

Abstract
The Crk SH2/SH3 adaptor and the Abl nonreceptor tyrosine kinase were first identified as oncoproteins, and both can induce tumorigenesis when overexpressed or mutationally activated. We previously reported the surprising finding that inhibition or knockdown of Abl family kinases enhanced transformation of mouse fibroblasts by CrkI. Abl family inhibitors are currently used or are being tested for treatment of human malignancies, and our finding raised concerns that such inhibitors might actually promote the growth of tumors overexpressing CrkI. Here, we identify the Dok1 adaptor as the key effector for the enhancement of CrkI transformation by Abl inhibition. We show that phosphorylation of tyrosines 295 and 361 of Dok1 by Abl family kinases suppresses CrkI transforming activity, and that upon phosphorylation these tyrosines bind the SH2 domains of the Ras inhibitor p120 RasGAP. Knockdown of RasGAP resulted in a similar enhancement of CrkI transformation, consistent with a critical role for Ras activity. Imaging studies using a FRET sensor of Ras activation revealed alterations in the localization of activated Ras in CrkI-transformed cells. Our results support a model in which Dok1 phosphorylation normally suppresses localized Ras pathway activity in Crk-transformed cells via recruitment and/or activation of RasGAP, and that preventing this negative feedback mechanism by inhibiting Abl family kinases leads to enhanced transformation by Crk.
Keywords: Crk, imatinib, Abl, Dok1, tyrosine phosphorylation, SH2/SH3 adaptors
Introduction
Crk was first identified as the oncogene of avian sarcoma virus CT10 (1). Later studies identified the cellular homologs of the viral oncoprotein (v-Crk): CrkI and CrkII are alternatively spliced forms of Crk (2), and CRKL is encoded by a different gene but is highly similar to CrkII in sequence and overall structure (3). Crk was the first example of an SH2/SH3 adaptor, a family of small proteins that lack catalytic domains but contain multiple modular protein binding domains. CrkI and v-Crk consist of one SH2 and one SH3 domain, while CrkII and CRKL both have an additional C-terminal SH3 domain (2, 4). CrkII also contains a regulatory tyrosine residue (Y221 in human CrkII) (4) that, when phosphorylated by Abl family kinases, binds intramolecularly to the SH2 domain and thus downregulates biological activity (5). This negative regulatory site is not present in CrkI and v-Crk. Crk overexpression has been reported in several types of human cancer (6-8), and also in established cancer cell lines (9, 10). Expression levels of Crk correlated with the aggressiveness of tumor cells, consistent with a positive role in cancer. In the laboratory, CrkI overexpression has been used as a model to study the transforming mechanisms of Crk (2, 11).
Like other adaptors, Crk proteins function in signaling by mediating the formation of multiprotein complexes through their modular protein binding domains (12), and transformation by v-Crk and CrkI requires the binding activity of both the SH2 and SH3 domains (11, 13). The Crk SH2 domain binds tyrosine-phosphorylated peptides with pY-x-x-P motifs (14), and its major binding partners include the focal adhesion proteins paxillin (15) and p130Cas (16). The N-terminal SH3 domain of Crk binds proline-rich peptides with P-x-x-P-x-K motifs (17, 18), and the major binding partners include Abl family tyrosine kinases (19, 20), and small G protein guanine nucleotide exchange factors (GEFs) including Sos (21), DOCK180 (22), and C3G (18).
In our previous work to identify the Crk SH3 binding partners essential for transformation of NIH3T3 cells by CrkI, we found that the activity of Abl family kinases antagonized the transforming activity of Crk (11). Knocking down expression of Abl and its close relative Arg, or inhibiting Abl family kinases with imatinib (a clinically prescribed Abl kinase inhibitor), both led to increased tumorigenicity of CrkI-overexpressing cells in vitro (assayed by anchorage independent growth) and in vivo (assayed by injection of cells into nude mice).
The Abl tyrosine kinase, originally identified in Abelson murine leukemia virus (23), causes Chronic Myelogenous Leukemia (CML) in humans through a chromosomal translocation resulting in a fusion protein, Bcr-Abl, with constitutively high kinase activity (24). Clinically, imatinib and similar compounds work by inhibiting Abl kinase activity and are effective in treating CML. Imatinib has also been shown to inhibit Platelet Derived Growth Factor Receptor (PDGFR) (25, 26) and c-Kit (27). Due to the efficacy of imatinib in CML treatment, it and other Abl inhibitors are now used to target Abl, PDGFR and c-Kit in various types of cancer (28-30). However, our recent observations raise concerns that Abl inhibitors have the potential to promote the growth and survival of tumor cells in some instances, particularly in those with CrkI overexpression. We therefore sought to understand the mechanism whereby Abl inhibition promotes transformation by Crk.
