hearts are more reliant on the CAC for the balance of NADH production to support electron transfer chain activity and oxidative phosphorylation. The significantly higher protein expression of OMC in the diabetic heart is related to the diabetic phenotype and not responsive to ischemia/reperfusion. The OMC content of diabetic hearts would appear to be a compensatory response to reduced malate-aspartate shuttle activity under conditions of low NADH:NAD+ in the cytosol mitochondria (Fig. 2). Despite a higher OMC content, flux through the M/A shuttle of the db/db hearts was significantly less than that of normal c57bl/6 hearts. Therefore, the upregulation of a M/A shuttle protein in diabetic hearts appears to be a compensatory response to insulin insensitivity in the diabetic heart, but in the current experiments did t offset the limited contributions no of cytosolic NADH to oxidative metabolism.
Conclusions
These data represent the first analysis of both CAC flux and malate aspartate contributions to the oxidative metabolism of the diabetic heart, and its susceptibility to ischemic reperfusion damage. Unlike remote measurements that rely on the released gases from the coronary effluent, the current experimental approach enabled direct detection of flux-dependent events within the cardiomyocytes of the intact, functioning diabetic heart to provide CAC flux and unidirectional flux through the malate aspartate shuttle [19, 53]. In doing so, the study indicates that the db/db heart exhibits limited glycolytic NADH contributions to oxidative energy production, normally transferred to the mitochondrial matrix through the OMC transporter of the malate-aspartate shuttle, with compensatory elevation of OMC. This finding is superimposed on the elevation of UCP3 in diabetic hearts, which renders the diabetic heart at greater risk of mitochondrial inefficiency than normal hearts following ISC/REP. The short-term response of diabetic hearts to ISC/REP included an increased content of UCP3 mRNA, suggesting an additional increase in the inefficiency of CAC flux in the post-ischemic db/db heart with continued reperfusion. Importantly, ISC/REP exacerbates pre-existing mitochondrial defects rendering db/db hearts more susceptible to mitochondrial inefficiencies in CAC flux, including uncoupling and ROS production. The observed inefficiency in carbon flux through the CAC during reperfusion in the postischemic db/db mouse heart is therefore a multifactorial result of elevated uncoupling protein levels and the limitations in availability of cytosolic NADH for oxidative metabolism due to insulin insensitivity for glucose uptake.
Supplementary Material
Table 1.
Hemodynamic function, as indexed by rate-pressure-product (RPP) from c57bl/6 and db/db hearts at baseline and following ischemia/reperfusion.
| Baseline RPP mmHg/min | RRP, ISC/REP mmHg/min | % RPP ISC/REP | |
|---|---|---|---|
| c57bl/6 | 27,206± 692 | NA | NA |
| c57bl/6 ISC/REP |
31,844± 1,517 | 21,268± 1,225* | 66%± 6% |
| db/db | 31,380± 1,812 | NA | NA |
| db/db ISC/REP |
26,910± 3,657 | 14,732± 1,342 †,‡ | 55%± 6 %† |
Note greater impairment in RPP in db/db than control, c57bl/6 hearts. C57b/6: N = 5; c67bl/6 ISC/REP: N = 7; db/db: N = 8; db/db ISC/REP: N = 7. NA, not available.
P < 0.05 vs. c57bl/6 pre-ischemic;
P < 0.05 vs.c57bl6 ISC/REP;
P < 0.05 vs. db/db pre-ischemic.
Highlights.
Carbon flux through oxidative pathways is inefficient in post-ischemic, diabetic hearts
Transporter expression does not fully compensate for limited cytosolic NADH contributions to electron transport in diabetic hearts.
Ischemia/reperfusion induces early UCP3 gene activation in diabetic hearts, suggesting reduced mitochondrial efficiency with prolonged reperfusion.
Acknowledgments
This work was supported by NIH grants R37HL49244, R01HL62702, and R01HL113057.
