

Abstract
Promoting angiogenesis is a key therapeutic target for protection from chronic ischemic cardiac injury. Endothelial-Monocyte-Activating-Polypeptide-II (EMAP II) protein, a tumor-derived cytokine having anti-angiogenic properties in cancer, is markedly elevated following myocardial ischemia. We examined whether neutralization of EMAP II induces angiogenesis and has beneficial effects on myocardial function and structure after chronic myocardial infarction (MI). EMAP II antibody (EMAP II AB), vehicle, or non-specific IgG (IgG) was injected ip at 30 min and 3, 6, and 9 days after permanent coronary artery occlusion in mice. EMAP II AB, compared with vehicle or non-specific antibody, significantly, p<0.05, improved the survival rate after MI, reduced scar size and attenuated the development of heart failure, i.e., left ventricular ejection fraction was significantly higher in EMAP II AB group, fibrosis was reduced by 24%, and importantly, more myocytes were alive in EMAP II AB group in the infarct area. In support of an angiogenic mechanism, capillary density (193/HPF vs. 172/HPF), doubling of the number of proliferating endothelial cells, and angiogenesis related biomarkers were upregulated in mice receiving EMAP II AB treatment as compared to IgG. Furthermore, EMAP II AB prevented EMAP II protein inhibition of in vitro tube formation in HUVECs. We conclude that blockade of EMAP II induces angiogenesis and improves cardiac function following chronic MI, resulting in reduced myocardial fibrosis and scar formation and increased capillary density and preserved viable myocytes in the infarct area.
Keywords: EMAP II inhibition, myocardial infarction, cardioprotection, angiogenesis
1. Introduction
Myocardial infarction (MI), caused by coronary artery disease, is the leading cause of morbidity and mortality in the United States. After coronary artery occlusion, the recovery of myocardial function is largely dependent on the heart’s ability to re-vascularize the peri-infarcted region. Growth factors such as vascular endothelial growth factor (VEGF) were found to be important in promoting angiogenesis within the peri-infarcted region[1–6]. Consequently, angiogenesis has been studied as a promising therapeutic target in ischemic cardiovascular disease for some time, but despite the huge amount of data supporting this mechanism in experimental settings, little has moved forward to clinical therapy, making the discovery of novel pro-angiogenic mechanisms critical.
Accordingly, we studied a novel candidate, an EMAP II antibody, which blocks the Endothelial-Monocyte-Activating-Polypeptide-II (EMAP II) mediated pathway. EMAP II is a tumor-derived pro-inflammatory cytokine with anti-angiogenic properties[7–13] and inhibits VEGF-induced pro-angiogenic signaling in human umbilical vein endothelial cells (HUVEC)[14], making it an important target for retarding tumor progression [15–18]. In the heart, EMAP II protein expression was increased 24hrs after MI and remained elevated for 6 weeks after MI [19]. If this intrinsic mechanism could be neutralized, it might provide a novel angiogenic mechanism, and protect ischemic myocardium. Indeed, after permanent CAO in hearts without preformed collaterals, angiogenesis is essentially the only mechanism to protect the ischemic myocardium. Therefore the goal of the present investigation was to determine in a mouse MI model, induced by permanent coronary artery occlusion (CAO), whether blocking the EMAPII pathway with an EMAP II neutralizing antibody induces angiogenesis and protects cardiac function after permanent CAO.
2. Materials and Methods
2.1 Surgical animal model of myocardial infarction
We performed left anterior descending (LAD) coronary artery occlusion (CAO) in 3–6 month-old male C57BL/6 mice for myocardial infarction (MI). Briefly, mice were anesthetized with pentobarbital sodium (60 mg/kg, ip). The left side of the chest was opened, and the LAD artery was permanently ligated with 8-0 prolene suture through the third intercostal space incision under proper ventilation. Regional ischemia was confirmed by visual inspection (color change) and ECG monitoring (ST elevation). Sham-operated control mice underwent the same procedure without CAO. Animals used in this study were maintained in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, Eighth Edition 2011). These studies were approved by the Institutional Animal Care and Use Committee in the New Jersey Medical School.
2.2 EMAP II Antibody (AB) treatment
We used a rabbit anti-EMAP II (rEMAP II) antibody (polyclonal monospecific antibody based on immunoblotting of plasma and cell extracts; anti-EMAP II IgG blocked the activity of rEMAP II in cell culture assays) [19]. EMAP II antibody was produced as previously described [20]. In brief, a peptide sequence of 13 amino acid residues located within a homologous region of the human and murine forms of mature EMAP II were used to generate an antibody. This peptide was synthesized and the antibody produced by Zymed Laboratories, Inc. The antibody is specific to EMAP II identified by producing a single band on a western blot that is blocked after being incubated with excess EMAP II. In addition, the antibody used in these publications and in these studies is from a single rabbit and was protein A purified and sterile filtered. EMAP II dosing and injection interval of EMAP II antibody treatment is based on 125I EMAP II antibody labeled clearance studies as previously described [20]. Functionality of the antibody from a single rabbit was validated in vitro in a lung co-culture system markedly increased endothelial cell growth and tube formation [21] and in vivo [20]. Mice were selected randomly for treatment with either vehicle (rabbit IgG) or EMAP II AB (2mg/kg) via intraperitoneal injection by an investigator who was not involved in the surgery. EMAP II AB was injected at 30 min, day 3, day 6, and day 9 after CAO surgery.