In this study, we show that Dok1 is responsible for the enhancement of CrkI transformation upon Abl kinase inhibition. Dok1 was first discovered as a substrate for Abl (31, 32), and is one of seven members to the Dok family (33). Dok family proteins lack catalytic domains, consisting of a Pleckstrin Homology (PH) domain, a phosphotyrosine binding PTB domain, and a C-terminal tail with multiple tyrosine residues that can be phosphorylated and thereby recruit proteins containing modular phosphotyrosine (pTyr) binding domains (33). Dok1 and Dok2 negatively regulate B-cell receptor (BCR) (34) and T-cell receptor (TCR) (35) signaling and modulate the proliferation of myeloid cells (36, 37). Dok1, 2 and 3 also have been shown to possess tumor suppressor activity in several studies (38, 39). Our results suggest the existence of a general feedback control mechanism whereby Abl, Dok family proteins, and RasGAP work together to locally downregulate Ras activity.
Results
Dok1 is the major Abl-dependent phosphoprotein in Crk-transformed cells
We first examined more closely how Abl inhibition affected the ability of CrkI-transformed NIH3T3 cells to grow in suspension, a hallmark of malignant transformation. Consistent with previous results (11), we found a significant increase (up to 10-fold) in the number of colonies in the soft agar growth assay when cells were treated with the Abl inhibitor imatinib (Fig. 1a). The stimulatory effect of imatinib increased proportionately with concentration up to 10μM then decreased slightly, presumably due to increased toxicity (the reported IC50 for imatinib falls within the range of 0.4 -1.5μM (40)).
Figure 1. Decreased phosphorylation of Dok1 in CrkI-transformed cells treated with imatinib.
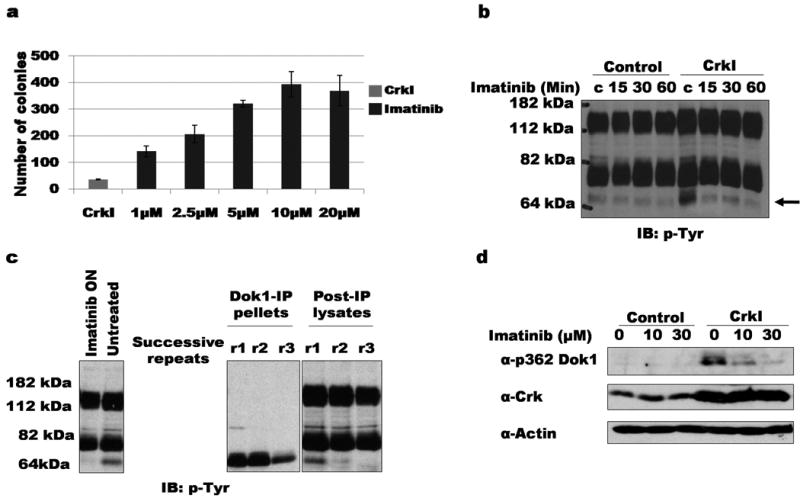
a) Soft agar colonies formed by Crk1-tranformed NIH3T3 cells treated continuously with the indicated concentrations of imatinib. b) Serum-starved CrkI-transformed NIH3T3 cells treated with 20 μM imatinib for indicated times were lysed and blotted with anti-pTyr. Phosphorylation of ∼64 kDa band is decreased upon imatinib treatment of CrkI-transformed cells (indicated by arrow). IB, immuno-blot; pTyr, anti-phosphotyrosine. c) Lysates of CrkI-transformed NIH3T3 cells were serially immunoprecipitated using anti-Dok1 antibody. Left panel, whole cell lystates treated with or without 2.5 μM imatinib; center and right panel, immunoprecipitate (IP) and supernatant (post-IP) fractions. ON: overnight incubation; successive rounds of immunoprecipitation indicated by r1, r2, and r3. d) CrkI-expressing or control NIH3T3 cells treated with indicated concentrations of imatinib were lysed and immunoblotted with phosphospecific Dok1 antibody (α-p362 Dok1). Immunoblotting with anti-Crk and anti-actin shown as controls.
We reasoned that Abl inhibition exerted its effects on Crk transformation by altering tyrosine phosphorylation. To identify Abl-dependent phosphoproteins, lysates of control and CrkI-transformed cells (with and without imatinib treatment) were immunoblotted with anti-phosphotyrosine (anti-pTyr) antibody. A prominent tyrosine-phosphorylated band of ∼64 kDa was seen in CrkI-overexpressing cells when compared to the controls, the phosphorylation of which was strongly reduced upon imatinib treatment (Fig. 1b). Based on known substrates of Abl and the apparent molecular weight, we surmised this phosphoprotein might be Dok1 (31). To test this, a lysate of Crk-transformed cells was serially immunoprecipitated with anti-Dok1 antibody. This treatment depleted the 64 kDa tyrosine-phosphorylated protein from the lysates, verifying its identity as Dok1 (Fig. 1c). Immunoblotting with a phosphospecific antibody recognizing pY362 of human Dok1 (pY361 in mouse Dok1) further confirmed the dependence of Dok1 phosphorylation on Abl activity (Fig. 1d).
