

Abstract
Background
Programmed necrosis (necroptosis) plays an important role in development, tissue homeostasis, and disease pathogenesis. The molecular mechanisms that regulate necroptosis in the heart and its physiological relevance in myocardial remodeling and heart failure remain largely unknown.
Methods and Results
Here we identified an obligate function for TAK1 (TGFβ-activated kinase 1, gene name Map3k7) in regulating necroptotic myocyte death, myocardial remodeling, and heart failure propensity. Cardiac-specific ablation of Map3k7 induced spontaneous apoptosis and necroptosis that led to adverse remodeling and heart failure, and these effects were abolished by ablation of tumor necrosis factor receptor 1 (TNFR1). Mechanistically, TAK1 functions as a “molecular switch” in TNFR1 signaling by regulating the formation of two cell death complexes, RIP1-FADD-caspase 8 and RIP1-RIP3, a process that is dependent on FADD and caspase 8 as scaffolding molecules. Importantly, ablation of RIP1 or RIP3 largely blocked necroptotic cell death, adverse remodeling, and heart failure in TAK1-deficient mice.
Conclusions
These results indicate that TAK1 functions as a key survival factor in the heart by directly antagonizing necroptosis, which is critical for the maintenance of myocardial homeostasis and the prevention of adverse myocardial remodeling.
Keywords: signal transduction, myocyte apoptosis and necrosis, remodeling, heart failure
Introduction
Prominent cell death, unmatched by new cell generation, is a hallmark of cardiomyopathy, acute myocardial injury, and end stage heart failure. The cumulative loss of myocytes in heart failure has been generally attributed to an apoptotic process1, which was thought to be the only form of programmed cell death. While considerably less studied, necrosis may also be a critical mechanism underlying heart failure or with aging2,3. Apoptosis and necrosis are two major types of cell death, each showing distinct morphological features. Apoptosis is an ATP- and caspase-dependent event characterized by chromatin condensation, cell shrinkage, and plasma membrane blebbing4. Necrosis, on the other hand, occurs at an energy-depleted setting and involves organelle swelling, membrane rupture, cell lysis and inflammation4. Necroptosis, a regulated form of necrosis, is mediated by death receptors such as tumor necrosis factor receptor 1 (TNFR1), and is executed through the induction of the RIP1-RIP3 (Receptor-interacting proteins 1 and 3) necroptotic complex, mitochondrial reactive oxygen species (ROS) production, and depletion of cellular energy5–8.
Death receptor ligands such as TNFα can trigger a variety of cellular responses including cell survival, apoptosis, and necrosis, depending on cellular context. Ligation of TNFR1 leads to the assembly of a plasma membrane-bound signaling complex, referred to as complex I, consisting of TNF receptor associated-protein with death domain (TRADD), TNF receptor associated protein 2 (TRAF2), RIP1, and cellular inhibitor of apoptosis protein 1 and 2 (cIAP1 and cIAP2)9. Complex I then recruits and activates TGFβ-activated kinase 1 (TAK1) and the IκB kinase (IKK) complex, leading to the activation of NFκB, which drives the transcription of pro-survival genes. Under certain conditions, complex I dissociates from the membrane and converts to an apoptosis-inducing complex, referred to as complex II, with further recruitment of FADD (Fas-associated protein with death domain) and caspase 89. Moreover, a third complex consisting of RIP1 and RIP3 (termed “necrosome”) can also be induced, which is critical for the initiation of necroptosis5–7. Despite recent progress, the precise molecular mechanisms that determine the distinct cellular outcomes of TNFR1 signaling remain not fully understood.
In this study, we identified TAK1 (encoded by Map3k7 gene) as a nodal regulator of necroptotic cell death by functioning as a “molecular switch” in TNFR1 signaling. TAK1 was first identified as a TGFβ-activated kinase, but recent studies suggest it is also activated by other inflammatory cytokines, such as TNFα, interleukin-1 (IL-1), IL-18, and receptor activator of nuclear factor κB ligands10,11. Importantly, TAK1 is activated in the mouse heart following pathological stress such as pressure overload and myocardial infarction, as well as in diseased human hearts12–14. Here we show that cardiac-specific ablation of Map3k7 in mice promoted pathological remodeling, heart failure, and spontaneous death through induction of apoptotic and necroptotic cell death. We also provide genetic evidence identifying TNFR1-mediated necroptotic signaling as the essential initiating event triggering adverse cardiac remodeling and heart failure in TAK1-deficient mice. Thus regulation of necroptosis by TAK1 is critical for the maintenance of myocardial homeostasis and the prevention of pathological remodeling and heart failure progression.
Methods
An expanded Methods section is available in the Online Data Supplement.
Animal studies
Map3k7 (TAK1) loxP-tageted mouse was described previously15. All procedures were approved and performed according to the guidelines of the Institutional Animal Care and Use Committees of the University of Washington.
Echocardiography, TAC, and plasma HMGB1
Echocardiography was performed with a VisualSonics Vevo 2100 imaging system. Transverse aortic constriction (TAC) was performed using a 27- or 28-gauge needle. Mouse plasma HMGB1 levels were measured using an enzyme-linked immunosorbent assay kit (Chondrex).
Cell lines
Tnfrsf1a−/−, Tnfrsf1b−/−, casp8−/− MEFs were provided by David Vaux (Walter and Eliza Hall Institute Biotechnology Centre, Australia). Ripk1−/− and Ripk3−/− MEFs were obtained from Zheng-gang Liu (National Institutes of Health) and Francis Chan (University of Massachusetts).
Statistics
Exact Wilcoxan Rank-sum or Kruskal-Wallis test was used for studies with small sample sizes as well as whenever feasible for large sample sizes. Data with normal distribution were evaluated by one-way ANOVA with the Bonferroni's post hoc test or repeated-measures ANOVA. P < 0.05 was considered statistically significant.