2.3 Measurement of cardiac function by echocardiography
To measure LV function, two-dimensional transthoracic echocardiography was performed in mice using ultrasonography (Accuson 256, Siemens Medical Solutions) with a 13-MHz linear ultrasound transducer under anesthesia with 1.25 % Tribromoethanol (Avertin, 290 mg/kg, ip) Measurement of LV end-diastolic and end systolic diameters and wall thickness and LV ejection fraction and fractional shortening were taken from the myocardium proximal to the infarct. Following echocardiography, the heart tissue was harvested after the mice were euthanized using CO2 inhalation.
2.4 Measurement of capillary density and proliferating endothelial cells (ECs)
Four weeks after CAO, the heart was immediately excised, fixed with 10% neutralized buffered formalin and embedded in paraffin for histological studies. 5μm thickness of serial heart tissue sections were used for histological assessment and immunohistochemical staining. In order to discriminate proliferation in non-myocytes and myocytes, tissue sections were counterstained with wheat germ agglutinin (WGA) Alexa Fluor-488 conjugate (Invitrogen) or troponin I, a myocyte marker, as described in our previous study [22].
For measurement of capillary density, ECs were stained with isolectin-IB4 from Griffonia simplicifolia (InVitrogen), and quantitative analysis was accomplished using Image-Pro Plus software after taking pictures under 40×magnification with an Olympus microscope. Relative capillary density was calculated as capillary numbers/HPF (high power field).
Tissue sections were double-immunostained with rabbit-anti Ki67 antibody (Neomaker) or anti-phosphohistone H3 (PHH3) (Cell Signaling) as a proliferation marker with isolectin-IB4 as an EC marker. Antigen retrieval was performed in 10mM citrate buffer (pH 6.0) with boiling under pressure for 10 min. After the blocking process with Protein Block Serum-Free solution (DAKO), the tissue sections were incubated with Ki67 or PHH3 for 1hr at 37°C followed by detection with goat anti-rabbit Alexa 594 conjugated (Invitrogen). The myocardium was further incubated with isolectin-IB4 Alexa Fluor-488 conjugated antibody (Invitrogen) overnight at 4°C. Sections were mounted using ProLong Gold antifade reagent with DAPI (Invitrogen). The proliferation of ECs were expressed as the number of Ki67+Isolectin+ cells per mm2 or PHH3+isolectin+ cells per mm2. Vessels in the infarct area were immunostained with α-smooth muscle actin (α-SMA) (Abcam). Only vessels with ring structure were counted to distinguish arterial vessels from myofibroblasts, which also stain with α-SMA.
2.5 In vitro EC tube formation assay
Human microvascular endothelial cells from the heart (HMVEC-Cs) were purchased from Lonza (Basel, Switzerland). Human umbilical vein endothelial cells (HUVECs) were purchased from ATCC (Manassas, VA, USA). Matrigel (reduced growth factor, BD Bioscience) was placed in 12-well tissue culture plates allowed to gel at 37°C for 30 min. Cells were cultured on the Matrigel with either vehicle (rabbit nonspecific IgG 1μM), EMAP II protein (1μM), or EMAP II AB (1μM) treatment for 20hrs. For an hypoxic condition, cells were incubated in a hypoxia chamber for 5hrs.
2.6 Measurement of interstitial fibrosis
To examine the collagen deposition after MI, tissue sections were stained with Picric acid Sirius Red (PSR) after 1 and 4 weeks with vehicle or EMAP II AB treatment. Quantitative analysis of interstitial fibrosis was accomplished using Image-Pro Plus software after taking pictures under an Olympus microscope at 20× magnification.
2.7 Measurement of scar size after chronic MI
For measuring scar size after 4weeks MI, LV was cross-sectioned at two levels below the coronary artery ligation position, and paraffin-embedded. Each level of tissue was divided by five sub-levels, and a 5μm serial cut was performed. Tissue slides were stained with PSR and scar size was measured with ImageJ software. For measuring viable myocytes in the infarcted area after 4wks MI, LV was stained with troponin I, with isolectin-IB4 and DAPI after longitudinal-sectioning of the heart with a four chamber-view. The number of viable myocytes in the infarct area was determined by counting troponin I + myocytes in the infarct area.
2.8 Myocyte size and number
To measure myocyte cross-sectional area, tissue sections were co-stained with WGA and DAPI, and quantitated using ImagePro-Plus software. The total number of myocytes of each group was measured by the method of Kajstura et al.[23].
2.9 Quantitative RT-PCR
Specific primers and probes (derived with FAM and TAMRA, ordered from IDT DNA Company) were designed for the transcripts of interest. The optimal combination of primers and probes for a qPCR assay was determined with the Primer Express software (Applied Biosystems). Following reverse transcription of the mRNA of interest from 50ng of total RNA, the cDNA was used for quantitative PCR (qPCR) (40 cycles of a 10-s step at 95°C and a 1-min step at 60°C) using the SybrGreen method on a 7700 ABI-Prizm Sequence Detector (Applied Biosystems, Foster City, CA). Values are reported per 18s rRNA transcript to correct for sample-to-sample RNA loading variations.