Dok1 corresponded to the only prominent tyrosine-phosphorylated band in CrkI-transformed cells that was Abl-dependent. Somewhat paradoxically, Dok1 phosphorylation was increased in CrkI-transformed cells compared to normal control cells (Fig. 1b). Together, these data suggest that tyrosine phosphorylation of Dok1 by Abl is induced by CrkI overexpression, and may act to partially suppress Crk-mediated transformation.
Phosphorylation of Dok1 regulates CrkI transformation
To test whether Dok1 is acting as a tumor suppressor protein in this system, we knocked down Dok1 expression in CrkI overexpressing cells using shRNA (Fig. 2b). We observed an enhancement of soft agar colony formation upon Dok1 knockdown, comparable to the effect of Abl knockdown or imatinib treatment (Fig. 2c), implicating Dok1 as a crucial regulator of CrkI transformation.
Figure 2. Phosphorylation of tyrosine 295 and 361 of Dok1 correlates with suppression of CrkI transformation.
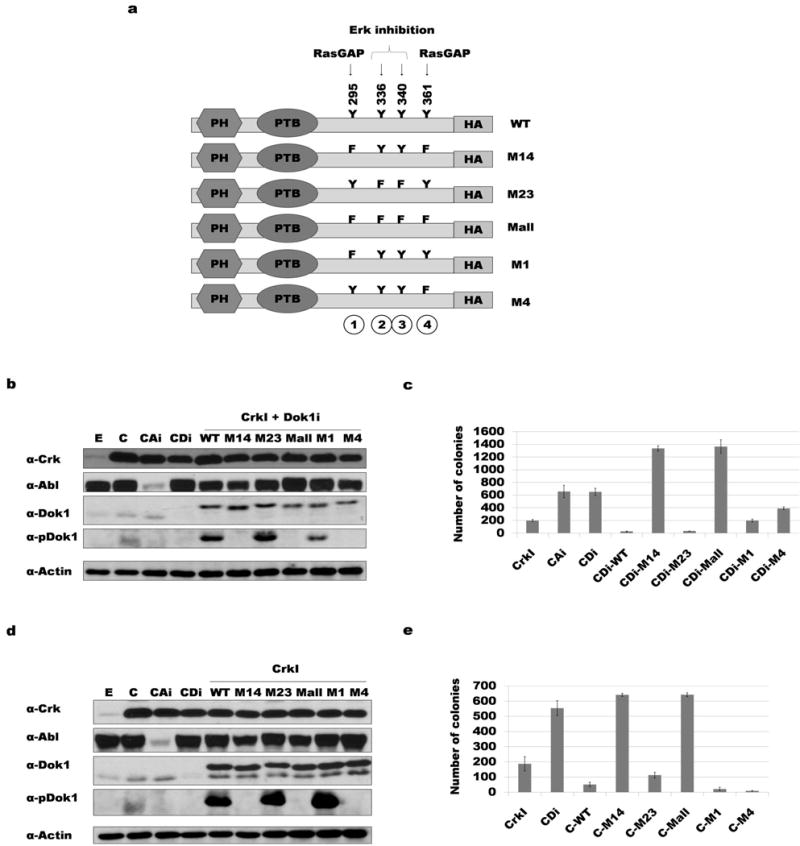
a) Diagram of the human Dok1 cDNA constructs used, indicating positions of tyrosine phosphorylation sites mutated. PH: Pleckstrin homology domain; PTB: phospho-tyrosine binding domain; Y: tyrosine; F: phenylalanine; HA: HA epitope tag. b) Dok1 knockdown and rescue with HA-tagged Dok1 constructs. NIH3T3 cell lysates were immunoblotted with antibodies indicated (αpDok1 = phosphospecific Dok1 antibody). E: empty vector control; C: CrkI transformed; CAi: CrkI-transformed, Abl knockdown; CDi: CrkI-transformed, Dok1 knockdown; WT: wild-type Dok1; M14: Y295F and Y361F mutant Dok1; M23: Y336F and Y340F mutant Dok1; M-all: Y295F, Y336F, Y340F and Y361F mutant Dok1; M1: Y295F mutant Dok1; M4: Y361F mutant Dok1. c) Soft agar colony formation results for cells in panel b. d) Over-expression of Dok1 in CrkI-transformed NIH3T3 cells; abbreviations as in panel b. e) Soft agar colony formation results for cells in panel d.
To further probe the significance of Dok1 in suppressing CrkI-mediated transformation, the human homolog of Dok1 (hDok1) was used for rescue experiments. We used human instead of mouse Dok1 to avoid its being targeted for shRNA-mediated knockdown. The human homolog of Dok1 has an insertion at position 271, which shifts the amino acid numbering C-terminal to the insertion by +1 compared to mouse. For simplicity, henceforth the mouse amino acid numbering will be used unless stated otherwise.