Results
TAK1 is essential for cardiac myocyte survival and myocardial homeostasis
To study the function of TAK1 in the heart, we generated mice with cardiomyocyte-specific deletion of Map3k7 using a Cre/loxP-dependent conditional gene targeting approach. Mice homozygous for the Map3k7-loxp (fl)-targeted allele (Map3k7fl/fl)15 were crossed with several cardiomyocyte-specific Cre lines including β-myosin heavy chain (MHC)-Cre16, αMHC-Cre17, and a tamoxifen inducible αMHC-MerCreMer18. Western blot analysis showed that TAK1 was efficiently deleted (> 90%) from the hearts of Map3k7fl/fl-βMHC-Cre mice (Figure 1A). Strikingly, these TAK1-deleted mice showed a very severe phenotype with greatly reduced body weight and premature death by postnatal day 8 (Figure 1B and 2D). Mason’s trichrome staining of cardiac sections revealed massive focal and interstitial fibrosis, myocyte dropout, and myofiber disorganization (Figure 1C and 1D). To study the function of TAK1 in the adult heart, we also crossed Map3k7fl/fl mice with the slightly later expressing αMHC-Cre line. Here, TAK1 was again efficiently deleted from the heart of Map3k7fl/fl-αMHC-Cre mice (Figure 1E), but these mice were viable as adults, although ~30% died before 6 months of age. Histological analysis of cardiac sections of Map3k7fl/fl-αMHC-Cre mice again revealed high levels of myocardial fibrosis and cardiomyocyte dropout (Figure 1F and 1G). In addition, a significant increase in TUNEL-positive cells, cleaved caspase 3, and ROS production was detected in cardiac sections of the TAK1-deficient mice (Figure 1H, Supplemental Figure 1A and 1B), as well as increased plasma levels of high mobility group box 1 (HMGB1, Figure 1I) that is a biomarker for necrotic cell death and myocardial infarction19,20. Western blot analysis of the cleaved forms of caspase 8, caspase 3, and PARP showed greater caspase activation in the Map3k7-deficient heart compared with Map3k7fl/fl controls (Supplemental Figure 1C). Intriguingly, Bnip3 (Bcl-2/adenovirus E1B 19 kDa protein-interacting protein 3), which has been implicated in regulating cardiac cell death and pathological remodeling21, was greatly upregulated in the absence of TAK1 (Supplemental Figure 1C). Taken together, these data indicate that loss of TAK1 promoted apoptotic and necrotic cell death in the heart.
Figure 1.

Ablation of TAK1 in the heart triggers pathological cardiac remodeling and dysfunction. (A) Immunoblots for TAK1 and α-tubulin (loading control) in cardiac extracts from Map3k7fl/fl and Map3k7fl/fl-βMHC-Cre mice. (B) Representative images and quantification of body weights of Map3k7fl/fl (n=5) and Map3k7fl/fl-βMHC-Cre (n=4) mice at 1 week of age. *P < 0.05 versus Map3k7fl/fl. (C) Masson’s trichrome-stained, paraffin-embedded sections from the hearts of Map3k7fl/fl and Map3k7fl/fl-βMHC-Cre mice. (D) Myocardial fibrosis as determined by MetaMorph software. *P < 0.01 versus Map3k7fl/fl. n=7 per group. (E) Immunoblots for TAK1 and α-tubulin in cardiac extracts from Map3k7fl/fl and Map3k7fl/fl-αMHC-Cre (Map3k7fl/fl-αCre) mice. (F) Masson’s trichrome-stained, paraffin-embedded sections from the hearts of Map3k7fl/fl and Map3k7fl/fl-αCre mice at 2 months of age. (G) Quantification of myocardial fibrosis by MetaMorph software. *P < 0.01 versus Map3k7fl/fl. n=7 per group. (H) TUNEL-positive nuclei of paraffin-embedded sections from the hearts of Map3k7fl/fl and Map3k7fl/fl-αCre mice. n=5 per group. (I) Plasma HMGB1 levels from Map3k7fl/fl and Map3k7fl/fl-αCre mice. n=4 per group. *P < 0.05 versus Map3k7fl/fl. (J, K, L) Fractional shortening (FS), left ventricular dimension in diastole (LVED), and systole (LVES) determined by echocardiography. (M) Heart weight to body weight ratio (HW/BW) of the indicated groups. *P < 0.05 versus Wt, αCre, or Map3k7fl/fl. Scale bars, 50µm.
Figure 2.

Genetic deletion of TNFR1 rescues pathological remodeling and cardiac dysfunction in TAK1-deficient mice. (A) Masson’s trichrome-stained, paraffin-embedded cardiac sections from mice of the indicated genotypes at 1 week of age. (B) Myocardial fibrosis quantified by MetaMorph software. *P < 0.01 versus Tnfrsf1a−/−Map3k7fl/fl-βCre. n=7 per group. (C) Representative images and quantification of body weights from mice of the indicated genotypes at 1 week of age. n=4–5 per group. *P < 0.05 vs. Tnfrsf1a−/−Map3k7fl/fl-βCre. (D) Survival curve of mice of the indicated genotypes. *P < 0.05 versus Tnfrsf1a−/−Map3k7fl/fl-βCre. (E) Heart weight to body weight ratio (HW/BW) of mice of the indicated genotypes. (F, G, H) Fractional shortening (FS), left ventricular dimension in diastole (LVED) and systole (LVES) determined by echocardiography in mice of the indicated genotypes. *P < 0.05 versus Tnfrsf1a+/+Map3k7fl/fl; #P < 0.05 versus Tnfrsf1a+/+Map3k7fl/fl-αCre. Scale bars, 50 µm.
Map3k7fl/fl-αMHC-Cre mice also showed significantly impaired cardiac function, ventricular dilation, and cardiac hypertrophy as compared to the littermate controls (Figure 1J–1M). These results indicate that ablation of TAK1 promotes adverse ventricular remodeling and cardiac dysfunction, further suggesting a homeostatic role for TAK1 in the adult myocardium. We observed that TAK1 expression in the mouse heart was up-regulated by mild pressure overload, but down-regulated by severe pressure overload (Supplemental Figure 1D and 1E). This led us to investigate the role of TAK1 in regulating myocardial remodeling and heart failure propensity following pathologic stimulation. Mice heterozygous for Map3k7-loxP allele with αMHC-Cre (Map3k7fl/+αCre) were used to evaluate the role of TAK1 in pressure overload-induced cardiomyopathy. In contrast to the Map3k7fl/fl-αCre mice, Map3k7fl/+αCre mice were overtly normal, with no signs of cardiac hypertrophy or cardiac dysfunction at baseline (Supplemental Figure 2). However, pressure overload stimulation uncovered a greater propensity toward cardiac dysfunction and failure in Map3k7fl/+αCre mice (Supplemental Figure 2). Thus, reduced TAK1 expression predisposed mice to cardiac dysfunction and failure after pressure-overload stimulation, suggesting a critical cardio-protective role for TAK1 in response to pathological stress.
