

Abstract
The NF-κB family of transcription factors regulates numerous cellular processes, including cell proliferation and survival responses. The constitutive activation of NF-κB has also emerged as an important oncogenic driver in many malignancies, such as activated B-cell like diffuse large B cell lymphoma, among others. In this study, we investigated the impact and mechanisms of action of Withaferin A, a naturally produced steroidal lactone, against both signal-inducible as well as constitutive NF-κB activities. We found that Withaferin A is a robust inhibitor of canonical and constitutive NF-κB activities, leading to apoptosis of certain lymphoma lines. In the canonical pathway induced by TNF, Withaferin A did not disrupt RIP1 polyubiquitination or NEMO-IKKβ interaction and was a poor direct IKKβ inhibitor, but prevented the formation of TNF induced NEMO foci which colocalized with TNF ligand. While GFP-NEMO efficiently formed TNF-induced foci, a GFP-NEMOY308S mutant that is defective in binding to polyubiquitin chains did not form foci. Our study reveals that Withaferin A is a novel type of IKK inhibitor which acts by disrupting NEMO reorganization into ubiquitin-based signaling structures in vivo.
Keywords: NF-κB, NEMO, IKK, ubiquitin, Withaferin A, NEMO foci, ABC type diffuse large B-cell lymphoma, apoptosis
INTRODUCTION
Nuclear Factor kappa B (NF-κB) is a family of transcription factors responsible for regulating numerous cellular processes including immunity, inflammation, and is heavily implicated in numerous types of cancer [1–5]. In the canonical pathway, NF-κB is sequestered in the cytoplasm by the inhibitor of kappa B (IκB), preventing its translocation into the nucleus and subsequent target gene expression [6]. Following stimulation, a series of events are triggered to facilitate protein-protein interactions and perpetuate the signalling cascade. In tumor necrosis factor (TNF) induced NF-κB signalling, receptor-interacting protein 1 (RIP1) undergoes polyubiquitination, serving as a scaffold to recruit TGFβ-activated kinase 1 (TAK1) via the adapter proteins TAB2/3 [7,8]. The polyubiquitin scaffold also allows for recruitment of the IκB kinase (IKK) complex with the main catalytic subunit being IKKβ by a process mediated by NEMO (NF-κB essential modulator), also known as IKKγ. After assembly of the signaling components to these polyubiquitin chains, TAK1 activates IKKβ, resulting in the phosphorylation of the IκB proteins. Phosphorylation of IκB results in its polyubiquitination, marking the protein for degradation by the proteasome which results in the liberation of functionally active NF-κB [9–12].
In addition to the canonical pathway, an alternative or non-canonical pathway for NF-κB activation has also been described. In this pathway, stimuli such as CD40L, B-cell activating factor (BAFF), and Lymphotoxin β receptor (LTβR) activate the NF-κB inducing kinase (NIK), which in conjunction with an IKKα homodimer, induces processing of the NF-κB family member p100 to its functional form, p52 [13,14]. While both pathways play a significant role in normal cellular physiology, the non-canonical pathway is heavily implicated in B-cell maturation and lymphoid tissue development.
In addition to stimulus dependent NF-κB activation, constitutive NF-κB has been reported in certain cell types and cancer tissues [15–17]. Interest in NF-κB is particularly high in hematological malignancies, where the link between cell survival and constitutive NF-κB activity has been well established. This has been described in multiple myeloma, acute myeloid leukemia (AML), as well as activated B cell-like (ABC) diffuse large B cell lymphoma (DLBCL) [18–20]. Numerous mechanisms have been reported that result in constitutive NF-κB activation, including loss of function mutations in deubiquitination enzymes such as A20 or CYLD as well as gain of function mutations in adapter proteins such as MyD88 that results in constitutive organization of NF-κB upstream signaling complexes [21–23]. Inhibition of NF-κB in these cancer types, through pharmacological interventions or by over-expression of the IκBα super-repressor, a mutant form of IκBα incapable of being phosphorylated by IKK, results in cell death via apoptosis. Among individuals with DLBCL, those with the ABC subgroup have the worst clinical prognosis with a 5 year survival of 30%, compared with 59% and 64% amongst those in different subgroups [24,25]. Therefore, targeting constitutive NF-κB activation remains an encouraging strategy in many hematological malignancies, especially in cancers like ABC-DLBCL that have poor prognoses. While several groups have sought to develop an NF-κB inhibitor for use in cancer and other diseases, questions over adverse effects and specificity have prevented mainstream clinical use [26,27].
One approach that has shown promise in pre-clinical trials is the NBD (NEMO binding domain) peptide, a small cell-permeable peptide which mimics the NEMO binding domain of IKKβ, serving to disrupt the interaction between NEMO and IKKβ and thus preventing phosphorylation of IκB [28–31]. However, peptide inhibitors can be costly to manufacture, and require additional permeabilization sequences for intracellular targets, all of which limit the potential clinical utility of the NBD peptide. Chemical inhibitors that disrupt NEMO-IKKβ interaction would be useful for such applications.
An in silico study identified a small molecule, Withaferin A (WA), which was proposed to function similarly to the NBD peptide [32]. WA, a steroidal lactone, is a metabolite from Withania somnifera (winter cherry) that has been reported to have a number of wide-ranging molecular effects [33,34]. In their in silico modeling study, Grover et. al hypothesized that Withaferin A was capable of interacting with NEMO at the site of interaction with IKKβ. Furthermore, WA has been described as an NF-κB inhibitor in several models [35,36]. WA has a number of advantages as a pharmacological inhibitor, mainly its high abundance from natural sources, high bioavailability, and favorable in vivo pharmacokinetics. In mouse studies, WA was observed to have an 82 minute plasma half-life, an advantage for controlling desirable plasma concentrations [37]. Although the above in silico modeling study suggested that disrupts the NEMO-IKKβ interface, this has yet to be experimentally confirmed. Furthermore, it was reported that WA induces hyperphosphorylation of IKKβ resulting in inactivation of its kinase activity [38].
There are several mechanisms by which Withaferin A is proposed to inhibit NF-κB signaling, therefore we first sought to evaluate the mechanisms that had been put forth by Grover et al. and Kaileh et al. We were able to verify robust inhibition of NF-κB by WA in a variety of cell systems and NF-κB stimuli, including genotoxic agents and those that are constitutively activated in ABC-DLBCL cell lines. Indeed, WA induced apoptotic cell death of ABC-DLBCL lines at concentrations that had limited impact on germinal center B-cell (GCB) like DLBCL cells that do not depend on constitutive NF-κB activity for survival. However, our mechanistic data were inconsistent with the concept that WA disrupts the NEMO-IKKβ interaction. Instead, we found that WA disrupts ubiquitin-dependent NEMO reorganization induced by TNF. WA disruption of NEMO reorganization was associated with the lack of IKKβ activation despite efficient activation of the upstream kinase, TAK1. Moreover, WA did not inhibit TNF-induced RIP1 polyubiquitination in vivo. Thus, our findings are consistent with the notion that WA disrupts NEMO’s ability to reorganize into ubiquitin-based signaling structures in vivo. This represents an unprecedented mechanism of NF-κB inhibition by a chemical inhibitor.
METHODS
Reagents
Withaferin A and gram-negative bacterial lipopolysaccharide (LPS) were purchased from Calbiochem (Darmstadt, Germany). Recombinant human and mouse TNFα and recombinant human IL-1β was purchased from EMD Millipore (Darmstadt, Germany). Etoposide (VP-16) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against NEMO (FL-419), Ubiquitin (P4D1), IκBα (C-21) and Actin (I-19) were purchased from Santa Cruz Biotechnology. Antibodies against RIP1 were purchased from BD Biosciences. Antibodies against γ-tubulin (GTU-88) were purchased from Sigma-Aldrich. Antibodies against p52/p100 (4882), Phospho-IκBα (9246), Phospho-IKK (2697), Phospho-JNK (4671), JNK (9251), Phospho-ERK (4377), ERK (4695), Phospho-TAK1 (4536), TAK1 (4505), TNF (11948), and p65 (8242) were purchased from Cell Signaling Technology (Beverly, MA). Anti-rabbit Alexa Fluor 488 or 594 conjugate secondary antibodies (4412 or 8889) and anti-mouse Alexa Fluor 594 conjugate secondary antibody (8890) were from Cell Signaling Technology. OCT1 consensus oligonucleotide was purchased from Promega (Madison, WI).