Genes and primer sequences used in this study:
| Gene | Forward (5′ 3′) → | Reverse (5′ 3′) → |
|---|---|---|
| Rn18s | GTAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGC |
| Ephb2 | GCCGTGGAAGAAACCCTGAT | GTTCATGTTCTCGTCGTAGCC |
| Ephb4 | GGAAACGGCGGATCTGAAATG | TGGACGCTTCATGTCGCAC |
| Cdh1 | CAGTTCCGAGGTCTACACCTT | CCAGGACATGGCCTCTCTC |
| Icam1 | CGCTGTGCTTTGAGAACTGTG | ATACACGGTGATGGTAGCGGA |
| Vcam1 | TTGGGAGCCTCAACGGTACT | GCAATCGTTTTGTATTCAGGGGA |
| Itga5 | TGCAGTGGTTCGGAGCAAC | TTTTCTGTGCGCCAGCTATAC |
| Vegfa | CTGCCGTCCGATTGAGACC | CCCCTCCTTGTACCACTGTC |
| Pecam1 | ACGCTGGTGCTCTATGCAAG | GTGAAGGAGTTTTCCTCAGCC |
| Kdr | TTTGGCAAATACAACCCTTCAGA | GCTCCAGTATCATTTCCAACCA |
2.10 Western blot
Western blot was performed to determine the time course of EMAP II expression after MI as previously described [1]. Briefly, tissue was obtained from the same region in the heart in every animal, rinsed in 50mM Tris pH 7.4, 0.9 N NaCl, and 0.01% NaN3, homogenized in the presence of proteinase inhibitors. The homogenates were cleared by centrifugation at 14,000 g for 20 min; the protein concentration was determined by Bradford analysis (Bio-Rad, CA), and the samples were normalized by protein content. Equal amounts of protein were electrophoresed on a 12% SDS–PAGE gel, transferred to a nitrocellulose membrane. After blocking with 5% milk for 1 hour, the membrane was probed with a rabbit anti-EMAP II antibody (1:200) at 4°C for 1 hour, rinsed with TBST X3, then probed with HRP conjugated anti-rabbit secondary antibody (1:10000, Bio-Rad, CA). Specific binding was detected using a chemiluminescence substrate (Pierce, Rockford, IL) and blue basic autorad film (BioExpress, Belgium). Quantitative analysis was accomplished using a GS-800 calibrated densitometer (Bio-Rad, CA).
2.11 Statistical analysis
All data are expressed as mean ±SE. Comparisons between vehicle and the EMAP II AB treated groups were performed by the Student’s t-test. For statistical analysis of data from multiple groups, we used ANOVA followed by the Student-Newman-Keuls or Bonferroni post hoc analysis. For data that did not meet the ANOVA assumptions, the Kruskal-Wallis test was applied using GraphPad-Prism 5.0 (GraphPad-Software, San Diego, CA). The data for uncensored survival time was analyzed using the Wilcoxon and the log-rank test. p<0.05 was taken as a minimal level of significance.
3. Results
3.1 EMAP II protein was increased in the heart after MI
Previous studies in the rat heart have shown that EMAP II protein expression was increased 24hrs after MI and remained elevated for 6 weeks post MI10. To confirm that EMAP II protein expression was also induced by MI in the mouse heart, we did a time course study of EMAP II expression after MI. As shown in Figure 1C, EMAP II protein expression was increased, p<0.05, by 1.9 fold 24 hours after MI in the mouse heart, and remained elevated, p<0.05, at 1 week (1.3 fold), 2 weeks (1.5 fold) and 4 weeks (1.2 fold).
Figure 1.

EMAP II AB promotes survival and preserves heart function after chronic myocardial infarction (MI). (A) Kaplan-Meyer survival curve of mice treated with either EMAP II AB or vehicle after 4 week chronic MI. *: p<0.05 compared with vehicle, n=30 for EMAP II AB group, n=28 for vehicle group. (B) EMAP II AB treated mice have preserved left ventricle ejection fraction after both 1 and 4 weeks chronic MI. *:p<0.05 compared with vehicle, n=10 for EMAP II AB group, n=7 for vehicle group. (C) Dynamics of EMAPII expression after chronic MI. EMAP II expression was upregulated one day after MI surgery and continues to be highly expressed at least two weeks after MI as shown with representative western blot (top panel) and quantification (bottom panel). **: p<0.01, *: p<0.05, all compared with Sham.
3.2 EMAP II AB significantly improved the survival rate after MI
To test whether EMAP II AB increased survival of mice after chronic myocardial infarction (MI), permanent CAO was induced in 3–6 month-old male C57BL/6 mice, either treated with vehicle (rabbit IgG) or EMAP II AB (2 mg/kg) via intraperitoneal injection. EMAP II AB was injected at 30 min, day 3, day 6 and day 9 after CAO. As shown in Figure 1A, the Kaplan-Meyer survival study indicated that the survival rate in mice treated with EMAP II AB was significantly improved after MI.