Ablation of TNFR1 rescued pathological remodeling and dysfunction of TAK1-deficient heart
The TNFR1-mediated cell death pathway has been implicated in the pathogenesis of myocardial remodeling and heart failure22. Previous studies showed that TAK1 can be recruited to the TNFR1 complex where it regulates downstream signaling23. Based on these observations, we hypothesized that the TNFR1-mediated cell death pathway might account for the development of pathological remodeling and heart failure in the Map3k7-deficient mice. To explore this, we crossed Tnfrsf1a−/− mice with Map3k7fl/fl-βCre mice. Remarkably, ablation of TNFR1 largely rescued the pathological phenotype of Map3k7fl/fl-βCre mice by preventing widespread myocyte drop-out and subsequent cardiac fibrosis, retarded growth, and premature death (Figure 2A–2D). In addition, we crossed Tnfrsf1a−/− mice with Map3k71fl/fl-αCre mice to generate the Tnfrsf1a−/−Map3k7fl/fl-αCre mice for functional analysis of the adult heart. Map3k7fl/fl-αCre mice in the Tnfsrf1a−/− background showed better contractile function, less cardiac hypertrophy, and less ventricular dilation compared with Map3k7fl/fl-αCre mice in the Tnfrsf1a+/+ background (Figure 2E–2H). These data suggest that TNFR1 signaling plays a critical role in the pathogenesis of adverse cardiac remodeling and dysfunction associated with TAK1-deficiency.
TAK1 is a nodal regulator of TNFR1-mediated necroptosis
Based on data presented above, we further hypothesized that deletion of TAK1 triggers adverse cardiac remodeling and heart failure by promoting TNFR1-mediated cell death signaling. To test this hypothesis, we assessed whether TAK1 directly regulates cardiac myocyte survival/death. Rat neonatal cardiomyocytes were infected with an adenovirus encoding the dominant negative TAK1 (AdTAK1-KW) to inhibit endogenous TAK1 activity, or with a control adenovirus expressing β-galactosidase (Ad-βgal), followed by TNFα stimulation. TNFα induced rapid cell death with propidium iodide (PI) uptake in Ad-TAK1-KW infected myocytes, but not in Ad-βgal infected myocytes (Figure 3A). This effect was largely blocked by the RIP1 kinase inhibitor necrostatin-1 (Nec-1)24, but not by z-VAD-fmk (zVAD), a broad-spectrum caspase inhibitor. Thus, inactivation of TAK1 promoted TNFα-induced myocyte death through a RIP1-dependent but caspase-independent mechanism, suggesting the induction of necroptosis. Moreover, Western blot analysis revealed that inactivation of TAK1 by AdTAK1-KW infection promoted the cleavage of PARP, caspase 8, and caspase 3 in cardiac myocytes following TNFα stimulation (Figure 3B). Importantly, HMGB1, a biomarker for necrosis, was readily detectable in the culture supernatants of Ad-TAK1-KW infected cells upon TNFα stimulation for 2 hours (Figure 3B). These results indicate that inhibition of TAK1 promotes both apoptotic and necroptotic signaling. Furthermore, inactivation of TAK1 also abolished TNFα-stimulated c-Jun N-terminal kinase (JNK) phosphorylation and transient IκBα degradation in cardiac myocytes (Figure 3B). These data suggest that TAK1 is essential for the activation of JNK and NFκB, both of which have been implicated in the regulation of cell survival and death.
Figure 3.

TAK1 is a nodal regulator of TNFα-mediated necrotic cell death signaling. (A) Cell death assessed by propidium iodide (PI) staining of cardiomyocytes infected with β-gal or TAK1-KW adenovirus, followed by 10 ng/ml TNFα or vehicle control for 4 hours, in the presence or absence of necrostatin-1 (Nec-1; RIP1 inhibitor) or zVAD-fmk (zVAD; pan caspase inhibitor). *P < 0.05 versus Control; #P < 0.05 versus Ad-TAK1-KW TNFα. (B) Immunoblots for the indicated proteins in cardiomyocytes infected with β-gal or TAK1-KW adenovirus, followed by 10 ng/ml TNF for the indicated times. Asterisks denote cleaved proteins. (C) PI staining of Map3k7+/+ and Map3k7−/− mouse embryonic fibroblasts (MEFs) infected with the indicated adenoviruses, followed by 10 ng/ml TNFα for 4 hours. *P < 0.05 versus Vehicle; #P < 0.05 versus Ad-βgal Map3k7−/− TNFα. (D) Immunoblots for the indicated proteins in wild-type MEFs stimulated with 10 ng/ml TNFα or vehicle control for 4 hours, with or without 5z-7-oxozeaenol (5z-7; TAK1 inhibitor), Nec-1, or zVAD. In A and C, n ≥ 600 cells were quantified from 4 independent experiments.
Consistent with our results in cardiomyocytes, deletion of TAK1 (Map3k7) in mouse embryonic fibroblasts (MEFs) also promoted cell death upon TNFα stimulation (Figure 3C). Importantly, reconstitution of Map3k7−/− MEFs with wild-type TAK1, but not TAK1-KW, prevented TNFα-induced cell death. These results further confirm that TAK1 kinase activity is essential for cell survival upon TNFα stimulation. To further investigate the role of TAK1 in TNFα signaling, wild-type MEFs were treated with 5Z-7-oxozeanol (5z-7)25, a selective inhibitor for TAK1, in the presence or absence of Nec-1 or zVAD. Western blot analysis showed that inhibition of TAK1 with 5z-7 promoted TNFα-induced cleavage of key regulators of cell death pathways, including PARP, caspase 3, RIP1, RIP3, FLIP, and CYLD (Figure 3D). These effects were reversed by the RIP1 inhibitor Nec-1, suggesting that RIP1 is a key regulator for both apoptotic and necroptotic signaling in the setting of TAK1 inhibition. Intriguingly, although zVAD efficiently blocked the cleavage of PARP, caspase 3, and CYLD, a significant loss of full-length RIP1, RIP3, and FLIP was still observed. Importantly, zVAD failed to block the necrotic HMGB1 release. Therefore, protein cleavage, a key event in apoptosis, doesn’t necessarily put necroptosis in check under certain conditions. Finally, we confirmed that TAK1 regulates TNFα-mediated cell death through TNFR1 (Tnfrsf1a), but not TNFR2 (Tnfrsf1b) (Supplemental Figure 3).