Abbreviations
- CAC
citric acid cycle
- DM2
type 2 diabetes mellitus
- db/db
db/db mouse
- FAO
fatty acid oxidation
- ISC/REP
ischemia/reperfusion
- LCFA
long chain fatty acid
- MA
malate-aspartate shuttle
- MHC-PPARα
myosin heavy chain-peroxisome proliferator activated receptor alpha
- MVO2
myocardial oxygen consumption
- NADHc
cytosolic NADH
- NMR
nuclear magnetic resonance
- NORM
normal c57bl/6 heart
- OMC
oxoglutarate malate carrier
- RPP
rate pressure product
- UCP3
uncoupling protein 3
- UCP2
uncoupling protein 2
Footnotes
Disclosures
No disclosures to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5.0 References
- 1.Belke DD, Severson DL. Diabetes in mice with monogenic obesity: the db/db mouse and its use in the study of cardiac consequences. Methods Mol Biol. 2012;933:47–57. doi: 10.1007/978-1-62703-068-7_4. [DOI] [PubMed] [Google Scholar]
- 2.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–23. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 3.Chambers KT, Leone TC, Sambandam N, Kovacs A, Wagg CS, Lopaschuk GD, et al. Chronic inhibition of pyruvate dehydrogenase in heart triggers an adaptive metabolic response. J Biol Chem. 2011;286:11155–62. doi: 10.1074/jbc.M110.217349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feuvray D, Lopaschuk GD. Controversies on the sensitivity of the diabetic heart to ischemic injury: the sensitivity of the diabetic heart to ischemic injury is decreased. Cardiovasc Res. 1997;34:113–20. doi: 10.1016/s0008-6363(97)00037-0. [DOI] [PubMed] [Google Scholar]
- 5.Welch MJ, Lewis JS, Kim J, Sharp TL, Dence CS, Gropler RJ, et al. Assessment of myocardial metabolism in diabetic rats using small-animal PET: a feasibility study. J Nucl Med. 2006;47:689–97. [PubMed] [Google Scholar]
- 6.Aasum E, Hafstad AD, Severson DL, Larsen TS. Age-dependent changes in metabolism, contractile function, and ischemic sensitivity in hearts from db/db mice. Diabetes. 2003;52:434–41. doi: 10.2337/diabetes.52.2.434. [DOI] [PubMed] [Google Scholar]
- 7.Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, et al. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–74. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 8.Neely JR, Grotyohann LW. Role of glycolytic products in damage to ischemic myocardium. Dissociation of adenosine triphosphate levels and recovery of function of reperfused ischemic hearts. Circ Res. 1984;55:816–24. doi: 10.1161/01.res.55.6.816. [DOI] [PubMed] [Google Scholar]
- 9.Bugger H, Boudina S, Hu XX, Tuinei J, Zaha VG, Theobald HA, et al. Type 1 diabetic akita mouse hearts are insulin sensitive but manifest structurally abnormal mitochondria that remain coupled despite increased uncoupling protein 3. Diabetes. 2008;57:2924–32. doi: 10.2337/db08-0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hafstad AD, Khalid AM, How OJ, Larsen TS, Aasum E. Glucose and insulin improve cardiac efficiency and postischemic functional recovery in perfused hearts from type 2 diabetic (db/db) mice. Am J Physiol Endocrinol Metab. 2007;292:E1288–94. doi: 10.1152/ajpendo.00504.2006. [DOI] [PubMed] [Google Scholar]
- 11.Wang P, Chatham JC. Onset of diabetes in Zucker diabetic fatty (ZDF) rats leads to improved recovery of function after ischemia in the isolated perfused heart. Am J Physiol Endocrinol Metab. 2004;286:E725–36. doi: 10.1152/ajpendo.00295.2003. [DOI] [PubMed] [Google Scholar]