3.3 EMAP II AB protected cardiac function after MI
Blockade of EMAP II improved LV systolic function after chronic MI induced by permanent CAO: LV ejection fraction and fractional shortening were not different in sham animals treated with vehicle or EMAPII AB. LV ejection fraction was reduced significantly more (p<0.05) in the vehicle group at 1 week (53±2.2%) and 4 weeks (35±2.4%) after MI than in the EMAP II AB group (61±1.2% at 1 week and 53±4.4% at 4 weeks, respectively) from baseline values in sham operated mice (72±0.5%, n=4) (Figure 1B and Table 1). Fractional shortening was also significantly improved in the EMAP II AB group compared to the vehicle group, by levels similar to those for LV ejection fraction after 4 weeks MI (Table 1). Heart weight/tibial length (HW/TL) was not different in sham animals treated with vehicle or EMAPII AB. At 4 wks after MI, HW/TL was slightly, but not significantly greater in the EMAPII treated mice (8.3±0.4) compared with those receiving vehicle (8.0±0.2), potentially due to the increased numbers of myocytes in the MI zone. Lung weight/TL was not different in either sham or after 4 wks MI in the two groups. It is important to recognize that the beneficial effects of EMAP II AB on LV function after chronic MI would have been even more dramatic, if all animals had survived, since those that died (more in the vehicle group) most likely had worse LV function.
Table 1.
Echocardiographic Data
| Sham | 4 wk MI | ||
|---|---|---|---|
| Vehicle + EMAPII | Vehicle | EMAP-II AB | |
| n | 8 | 7 | 10 |
| LV EF (%) | 72 ± 1.0 | 35 ± 2.4* | 53 ± 4.4*† |
| LV FS (%) | 35 ± 0.8 | 13 ± 1.1* | 23 ± 2.4*† |
| LVEDD(mm) | 3.84 ± 0.05 | 5.51 ± 0.31* | 5.25 ± 0.22* |
| LVESD(mm) | 2.51 ± 0.05 | 4.78 ± 0.29* | 4.08 ± 0.25* |
| Heart Rate (bpm) | 440 ± 5 | 400 ± 18 | 375 ± 13* |
p<0.05 vs. Sham
p<0.05 vs. Vehicle after 4wk MI
(One way ANOVA with Bonferroni post hoc analysis)
3.4 EMAP II AB reduced fibrosis and scar size and preserved myocytes after MI
We then explored the underlying histological changes after EMAP II AB treatment. The scar size was decreased, p<0.05, after 4wks MI with the EMAP II AB (20.8±6.2%, n=6) compared with the vehicle group (32±7.9%, n=6) as shown by PSR staining (Figure 2A). Fibrosis adjacent to the infarct was also reduced in the EMAP II AB group, compared with the vehicle group at both 1 and 4 weeks after MI (Figure 2B). The number of viable myocytes in the infarct area was determined by counting troponin I+ myocytes in the infarct area. More preserved myocytes were seen in the EMAP II AB group in the infarct area, shown with H&E staining, after 4wk MI (Figure 3A). We used double immunostaining of Ki67 as a proliferation marker, and WGA as a myocyte marker, and DAPI to identify the nucleus, to study whether blockade of EMAP II affects the number of proliferating myocytes after MI. Proliferating myocytes were increased with EMAP II AB as shown in Figure 3B.
Figure 2.

EMAP II AB improved LV structure with reduced scar size and collagen deposition. (A) The scar size was decreased after EMAP II AB treatment for 4 weeks. Upper panels: Representative images of infarct rings after 4wk MI as shown by PSR staining; lower panel: quantitative data. (B) The interstitial fibrosis (%) in the adjacent area was less in the EMAP II AB group after both 1 and 4 week MI as shown by PSR staining. Upper panels: representative images after 1 wk MI; lower panel: quantitative data.
Figure 3.

EMAP II AB preserved myocytes and increased proliferating myocytes after chronic MI. (A) More preserved myocytes were seen in EMAP II AB as shown with H&E staining of infarct area after 4 week MI. Left panels: H&E staining images (original magnification, ×10); right panel: quantitative data. (B) Proliferating myocytes were increased with EMAP II AB as shown by Ki67-WGA-DAPI triple staining (left panel) and quantification (right panel). *: p<0.05 compared with vehicle. n=6 for each group.
3.5 EMAP II AB increased angiogenesis after chronic MI
We then used double immunostaining of Ki67 or phosphohistone H3 (PHH3) as proliferation markers, and isolectin-IB4 as an endothelial cell (EC) marker to study whether blockade of EMAP II affects angiogenesis after MI. Using immunostaining of CD 34 as an endothelial progenitor cell marker, vWF as a vessel endothelial cell marker, we found that the progenitor endothelial cells were increased after EMAP II AB treatment (Figure 4A). The number of Ki67-positive EC was also increased in the EMAP II AB group compared to vehicle after 4 week MI (Figure 4B). The EMAP II AB group showed increased capillary density in the non-ischemic zone of the heart compared to vehicle (193±5/HPF vs. 172±4/HPF) (n=5), p<0.05 (Figure 4C). EMAP II AB treatment also increased the number of blood vessel smooth muscle cells after chronic MI (Figure 4D).
Figure 4.