TAK1 regulates TNFR1-mediated necroptosis via RIP1-Fadd-caspase 8
Several signaling molecules of extrinsic and intrinsic cell death pathways, such as RIP1, caspase 8, Fadd, and cyclophilin D, have been implicated as key mediators in TNFα-induced cell death26–29. Here we assessed their roles in TAK1 regulated cell death using specific gene-deleted MEFs. Co-stimulation with TNFα and 5z-7 induced rapid necroptotic cell death in wild-type MEFs, which was largely blocked by deletion of RIP1 (Ripk1), FADD (Fadd), or caspase 8 (casp8), but not by cyclophilin D (Ppif) (Figure 4A). These results suggest TAK1 regulates necroptosis through a RIP1/FADD/caspase 8-dependent, but cyclophillin D-independent mechanism. To further assess the role of caspase 8 in necroptosis associated with TAK1 inhibition, we reconstituted casp8−/− MEFs with wild-type caspase 8, a C360A mutant (without proteolytic activity), or an empty vector. Importantly, reconstitution of casp8−/− MEFs with wild-type caspase 8 or C360A mutant restored the necroptotic response, whereas casp8−/− cells containing the empty vector were unable to undergo necroptosis (Supplemental Figure 4). These data suggest a new role for caspase 8 by functioning as a scaffolding molecule in necroptotic signaling.
Figure 4.

TAK1 regulates TNFα-induced cell death through a RIP1-Fadd-caspase 8 dependent mechanism. (A) Cell death assessed by PI staining of wild-type (Wt), Ripk1−/−, Fadd−/−, Casp8−/−, and Ppif −/− MEFs treated with vehicle control or 10 ng/ml TNFα for 4 hours with or without the TAK1 inhibitor 5z-7. Results represent 4 independent experiments. *P < 0.05 vs. vehicle control (Con); #P < 0.05 vs. Wt TNF+5z-7. (B, C, D) Immunoblots with the indicated antibodies from cellular extracts of wild-type and the indicated gene-deleted MEFs treated with TNFα for 0, 1, and 2 hours in the presence or absence of 5z-7. (E) Immunoblots following immunoprecipitation (IP) with an anti-FADD antibody from extracts of cardiomyocytes treated with vehicle control or 10 ng/ml TNFα for 1.5 hours in the presence or absence of 5z-7. (F) Immunoblots following IP with an anti-FADD antibody from extracts of cardiomyocytes infected with βgal, TAK1-ΔN, or TAK1 and TAB1 adenoviruses, followed by treatment with vehicle control or a combination of TNFα, cycloheximide (CHX), and zVAD for 4 hours. (G) Immunoblots for the indicated proteins following IP with an anti-RIP1 antibody from extracts of cardiomyocytes treated with vehicle control or 10 ng/ml TNFα for 10 min in the presence or absence of 5z-7. (H) Immunoblots for phospho-TAK1 at Thr187 (pTAK1), ubiquitin (Ub), and TAK1 following IP with an anti-TAK1 antibody from extracts of cardiomyocytes treated with vehicle control or 10 ng/ml TNFα for 10 min in the presence or absence of 5z-7.
Next, we assessed how FADD, caspase 8, and RIP1 affect apoptotic and necroptotic signaling. Co-stimulation with TNFα and 5z-7, but not TNFα alone, induced PARP and caspase 8 cleavage as well as HMGB1 release in wild-type MEFs (Figure 4B–4D). Importantly, these effects were largely abolished in Fadd−/− or casp8−/− MEFs, suggesting that both FADD and caspase 8 are required for apoptotic and necroptotic signaling in the setting of TAK1 inhibition (Figure 4B and 4C). Interestingly, deletion of RIP1 blocked HMGB1 release, but not PARP or caspase 8 cleavage (Figure 4D). Of note, TNFα alone was able to induce PARP and caspase 8 cleavage in Ripk1−/− MEFs, but not HMGB1 release. Thus, deletion of RIP1 inhibits necroptosis but promotes apoptosis, indicating that RIP1 plays distinct roles in apoptotic and necroptotic signaling.
To initiate cell death, RIP1 must dissociate from the TNFR1 complex and bind to FADD and caspase 8 to form a cell death-inducing complex (termed “complex II”). We next determined whether and how TAK1 regulates complex II formation by immunoprecipitation. As expected, no association of FADD with either RIP1 or caspase 8 was detected in cardiomyocytes treated with vehicle control, 5z-7 alone, or TNFα alone (Figure 4E). However, co-immunoprecipitation of both RIP1 and caspase 8 with FADD was observed in cells treated with 5z-7 plus TNFα, suggesting that inhibition of TAK1 promotes RIP1-FADD-caspase 8 complex formation (Figure 4E). An anti-cleaved caspase 8 antibody was used to recognize mainly the p43 fragment, because pro-caspase 8 migrated at the same mobility as the IgG heavy chain from the FADD immunoprecipitates, precluding the detection of uncleaved pro-caspase 8 by western blotting. Also of note, cleavage of RIP1, caspase 8, and FADD was detected upon treatment with 5z-7 plus TNFα, suggesting the induction of an active complex II (Figure 4E).
We then asked whether activation of TAK1 could inhibit the RIP1/FADD/caspase 8 interaction under necroptosis-inducing conditions. Cardiomyocytes were infected with adenoviruses encoding a constitutively active mutant TAK1ΔN, TAK1 plus its activator TAB1, or Adβgal control, followed by stimulation with TNFα, cycloheximide (CHX), and zVAD. Consistent with previous observations30, cleaved caspase 8 was detectable in the Fadd immunoprecipitates because zVAD failed to block the cleavage of caspase 8 into the p43 fragment (Figure 4F). Importantly, overexpression of TAK1ΔN, or TAK1 plus TAB1, largely blocked the Fadd-RIP1-caspase 8 interaction induced by the combination of TNFα, CHX, and zVAD (Figure 4F). These data strongly suggest that TAK1 functions as a key regulator in TNFα-mediated cell death signaling by specifically blocking the RIP1-FADD-caspase 8 cell death-inducing complex.