Cell Culture and Generation of Stable Cell Lines
Cell culture and stable cell conditions were performed as described previously [39]. Briefly, mouse pre-B cell line 70Z/3 and its NEMO-deficient derivative,1.3E2 cells, were cultured in RPMI1640 medium (Thermo Scientific) supplemented with 10% fetal bovine serum (FBS) (HyClone, Thermo Fisher) and 0.05 mM β-mercaptoethanol, penicillin/streptomycin (100 IU/ml and 100 µg/ml) per ml in a 5% CO2 humidified incubator (Forma). Human embryo kidney (HEK) 293 cells were cultured in DMEM medium (Corning Cellgro) supplemented with 10% fetal bovine serum, penicillin/streptomycin (100 IU/ml and 100 µg/ml) per ml in a 10% CO2 humidified incubator (Forma). HEK293 cells were grown on tissue culture plates coated with 0.1% gelatin. Human retinal pigment epithelial (RPE) cells were cultured in DMEM:F12 supplemented with 10% FBS, penicillin/streptomycin (100 IU/ml and 100 µg/ml) per ml in a 5% CO2 humidified incubator. NEMO- and IKKα/β-null MEF cell lines were cultured in DMEM supplemented with 10% FBS, penicillin/streptomycin (100 IU/ml and 100 µg/ml) per ml in a 5% CO2 humidified incubator. GFP-tagged NEMO MEF and RPE lines were generated via retroviral infection using the pBABE-Puro retroviral system. To produce virus, pBABE-Puro WT NEMO, GFP-NEMO, GFP-NEMOY308S, GFP-NEMOΔZF, or GFP-only and p-VSV-G were transiently transfected into Phoenix 293 cells via calcium phosphate and incubated for 8 hours, then media was changed. Virus was harvested after 48 h and filtered through a 0.4 µm membrane. To infect cells, virus was incubated with culture media containing 6 µg/ml polybrene for 12 h. Selection was performed in 1.5 µg/ml and 3 µg/ml puromycin for MEF and RPE cells, respectively. ABC- and GCB-DLBCL cells were grown in RPMI media supplemented with 20% FBS in a 10% CO2 incubator. 1.3E2 stable pools and clones were generated via electroporation and selected with G418 (1 mg/ml) [40].
Electrophoretic Mobility Shift Assay (EMSA)
The EMSA was performed as described previously [41,42]. Briefly, cell extracts were made using TOTEX buffer (20 mM HEPES [pH 7.9], 350 mM NaCl, 1 mM MgCl2, 0.5 mM EDTA, 0.1 mM EGTA, 0.5 mM DTT, 20% glycerol, and 1% NP-40) and 10 µg of extracts were incubated with in gel shift reaction buffer containing poly(dI-dC) in a total volume of 9 µl for 20 minutes. One µl of radiolabeled κB or Oct oligonucleotide probe was added and further incubated for 20 minute at room temperature. For all EMSA reactions, the final NaCl concentration was calculated to be 200 mM or less and was consistent between samples in any one experiment. The samples were separated on a 4% native polyacrylamide gel, dried, and exposed to film and/or a storage phosphor screen.
Luciferase Reporter Assay
The luciferase assay was performed as described previously [43]. Briefly, HEK-293 cells were transfected by calcium phosphate with 25 ng of 3× κB-Luc and 50 ng pRSV-LacZ. The following day, cells were treated with 5 µM WA (co-treatment), 10 ng/ml TNF (8 hours), or 10 µM VP-16 (10 hours). Cells were lysed in lysis buffer (Promega, Madison, WI) containing 1× Halt protease inhibitor cocktail (Thermo Fisher Scientific, Rockford, IL) for 30 minutes while rocking. Cellular debris was pelleted in a microcentrifuge and lysate and 5 µl of lysate was added to a luminometer cuvette for luciferase detection and 5 µl of lysate was added to 70 µl of appropriately diluted Galacton-Plus substrate (Life Technologies, Carlsbad, California). Samples were run with 3 biological replicates and 3 technical replicates on a Monolight 3100 (PharMingen, San Diego, CA).
Flow Cytometry and Cell Viability Assays
Apoptosis was detected by flow cytometry using the FITC Annexin V Apoptosis Detection Kit (BD Pharmigen, San Diego, CA). Cells were then processed on the BD Accuri C6 flow cytometer. Cell viability was measured by incubating cells with trypan blue to a final concentration of 0.04% and determining the ratio of unstained/stained cells.
Co-immunoprecipitation and IKK kinase assay
Co-immunoprecipitation, IKK kinase assay, and western blot analysis were performed as described previously [44].
Immunofluorescence
Cells were cultured on #1.5 coverslips, treated, then rinsed 1× with PBS, fixed in 4% paraformaldehyde for 10 minutes, and rinsed 2× with PBS and stored in PBS at 4°C. A 0.2% triton-X 100 solution was used to permeablize the cells for 5 minutes and a 10% goat serum/PBS solution was used to block for 20 minutes. Primary antibodies were diluted in 1.5% goat serum/PBS and applied for 1 hour. Anti-rabbit Alexa Fluor 488 conjugate secondary antibody or anti-rabbit Alexa Fluor 594 conjugate secondary antibody was diluted 1:1000 (Cell Signaling 4412 or 8889) with 1.5% goat serum/PBS. Slides were washed in PBS, twice rapidly, followed by three 5 minute washes (total of 5 PBS washes) after each antibody incubation. Hoechst (1:7,500) was used as a nuclear stain and coverslips were mounted with Vectashield mounting medium (Vector Labs) or ProLong Gold (Life Technologies). Primary antibody dilutions were 1:50 for NEMO rabbit pAb and 1:200 for p65 rabbit mAb for γ-tubulin mouse mAb. Confocal videos were acquired on a swept-field confocal microscope (Nikon Ti-E) equipped with a Roper CoolSnap HQ2 CCD camera using a Nikon 60×, 1.4 NA plan apo oil objective lens. Widefield images were acquired on a Nikon Ti-Eu Epifluorescence with Nikon DS-Qi1 camera with CCTB 0.7× relay adaptor using either Nikon 40x/0.75 or 60x/0.50–1.25 oil PlanFluor objectives. Nikon Elements was used for image post-processing.
Quantification of NEMO foci
Images were randomly acquired for each condition. For each condition, ten 600× images (roughly 25 RPE nuclei per field) were acquired. For each image, the number of foci per field was measured using the “Find Maxima…” process on ImageJ (NIH). The noise tolerance was set to the nearest value that corresponded to approximately 5 NEMO foci per nuclei in the same field for unstimulated cells. This noise tolerance was then fixed for all subsequent analysis within an experiment (generally around 80). The number of nuclei was scored per field using the ImageJ plugin “Count Cells.” The average number of NEMO foci per nuclei in the same field and one standard deviation was calculated for each condition.
Statistical Analysis
Two-sided Wilcoxon Rank Sum (WRS) [45] was used to determine significant difference for all foci quantification. The Mstat software package (Dr. Norman Drinkwater, University of Wisconsin-Madison, WI, http://www.mcardle.wisc.edu/mstat/) was used for all statistical analysis.
RESULTS
Withaferin A inhibits canonical NF-κB activation and induces apoptotic cell death of ABC-DLBCL cells
While WA has been reported to inhibit NF-κB through the canonical tumor necrosis factor (TNF) pathway [36], we sought to test whether WA could also effectively inhibit activation induced by other inducers as well as the constitutive activity present in certain cancer cell lines. Following WA pre-treatment, HEK293 cells stimulated with either TNF or a topoisomerase II poison, VP-16 (etoposide), exhibited diminished NF-κB activation in a dose-dependent manner as measured by EMSA (Fig. 1A). WA also effectively inhibited ionizing radiation-induced NF-κB activity in a head and neck squamous cell carcinoma cell line, SCC-1483 (Fig. 1B), and that induced by bacterial lipopolysaccharide (LPS) (Fig. 1C). Consistent with the EMSA data above, WA inhibited NF-κB dependent luciferase reporter activity induced by TNF or VP-16 in HEK293 cells (Fig. 1D) and nuclear translocation of p65 after TNF treatment in RPE cells (Fig. 1E). Thus, WA is an effective inhibitor of NF-κB activation induced by multiple canonical inducers in a variety of cell systems.
Figure 1. Withaferin A inhibits canonical and DNA damage induced NF-κB signaling in a variety of cell types.

(A) HEK293 cells were treated with etoposide (VP-16) at 10 µM for 120 minutes or TNF at 10 ng/ml for 30 minutes. Cells were pre-treated with the indicated dose of WA for 30 minutes prior to stimulation with TNF or VP-16. NF-κB activity was measured by EMSA
(B) NF-κB Luciferase assay performed in HEK293 cells, with and without 5 µM WA pre-treatment. Luciferase readings were normalized to β-gal activity.
(C) SCC-1483 cells were irradiated with 20 Gy of ionizing radiation after 30 minutes of pre-treatment with the indicated dose of WA.
(D) RPE cells were pre-treated with WA and treated with TNF. Cells were stained for p65 (red).
We next evaluated if WA could block constitutive NF-κB activity present in the ABC-DLBCL cell lines, OCI-Ly10 and HBL1. Both of these lines contain a mutation in MyD88 (L265P) that results in constitutive canonical NF-κB activation [21]. In addition, we tested a GCB-DLBCL cell line, HT, that does not harbor constitutive NF-κB activity as a control. When the ABC-DLBCL cells were exposed to WA, we observed a decrease in the constitutive NF-κB activity as measured by EMSA (Fig. 2A). In addition, treatment with WA resulted in apoptosis-induced cell death in the ABC-DLBCL cells as measured by flow cytometry (Fig. 2B) and a decrease in cell viability and proliferation (Fig. 2C–D) when compared with the control HT cell line. Thus, WA inhibited constitutive NF-κB activity and induced apoptotic cell death in ABC-DLBCL cell lines at concentrations that had little impact on GCB-DLBCL cells.
Figure 2. Withaferin A induces apoptosis of ABC-DLBCL cells.

(A) NF-κB activity in the ABC DLBCL cell lines OCI-Ly10 and HBL1, both of which harbor the MYD88 L265P mutation, were treated with WA for 3 hours at the time indicated, as measured by EMSA.