EMAP II AB increased angiogenesis after chronic MI. (A) EMAP II AB increased progenitor ECs as shown by increased numbers of vWF+ and CD34+ endothelial cells. Left panels: vWF and CD34+ staining images; right panel: quantitative data. (B) EMAP II AB increased proliferating ECs as shown by representative photographs of Ki67+ endothelial cells (left panel) and quantification (right panel). (C) EMAP II AB increased capillary density. (D) EMAP II AB also increased blood vessel smooth muscle cells after chronic MI. *: p<0.05 compared with vehicle. n=5 for each group.
3.6 EMAP II AB promotes angiogenesis in vitro
Finally, we used an in vitro tube formation model to confirm that EMAP II AB could promote angiogenesis. HMVEC-Cs were cultured on the Matrigel with vehicle or EMAP II AB treatment for 20hrs. Hypoxia, where cells were incubated in a hypoxia chamber for 5 hours, was used to mimic in vivo MI. As shown in Figure 5A, EMAP II AB significantly increased tube formation as compared with vehicle. Next, to confirm this and also to more closely mimic the in vivo condition, we used EMAP II protein to see whether it could inhibit in vitro tube formation, and whether EMAP II AB could reverse the anti-angiogenic effect of EMAP II protein. As shown in Figure 5B, EMAP II protein did inhibit the in vitro tube formation, and this phenomenon was reversed by EMAP II AB, indicating that the protective role of EMAP II AB was through its neutralization of the anti-angiogenic effect by EMAP II protein. Most of angiogenesis related biomarkers were also upregulated after EMAP II AB treatment (Figure 6). These results strongly suggest that the beneficial effects of EMAP II AB after chronic CAO are due to enhanced angiogenesis.
Figure 5.

EMAP II AB promotes angiogenesis demonstrated with in vitro tube formation assay. (A) EMAP II AB stimulates more tube formation in cultured HMVEC-Cs under hypoxia as shown by representative photographs (left panel; original magnification, ×10) and quantification (right panel). *:p<0.05 compared with vehicle (B) EMAP II protein inhibited tube formation in culture HUVECs which could be rescued by EMAP II AB. as shown by representative photographs (upper panel) and quantification (lower panel). **:p<0.01 compared with no treatment; #: p<0.05 compared with EMAP II protein; +:p<0.05 compared with EMAP II protein + vehicle. Data presented are representative of experiments repeated independently three times.
Figure 6.
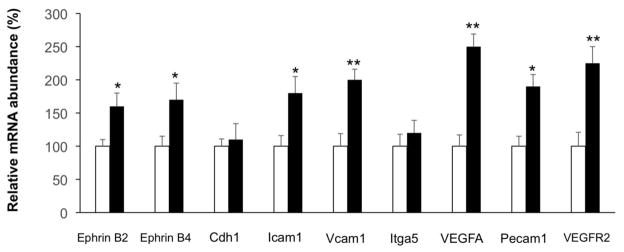
EMAP II AB upregulated angiogenesis related biomarkers after chronic MI as determined by quantitative PCR. Results are expressed as the mean ± SEM. *: p<0.05 compared with vehicle, **: p<0.01 compared with vehicle. n=5 per group.
4. Discussion
In hearts without preformed collateral vessels, it is almost impossible to protect the ischemic myocardium in the absence of angiogenesis. In this study, we found that EMAP II AB reduced MI scar size and significantly improved the survival rate and cardiac function after chronic MI through an EMAP II angiogenic mechanism. EMAP II is an anti-angiogenic protein that has been identified in the myocardium during fetal development [24], and has been shown to inhibit metastatic tumor growth with its anti-angiogenic properties [15, 17, 18, 20]. Furthermore, EMAP II was shown to be expressed with transcription products localizing predominantly to the perivascular stroma region in the normal rat myocardium and was upregulated 2-fold in the heart 6 hours following MI and remained elevated following MI in the peri-infarct region [19].
The underlying hypothesis of this study was that blocking the anti-angiogenic properties of EMAP II after MI would result in increased angiogenesis, which would in turn protect the heart against the development of heart failure after MI. Indeed, the mice treated with EMAP II antibody exhibited reduced mortality from MI (Figure 1A) and protected the ischemic heart, as reflected by reduced infarct scar and adjacent fibrosis resulting in a significant preservation of cardiac function.
The reduced scar and preservation of cardiac function were most likely due to the angiogenesis that was induced by the EMAP II antibody, as demonstrated by increased capillary density, upregulated angiogenesis related biomarkers and increased vessel tube formation in vitro, which has also been demonstrated in developmental models [20, 21]. Furthermore, we have previously reported that inhibition of VEGF signaling is one possible anti-angiogenic mechanism of EMAP II [14]. In that report, we showed that EMAP II protein efficiently and dose dependently binds to VEGF receptor 1 (VEGFR1) and VEGF receptor 2 (VEGFR2). In addition, EMAP II protein inhibited binding of VEGF to VEGFR1 and VEGFR2. In the current study, we did a comprehensive qPCR study of angiogenesis markers and determined that most of these markers, such as VEGFA and VEGFR2 (KDR), were significantly up-regulated after treatment with a neutralizing EMAP II AB treatment, but not nonspecific IgG, as shown in Figure 6. An in vitro vessel tube formation study with HMVEC-Cs cultured under hypoxia, showed that EMAP II AB significantly increased tube formation as compared with vehicle (Figure 5A). In addition, we examined in vitro vessel formation with HUVEC cells treated with either EMAP II protein, EMAP II AB, EMAP II protein + EMAP II AB, or EMAP II protein + nonspecific IgG. We found that EMAP II protein significantly inhibited vessel formation, and this inhibition could be neutralized and rescued by EMAP II AB, but not by nonspecific IgG, as shown in Figure 5B. This supports the concept that VEGF mediated angiogenesis is a key mechanism of action of EMAP II AB neutralization of EMAP II protein.