- 12.Cole MA, Murray AJ, Cochlin LE, Heather LC, McAleese S, Knight NS, et al. A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Res Cardiol. 2011;106:447–57. doi: 10.1007/s00395-011-0156-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, et al. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. 2005;146:5341–9. doi: 10.1210/en.2005-0938. [DOI] [PubMed] [Google Scholar]
- 14.Murray AJ, Anderson RE, Watson GC, Radda GK, Clarke K. Uncoupling proteins in human heart. Lancet. 2004;364:1786–8. doi: 10.1016/S0140-6736(04)17402-3. [DOI] [PubMed] [Google Scholar]
- 15.Yu X, White LT, Alpert NM, Lewandowski ED. Subcellular metabolite transport and carbon isotope kinetics in the intramyocardial glutamate pool. Biochemistry. 1996;35:6963–8. doi: 10.1021/bi960199l. [DOI] [PubMed] [Google Scholar]
- 16.Lewandowski ED, Yu X, LaNoue KF, White LT, Doumen C, O’Donnell JM. Altered metabolite exchange between subcellular compartments in intact postischemic rabbit hearts. Circulation research. 1997;81:165–75. doi: 10.1161/01.res.81.2.165. [DOI] [PubMed] [Google Scholar]
- 17.O’Donnell JM, Doumen C, LaNoue KF, White LT, Yu X, Alpert NM, et al. Dehydrogenase regulation of metabolite oxidation and efflux from mitochondria in intact hearts. Am J Physiol. 1998;274:H467–76. doi: 10.1152/ajpheart.1998.274.2.H467. [DOI] [PubMed] [Google Scholar]
- 18.Griffin JL, O’Donnell JM, White LT, Hajjar RJ, Lewandowski ED. Postnatal expression and activity of the mitochondrial 2-oxoglutarate-malate carrier in intact hearts. Am J Physiol Cell Physiol. 2000;279:C1704–9. doi: 10.1152/ajpcell.2000.279.6.C1704. [DOI] [PubMed] [Google Scholar]
- 19.Griffin JL, White LT, Lewandowski ED. Substrate-dependent proton load and recovery of stunned hearts during pyruvate dehydrogenase stimulation. Am J Physiol Heart Circ Physiol. 2000;279:H361–7. doi: 10.1152/ajpheart.2000.279.1.H361. [DOI] [PubMed] [Google Scholar]
- 20.O’Donnell JM, Kudej RK, LaNoue KF, Vatner SF, Lewandowski ED. Limited transfer of cytosolic NADH into mitochondria at high cardiac workload. Am J Physiol Heart Circ Physiol. 2004;286:H2237–42. doi: 10.1152/ajpheart.01113.2003. [DOI] [PubMed] [Google Scholar]
- 21.Bugger H, Abel ED. Rodent models of diabetic cardiomyopathy. Dis Model Mech. 2009;2:454–66. doi: 10.1242/dmm.001941. [DOI] [PubMed] [Google Scholar]
- 22.Boquist L, Hellman B, Lernmark A, Taljedal IB. Influence of the mutation “diabetes” on insulin release and islet morphology in mice of different genetic backgrounds. J Cell Biol. 1974;62:77–89. doi: 10.1083/jcb.62.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belke DD, Larsen TS, Severson DL. Cardiac function in perfused hearts from diabetic mice. Adv Exp Med Biol. 2001;498:241–5. doi: 10.1007/978-1-4615-1321-6_30. [DOI] [PubMed] [Google Scholar]
- 24.Harmancey R, Lam TN, Lubrano GM, Guthrie PH, Vela D, Taegtmeyer H. Insulin resistance improves metabolic and contractile efficiency in stressed rat heart. FASEB J. 2012;26:3118–26. doi: 10.1096/fj.12-208991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harmancey R, Vasquez HG, Guthrie PH, Taegtmeyer H. Decreased long-chain fatty acid oxidation impairs postischemic recovery of the insulin-resistant rat heart. FASEB J. 2013;27:3966–78. doi: 10.1096/fj.13-234914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belke DD, Swanson EA, Dillmann WH. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes. 2004;53:3201–8. doi: 10.2337/diabetes.53.12.3201. [DOI] [PubMed] [Google Scholar]