(B) Flow cytometry analysis of WA-induced apoptosis by measuring Annexin V and propidium iodide were 2 days after treatment. The percentage of apoptotic cells is shown.
(C,D) Cell viability of ABC and GCL DLBCL cell lines as measured by trypan blue exclusion assay. Data of time and dose course experiments are shown. Bars represent mean ± SD (N = 3).
Withaferin A disrupts canonical NF-κB signaling upstream of IKK activation
To dissect the step (or steps) that WA acts on the canonical NF-κB signaling pathway, we examined intermediate events induced by TNF leading to the release of NF-κB from IκBα by Western blot analysis. Interestingly, WA was able to increased phosphorylation of the major upstream IKKβ kinase TAK1, which indicates its activation (Fig. 3A). Despite an increase in the detection of phospho-TAK1, phosphorylation of IKKβ on S177/181 (IKKα on S176/180) as well as IκBα on S32/36 (Fig. 3A) were both reduced. A time course analysis further indicated that WA could induce phosphorylation of TAK1 and its downstream phosphorylation of two downstream substrates, JNK and ERK, without inducing phosphorylation of IKKβ (Fig. 3B). If WA was an IKKβ inhibitor as suggested by Kaileh et al., it could explain the above observations. Thus, to test whether WA is a direct inhibitor of IKK kinase activity or not, we performed an in vitro IKK kinase assay whereby endogenous IKK was immunoprecipitated with an anti-NEMO antibody and mixed with recombinant GST-IκBα (residues 1–56) in the absence and presence of WA. We observed a robust increase in TNF induced IKK kinase activity, which was inhibited by pre-treatment of cells with WA. However, immunoprecipitated IKK treated directly with WA in vitro still maintained kinase activity at concentrations that inhibited IKK kinase activity when cells were treated in culture (Fig. 3A and 3C). While we observed a small decrease in activity at the highest in vivo concentration tested, the same concentration of WA used during the treatment of cells resulted in total inhibition of IKK catalytic activity. Thus, inhibition of the IKK kinase activity observed in WA-treated cells is likely related to inhibition of a step (or steps) upstream of IKKβ activation as opposed to targeted direct inhibition of IKKβ.
Figure 3. NF-κB inhibition by Withaferin A is upstream of IKK phosphorylation, but downstream of TAK1 activation.

(A) HEK293 cells were treated with TNF at 10 ng/ml and pre-treated with WA at the dose indicated 30 minutes prior to TNF treatment. Cells were lysed and assayed by western blot.
(B) HEK293 cells were treated with 5 µM WA or 10 ng/ml TNF at the time course indicated. Cells were lysed and immunoblotted as indicated.
(C) IKK in vitro kinase assay. HEK293 cells were pre-treated with WA for 30 minutes and treated as indicated. Cells were lysed and the IKK complex was immunoprecipitated using an α-NEMO antibody. Kinase activity was measured using GST-IκBα substrate and quantified by phosphorimager.
Withaferin A does not disrupt NEMO-IKK interaction
A previous in silico modeling study has suggested that WA could be capable of interacting with NEMO at the IKKβ binding site, thus potentially disrupting the protein-protein interaction similar to NBD peptide [32]. If this were the case, active TAK1 would be unable to activate IKKβ because NEMO is required to recruit polyubiquitin chain scaffolds to mediate TAK1-dependent IKK activation [7]. We tested this model by co-immunoprecipitation of NEMO-IKKβ complex with α-NEMO antibodies from WA-treated cells. We also added WA in all immunoprecipitation and wash buffers to maintain its effective concentration. However, we were unable to observe any disruption of the NEMO and IKKα/β interaction upon treatment of the cells or the lysate with WA (Fig. 4A). We repeated the experiment in 70Z/3 cells, a mouse pre-B cell line, which revealed an identical result (Fig. 4B).
Figure 4. Withaferin A does not disrupt NEMO-IKK complex.

(A/B) HEK293 cells (A) or 70Z/3 cells (B) were treated for 30 minutes with the indicated dose of WA. Cells were lysed in the continued presence of the indicated dose of WA and subjected to NEMO immunoprecipitation.
(C) NEMO deficient 1.3E2 cells were stably reconstituted with 6× Myc NEMO wild-type or 6× Myc tagged NEMO. Cells were pre-treated with DMSO or WA for 30 minutes (dose indicated) and treated with LPS for 30 minutes (10 ng/µl). Cells were harvested and NF-κB activity was measured by EMSA.
(D) NEMO deficient 1.3E2 cells were stably reconstituted with untagged NEMO wild-type, NEMO E89/99A, or NEMO 5A (E89A, F92A, L93A, M94A, F97A) and treated with VP-16 for 120 minutes (10 µM), LPS for 30 minutes (10 ng/µl), or LPS after 30 minute WA pre-tx (10 ng/µl and 3 µM, respectively) as indicated. NF-κB activity was measured via EMSA.
In the previous modeling study [32], several key amino acids were identified as being critical for NEMO to interact with WA. Among the most important were glutamic acid residues at position 89 and 99 of NEMO, which were thought to bind WA via ionic interactions. We theorized that mutating the residues thought to interact with WA would prevent WA from interacting with NEMO and thus create a WA ‘resistant’ version of NEMO. Using 1.3E2 cells, a NEMO-deficient derivative of 70Z/3, we created cell lines expressing wild-type NEMO or NEMO E89A and an E89A/E99A double mutant. When treated with WA and either LPS or VP-16, both cell lines were still equally inhibited by WA (Fig. 4C). Even when additional residues implicated in WA-NEMO interaction were mutated (5A mutant), we still observed inhibition of NF-κB activation by WA (Fig. 4C). Together with the co-immunoprecipitation experiments above, our results are inconsistent with the hypothesis that WA disrupts the protein-protein interaction between NEMO and IKKβ. Thus, WA must act on another mechanism to prevent TAK1-dependent IKKβ activation.
TNF-induced NEMO structures detected and quantified by widefield immunofluorescence
As TAK1 activation and subsequent phosphorylation of JNK and ERK were induced by WA but IKKβ activation was prevented (Fig. 3B), we hypothesized that WA could be effecting the polyubiquitin chain-related events that are known to mediate TAK1-dependent IKK activation [46–48]. In contrast, polyubiquitin chains are not implicated in TAK1-dependent phosphorylation of JNK and ERK. We next tested whether WA could prevent changes in NEMO localization induced by TNF stimulation by immunofluorescence analysis. Prior to testing the impacts of WA, we first characterized the induction of NEMO reorganization following TNF stimulation and the requirement of NEMO's ability to bind ubiquitin.
A TNF time course analysis using RPE cells demonstrated a time-dependent induction of NEMO foci (Fig. 5A). We employed RPE cells because of their flat morphology which aided the analysis of NEMO reorganization by immunofluorescence. TNF-induced NEMO foci were readily detected four minutes after TNF treatment and maximum foci formation was observed 8 minutes after TNF stimulation. The NEMO foci were generally uniform in size (roughly 250–500 nm) and foci size did not appear to change appreciably over time. NEMO foci were generally evenly distributed, but were localized near the cell surface, as observed in a video of a confocal Z-stack series (Fig. SV1A–B). Next, we treated RPE cells with increasing doses of TNF for 8 minutes, then examined their NEMO foci by NEMO immunofluorescence as above. We found that increasing the dose of TNF used for treatment did not affect the size of the NEMO foci induced at 8 minutes, but rather induced more NEMO foci (Fig. 5B). We used the ImageJ process “Find Maxima…” to quantify the number of NEMO foci per nuclei in the same field to estimate the number of NEMO foci per cell (Fig. 5C and 5D). The number of NEMO foci increased up to 8 minutes after TNF stimulation, and began to decrease by 10 minutes (Fig. 5C). We observed an increase in the number of NEMO foci from 10 to 20 ng/ml, with the number of foci reaching saturation around 20 ng/ml TNF (Fig. 5D). Every dose of TNF, from 10 to 80 ng/ml, had significantly more NEMO foci than untreated RPE cells (P < 0.0001). We confirmed the specificity of the anti-NEMO polyclonal rabbit antibody by evaluating NEMO-null mouse embryonic fibroblasts (MEFs) which showed no signals whereas those reconstituted with wild-type NEMO exhibit robust NEMO immunofluorescence signal (Fig. S1).
Figure 5. In situ detection and quantification of TNF-induced NEMO structures.