In addition to the wild-type (WT) hDok1, we also generated mutant constructs to test the importance of several potential tyrosine phosphorylation sites. Shinohara et al. (41) reported that phosphorylation of tyrosines 259 and 361 was required for RasGAP binding, while phosphorylation of tyrosines 336 and 340 inhibited Erk activation through unidentified mechanism(s). Both Ras and its activator Sos1 were previously shown to play essential roles in Crk transformation (11, 42), and the MEK/ERK pathway downstream of Ras is well known to promote cell proliferation (43).
Using site-directed mutagenesis, putative tyrosine phosphorylation sites were changed to phenylalanine. The resulting constructs were named according to the tyrosines mutated (Fig. 2a): M14 for mutation of sites 1 and 4 (Y295 and Y361), M23 for mutation of sites 2 and 3 (Y336 and Y340), M-all for mutation of all four sites, M1 for mutation of Y295, and M4 for mutation of Y361. These constructs were re-expressed in the CrkI-transformed Dok1 knockdown cells (Fig. 2b) and cells assayed for anchorage-independent growth in soft agar (Fig. 2c). Expression of the WT Dok1 or the M23 mutant rescued the phenotype (enhanced CrkI transformation) caused by Dok1 knockdown; in fact, transforming activity of rescued cells was even lower than cells expressing endogenous amounts of Dok1. On the other hand, knockdown cells expressing the M14 and M-all mutants showed an even greater increase in colony formation than seen with Dok1 knockdown alone. Expression of single mutants (M1 and M4) partially rescued the Dok1 knockdown phenotype. These results suggested that Y295 and Y361 work together to suppress CrkI tumorigenesis when phosphorylated. Dok1 knockdown followed by re-expression of WT or mutant Dok1 in control NIH3T3 cells yielded no colonies (data not shown), consistent with the effect of Dok1 tumor suppression being specific to cells transformed by Crk.
We also tested the effect of simple Dok1 overexpression in CrkI-transformed NIH3T3 cells (Fig. 2d). Consistent with its role as a putative tumor suppressor, over-expression of WT Dok1 and the M23 Dok1 mutant both suppressed CrkI transformation in the soft agar assay; by contrast, the M14 and M-all mutants enhanced CrkI transformation (Fig. 2e). This demonstrates that expression of Dok1 mutants that cannot be phosphorylated at sites 295 and 361 exerts a dominant-negative (pro-oncogenic) effect over endogenous Dok1. Surprisingly, overexpression of the M1 and M4 single mutants showed a greater suppression of transformation than seen for WT Dok1. Once again, these results were CrkI-dependent, as no colonies were seen in control 3T3 cells overexpressing Dok1 mutants (data not shown).
SH2 domain binding partners of tyrosine-phosphorylated Dok1
Most tyrosine-phosphorylated sites function in signaling by binding to the SH2 or PTB domains of effector proteins (44). To assess what SH2 domains bind to the Dok1 sites implicated in suppressing CrkI transformation, we carried out a dot-blot SH2 profiling assay (45) using purified glutathione S-transferase (GST)-SH2 or GST-PTB domain fusion proteins to probe synthetic tyrosine-phosphorylated peptides corresponding the four sites of interest in Dok1 (Fig. 3a).
Figure 3. Binding of SH2 domains to Dok1 phosphorylation sites.
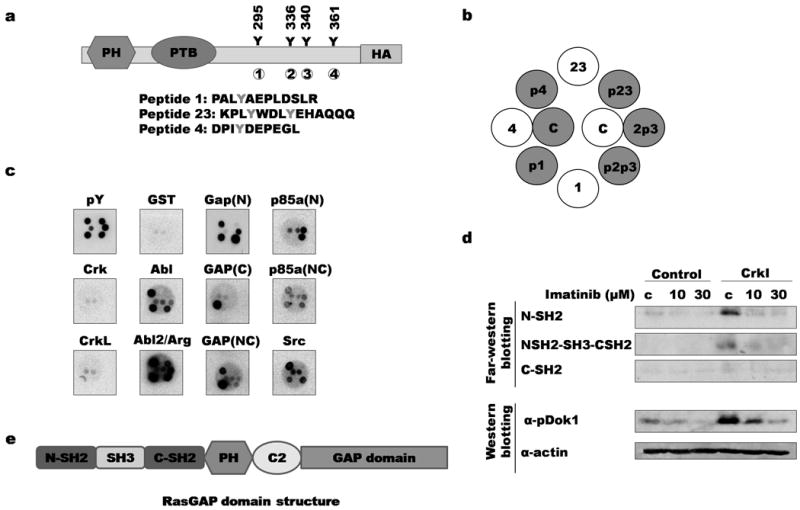
a) Phosphorylation sites on Dok1 are indicated; each site is numbered from 1 to 4. Sequences of corresponding synthetic peptides are indicated below. b) Synthetic peptides were spotted to filters in the pattern shown. Grey circles and “p” indicate tyrosine phosphorylation of corresponding site (for example, “p1” denotes peptide in which site 1 is phosphorylated); c: control (whole cell lysates) c) Peptide-spotted filters were probed with purified SH2 and PTB domains. Binding results for selected domains are shown (for data for all domains, see Supplementary Figure 1). GAP(N): RasGAP N-terminal SH2 domain; GAP(C): RasGAP C-terminal SH2 domain; GAP(NC): Both SH2 domains and SH3 domain of RasGAP; p85a(N): PI3K regulatory subunit 1 (α) N-terminal SH2 domain; p85a(NC): PI3Kα N- and C-terminal SH2 domains; GST: negative control; pY: anti-pTyr antibody. d) Far-Western and immunoblotting of lysates from imatinib-treated control and CrkI-transformed NIH3T3 cells. Top, lysates were probed with GST-RasGAP SH2 domain fusions; bottom, same lysates were probed with phosphospecific Dok1 antibody. C: untreated control lysates. e) RasGAP domain structure. C2: C2 domain.