- 27.Banke NH, Wende AR, Leone TC, O’Donnell JM, Abel ED, Kelly DP, et al. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPARalpha. Circulation research. 2010;107:233–41. doi: 10.1161/CIRCRESAHA.110.221713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopaschuk GD, Hansen CA, Neely JR. Fatty acid metabolism in hearts containing elevated levels of CoA. Am J Physiol. 1986;250:H351–9. doi: 10.1152/ajpheart.1986.250.3.H351. [DOI] [PubMed] [Google Scholar]
- 29.Lewandowski ED, Doumen C, White LT, LaNoue KF, Damico LA, Yu X. Multiplet structure of 13C NMR signal from glutamate and direct detection of tricarboxylic acid (TCA) cycle intermediates. Magn Reson Med. 1996;35:149–54. doi: 10.1002/mrm.1910350203. [DOI] [PubMed] [Google Scholar]
- 30.O’Donnell JM, Zampino M, Alpert NM, Fasano MJ, Geenen DL, Lewandowski ED. Accelerated triacylglycerol turnover kinetics in hearts of diabetic rats include evidence for compartmented lipid storage. American journal of physiology Endocrinology and metabolism. 2006;290:E448–55. doi: 10.1152/ajpendo.00139.2005. [DOI] [PubMed] [Google Scholar]
- 31.O’Donnell JM, Alpert NM, White LT, Lewandowski ED. Coupling of mitochondrial fatty acid uptake to oxidative flux in the intact heart. Biophys J. 2002;82:11–8. doi: 10.1016/S0006-3495(02)75369-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Donnell JM, Fields A, Xu X, Chowdhury SA, Geenen DL, Bi J. Limited functional and metabolic improvements in hypertrophic and healthy rat heart overexpressing the skeletal muscle isoform of SERCA1 by adenoviral gene transfer in vivo. Am J Physiol Heart Circ Physiol. 2008;295:H2483–94. doi: 10.1152/ajpheart.01023.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lehman JJ, Boudina S, Banke NH, Sambandam N, Han X, Young DM, et al. The transcriptional coactivator PGC-1alpha is essential for maximal and efficient cardiac mitochondrial fatty acid oxidation and lipid homeostasis. Am J Physiol Heart Circ Physiol. 2008;295:H185–96. doi: 10.1152/ajpheart.00081.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bowyer DE. Recovery of lipids from aortas stained with Sudan IV. Atherosclerosis. 1977;26:387–8. doi: 10.1016/0021-9150(77)90093-4. [DOI] [PubMed] [Google Scholar]
- 35.Williamson J, Corkey B. Assays of intermediates of the citric acid cycle and related compounds of fluorometric enzyme methods. New York, NY: Colowick SP Kaplan NO; 1969. [Google Scholar]
- 36.Lewandowski ED, Fischer SK, Fasano M, Banke NH, Walker LA, Huqi A, et al. Acute liver carnitine palmitoyltransferase I overexpression recapitulates reduced palmitate oxidation of cardiac hypertrophy. Circulation research. 2013;112:57–65. doi: 10.1161/CIRCRESAHA.112.274456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu X, White LT, Doumen C, Damico LA, LaNoue KF, Alpert NM, et al. Kinetic analysis of dynamic 13C NMR spectra: metabolic flux, regulation, and compartmentation in hearts. Biophys J. 1995;69:2090–102. doi: 10.1016/S0006-3495(95)80080-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malloy CR, Sherry AD, Jeffrey FM. Evaluation of carbon flux and substrate selection through alternate pathways involving the citric acid cycle of the heart by 13C NMR spectroscopy. J Biol Chem. 1988;263:6964–71. [PubMed] [Google Scholar]