To gain further insight into the regulatory mechanism underlying this observation, we determined that TNFα stimulation promoted rapid association of TAK with RIP1 (Figure 4G). Intriguingly, this association was abolished by inhibition of TAK1 with 5z-7 (Figure 4G). Moreover, TAK1 phosphorylation and ubiquitination induced by TNFα was also blocked by 5z-7 (Figure 4H). These data suggest that TAK1 phosphorylation and ubiquitination are critical for the RIP1-TAK1 interaction. Based on these data we propose that activation of TAK1 promotes its association with RIP1, thus preventing the interaction of RIP1 with other cell death signaling proteins, including FADD and caspase 8. On the other hand, inhibition of TAK1 disrupts its association with RIP1, leading to a switch of RIP1-binding partners and the induction of RIP1-FADD-caspase 8 complex. Therefore, TAK1 functions as a “molecular switch” in TNFR1-mediated cell survival and death signaling.
TAK1 regulates the RIP1-RIP3 interaction through Fadd and caspase 8
Induction of necroptosis relies on the interaction of RIP1 and RIP3 through the homotypic interaction motif and the formation of the necroptosis complex5–7. Here we assessed whether TAK1 exerts its effect on necroptotic signaling by regulating the RIP1-RIP3 interaction. Indeed, the RIP1-RIP3 interaction was detected in cells treated with TNFα and 5z-7 (Figure 5A). Nec-1, but not zVAD, efficiently blocked the RIP1-RIP3 interaction, suggesting that RIP1 kinase activity, but not caspase activity, is required in this process (Figure 5A). Importantly, no RIP1-RIP3 interaction was detected in Fadd−/− or casp8−/− MEFs following treatment with 5z-7 plus TNFα (Figure 5B and 5C). Therefore, both FADD and caspase 8 are required in TAK1-mediated molecular regulation of RIP1-RIP3 necrosome formation.
Figure 5.

Inhibition of TAK1 promotes necroptosis by regulating the RIP1-RIP3 interaction. (A, B, C) Immunoblots following IP with an anti-RIP1 antibody from extracts of wild-type (A), Fadd−/− (B), and Casp8−/− (C) MEFs treated with vehicle control or 10 ng/ml TNFα for 1.5 hours in the presence or absence of 5z-7, Nec-1, or zVAD. (D) Immunoblots for the indicated proteins from extracts of cardiomyocytes infected with adenoviruses encoding RIP3 shRNA (Ad-shRIP3) or a scrambled sequence (Ad-shScram), followed by treatment with vehicle control or 10 ng/ml TNFα for 2 hours in the presence or absence of 5z-7, Nec-1, or zVAD. (E) Representative images of cardiomyocytes infected with Ad-shRIP3 or Ad-shScram, followed by treatment with vehicle control or 10 ng/ml TNFα for 4 hours in the presence or absence of 5z-7, Nec-1, or zVAD. Cells were stained with Hoechst 33342 (blue) and PI (red). (F) Quantification of necroptosis (PI positive without chromatin condensation) from cells treated as in e. *P < 0.05 versus vehicle control; #P < 0.05 versus Ad-shScram in the corresponding group; §P < 0.05 versus Ad-shScram 5z-7 plus TNF. (G) Quantification of apoptosis (PI negative with chromatin condensation) from cells treated as in e. *P < 0.05 versus vehicle control; #P < 0.05 versus Ad-shScram 5z-7 plus TNF; §P < 0.05 versus Ad-shRIP3 5z-7 plus TNF. Scale bars, 10 µm. In F and G, n ≥ 600 cells were quantified from 4 independent experiments.
To determine the role of RIP3 in TNFα-induced cell death in the setting of TAK1 inhibition, we infected cardiomyocytes with adenoviral vectors encoding RIP3 shRNA (Ad-shRIP3) or a scrambled sequence (Ad-shScram). RIP3 silencing largely abolished HMGB1 release and necroptic cell death induced by 5z-7 and TNFα, but had no effects on the cleavage of PARP and caspase 8 (Figure 5D– 5F). Similar effects were observed in Ripk3−/− MEFs (Supplemental Figure 5). These data suggest that RIP3 is a dedicated regulator of TNFα-induced necroptosis, but not apoptosis, in the setting of TAK1 inhibition. Interestingly, loss of RIP3 mildly enhanced apoptosis, which was blocked by zVAD (Figure 5G). Moreover, zVAD further increased HMGB1 release and necroptosis, which were blocked by RIP3 silencing (Figure 5D–5F). Taken together, these data suggest that RIP1 is involved in both apoptosis and necroptosis in the setting of TAK1 inhibition, whereas RIP3 is a dedicated regulator of necroptosis.
Blockade of necroptosis prevents pathological remodeling and heart failure in TAK1-deficient mice
To fully assess the role of TAK1 in adult mice, we utilized a tamoxifen inducible Cre-mediated recombination system for acute deletion of Map3k7 in the heart. Map3k7fl/fl mice were crossed with MHC-MerCreMer (MCM) transgenic mice, which in the absence of tamoxifen were overtly normal with no detectable phenotype at baseline (data not shown). After tamoxifen treatment, TAK1 was efficiently deleted from the hearts of Map3k7fl/fl-MCM mice, but not Map3k7fl/fl or MCM control mice (Figure 6A). Strikingly, Map3k7fl/fl-MCM mice developed severe cardiac dysfunction, ventricular dilation, and hypertrophy 2 week after tamoxifen administration (Figure 6B–6D). This was associated with a marked increase in plasma HMGB1 and cardiac fibrosis, indicating that cardiac necrosis and pathological remodeling were induced by acute deletion of Map3k7 in the adult myocardium (Figure 6E–6G). To test the hypothesis that the pathological cardiac phenotype associated with TAK1 deficiency is due to necroptotic myocyte death, Map3k7fl/fl-MCM and control mice were treated with the RIP1 inhibitor Nec-1 or vehicle control along with tamoxifen. Remarkably, administration of Nec-1 largely rescued cardiac dysfunction, ventricular dilation, and hypertrophy in Map3k7fl/fl-MCM mice (Figure 6B–6D). Necrotic HMGB1 release and cardiac fibrosis in the TAK1-deficient mice were also largely abolished by Nec-1 (Figure 6E–6G). These data further suggest that myocardial necroptosis is a critical initiating event in the pathogenesis of cardiac dysfunction and remodeling in Map3k7-deficient mice. Moreover, these results also validate RIP1 as a therapeutic target for heart failure by demonstrating that inhibition of RIP1 by Nec-1 prevented myocyte death, pathological remodeling, and heart failure progression in a mouse model of myocardial necroptosis in vivo.