Of the 123 SH2 or PTB probes tested (supplemental figure S1), a few showed strong binding to Dok1-derived phosphopeptides. Abl and Arg (Abl2) SH2 domains strongly bound to all four phosphorylated sites, but not the unphosphorylated control peptides (Fig. 3c). As expected, the SH2 domains of RasGAP bound strongly to pTyr 295 and 361. The C-terminal RasGAP SH2 domain bound specifically to pTyr 295, while the N-terminal domain was less specific and bound to phosphopeptides corresponding to tyrosines 295, 340 and 361. SH2 domains of p85α, a subunit of phosphatidylinositol 3-kinase, also bound to phosphorylated sites in Dok1. The N-terminal SH2 domain of p85α bound pTyr 336, while a construct encompassing both SH2 domains bound pTyr 295, 336, and 361. Notably, none of the phosphopeptides tested bound strongly to the Crk SH2 domain (Fig. 3c). The unphosphorylated peptides did not bind appreciably to any of the SH2 domain probes, highlighting the specificity of our SH2 probes for tyrosine-phosphorylated peptides.
We also performed far-Western blotting to probe imatinib-treated cell lysates with RasGAP SH2 domains. The RasGAP SH2 probes bound to a band in Crk-transformed cells corresponding to phosphorylated Dok1; binding was abolished when Abl-mediated phosphorylation was inhibited with imatinib (Fig. 3d). The relatively less specific RasGAP N-terminal SH2 domain showed the strongest difference in binding to Dok1 when cells were treated with imatinib. This suggests that in vivo, Abl preferentially phosphorylates site 4 (Y361) relative to site 1 (Y295), since little binding was seen with the RasGAP C-terminal SH2 probe which is highly specific to site 1. A similar pattern of binding was also observed in the control 3T3 cell lysates, albeit at a much lower intensity.
Role of RasGAP in regulating Crk transformation
We next sought to confirm that RasGAP binds to phosphorylated Dok1 in our cell system by expressing HA-tagged hDok1 and immunoprecipitating with anti-HA antibody. To maximize the detection of phosphotyrosine-dependent interactions, cells were treated briefly with pervanadate (POV) before lysis to inhibit endogenous tyrosine phosphatases. Immunoblotting of anti-HA immunoprecipitates with anti-RasGAP antibody demonstrated association between RasGAP and WT Dok1; this binding was decreased, however, when tyrosines 295 and 361 were mutated (mutants M14 and M-all) (Fig. 4a). These results are consistent with the results of phosphopeptide binding experiments (Fig. 3). The M1 and M4 mutants of Dok1 both associated with RasGAP to a similar extent as WT Dok1, demonstrating that, under these conditions, phosphorylation of either Y295 or Y361 alone is sufficient to mediate RasGAP binding.
Figure 4. RasGAP binds to phosphorylated Dok1 and is involved in suppressing Crk transformation.
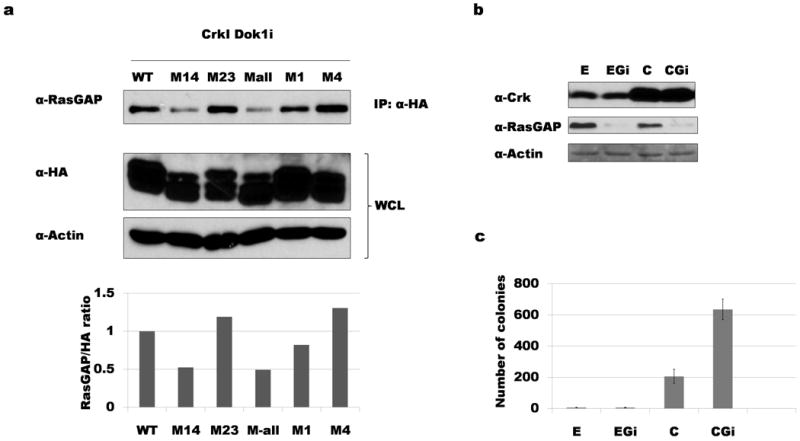
a) Lysates of CrkI-expressing Dok1 knockdown cells rescued with HA-tagged Dok1 mutants indicated were immunoprecipitated with anti-HA antibody before immunoblotting with anti-RasGAP. RasGAP binding to Dok1 was decreased when tyrosines 295 and 361 were both mutated (M14 and M-all). Cells were treated with pervanadate prior to lysis to increase total pTyr levels. Bottom: bands from two independent experiments were quantified; average RasGAP/HA ratio was normalized to WT Dok1. b) Western blotting demonstrating knockdown of RasGAP. E: empty vector control NIH3T3 cells; EGi: RasGAP knockdown control cells; C: CrkI-transformed cells; CGi: CrkI-transformed, RasGAP knockdown cells. c) Soft agar colony formation results for cells shown in panel b. RasGAP knockdown increases the number of colonies in CrkI-transformed cells but has no effect on the empty vector control cells.