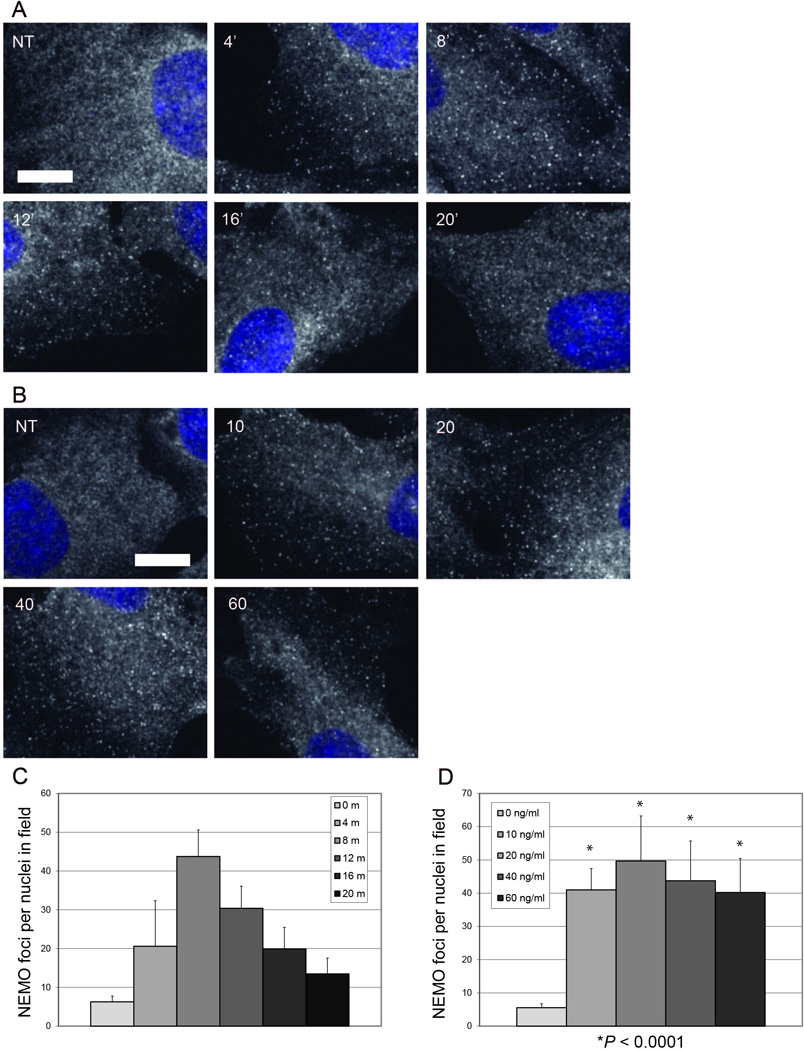
(A) NEMO immunofluorescence in WT RPE cells. Cells were either untreated (NT) or treated with 10 ng/ml TNF for 4, 8, 12, 16 or 20 minutes before fixation. Each image is a merge of NEMO immunofluorescence and Hoechst nuclear stain. Size bar represents 10 µm.
(B) NEMO immunofluorescence in WT RPE cells. Cells were either untreated (NT) or treated with 10, 20, 40, or 60 ng/ml TNF for 8 minutes before fixation. Each image is a merge of NEMO immunofluorescence and Hoechst nuclear stain. Size bar represents 10 µm.
(C) The number of NEMO foci per nuclei in the same field were plotted for the TNF time course in (A) (mean + SD). N = 10 random 600× images for all time points.
(D) The number of NEMO foci per nuclei in the same field were plotted for the TNF dose course in (B) (mean + SD). N = 10 random 600× images for untreated and all TNF doses. * = P < 0.01 versus untreated.
TNF-induced NEMO foci colocalize with TNF ligand and require polyubiquitin binding, but not IKKα/β
To test whether the pattern of endogenous NEMO reorganization in response to TNF stimulation could be recapitulated with exogenous protein, we stably expressed GFP-fused NEMO in NEMO-null MEF cells and stimulated these cells with TNF. Consistent with the endogenous NEMO staining described above, GFP-NEMO also formed TNF-induced foci (Fig. 6A). Next, to test whether these NEMO foci are associated with TNF receptor complex, we subjected TNF-treated GFP-NEMO MEFs to TNF immunofluorescence. GFP-NEMO MEFs were treated with 40 ng/ml mouse TNF for 8 minutes, fixed, then incubated with anti-TNF monoclonal antibody followed by incubation with anti-rabbit Alexa Fluor 594-conjugated secondary antibody. Indeed, TNF immunofluorescence co-localized with GFP-NEMO foci (Fig. 6A). This result provides strong evidence that the TNF-induced NEMO foci are accumulations of NEMO at activated TNF receptor complex.
Figure 6. TNF-induced NEMO structures colocalize with TNF and require NEMO residue Y308, but not IKKα/β.
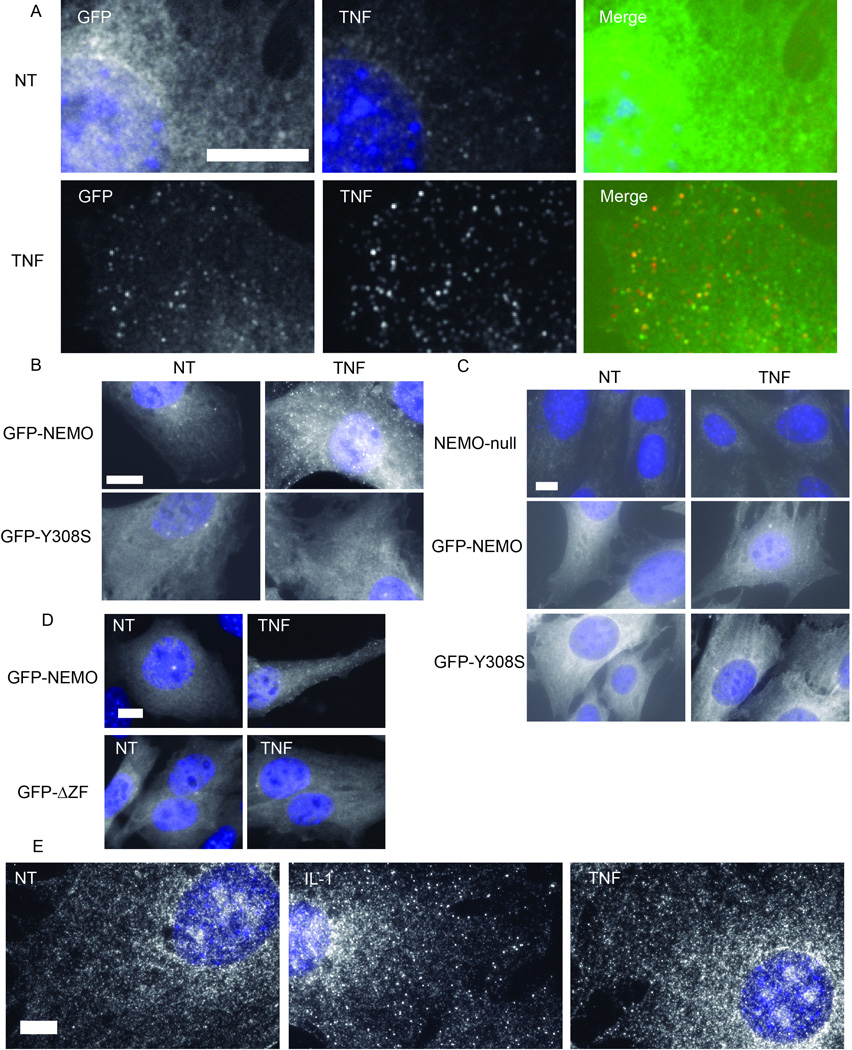
(A) GFP-NEMO and TNF immunofluorescence in reconstituted NEMO-null MEFs. Stably reconstituted GFP-NEMO MEFs were either untreated (NT) or treated with 40 ng/ml TNF (TNF). From left to right, GFP-NEMO and Hoechst merge, TNF immunofluorescence with Hoechst merge, and a merge of GFP-NEMO, TNF, and Hoechst channels. Size bars represent 10 µm.
(B) Fluorescent microscopy of stably expressed GFP-NEMO and GFP-NEMOY308S RPE cells. Cells were either untreated (NT) or treated with 20 ng/ml TNF. Each image is a merge of the GFP channel with the Hoechst channel. Size bar represents 10 µm.
(C) Fluorescent microscopy of NEMO-null MEFs reconstituted with GFP-NEMO or GFP-NEMOY308S. Cells were either untreated (NT) or treated with 20 ng/ml TNF for 8 minutes. Each image is a merge of the GFP channel with the Hoechst channel. Size bar represents 10 µm.
(D) Fluorescent microscopy of NEMO-null MEFs reconstituted with GFP-NEMO or GFP-NEMOΔZF. Cells were either untreated (NT) or treated with 10 ng/ml TNF for 8 minutes. Each image is a merge of the GFP channel with the Hoechst channel. Size bar represents 10 µm.
(E) NEMO immunofluorescence in IKKα/β-null MEFs. Cells were either untreated (NT) or treated with 10 ng/ml IL-1 (IL-1) or 40 ng/ml TNF (TNF). Each image is a merge of NEMO immunofluorescence with the Hoechst channel. Size bar represents 10 µm.