Figure 6.
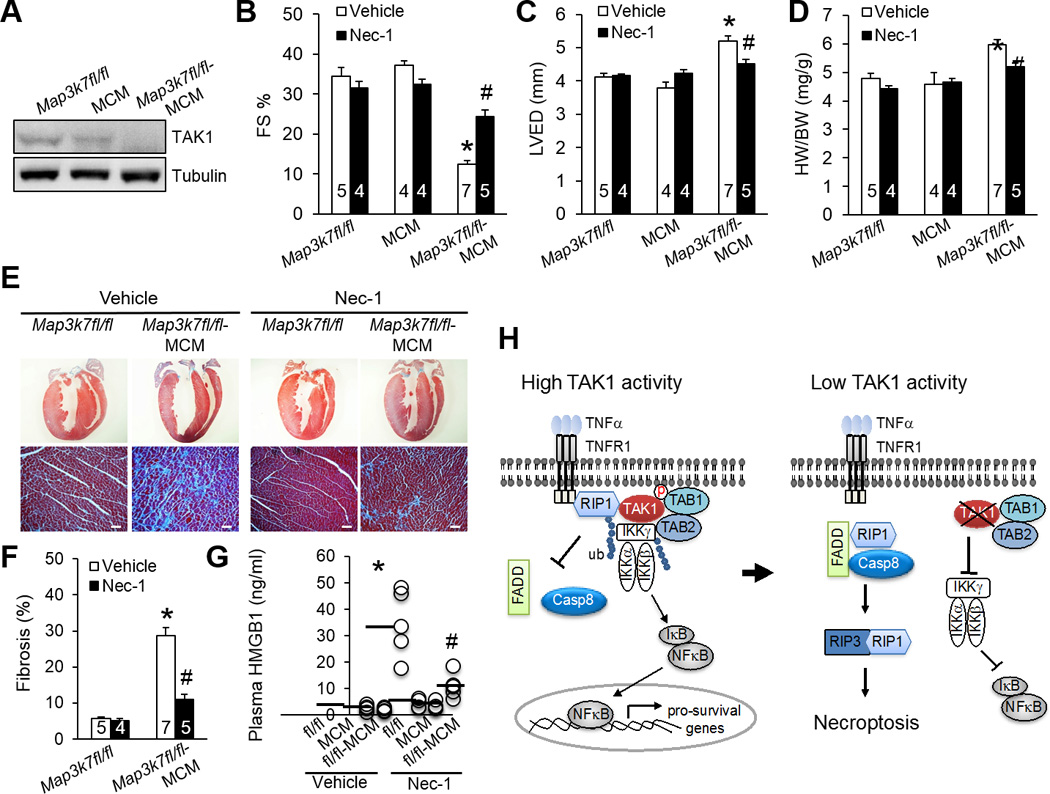
Inhibition of necroptosis with necrostatin-1 rescues pathological cardiac remodeling and dysfunction in TAK1-deficient mice. (A) Immunoblots for TAK1 and α-tubulin in cardiac extracts from Map3k7fl/fl, αMHC-MerCreMer (MCM), and Map3k7fl/fl-MCM mice 2 weeks after treatment with tamoxifen as described in Methods. (B, C, D) FS, LVED, and HW/BW in mice of the indicated genotypes 2 weeks after treatment with tamoxifen in the presence of Nec-1 or vehicle control. *P < 0.01 versus Vehicle Map3k7fl/fl; #P < 0.05 versus Vehicle Map3k7fl/fl-MCM. (E) Masson’s trichrome-stained cardiac sections from mice as described in in B, C, and D. Scale bars, 50 µm. (F) Myocardial fibrosis quantified by MetaMorph software. *P < 0.01 versus Vehicle Map3k7fl/fl; #P < 0.05 versus Vehicle Map3k7fl/fl-MCM. (G) Plasma HMGB1 levels from mice as described in B, C, and D. n=4–5 per group. *P < 0.01 versus Vehicle Map3k7fl/fl; #P < 0.05 versus Vehicle Map3k7fl/fl-MCM. (H) Proposed model: TAK1 functions as a “molecular switch” in TNFR1-mediated cell survival/death signaling.
Discussion
TAK1 signaling is essential for regulating a number of important biological processes, including immune cell activation, inflammation, cell differentiation, and cardiac hypertrophic growth6,31. The present study uncovers a previously unrecognized biological function for TAK1 in regulating myocardial survival/death and cardiac homeostasis. Consistent with our findings, ablation of TAK1 in non-cardiac tissues including skin, intestine, and liver, resulted in spontaneous cell death, inflammation, and fibrosis32–34. However, the mechanism underlying this protective role of TAK1 was not identified in those earlier studies. Here we demonstrate that TAK1 functions as nodal regulator of TNFR1-mediated apoptotic and necroptotic cell death signaling pathways (Figure 6H). Under normal conditions, ligation of TNFR1 promotes the association of TAK1 with RIP1, which prevents RIP1-FADD-caspase8 cell death complex formation. This is associated with the activation of the downstream IKK-NFκB pro-survival pathway (Figure 6H). However, with low or inhibited TAK1 activity, RIP1 dissociates from TAK1 and switches its interacting partners to bind caspase 8 and FADD. The induction of the RIP1-FADD-caspase 8 and the RIP1-RIP3 complexes leads to necroptotic cell death (Figure 6H). In addition, blockade of the IKK-NFκB pro-survival pathway may represent another switch for cell death induction at low TAK1 activity.