- 39.Yu X, Alpert NM, Lewandowski ED. Modeling enrichment kinetics from dynamic 13C-NMR spectra: theoretical analysis and practical considerations. Am J Physiol. 1997;272:C2037–48. doi: 10.1152/ajpcell.1997.272.6.C2037. [DOI] [PubMed] [Google Scholar]
- 40.O’Donnell JM, White LT, Lewandowski ED. Mitochondrial transporter responsiveness and metabolic flux homeostasis in postischemic hearts. Am J Physiol. 1999;277:H866–73. doi: 10.1152/ajpheart.1999.277.3.H866. [DOI] [PubMed] [Google Scholar]
- 41.Banke NH, Wende AR, Leone TC, O’Donnell JM, Abel ED, Kelly DP, et al. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPARalpha. Circ Res. 2010;107:233–41. doi: 10.1161/CIRCRESAHA.110.221713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riehle C, Wende AR, Zaha VG, Pires KM, Wayment B, Olsen C, et al. PGC-1beta deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circ Res. 2011;109:783–93. doi: 10.1161/CIRCRESAHA.111.243964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–66. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 44.Mailloux RJ, Seifert EL, Bouillaud F, Aguer C, Collins S, Harper ME. Glutathionylation acts as a control switch for uncoupling proteins UCP2 and UCP3. J Biol Chem. 2011;286:21865–75. doi: 10.1074/jbc.M111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boudina S, Abel ED. Mitochondrial uncoupling: a key contributor to reduced cardiac efficiency in diabetes. Physiology (Bethesda) 2006;21:250–8. doi: 10.1152/physiol.00008.2006. [DOI] [PubMed] [Google Scholar]
- 46.Carley AN, Severson DL. Fatty acid metabolism is enhanced in type 2 diabetic hearts. Biochim Biophys Acta. 2005;1734:112–26. doi: 10.1016/j.bbalip.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 47.Boudina S, Han YH, Pei S, Tidwell TJ, Henrie B, Tuinei J, et al. UCP3 regulates cardiac efficiency and mitochondrial coupling in high fat-fed mice but not in leptin-deficient mice. Diabetes. 2012;61:3260–9. doi: 10.2337/db12-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murray AJ, Panagia M, Hauton D, Gibbons GF, Clarke K. Plasma free fatty acids and peroxisome proliferator-activated receptor alpha in the control of myocardial uncoupling protein levels. Diabetes. 2005;54:3496–502. doi: 10.2337/diabetes.54.12.3496. [DOI] [PubMed] [Google Scholar]
- 49.Gerber LK, Aronow BJ, Matlib MA. Activation of a novel long-chain free fatty acid generation and export system in mitochondria of diabetic rat hearts. Am J Physiol Cell Physiol. 2006;291:C1198–207. doi: 10.1152/ajpcell.00246.2006. [DOI] [PubMed] [Google Scholar]
- 50.Rame JE, Barouch LA, Sack MN, Lynn EG, Abu-Asab M, Tsokos M, et al. Caloric restriction in leptin deficiency does not correct myocardial steatosis: failure to normalize PPAR{alpha}/PGC1{alpha} and thermogenic glycerolipid/fatty acid cycling. Physiol Genomics. 2011;43:726–38. doi: 10.1152/physiolgenomics.00088.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou YT, Shimabukuro M, Koyama K, Lee Y, Wang MY, Trieu F, et al. Induction by leptin of uncoupling protein-2 and enzymes of fatty acid oxidation. Proc Natl Acad Sci U S A. 1997;94:6386–90. doi: 10.1073/pnas.94.12.6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–58. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 53.Lewandowski ED, Johnston DL, Roberts R. Effects of inosine on glycolysis and contracture during myocardial ischemia. Circulation research. 1991;68:578–87. doi: 10.1161/01.res.68.2.578. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.