As a further result of the angiogenesis, the infarct area after 4 week MI demonstrated significantly more viable myocytes in the EMAP II AB group than in the vehicle group, as assessed by dual staining with troponin I and isolectin-IB4. The numbers of proliferating myocytes were also increased at both 1 and 4 weeks after MI, as assessed by Ki67 [25, 26]. Myocyte regeneration and proliferation are controversial topics [27–32]. On one hand several investigators report that this mechanism is important to repairing the infarcted heart [33–38]. On the other hand others have reported that this mechanism does not occur [39, 40]. One other point of view is that it occurs but just at lower levels [41, 42]. Although it could be argued whether or not myocyte regeneration or proliferation occurred in the present study, it is clear that the numbers of Ki67 positive myocytes did increase in the ischemic zone of the mice treated with EMAP II antibody, and most importantly, the numbers of myoctes within the ischemic zone were significantly greater in those animals with antibody treatment.
In summary, neutralization of the anti-angiogenic cytokine, EMAP II, preserved LV function by promoting angiogenesis with reduction of scar size and fibrosis, and preservation of myocytes in the infarct area after chronic MI. Since EMAP II increases naturally during chronic MI, it could play a role in limiting angiogenesis with chronic myocardial ischemia and conversely, blocking this adverse effect could be a potential novel therapeutic target for chronic myocardial ischemic disease.
5. Conclusion
Neutralization of EMAP II induces angiogenesis and improves cardiac function following chronic MI, resulting in reduced myocardial fibrosis and scar formation and increased capillary density and most importantly, preserved viable myocytes in the infarct area.
Highlights.
First in vivo study showing that blockade of EMAP II by a neutralizing antibody significantly improved survival rate after chronic myocardial infarction
Blockade of EMAP II improves cardiac function following chronic myocardial infarction, resulting in reduced myocardial fibrosis and scar formation.
The underlying EMAP II mechanism involves angiogenesis as demonstrated both in vitro and in vivo.
Acknowledgments
Sources of Funding: This study was supported by National Institutes of Health grants HL093481, HL106511, HL033107, HL095888, HL69020, HL60665, AG27211, HL60061, HL75764, HL114977, 0150140N (AHA), 0050782Y (AHA).
Abbreviations
- EMAP II
Endothelial Monocyte Activating Polypeptide II
- CAO
Coronary Artery Occlusion
- MI
Myocardial Infarction
- EF (%)
Ejection Fraction (%)
- HMVEC-Cs
Human Microvascular Endothelial Cells from the Heart
- HUVECs
Human Umbilical Vein Endothelial Cells
Footnotes
Disclosure: The authors declare that there is no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Li J, Brown LF, Hibberd MG, Grossman JD, Morgan JP, Simons M. VEGF, flk-1, and flt-1 expression in a rat myocardial infarction model of angiogenesis. The American journal of physiology. 1996;270:H1803–11. doi: 10.1152/ajpheart.1996.270.5.H1803. [DOI] [PubMed] [Google Scholar]
- 2.Shinohara K, Shinohara T, Mochizuki N, Mochizuki Y, Sawa H, Kohya T, et al. Expression of vascular endothelial growth factor in human myocardial infarction. Heart and vessels. 1996;11:113–22. doi: 10.1007/BF01745169. [DOI] [PubMed] [Google Scholar]
- 3.Miyataka M, Ishikawa K, Katori R. Basic fibroblast growth factor increased regional myocardial blood flow and limited infarct size of acutely infarcted myocardium in dogs. Angiology. 1998;49:381–90. doi: 10.1177/000331979804900507. [DOI] [PubMed] [Google Scholar]
- 4.Tomanek RJ, Zimmerman MB, Suvarna PR, Morkin E, Pennock GD, Goldman S. A thyroid hormone analog stimulates angiogenesis in the post-infarcted rat heart. Journal of molecular and cellular cardiology. 1998;30:923–32. doi: 10.1006/jmcc.1998.0671. [DOI] [PubMed] [Google Scholar]
- 5.Losordo DW, Vale PR, Symes JF, Dunnington CH, Esakof DD, Maysky M, et al. Gene therapy for myocardial angiogenesis: initial clinical results with direct myocardial injection of phVEGF165 as sole therapy for myocardial ischemia. Circulation. 1998;98:2800–4. doi: 10.1161/01.cir.98.25.2800. [DOI] [PubMed] [Google Scholar]
- 6.Horrigan MC, Malycky JL, Ellis SG, Topol EJ, Nicolini FA. Reduction in myocardial infarct size by basic fibroblast growth factor following coronary occlusion in a canine model. International journal of cardiology. 1999;68 (Suppl 1):S85–91. doi: 10.1016/s0167-5273(98)00296-4. [DOI] [PubMed] [Google Scholar]