In mice, TAK1 expression is relatively high in embryonic and neonatal myocardium and gradually becomes downregulated toward adulthood6. We observed that TAK1 expression in the mouse heart was upregulated by mild pressure overload, but downregulated by severe pressure overload (Supplemental Figure 1). Upregulation of TAK1 seems to represent an important adaptive mechanism under pathological conditions, as TAK1-deficient mice are predisposed to adverse cardiac remodeling and heart failure after pressure overload. In addition, by using an inducible transgenic mouse model expressing active TAK1ΔN, we observed that activation of TAK1 in the adult mice prevented heart failure progression following myocardial infarction (Y Chen, L Li, and Q Liu, unpublished observations). However, conventional transgenic mice with extensive overexpression of TAK1ΔN died within 2 weeks of birth with hypertrophic cardiomyopathy6, suggesting that the timing and extent of TAK1 activation are critical in determining its biological effects in the heart.
Our data indicate that TAK1 also regulates the IKK-NFκB pro-survival pathway in cardiac myocytes. In contrast to the severe cardiac phenotype observed in our TAK1-deficient mice, cardiac-specific ablation of RelA (NFκB-p65) did not show cardiac abnormalities35. Similarly, mice lacking Nfkb1 (NFκB-p50) exhibited no detectable cardiac phenotype at baseline and even showed protection against myocardial ischemic injury36. In line with this, it has been shown that TAK1 can regulate cell death in a NFκB-independent manner37. These observations suggest that although NFκB is potentially important in regulating myocardial survival and death, TAK1 is more critical in determining cell fate by functioning as a molecular switch in determining outcomes of TNFα signaling (pro-survival or pro-death effects22,38).
Although initially thought to constitute mutually exclusive cellular states, we provide evidence that apoptosis and necroptosis can co-exist under certain cell death-inducing conditions. Moreover, we showed that death receptor-mediated apoptosis and necroptosis share some common signaling components. Our data revealed that FADD, RIP1, and caspase-8 are common signaling components for apoptosis and necroptosis under certain conditions. Indeed, we observed that genetic deletion of Fadd or caspase 8 prevented RIP1-RIP3 interaction and necroptotic cell death in TAK1-deficient cells. Whether Fadd is strictly required for the formation of the necroptotic complex has been controversial28,39–41. We demonstrate that Fadd is crucial for both apoptotic and necroptotic signaling through regulation of two cell death-inducing complexes (i.e., RIP1-FADD-caspase 8 and RIP1-RIP3). Similarly, the role of caspase 8 in necroptosis has not been unequivocally established28, 42–45. Our results suggest a new model whereby caspase 8 functions as a scaffold molecule in transducing necroptotic signaling independent of its proteolytic activity under certain cell death-inducing conditions, such as TAK1 inhibition.
There is a lack of mechanistic data implicating necroptotic cell death as a significant contributor to heart failure, although correlative data have been shown. Here we provide an in vivo experimental paradigm that regulation of necroptosis by TAK1 is critical for the maintenance of myocardial homeostasis and the prevention of pathological remodeling and heart failure progression. Our data also suggest that the TAK1 signaling pathway and its effectors may serve as new therapeutic targets for treating heart failure. For instance, we showed that overexpression of TAK1N or TAK1-TAB1 prevented necroptosis signaling in cardiac myocytes. Moreover, based on our observations that FADD and caspase 8 are critical for both apoptotic and necroptotic signaling, it will be important to further evaluate the therapeutic potential of FADD or caspase 8 in experimental models of heart disease. Finally, we demonstrated that inhibition of RIP1 by Nec-1 blocked necroptotic cell death in cardiac myocytes and rescued pathological remodeling and heart failure in a mouse model of myocardial necroptosis in vivo. Nec-1 has also been shown to reduce ischemic injuries in the heart and brain24,46. These results suggest that inhibition of RIP1 may have widespread clinical utility for a range of pathological conditions involving cell death.
Supplementary Material
Acknowledgments
We are very grateful to Jun Ninomiya-Tsuji for providing the Map3k7fl/fl mice originated from Akira laboratory.
Funding Sources: This work was supported by grants from the National Institutes of Health R00HL0908076 and R01HL116507 (to Q.L.) and the Howard Hughes Medical Institute (to J.D.M.).
Footnotes
Disclosures: None.
References
- 1.Foo RS, Mani K, Kitsis RN. Death begets failure in the heart. J Clin Invest. 2005;115:565–571. doi: 10.1172/JCI24569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldspink DF, Burniston JG, Tan LB. Cardiomyocyte death and the ageing and failing heart. Exp Physiol. 2003;88:447–458. doi: 10.1113/eph8802549. [DOI] [PubMed] [Google Scholar]
- 3.Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010;72:19–44. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PubMed] [Google Scholar]
- 4.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 5.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 7.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 8.Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833:3448–3459. doi: 10.1016/j.bbamcr.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 9.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 10.Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 11.Besse A, Lamothe B, Campos AD, Webster WK, Maddineni U, Lin SC, Wu H, Darnay BG. TAK1-dependent signaling requires functional interaction with TAB2/TAB3. J Biol Chem. 2007;282:3918–3928. doi: 10.1074/jbc.M608867200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang D, Gaussin V, Taffet GE, Belaguli NS, Yamada M, Schwartz RJ, Michael LH, Overbeek PA, Schneider MD. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat Med. 2000;6:556–563. doi: 10.1038/75037. [DOI] [PubMed] [Google Scholar]
- 13.Matsumoto-Ida M, Takimoto Y, Aoyama T, Akao M, Takeda T, Kita T. Activation of TGF-beta1-TAK1-p38 MAPK pathway in spared cardiomyocytes is involved in left ventricular remodeling after myocardial infarction in rats. Am J Physiol Heart Circ Physiol. 2006;290:H709–H715. doi: 10.1152/ajpheart.00186.2005. [DOI] [PubMed] [Google Scholar]