Next we knocked down RasGAP in both the control and CrkI overexpressing cells (Fig. 4b). Loss of endogenous RasGAP in CrkI-transformed NIH3T3 increased the number of colonies (Fig. 4c), similar to what was observed in Abl and Dok1 knockdowns (Fig. 2c). This observation is consistent with a model in which Abl works through Dok1 and RasGAP to regulate CrkI transformation.
Ras activation in CrkI-transformed cells
While the previous results all suggest a role for elevated Ras activity in Crk transformation, over many experiments we did not detect significant differences in total Ras or Erk activity in CrkI-transformed cells relative to control, or under conditions where Abl or Dok1 activity was manipulated (see supplemental figure S2). We therefore considered whether CrkI overexpression might induce localized differences in Ras activity that were not evident in total cell lysates. To study the spatiotemporal aspects of Ras activation, we employed a newly developed FRET sensor for activated Ras (Dora-Ras) and conducted live cell imaging of Ras activation during cell spreading on fibronectin. Since Ras is activated on the plasma membrane, we used total internal reflection fluorescence (TIRF) excitation in our imaging studies to reduce imaging artifacts typically associated with wide-field imaging. Fifteen minutes after plating, cells transiently transfected with Dora-Ras sensor showed elevated ratio (FRET/CFP) values, indicative of Ras activation, in lamellipodial protrusions (Fig. 5a). In contrast, cells transfected with a control construct (with a point mutation in the Ras binding domain that abolishes sensor response), showed no polarized distribution of ratio values (Fig. 5b).
Figure 5. Ras activation is partially uncoupled from the turnover of adhesions in CrkI-transformed cells.
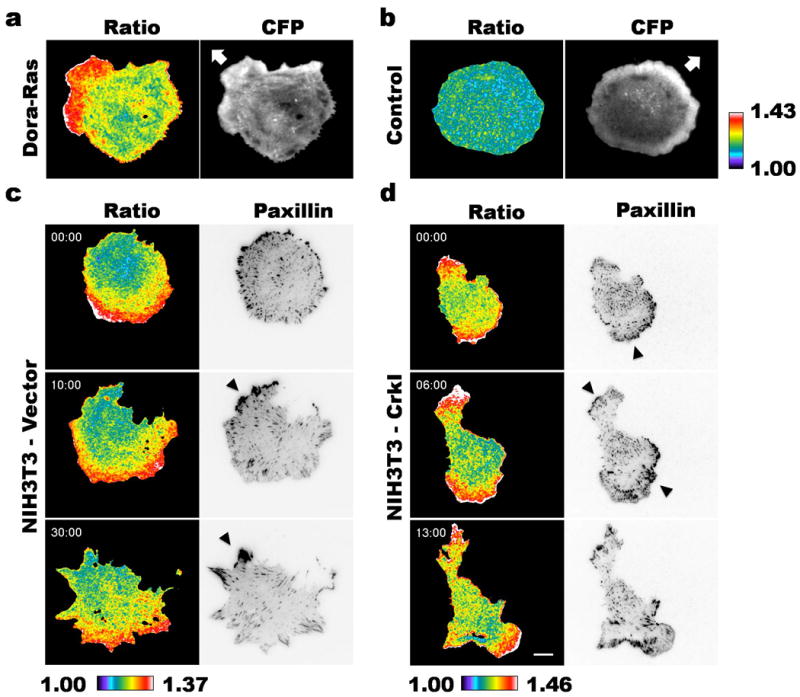
NIH3T3-vector (a-c, n = 18 cells) or -CrkI (d, n = 18 cells) cells were co-transfected with the Dora-Ras FRET sensor (a, c and d) or sensor control (b, n = 9 cells) and mCherry-tagged paxillin. The cells were seeded onto fibronectin-coated coverslips and imaged with TIRF excitation 15 minutes after plating. The ratio (FRET/CFP) images were calculated and presented in pseudocolor based on the lookup table provided (a and b, right; c and d, bottom). CFP and mCherry intensities were illustrated in grayscale and inverted grayscale images, respectively. The arrows (a and b) indicate directions of cell movement and the arrowheads (c and d), clusters of focal adhesions. Scale bar, 10 μm.