Biochemical and genetic reconstitution experiments support the hypothesis that NEMO’s ability to bind polyubiquitin chains is required for NEMO recruitment to TNF receptor complex and subsequent activation of IKK and NF-κB [49–54]. In particular, mutations within the NOA/UBAN/NUB domain (NEMO optineurin ABIN; ubiquitin binding in ABIN and NEMO; NEMO ubiquitin binding (NUB)) domain of NEMO, such as Y308S, disrupt NEMO-IKK recruitment and subsequent NF-κB activation [49,53]. Additionally, NEMO’s zinc finger has been shown to bind ubiquitin and functional data indicates its ubiquitin-binding zinc finger is critical to NF-κB activation by TNF [54]. To test whether TNF-induced NEMO foci formation also requires NEMO’s ubiquitin binding activity, we first stably expressed in RPE cells a GFP-fused polyubiquitin binding-defective NEMO mutant, GFP-NEMOY308S. Unlike GFP-NEMO wild-type protein, which formed foci in response to TNF stimulation, GFP-NEMOY308S did not (Fig. 6B). As with RPE cells, stable reconstitution of NEMO-null MEFs with GFP-NEMO resulted in TNF-induced foci, but stable reconstitution with GFP-Y308S did not (Fig. 6C). Similar to GFP-Y308S, stable reconstitution of NEMO-null MEFs with GFP-NEMO resulted in TNF-induced foci, but stable reconstitution with GFP-NEMOΔZF (NEMO AA 1–394) did not (Fig. 6D). In general, detection of TNF-induced NEMO foci in reconstituted MEF cells was not as robust as seen in RPE cells. The reason for this difference remains unclear. Nevertheless, these results are consistent with the established model in which NEMO is recruited to activated TNF receptor signaling complexes by binding to polyubiquitinated proteins, including RIP1 [49,55].
Biochemical studies indicate that NEMO at the activated TNF receptor is in complex with IKKα/β [56]. IKKβ is brought to activated receptor complex by NEMO’s polyubiquitin binding abilities, and NEMO-null cells fail to activate IKKβ after TNF stimulation [10,12,49,57–59]. To test whether IKKα/β is required for TNF-induced NEMO foci formation, we stimulated IKKα/β-null MEFs with TNF for 8 minutes and performed NEMO immunofluorescence as above. Surprisingly, IKKα/β-null MEFs formed TNF-induced NEMO foci (Fig. 6E). As mentioned, TNF-induced NEMO foci were generally more difficult to observe in MEFs than in RPE cells. Further, NEMO immunofluorescence is generally grainier in appearance in IKKα/β-null MEFs than other cell types we examined, for unclear reasons. As with TNF, we observed robust induction of NEMO foci 8 minutes after IL-1 stimulation (Fig. 6E). Interestingly, IL-1-induced NEMO foci were easier to detect in IKKα/β-null MEFs than the foci induced by TNF. Therefore, we found induction of NEMO foci by cytokines TNF or IL-1 does not require IKKα/β, which is consistent with the biochemical evidence that NEMO does not depend on IKK to bind polyubiquitin chains in vitro.
Withaferin A blocks the formation of TNF- and IL-1-induced NEMO structures
Next, we tested the hypothesis that WA prevents recruitment of the NEMO to TNF-induced foci. RPE cells were stimulated with 10 ng/ml TNF for 8 minutes with or without pre-treatment with WA. RPE cells pre-treated with WA had similar NEMO distribution as untreated cells, except for the induction of large perinuclear NEMO structure (Fig. 7A and 7B). These structures colocalize with γ-tubulin at the centrosome, indicating WA causes NEMO accumulation at the centrosome (Fig. 7A). WA pre-treatment followed by TNF stimulation resulted in a marked reduction in the number of TNF-induced NEMO foci (Fig. 7B and 7C). As with WA treatment alone, centrosomal NEMO localization was observed in cells treated with both WA and TNF. Quantification of NEMO foci demonstrated that while TNF stimulation induced roughly 9-fold more NEMO foci (P < 0.0001) in TNF treated cells than untreated controls, pre-treatment with 3 µM or 10 µM WA significantly reduced the number of TNF-induced NEMO foci by 2-fold (P < 0.01) and 11-fold (P < 0.0001), respectively (Fig.7C). If NEMO was not recruited to the TNF receptor complex due to WA treatment, the inability of TAK1 to activate the IKK complex observed above could be explained. However, WA did not prevent RIP1 ubiquitination (Fig. 7D). Thus, these results revealed that WA inhibits IKKβ and NF-κB signaling via a novel mechanism involving disruption of NEMO recruitment to ubiquitin-dependent signaling complexes without inhibiting RIP1 ubiquitination.
Figure 7. Withaferin A blocks the formation of TNF-induced NEMO structures.
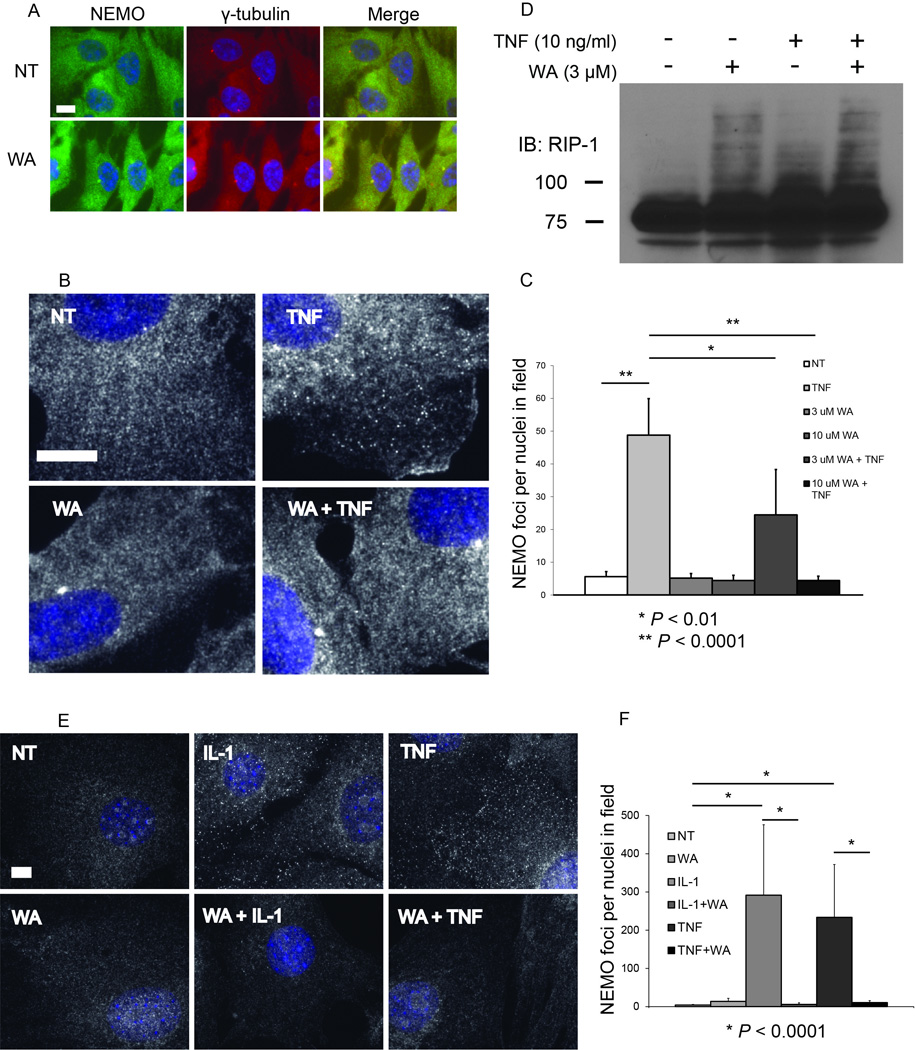
(A) Immunofluorescence for NEMO and γ-tubulin in WT RPE cells. Cells were either untreated or treated with 10 µM WA for 30 minutes. From left to right, NEMO immunofluorescence and Hoechst merge, γ-tubulin immunofluorescence with Hoechst merge, and a merge of NEMO and γ-tubulin immunofluorescence with Hoechst. Size bars represent 10 µm.
(B) Immunofluorescence for NEMO in WT RPE cells following WA pre-treatment and subsequent TNF stimulation. Cells were either untreated (NT), treated with 10 µM WA for 38 minutes (WA), treated with 10 ng/ml TNF for 8 minutes (TNF), or pre-treated with 10 µM WA for 30 minutes then treated with 10 ng/ml TNF for 8 minutes (WA + TNF). Each image is a merge of NEMO immunofluorescence with the Hoechst channel. Size bar represents 10 µm.
(C) The number of NEMO foci per nuclei in the same field was quantified and plotted for the NEMO immunofluorescence in (C) (mean + SD). N = 10 random 600× images per treatment.
(D) HEK293 cells were treated with 10ng/ml of TNF for 7.5 minutes and/or pre-treated with 5 uM WA for 30 minutes. Cells were lysed and immunoblotted with anti-RIP1 antibody.
(E) Immunofluorescence for NEMO in IKKα/β-null MEFs following WA pre-treatment and subsequent IL-1 or TNF stimulation. Cells were either untreated (NT), treated with 10 µM WA for 38 minutes (WA), treated with 20 ng/ml IL-1 or 20 ng/ml TNF for 8 minutes (TNF), or pre-treated with 10 µM WA for 30 minutes then treated with 20 ng/ml IL-1 or 20 ng/ml TNF for 8 minutes (WA + TNF). Each image is a merge of NEMO immunofluorescence with the Hoechst channel. Size bar represents 10 µm.
(F) The number of NEMO foci per nuclei in the same field was quantified and plotted for the NEMO immunofluorescence in (C) (mean + SD). N = 10 random 600× images per treatment.