- 14.Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, Mankowski J, Zhang D, Nakamura T, Takimoto E, Kass DA. Pivotal role of cardiomyocyte TGF-β signaling in the murine pathological response to sustained pressure overload. J Clin Invest. 2011;121:2301–2312. doi: 10.1172/JCI44824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, Akira S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol. 2005;6:1087–1095. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- 16.Parsons SA, Millay DP, Wilkins BJ, Bueno OF, Tsika GL, Neilson JR, Liberatore CM, Yutzey KE, Crabtree GR, Tsika RW, Molkentin JD. Genetic loss of calcineurin blocks mechanical overload-induced skeletal muscle fiber type switching but not hypertrophy. J Biol Chem. 2004;279:26192–261200. doi: 10.1074/jbc.M313800200. [DOI] [PubMed] [Google Scholar]
- 17.Palermo J, Gulick J, Colbert M, Fewell J, Robbins J. Transgenic remodeling of the contractile apparatus in the mammalian heart. Circ Res. 1996;78:504–509. doi: 10.1161/01.res.78.3.504. [DOI] [PubMed] [Google Scholar]
- 18.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20–25. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
- 19.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 20.Andrassy M, Volz HC, Riedle N, Gitsioudis G, Seidel C, Laohachewin D, Zankl AR, Kaya Z, Bierhaus A, Giannitsis E, Katus HA, Korosoglou G. HMGB1 as a predictor of infarct transmurality and functional recovery in patients with myocardial infarction. J Intern Med. 2011;270:245–253. doi: 10.1111/j.1365-2796.2011.02369.x. [DOI] [PubMed] [Google Scholar]
- 21.Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, Li H, Kirshenbaum LA, Hahn HS, Robbins J, Jones WK, Dorn GW. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest. 2007;117:2825–2833. doi: 10.1172/JCI32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamid T, Gu Y, Ortines RV, Bhattacharya C, Wang G, Xuan YT, Prabhu SD. Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: role of nuclear factor-kappaB and inflammatory activation. Circulation. 2009;119:1386–1397. doi: 10.1161/CIRCULATIONAHA.108.802918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan Y, Yu Y, Shi Y, Sun W, Xie M, Ge N, Mao R, Chang A, Xu G, Schneider MD, Zhang H, Fu S, Qin J, Yang J. Lysine 63-linked Polyubiquitination of TAK1 at Lysine 158 Is Required for Tumor Necrosis Factor α- and Interleukin-1β-induced IKK/NF-κB and JNK/AP-1 Activation. J Biol Chem. 2010;285:5347–5360. doi: 10.1074/jbc.M109.076976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ninomiya-Tsuji J, Kajino T, Ono K, Ohtomo T, Matsumoto M, Shiina M, Mihara M, Tsuchiya M, Matsumoto K. A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J Biol Chem. 2003;278:18485–18490. doi: 10.1074/jbc.M207453200. [DOI] [PubMed] [Google Scholar]
- 26.O'Donnell MA, Legarda-Addison D, Skountzos P, Yeh WC, Ting AT. Ubiquitination of RIP1 regulates an NF-κB-independent cell death switch in TNF signaling. Curr Biol. 2007;17:418–424. doi: 10.1016/j.cub.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Günther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, Becker C. Caspase-8 regulates TNF-alpha induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–339. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Irrinki KM, Mallilankaraman K, Thapa RJ, Chandramoorthy HC, Smith FJ, Jog NR, Gandhirajan RK, Kelsen SG, Houser SR, May MJ, Balachandran S, Madesh M. Requirement of FADD, NEMO, and BAX/BAK for Aberrant Mitochondrial Function in Tumor Necrosis Factor Alpha-Induced Necrosis. Mol Cell Biol. 2011;31:3745–3758. doi: 10.1128/MCB.05303-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 30.Biton S, Ashkenazi A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-α feedforward signaling. Cell. 2011;145:92–103. doi: 10.1016/j.cell.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 31.Liu Q, Busby JC, Molkentin JD. Interaction between TAK1-TAB1-TAB2 and RCAN1-calcineurin defines a signalling nodal control point. Nat Cell Biol. 2009;11:154–161. doi: 10.1038/ncb1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Omori E, Matsumoto K, Sanjo H, Sato S, Akira S, Smart RC, Ninomiya-Tsuji J. Tak1 is a Master Regulator of Epidermal Homeostasis Involving Skin Inflammation and Apoptosis. J Biol Chem. 2006;281:19610–19617. doi: 10.1074/jbc.M603384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inokuchi S, Aoyama T, Miura K, Osterreicher CH, Kodama Y, Miyai K, Akira S, Brenner DA, Seki E. Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc Natl Acad Sci USA. 2010;107:844–849. doi: 10.1073/pnas.0909781107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kajino-Sakamoto R, Inagaki M, Lippert E, Akira S, Robine S, Matsumoto K, Jobin C, Ninomiya-Tsuji J. Enterocyte-derived TAK1 signaling prevents epithelium apoptosis and the development of ileitis and colitis. J Immunol. 2008;181:1143–1152. doi: 10.4049/jimmunol.181.2.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Q, Chen Y, Auger-Messier M, Molkentin JD. Interaction between NFκB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ Res. 2012;110:1077–1086. doi: 10.1161/CIRCRESAHA.111.260729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frantz S, Hu K, Bayer B, Gerondakis S, Strotmann J, Adamek A, Ertl G, Bauersachs J. Absence of NF-kappaB subunit p50 improves heart failure after myocardial infarction. FASEB J. 2006;20:1918–1920. doi: 10.1096/fj.05-5133fje. [DOI] [PubMed] [Google Scholar]
- 37.Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, Vandenabeele P, Bertrand MJ. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 2013;20:1381–1392. doi: 10.1038/cdd.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res. 2002;91:988–998. doi: 10.1161/01.res.0000043825.01705.1b. [DOI] [PubMed] [Google Scholar]
- 39.Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, Lenardo MJ. A role for tumor necrosis factor receptor 2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003;278:51613–51621. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- 40.Bonnet MC, Preukschat D, Welz PS, van Loo G, Ermolaeva MA, Bloch W, Haase I, Pasparakis M. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity. 2011;35:572–582. doi: 10.1016/j.immuni.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 41.Lin Y, Choksi S, Shen HM, Yang QF, Hur GM, Kim YS, Tran JH, Nedospasov SA, Liu ZG. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J Biol Chem. 2004;279:10822–10828. doi: 10.1074/jbc.M313141200. [DOI] [PubMed] [Google Scholar]
- 42.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 43.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lluis JM, Nachbur U, Cook WD, Gentle IE, Moujalled D, Moulin M, Wong WW, Khan N, Chau D, Callus BA, Vince JE, Silke J, Vaux DL. TAK1 is required for survival of mouse fibroblasts treated with TRAIL, and does so by NF-kappaB dependent induction of cFLIPL. PLoS One. 2010;5:e8620. doi: 10.1371/journal.pone.0008620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM. Necrostatin: a potentially novel cardioprotective agent? Cardiovasc Drugs Ther. 2007;21:227–233. doi: 10.1007/s10557-007-6035-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.