Crk is known to localize to adhesions by binding to phosphorylated paxillin and p130Cas(46-49), and we showed that CrkI was enriched at paxillin-containing adhesions in CrkI-overexpressing cells (supplemental movie S3). In order to relate sites of Ras activation to the location of adhesions, we co-expressed mCherry-tagged paxillin with the Dora-Ras sensor. In control NIH3T3 cells, Ras activity co-localized with nascent adhesions at the leading front, but diminished in the vicinity of mature focal adhesions as the cells spread (Fig. 5c and supplemental movie S4). In contrast, CrkI-transformed cells maintained elevated Ras activation while adhesions matured (Fig. 5d and supplemental movie S5). Strikingly, the spatial distribution of Ras activation often appeared bipolar or multipolar, and the cells developed multiple protrusions at an early stage of cell spreading. Thus we conclude that the localized distribution of activated Ras is dramatically different in CrkI-transformed cells compared to their normal controls.
Discussion
Unregulated Abl activity causes CML and other malignancies in humans, and small molecule Abl inhibitors are now widely used to treat cancer. In this context, our previous finding that Abl downregulation enhanced transformation by Crk (11) was both surprising and potentially disturbing, as it raised the possibility that Abl inhibitors could promote instead of inhibit tumor growth in some situations. In fact, such a tumor-promoting effect of Abl inhibition had been reported in several previous studies, where Abl family kinases acted downstream of Epithelial Growth Factor (EGF) (49), Transforming Growth Factor-β (TGF-β) (50) or EphrinB (EphB) (51). In each case, enhanced xenograft growth was reported upon downregulation of active Abl and/or Arg. Our current results add to our understanding of the complex mechanisms whereby Abl family kinases can both positively and negatively impact different aspects of cell transformation.
We have now identified Dok1 as the downstream effector responsible for enhancing CrkI transformation upon Abl inhibition. Dok1, Dok2 and Dok3 knockout mice were shown in two independent studies to develop lung tumors (38) and histiocytic sarcomas (39), and the Dok1 gene maps to human chromosome 2p13, a frequent site of translocations in leukemia (52). Frameshift mutation (53) and aberrant DNA methylation in the promoter region (54, 55) were reported for Dok1 in various human cancers. Taken together, these results are consistent with our current observation that Dok1 plays an important role in downregulating CrkI-induced cell transformation.
In Fig. 6 we propose a model for how Abl regulates transformation through Dok1 phosphorylation. The overexpression of CrkI leads to its recruitment and clustering at membrane sites rich in tyrosine-phosphorylated proteins, such as focal adhesions. Crk in turn recruits its SH3-binding effectors such as Sos to these sites, leading to localized activation of Ras and ultimately resulting in cell transformation. At the same time, however, Crk triggers a negative feedback loop by recruiting Abl family kinases, which associate with and phosphorylate Dok1. Phosphorylated DokI recruits RasGAP, which counteracts the stimulation of Ras by Sos and thereby blunts Crk transformation. When Abl-mediated phosphorylation is inhibited (either by knockdown or imatinib), the resulting decrease in Dok1 phosphorylation prevents it from recruiting RasGAP, thereby increasing local Ras activation and enhancing transformation. Interestingly, we have noticed that CrkI overexpression reproducibly causes a modest increase in endogenous Dok1 expression in addition to increased Dok1 phosphorylation (see Fig. 2b and 2d), consistent with a negative feedback response to CrkI transformation.
Figure 6. Phosphorylated Dok1 regulates CrkI-transformation.
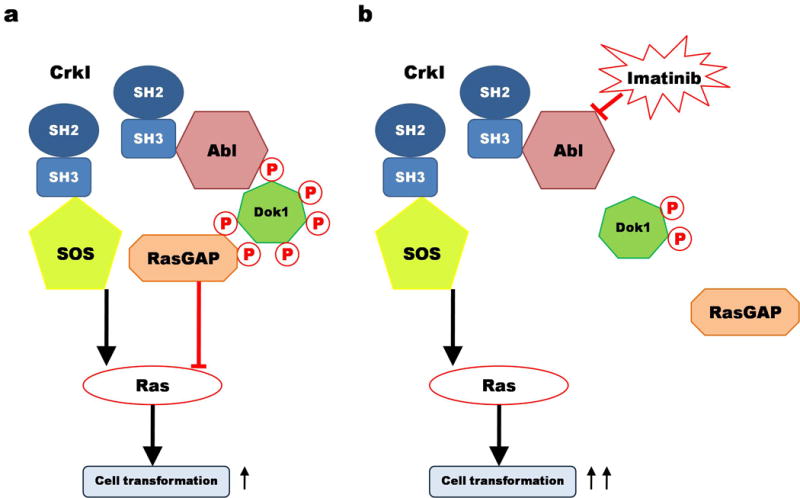
A) CrkI (blue) overexpression nucleates formation of localized protein complexes, including those directly bound (Sos and Abl shown here) and indirectly bound (Dok1 and RasGAP). While Sos serves as a crucial positive effector for CrkI-transformation, phosphorylated Dok1 acts as a negative regulator, likely by inhibiting the Ras pathway by recruiting RasGAP. The net output of the complex is relatively weak transformation. B) When Abl family kinase activity is inhibited (by knockdown or inhibition), Dok1 is no longer highly phosphorylated and thus loses its ability to repress cell transformation, shifting the balance further toward strong transformation.