- 7.Schwarz MA, Kandel J, Brett J, Li J, Hayward J, Schwarz RE, et al. Endothelial-monocyte activating polypeptide II, a novel antitumor cytokine that suppresses primary and metastatic tumor growth and induces apoptosis in growing endothelial cells. The Journal of experimental medicine. 1999;190:341–54. doi: 10.1084/jem.190.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kao J, Ryan J, Brett G, Chen J, Shen H, Fan YG, et al. Endothelial monocyte-activating polypeptide II. A novel tumor-derived polypeptide that activates host-response mechanisms. The Journal of biological chemistry. 1992;267:20239–47. [PubMed] [Google Scholar]
- 9.Kao J, Fan YG, Haehnel I, Clauss M, Stern D. Endothelial-monocyte activating polypeptides (EMAPs): tumor derived mediators which activate the host inflammatory response. Behring Institute Mitteilungen; 1993. pp. 92–106. [PubMed] [Google Scholar]
- 10.Schwarz M, Lee M, Zhang F, Zhao J, Jin Y, Smith S, et al. EMAP II: a modulator of neovascularization in the developing lung. The American journal of physiology. 1999;276:L365–75. doi: 10.1152/ajplung.1999.276.2.L365. [DOI] [PubMed] [Google Scholar]
- 11.Berger AC, Alexander HR, Wu PC, Tang G, Gnant MF, Mixon A, et al. Tumour necrosis factor receptor I (p55) is upregulated on endothelial cells by exposure to the tumour-derived cytokine endothelial monocyte-activating polypeptide II (EMAP-II) Cytokine. 2000;12:992–1000. doi: 10.1006/cyto.2000.0687. [DOI] [PubMed] [Google Scholar]
- 12.Kao J, Houck K, Fan Y, Haehnel I, Libutti SK, Kayton ML, et al. Characterization of a novel tumor-derived cytokine. Endothelial-monocyte activating polypeptide II. The Journal of biological chemistry. 1994;269:25106–19. [PubMed] [Google Scholar]
- 13.Berger AC, Alexander HR, Tang G, Wu PS, Hewitt SM, Turner E, et al. Endothelial monocyte activating polypeptide II induces endothelial cell apoptosis and may inhibit tumor angiogenesis. Microvascular research. 2000;60:70–80. doi: 10.1006/mvre.2000.2249. [DOI] [PubMed] [Google Scholar]
- 14.Awasthi N, Schwarz MA, Verma V, Cappiello C, Schwarz RE. Endothelial monocyte activating polypeptide II interferes with VEGF-induced proangiogenic signaling. Laboratory investigation; a journal of technical methods and pathology. 2009;89:38–46. doi: 10.1038/labinvest.2008.106. [DOI] [PubMed] [Google Scholar]
- 15.Schwarz RE, Schwarz MA. In vivo therapy of local tumor progression by targeting vascular endothelium with EMAP-II. The Journal of surgical research. 2004;120:64–72. doi: 10.1016/j.jss.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 16.Schwarz RE, Awasthi N, Konduri S, Caldwell L, Cafasso D, Schwarz MA. Antitumor effects of EMAP II against pancreatic cancer through inhibition of fibronectin-dependent proliferation. Cancer biology & therapy. 2010;9:632–9. doi: 10.4161/cbt.9.8.11265. [DOI] [PubMed] [Google Scholar]
- 17.Reznikov AG, Chaykovskaya LV, Polyakova LI, Kornelyuk AI. Antitumor effect of endothelial monocyte-activating polypeptide-II on human prostate adenocarcinoma in mouse xenograft model. Experimental oncology. 2007;29:267–71. [PubMed] [Google Scholar]
- 18.Schwarz RE, Awasthi N, Konduri S, Cafasso D, Schwarz MA. EMAP II-based antiangiogenic-antiendothelial in vivo combination therapy of pancreatic cancer. Annals of surgical oncology. 2010;17:1442–52. doi: 10.1245/s10434-009-0879-5. [DOI] [PubMed] [Google Scholar]
- 19.Thompson JL, Ryan JA, Barr ML, Franc B, Starnes VA, Schwarz MA. Potential role for antiangiogenic proteins in the myocardial infarction repair process. The Journal of surgical research. 2004;116:156–64. doi: 10.1016/j.jss.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Schwarz MA, Zhang F, Gebb S, Starnes V, Warburton D. Endothelial monocyte activating polypeptide II inhibits lung neovascularization and airway epithelial morphogenesis. Mechanisms of development. 2000;95:123–32. doi: 10.1016/s0925-4773(00)00361-0. [DOI] [PubMed] [Google Scholar]
- 21.Schwarz MA, Wan Z, Liu J, Lee MK. Epithelial-mesenchymal interactions are linked to neovascularization. American journal of respiratory cell and molecular biology. 2004;30:784–92. doi: 10.1165/rcmb.2003-0145OC. [DOI] [PubMed] [Google Scholar]