We then looked to see if WA could inhibit IL-1 and TNF-induced NEMO foci formation in the absence of IKKβ using IKKα/β-null MEFs. As with WT RPE cells, IKKα/β-null MEFs pre-treated with WA form significantly fewer NEMO foci in response to either with IL-1 or TNF stimulation (Fig. 7E and 7F). Quantification of NEMO foci demonstrated that while TNF stimulation induced roughly 50-fold more NEMO foci (P < 0.0001) in TNF treated cells than untreated controls, pre-treatment with 10 µM WA reduced the number of TNF-induced NEMO foci by 20-fold (P < 0.0001) (Fig. 7F). Further, while IL-1 stimulation induced 70-fold more NEMO foci (P < 0.0001), pre-treatment with 10 µM WA reduced the number of IL-1-induced 50-fold fewer NEMO foci (P < 0.0001). Therefore, WA can inhibit stimulus-induced NEMO foci formation in a manner independent of the IKK/NEMO interaction.
DISCUSSION
NF-κB is a key regulator of cellular homeostasis, controlling the regulation of genes important in inflammations, immunity, development, and cell survival. As such, aberrant regulation of NF-κB signaling is known to play a role in a variety of human pathologies, including several solid and hematological malignancies [2,60]. As a result, NF-κB is considered to be a highly desirable drug target. While many drugs are known to have an effect on NF-κB signaling [26], a safe, specific, and robust inhibitor has yet to make it to clinical use. In this context, we were initially interested in WA because (i) it is reported as a robust inhibitor of NF-κB activation [35,36]; (ii) it shows no overt toxicity in animal studies [37], (iii) it has not demonstrated side effects beyond placebo in two randomized-clinical trials [61,62] and (iv) it has been used in India for over 3000 years in Ayurvedic medicine [63]. Thus, WA demonstrates safety and favorable pharmacokinetic properties with potent NF-κB inhibitory activity. Indeed, we found that this chemical was a robust inhibitor of NF-κB signaling induced by inflammatory stimuli (TNF and LPS) and anticancer genotoxic agents (etoposide and ionizing radiation) in multiple cell systems. Moreover, the constitutive activity present in ABC-DLBCL cells was also efficiency inhibited, correlating with induction of apoptotic death in this clinically unfavorable subtype of lymphoma cells. In contrast, GCB-DLBCL cells that do not harbor constitutive NF-κB activity were less sensitive to WA-induced death. Therefore, WA, its derivatives, or compounds that act via the same mechanism, could be useful as anticancer and/or anti-inflammatory agents, as previously proposed by other investigators [32,38].
Prior reports that pertain to NF-κB signaling proposed WA as an IKKβ inhibitor [38] or through the same mechanism as the NBD peptide: inhibition of the protein-protein interaction between NEMO and IKKβ [32]. Consistent with these published reports, we found that the step at which WA interferes with canonical NF-κB signaling is between the downstream of TAK1 activation. Indeed, Withania somnifera extract has been shown to induce phosphorylation of the TAK1-substrates, JNK and ERK, in previous studies [38]. We were able to further show that this is, at least in part, secondary to WA induced TAK1 activation, allowing for further downstream application of TAK1 substrates. However, despite showing that WA disrupts NF-κB signaling between TAK1 and IKK, we were unable to generate data that supported the previously proposed model that WA is an IKKβ inhibitor. An in vitro IKK kinase assay in the presence of increasing doses of WA did not correspond to complete inhibition of IKKβ inhibition observed in vivo. Both co-immunoprecipitation study and functional mutation analysis of the putative WA-NEMO interaction residues did not support the involvement of NEMO-IKKβ dissociation in the presence of this inhibitor. Instead, we found that WA prevents NEMO recruitment to TNF-induced supramolecular foci in an ubiquitin binding domain-dependent manner. Further, we found that WA can prevent NEMO foci formation in the absence of IKKβ and IKKα, indicating this inhibition is independent of IKK/NEMO interaction.
Accumulated experimental evidence indicates that NEMO’s ability to bring IKK to the activated receptor complex is a critical step in canonical NF-κB activation. Current dogma postulates that TAK1 must be in close proximity to its kinase target IKKβ in order for IKKβ activation and subsequent NF-κB activation [48]. The IKK complex is directed to receptor complex containing activated TAK1 by NEMO’s strong affinity for specific polyubiquitin chains that are highly enriched at the receptor complex. This recruitment is thought to require NEMO binding to polyubiquitinated RIP1 and/or NEMO binding to other polyubiquitinated NEMO molecules [49,53,57]. However, most of the work characterizing these interactions has been performed by biochemical analysis. How these events are organized in situ remains poorly understood. Here, we provided evidence that NEMO reorganizes into distinct, cell surface-associated TNF-bound receptor complexes. These foci are distributed roughly equally throughout the cell surface, are generally homogenous in size, and the number of NEMO foci increases in a TNF dose-dependent manner. TNF immunofluorescence revealed that TNF ligand also forms foci, and this signal was similar in size to NEMO foci. Together, these results suggest that activated TNF receptor clusters and forms a complex that is also associated with a cluster of NEMO molecules.
While we were in the process of characterizing TNF-induced NEMO foci, another group published a paper describing proinflammatory cytokine-induced NEMO-IKK activation structures (Tarantino et al. 2014). Their work demonstrated that NEMO forms TNF-induced NEMO structures, and that these structures colocalize with labeled TNF ligand and phospho-IKK. This finding indicates that the formation of TNF-induced NEMO structures is associated with IKK activation. In their report, detection of TNF-induced NEMO foci required saponin permeabilization to purge the cytoplasm of unanchored NEMO prior to fixation. Further, the development of custom image-processing algorithms in ImageJ to reduce background noise, calibrate detection filters, and segment foci and nuclei were implemented to quantify NEMO structures. In contrast, we were able to detect and quantify NEMO foci in situ without cell permeabilization by saponin prior to fixation, image processing to reduce noise, or custom ImageJ algorithms. Quantification of NEMO foci was accomplished using only the ImageJ process “Find Maxima….” Thus, our work provides a basic method for the detection and quantification of TNF-inducible NEMO foci that is accessible to any lab equipped only with a widefield epifluorescent microscope.
Despite these differences in TNF-induced foci visualization, the results of our characterization were strikingly similar to those of Tarantino et al. We observed rapid induction of NEMO structures in TNF-treated RPE cells by NEMO immunofluorescence, with NEMO foci easily detected four minutes after TNF treatment, with peak foci formation at 8 minutes, followed by a decline in foci number by 12 minutes. Similarly, Tarantino et al. also observed rapid induction of NEMO foci, with peak foci formation roughly 7 minutes after TNF-stimulation of WT U2-OS cells [64]. Using 10 ng/ml TNF, our peak NEMO foci value at 8 minutes was roughly 45 NEMO foci per RPE nuclei in the same field, similar to their peak value with the same TNF dose of approximately 45 NEMO foci per U2-OS cell. NEMO foci we observed in RPE cells were generally uniform in distribution and size (roughly 250–500 nm), also consistent with their report. We were both able to visualize TNF-induced endogenous NEMO foci by NEMO immunofluorescence and using exogenous GFP-fused NEMO, indicating these foci are not an antibody artifact, such as a TNF-induced change in NEMO epitope availability. Further, since we observed endogenous NEMO foci formation in different cell systems, these results are likely not a cell type artifact. Finally, GFP-NEMOY308S and GFP-NEMOΔZF mutants defective in polyubiquitin binding failed to form these TNF-induced NEMO foci. Thus, our results are consistent with the current ubiquitin-based NEMO recruitment model for explaining IKK activation in canonical NF-κB signaling [49,53,57].
WA induces a relatively small portion of NEMO to the centrosome (Figure 7A), as similar levels of cytoplasmic NEMO are observed in WA treated cells and untreated cells. Therefore, it is unlikely that NEMO foci formation is inhibited directly by the sequestration of NEMO to the centrosome. There are a few other reports of IKK/NEMO at the centrosome. IKKα, but not IKKβ, has been shown to regulate the cell cycle through association with Aurora A at the centrosome [65]. IKK/NEMO complex binds to polyubiquitinated human T cell leukemia virus type 1 (HTLV-1) Tax (Tax1) viral transactivator at the centrosome, driving constitutive NF-κB activation [66,67]. Presently, it is unclear why WA induces centrosomal NEMO, though we hypothesize that it might be related to a change in NEMO’s ability to bind ubiquitin.
While our data strongly suggests that WA disrupts NF-κB signaling through disruption of TNF- and IL-1-induced NEMO foci, the mechanism of disruption is not clearly defined. One likely mechanism involves polyubiquitin chains that serve as critical scaffolds during NF-κB signaling. One hypothesis is that WA could be disrupting the formation of such polyubiquitin chains while an alternative hypothesis is that WA is affecting the ability for NEMO to interact with signaling induced polyubiquitin chains. We did not find inhibition of TNF-induced RIP1 ubiquitination in WA treated cells. Thus, further study is required to define precise mechanism of inhibition of NEMO foci formation by WA treatment, and if this mechanism is relevant to other stimulators, such as genotoxic agents that do not generally rely on cell surface receptor-mediated IKK activation.