- 22.Park M, Vatner SF, Yan L, Gao S, Yoon S, Lee GJ, et al. Novel mechanisms for caspase inhibition protecting cardiac function with chronic pressure overload. Basic research in cardiology. 2013;108:324. doi: 10.1007/s00395-012-0324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kajstura J, Zhang X, Reiss K, Szoke E, Li P, Lagrasta C, et al. Myocyte cellular hyperplasia and myocyte cellular hypertrophy contribute to chronic ventricular remodeling in coronary artery narrowing-induced cardiomyopathy in rats. Circulation research. 1994;74:383–400. doi: 10.1161/01.res.74.3.383. [DOI] [PubMed] [Google Scholar]
- 24.Zhang F, Schwarz MA. Temporo-spatial distribution of endothelial-monocyte activating polypeptide II, an anti-angiogenic protein, in the mouse embryo. Developmental dynamics : an official publication of the American Association of Anatomists. 2000;218:490–8. doi: 10.1002/1097-0177(200007)218:3<490::AID-DVDY1005>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 25.Tamamori-Adachi M, Takagi H, Hashimoto K, Goto K, Hidaka T, Koshimizu U, et al. Cardiomyocyte proliferation and protection against post-myocardial infarction heart failure by cyclin D1 and Skp2 ubiquitin ligase. Cardiovascular research. 2008;80:181–90. doi: 10.1093/cvr/cvn183. [DOI] [PubMed] [Google Scholar]
- 26.Novoyatleva T, Diehl F, van Amerongen MJ, Patra C, Ferrazzi F, Bellazzi R, et al. TWEAK is a positive regulator of cardiomyocyte proliferation. Cardiovascular research. 2010;85:681–90. doi: 10.1093/cvr/cvp360. [DOI] [PubMed] [Google Scholar]
- 27.Akhmedov AT, Marin-Garcia J. Myocardial regeneration of the failing heart. Heart failure reviews. 2013;18:815–33. doi: 10.1007/s10741-012-9348-5. [DOI] [PubMed] [Google Scholar]
- 28.LaPar DJ, Kron IL, Yang Z. Stem cell therapy for ischemic heart disease: where are we? Current opinion in organ transplantation. 2009;14:79–84. doi: 10.1097/MOT.0b013e328320d2e2. [DOI] [PubMed] [Google Scholar]
- 29.Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiological reviews. 2007;87:521–44. doi: 10.1152/physrev.00032.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pavo N, Charwat S, Nyolczas N, Jakab A, Murlasits Z, Bergler-Klein J, et al. Cell therapy for human ischemic heart diseases: Critical review and summary of the clinical experiences. Journal of molecular and cellular cardiology. 2014;75C:12–24. doi: 10.1016/j.yjmcc.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 31.Passier R, van Laake LW, Mummery CL. Stem-cell-based therapy and lessons from the heart. Nature. 2008;453:322–9. doi: 10.1038/nature07040. [DOI] [PubMed] [Google Scholar]
- 32.Rota M, Leri A, Anversa P. Human heart failure: is cell therapy a valid option? Biochemical pharmacology. 2014;88:129–38. doi: 10.1016/j.bcp.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kajstura J, Urbanek K, Perl S, Hosoda T, Zheng H, Ogorek B, et al. Cardiomyogenesis in the adult human heart. Circulation research. 2010;107:305–15. doi: 10.1161/CIRCRESAHA.110.223024. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Kajstura J, Rota M, Cappetta D, Ogorek B, Arranto C, Bai Y, et al. Cardiomyogenesis in the aging and failing human heart. Circulation. 2012;126:1869–81. doi: 10.1161/CIRCULATIONAHA.112.118380. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Laflamme MA, Zbinden S, Epstein SE, Murry CE. Cell-based therapy for myocardial ischemia and infarction: pathophysiological mechanisms. Annual review of pathology. 2007;2:307–39. doi: 10.1146/annurev.pathol.2.010506.092038. [DOI] [PubMed] [Google Scholar]
- 36.Chong JJ, Yang X, Don CW, Minami E, Liu YW, Weyers JJ, et al. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature. 2014;510:273–7. doi: 10.1038/nature13233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nature biotechnology. 2007;25:1015–24. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 38.Anversa P, Kajstura J. Ventricular myocytes are not terminally differentiated in the adult mammalian heart. Circulation research. 1998;83:1–14. doi: 10.1161/01.res.83.1.1. [DOI] [PubMed] [Google Scholar]
- 39.Zak R. Development and proliferative capacity of cardiac muscle cells. Circulation research. 1974;35(suppl II):17–26. [PubMed] [Google Scholar]
- 40.Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. The American journal of physiology. 1996;271:H2183–9. doi: 10.1152/ajpheart.1996.271.5.H2183. [DOI] [PubMed] [Google Scholar]
- 41.Laflamme MA, Murry CE. Heart regeneration. Nature. 2011;473:326–35. doi: 10.1038/nature10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oyama K, El-Nachef D, MacLellan WR. Regeneration potential of adult cardiac myocytes. Cell research. 2013;23:978–9. doi: 10.1038/cr.2013.78. [DOI] [PMC free article] [PubMed] [Google Scholar]