Although WA inhibits NEMO foci formation and downstream NF-κB inhibition, it is important to note that WA is not specific to NEMO. WA is also a reported inhibitor of the proteasome, so although it inhibits NEMO foci formation and thus IKK activation, it also likely inhibits NF-κB downstream from IKK activation, e.g., proteasome-mediated IκBα degradation, depending on the amount of drug used [68–71]. Further, WA has been shown to affect other factors unrelated to NF-κB signaling, such as inhibiting vascular endothelial growth factor (VEGF) and covalent binding to vimentin, and thus inhibiting angiogenesis and epithelial-mesenchymal transition, among its well-documented anti-cancer properties [72–77]. Though WA might not be suitable to selectively target cytokine-induced NEMO foci formation, it provides a proof-of-principle that a small cell-permeable molecule can be used to target NF-κB activation at the level of NEMO recruitment to receptor complex. Considering the central role of NF-κB in tumorigenesis, aging, cell survival, autoimmunity, inflammatory pathologies, and various other pathologies, these findings could lead to new tools to combat human disease.
Supplementary Material
{kind=link}
Highlights.
-
-
Withaferin A, a NF-κB inhibitor, disrupts signaling-induced NEMO localization, a novel point of NF-κB inhibition
-
-
NEMO can be localized to distinct signaling foci after treatment with TNF
-
-
ABC-type DLCBL cells can be sensitized to apoptosis after treatment with Withaferin A
ACKNOWLEDGEMENT
This work was funded by F30CA171840 (SSJ), NIH R01CA077474 (SM), NIH R01GM083681 (SM), UL1TR0000427 (LR), KL2TR000428 (LR) and T32GM008692 (University of Wisconsin MSTP)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 2.Karin M, Cao Y, Greten FR, Li Z-W. NF-kappaB in cancer: from innocent bystander to major culprit. Nat. Rev. Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 3.Pacifico F, Leonardi A. NF-kappaB in solid tumors. Biochem. Pharmacol. 2006;72:1142–1152. doi: 10.1016/j.bcp.2006.07.032. [DOI] [PubMed] [Google Scholar]
- 4.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 5.Perkins ND. The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer. 2012;12:121–132. doi: 10.1038/nrc3204. [DOI] [PubMed] [Google Scholar]
- 6.Baeuerle PA, Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science. 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- 7.Adhikari A, Xu M, Chen ZJ. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene. 2007;26:3214–3226. doi: 10.1038/sj.onc.1210413. [DOI] [PubMed] [Google Scholar]
- 8.Kanayama A, Seth RB, Sun L, Ea C-K, Hong M, Shaito A, et al. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol. Cell. 2004;15:535–548. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 9.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 10.Rothwarf DM, Zandi E, Natoli G, Karin M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature. 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- 11.Spencer E, Jiang J, Chen ZJ. Signal-induced ubiquitination of IkappaBalpha by the F-box protein Slimb/beta-TrCP. Genes Dev. 1999;13:284–294. doi: 10.1101/gad.13.3.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, et al. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell. 1998;93:1231–1240. doi: 10.1016/s0092-8674(00)81466-x. [DOI] [PubMed] [Google Scholar]
- 13.Senftleben U, Cao Y, Xiao G, Greten FR, Krähn G, Bonizzi G, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 14.Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 15.Frankenberger M, Pforte A, Sternsdorf T, Passlick B, Baeuerle PA, Ziegler-Heitbrock HW. Constitutive nuclear NF-kappa B in cells of the monocyte lineage. Biochem. J. 1994;304(Pt 1):87–94. doi: 10.1042/bj3040087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyamoto S, Chiao PJ, Verma IM. Enhanced I kappa B alpha degradation is responsible for constitutive NF-kappa B activity in mature murine B-cell lines. Mol. Cell. Biol. 1994;14:3276–3282. doi: 10.1128/mcb.14.5.3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ, Jr, Sledge GW., Jr Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Mol. Cell. Biol. 1997;17:3629–3639. doi: 10.1128/mcb.17.7.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bharti AC, Shishodia S, Reuben JM, Weber D, Alexanian R, Raj-Vadhan S, et al. Nuclear factor-kappaB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood. 2004;103:3175–3184. doi: 10.1182/blood-2003-06-2151. [DOI] [PubMed] [Google Scholar]
- 19.Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med. 2001;194:1861–1874. doi: 10.1084/jem.194.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grosjean-Raillard J, Tailler M, Adès L, Perfettini J-L, Fabre C, Braun T, et al. ATM mediates constitutive NF-kappaB activation in high-risk myelodysplastic syndrome and acute myeloid leukemia. Oncogene. 2009;28:1099–1109. doi: 10.1038/onc.2008.457. [DOI] [PubMed] [Google Scholar]
- 21.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim K-H, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470:115–119. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science. 2010;327:1135–1139. doi: 10.1126/science.1182364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424:793–796. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- 24.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 25.Rosenwald A, Wright G, Leroy K, Yu X, Gaulard P, Gascoyne RD, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J. Exp. Med. 2003;198:851–862. doi: 10.1084/jem.20031074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gilmore TD, Herscovitch M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene. 2006;25:6887–6899. doi: 10.1038/sj.onc.1209982. [DOI] [PubMed] [Google Scholar]
- 27.Ivanenkov YA, Balakin KV, Lavrovsky Y. Small Molecule Inhibitors of NF-κB and JAK/STAT Signal Transduction Pathways as Promising Anti-Inflammatory Therapeutics. Mini-Rev. Med. Chem. 2011;11:55–78. doi: 10.2174/138955711793564079. [DOI] [PubMed] [Google Scholar]
- 28.Dai S, Hirayama T, Abbas S, Abu-Amer Y. The IkappaB kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks osteoclastogenesis and bone erosion in inflammatory arthritis. J. Biol. Chem. 2004;279:37219–37222. doi: 10.1074/jbc.C400258200. [DOI] [PubMed] [Google Scholar]
- 29.Gaurnier-Hausser A, Patel R, Baldwin AS, May MJ, Mason NJ. NEMO-binding domain peptide inhibits constitutive NF-κB activity and reduces tumor burden in a canine model of relapsed, refractory diffuse large B-cell lymphoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011;17:4661–4671. doi: 10.1158/1078-0432.CCR-10-3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.May MJ, D’Acquisto F, Madge LA, Glöckner J, Pober JS, Ghosh S. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science. 2000;289:1550–1554. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- 31.Strickland I, Ghosh S. Use of cell permeable NBD peptides for suppression of inflammation. Ann. Rheum. Dis. 2006;65(Suppl 3):iii75–iii82. doi: 10.1136/ard.2006.058438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grover A, Shandilya A, Punetha A, Bisaria VS, Sundar D. Inhibition of the NEMO/IKKβ association complex formation, a novel mechanism associated with the NF-κB activation suppression by Withania somnifera’s key metabolite withaferin A. BMC Genomics. 2010;11(Suppl 4):S25. doi: 10.1186/1471-2164-11-S4-S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chowdhury K, Neogy RK. Mode of action of Withaferin A and Withanolide D. Biochem. Pharmacol. 1975;24:919–920. doi: 10.1016/0006-2952(75)90165-3. [DOI] [PubMed] [Google Scholar]
- 34.Shohat B, Gitter S, Abraham A, Lavie D. Antitumor activity of withaferin A (NSC-101088) Cancer Chemother. Rep. 1967;51:271–276. [PubMed] [Google Scholar]
- 35.Ichikawa H, Takada Y, Shishodia S, Jayaprakasam B, Nair MG, Aggarwal BB. Withanolides potentiate apoptosis, inhibit invasion, and abolish osteoclastogenesis through suppression of nuclear factor-kappaB (NF-kappaB) activation and NF-kappaB-regulated gene expression. Mol. Cancer Ther. 2006;5:1434–1445. doi: 10.1158/1535-7163.MCT-06-0096. [DOI] [PubMed] [Google Scholar]
- 36.Oh JH, Kwon TK. Withaferin A inhibits tumor necrosis factor-alpha-induced expression of cell adhesion molecules by inactivation of Akt and NF-kappaB in human pulmonary epithelial cells. Int. Immunopharmacol. 2009;9:614–619. doi: 10.1016/j.intimp.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 37.Thaiparambil JT, Bender L, Ganesh T, Kline E, Patel P, Liu Y, et al. Withaferin A inhibits breast cancer invasion and metastasis at sub-cytotoxic doses by inducing vimentin disassembly and serine 56 phosphorylation. Int. J. Cancer J. Int. Cancer. 2011;129:2744–2755. doi: 10.1002/ijc.25938. [DOI] [PubMed] [Google Scholar]
- 38.Kaileh M, Vanden Berghe W, Heyerick A, Horion J, Piette J, Libert C, et al. Withaferin a strongly elicits IkappaB kinase beta hyperphosphorylation concomitant with potent inhibition of its kinase activity. J. Biol. Chem. 2007;282:4253–4264. doi: 10.1074/jbc.M606728200. [DOI] [PubMed] [Google Scholar]
- 39.Jackson SS, Coughlin EE, Coon JJ, Miyamoto S. Identifying post-translational modifications of NEMO by tandem mass spectrometry after high affinity purification. Protein Expr. Purif. 2013;92:48–53. doi: 10.1016/j.pep.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang TT, Feinberg SL, Suryanarayanan S, Miyamoto S. The zinc finger domain of NEMO is selectively required for NF-kappa B activation by UV radiation and topoisomerase inhibitors. Mol. Cell. Biol. 2002;22:5813–5825. doi: 10.1128/MCB.22.16.5813-5825.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miyamoto S, Seufzer BJ, Shumway SD. Novel IkappaB alpha proteolytic pathway in WEHI231 immature B cells. Mol. Cell. Biol. 1998;18:19–29. doi: 10.1128/mcb.18.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miyamoto S, Schmitt MJ, Verma IM. Qualitative changes in the subunit composition of kappa B-binding complexes during murine B-cell differentiation. Proc. Natl. Acad. Sci. U. S. A. 1994;91:5056–5060. doi: 10.1073/pnas.91.11.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang TT, Kudo N, Yoshida M, Miyamoto S. A nuclear export signal in the N-terminal regulatory domain of IkappaBalpha controls cytoplasmic localization of inactive NF-kappaB/IkappaBalpha complexes. Proc. Natl. Acad. Sci. U. S. A. 2000;97:1014–1019. doi: 10.1073/pnas.97.3.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu Z-H, Wong ET, Shi Y, Niu J, Chen Z, Miyamoto S, et al. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol. Cell. 2010;40:75–86. doi: 10.1016/j.molcel.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hollander M, Wolfe DA, Chicken E. Nonparametric Statistical Methods. John Wiley & Sons; 2013. [Google Scholar]
- 46.Chen ZJ, Bhoj V, Seth RB. Ubiquitin, TAK1 and IKK: is there a connection? Cell Death Differ. 2006;13:687–692. doi: 10.1038/sj.cdd.4401869. [DOI] [PubMed] [Google Scholar]
- 47.Takaesu G, Surabhi RM, Park K-J, Ninomiya-Tsuji J, Matsumoto K, Gaynor RB. TAK1 is critical for IkappaB kinase-mediated activation of the NF-kappaB pathway. J. Mol. Biol. 2003;326:105–115. doi: 10.1016/s0022-2836(02)01404-3. [DOI] [PubMed] [Google Scholar]
- 48.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 49.Ea C-K, Deng L, Xia Z-P, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 50.Fujita H, Rahighi S, Akita M, Kato R, Sasaki Y, Wakatsuki S, et al. Mechanism underlying IκB kinase activation mediated by the linear ubiquitin chain assembly complex. Mol. Cell. Biol. 2014;34:1322–1335. doi: 10.1128/MCB.01538-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laplantine E, Fontan E, Chiaravalli J, Lopez T, Lakisic G, Véron M, et al. NEMO specifically recognizes K63-linked poly-ubiquitin chains through a new bipartite ubiquitin-binding domain. EMBO J. 2009;28:2885–2895. doi: 10.1038/emboj.2009.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell. 2009;136:1098–1109. doi: 10.1016/j.cell.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 53.Wu C-J, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected] Nat. Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- 54.Cordier F, Grubisha O, Traincard F, Véron M, Delepierre M, Agou F. The Zinc Finger of NEMO Is a Functional Ubiquitin-binding Domain. J. Biol. Chem. 2009;284:2902–2907. doi: 10.1074/jbc.M806655200. [DOI] [PubMed] [Google Scholar]
- 55.Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471:591–596. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 56.Devin A, Cook A, Lin Y, Rodriguez Y, Kelliher M, Liu Z. The distinct roles of TRAF2 and RIP in IKK activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP mediates IKK activation. Immunity. 2000;12:419–429. doi: 10.1016/s1074-7613(00)80194-6. [DOI] [PubMed] [Google Scholar]
- 57.Li H, Kobayashi M, Blonska M, You Y, Lin X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J. Biol. Chem. 2006;281:13636–13643. doi: 10.1074/jbc.M600620200. [DOI] [PubMed] [Google Scholar]
- 58.Mercurio F, Murray BW, Shevchenko A, Bennett BL, Young DB, Li JW, et al. IkappaB kinase (IKK)-associated protein 1, a common component of the heterogeneous IKK complex. Mol. Cell. Biol. 1999;19:1526–1538. doi: 10.1128/mcb.19.2.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rudolph D, Yeh WC, Wakeham A, Rudolph B, Nallainathan D, Potter J, et al. Severe liver degeneration and lack of NF-kappaB activation in NEMO/IKKgamma-deficient mice. Genes Dev. 2000;14:854–862. [PMC free article] [PubMed] [Google Scholar]
- 60.Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009;8:33–40. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chopra A, Lavin P, Patwardhan B, Chitre D. A 32-week randomized, placebo-controlled clinical evaluation of RA-11, an Ayurvedic drug, on osteoarthritis of the knees. J. Clin. Rheumatol. Pract. Rep. Rheum. Musculoskelet. Dis. 2004;10:236–245. doi: 10.1097/01.rhu.0000138087.47382.6d. [DOI] [PubMed] [Google Scholar]
- 62.Cooley K, Szczurko O, Perri D, Mills EJ, Bernhardt B, Zhou Q, et al. Naturopathic care for anxiety: a randomized controlled trial ISRCTN78958974. PloS One. 2009;4:e6628. doi: 10.1371/journal.pone.0006628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Werneke U. Withania somnifera: From Ayurveda towards Western medical practice: a systematic review. Studium Press LLC; 2009. [accessed June 13, 2014]. pp. 237–256. http://umu.diva-portal.org/smash/record.jsf?pid=diva2:526279. [Google Scholar]
- 64.Tarantino N, Tinevez J-Y, Crowell EF, Boisson B, Henriques R, Mhlanga M, et al. TNF and IL-1 exhibit distinct ubiquitin requirements for inducing NEMO-IKK supramolecular structures. J. Cell Biol. 2014;204:231–245. doi: 10.1083/jcb.201307172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Prajapati S, Tu Z, Yamamoto Y, Gaynor RB. IKKalpha regulates the mitotic phase of the cell cycle by modulating Aurora A phosphorylation. Cell Cycle Georget. Tex. 2006;5:2371–2380. doi: 10.4161/cc.5.20.3359. [DOI] [PubMed] [Google Scholar]
- 66.Journo C, Bonnet A, Favre-Bonvin A, Turpin J, Vinera J, Côté E, et al. Human T cell leukemia virus type 2 tax-mediated NF-κB activation involves a mechanism independent of Tax conjugation to ubiquitin and SUMO. J. Virol. 2013;87:1123–1136. doi: 10.1128/JVI.01792-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kfoury Y, Setterblad N, El-Sabban M, Zamborlini A, Dassouki Z, El Hajj H, et al. Tax ubiquitylation and SUMOylation control the dynamic shuttling of Tax and NEMO between Ubc9 nuclear bodies and the centrosome. Blood. 2011;117:190–199. doi: 10.1182/blood-2010-05-285742. [DOI] [PubMed] [Google Scholar]
- 68.Khan S, Rammeloo AW, Heikkila JJ. Withaferin A induces proteasome inhibition, endoplasmic reticulum stress, the heat shock response and acquisition of thermotolerance. PloS One. 2012;7:e50547. doi: 10.1371/journal.pone.0050547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khedgikar V, Kushwaha P, Gautam J, Verma A, Changkija B, Kumar A, et al. Withaferin A: a proteasomal inhibitor promotes healing after injury and exerts anabolic effect on osteoporotic bone. Cell Death Dis. 2013;4:e778. doi: 10.1038/cddis.2013.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang H, Wang Y, Cheryan VT, Wu W, Cui CQ, Polin LA, et al. Withaferin A inhibits the proteasome activity in mesothelioma in vitro and in vivo. PloS One. 2012;7:e41214. doi: 10.1371/journal.pone.0041214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang H, Shi G, Dou QP. The tumor proteasome is a primary target for the natural anticancer compound Withaferin A isolated from “Indian winter cherry,”. Mol. Pharmacol. 2007;71:426–437. doi: 10.1124/mol.106.030015. [DOI] [PubMed] [Google Scholar]
- 72.Bargagna-Mohan P, Hamza A, Kim Y, Khuan Abby Ho Y, Mor-Vaknin N, Wendschlag N, et al. The tumor inhibitor and antiangiogenic agent withaferin A targets the intermediate filament protein vimentin. Chem. Biol. 2007;14:623–634. doi: 10.1016/j.chembiol.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee J, Hahm E-R, Marcus AI, Singh SV. Withaferin A inhibits experimental epithelial-mesenchymal transition in MCF-10A cells and suppresses vimentin protein level in vivo in breast tumors. Mol. Carcinog. 2013 doi: 10.1002/mc.22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee J, Hahm E-R, Singh SV. Withaferin A inhibits activation of signal transducer and activator of transcription 3 in human breast cancer cells. Carcinogenesis. 2010;31:1991–1998. doi: 10.1093/carcin/bgq175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mohan R, Hammers HJ, Bargagna-Mohan P, Zhan XH, Herbstritt CJ, Ruiz A, et al. Withaferin A is a potent inhibitor of angiogenesis. Angiogenesis. 2004;7:115–122. doi: 10.1007/s10456-004-1026-3. [DOI] [PubMed] [Google Scholar]
- 76.Saha S, Islam MK, Shilpi JA, Hasan S. Inhibition of VEGF: a novel mechanism to control angiogenesis by Withania somnifera’s key metabolite Withaferin A. Silico Pharmacol. 2013;1:1–9. doi: 10.1186/2193-9616-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stan SD, Hahm E-R, Warin R, Singh SV. Withaferin A causes FOXO3a- and Bim-dependent apoptosis and inhibits growth of human breast cancer cells in vivo. Cancer Res. 2008;68:7661–7669. doi: 10.1158/0008-5472.CAN-08-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.