While our data demonstrate a strong negative correlation between phosphorylated Dok1 and CrkI transformation, it is unclear what role this pathway plays in other types of cell transformation. On the one hand, we previously showed that imatinib had little effect on the anchorage-independent growth of cells transformed by Src or Ras (11), and preliminary experiments with a panel of human cancer cell lines showed that Abl inhibitors do not generally stimulate the growth of tumor cells (KN and BJM, unpublished observations). However, previous results showing that Dok proteins can negatively regulate signaling from the BCR and TCR, and that they exhibit tumor suppressor activity, suggest a more general role in feedback regulation of the Ras pathway. It is possible that the repressive role of Dok is particularly evident in Crk transformation because Crk is a relatively weak oncogene in mammalian fibroblasts.
Greulich and Hanafusa previously showed that dominant-negative Ras mutants blocked transformation by v-Crk (42), and we later showed that Sos1, the major guanine nucleotide exchange factor (GEF) for Ras, is by far the most critical SH3 binding protein of those tested for CrkI transformation (11). However, the specific role of Ras activation in Crk transformation remains enigmatic, as we and others have not seen a consistent or significant increase in the activity of Ras or its downstream effectors in Crk-transformed cells relative to controls (11, 42, 56). Our current results further implicate Ras activity in Crk transformation by demonstrating that phosphorylation of two sites in Dok1 that bind the Ras inhibitor RasGAP (tyrosines 295 and 361) is critical for suppressing Crk transformation. We show that Dok1 is highly phosphorylated in Crk-transformed cells, and that this phosphorylation is abrogated by the Abl inhibitor imatinib, which stimulates Crk transformation. We also show that overexpression of Dok1 mutants that cannot be phosphorylated at the Ras-GAP binding sites strongly stimulates Crk transformation, while expression of WT Dok1 or Dok1 mutants in which these sites are intact strongly inhibits anchorage independent growth induced by CrkI overexpression. Finally, we also show that RasGAP knockdown enhances CrkI transformation (Fig. 4c).
Interestingly, Dok1 knockdown cells rescued with Dok1 mutated at tyrosines 295 and 361 (M14 and M-all) were even more transformed than the Dok1 knockdown alone (Fig. 2). A likely explanation is that the Dok1 mutants have a dominant-negative effect on the residual, endogenous Dok1 present in knockdown cells. The fact that simple overexpression of these Dok1 mutants had similar effects, even in the presence of unaltered endogenous Dok1, is also consistent with this idea. Phospho-dependent homotypic and heterotypic oligomerization between Dok1 and Dok2 is reported to be critical for their function (57), so the presence of mutated Dok1 would likely disrupt the integrity of oligomers and compromise their normal ability to downregulate signaling.
One way to reconcile the apparent importance of Ras in Crk transformation with the lack of obvious elevation in total Ras activity in Crk-transformed cells is to suppose that Ras activation by CrkI is highly localized and therefore not apparent in whole cell lysates. Indeed, during cell spreading we noted striking differences in the localization of activated Ras in Crk-transformed cells relative to controls (Fig. 5). In general, upon spreading control cells rapidly formed a single leading edge where Ras activity was highest, while mature focal adhesions with the highest concentration of paxillin and Crk were mostly found along the sides and trailing edge of the cell, away from the areas of highest Ras activity. Others have previously noted that activated Ras is enriched in lamellipodia and the leading edge of polarized cells (58, 59). In CrkI-transformed cells, however, most cells formed multiple highly protrusive fronts with high Ras activity, which in some cases coincided with mature focal adhesions. This phenotype is consistent with the partial loss of a negative feedback loop that normally functions to repress Ras at focal adhesions after a brief burst of activity.
The predominant form of Crk in normal cells, CrkII, contains a C-terminal region with a negative regulatory Abl phosphorylation (60, 61). The highly related CRKL protein also contains a homologous tyrosine phosphorylation site (62, 63). In normal cells, CrkII or recruited to focal adhesions is likely to be rapidly downregulated by Abl, and thus can only transiently recruit effectors such as Sos to adhesions. CrkI, by contrast, lacks this negative regulatory site. Thus in cells overexpressing CrkI, one might expect relatively sustained Ras activation at sites where Crk is localized, such as focal adhesions. In these cells however, Dok1 phosphorylation appears to trigger a slower, less efficient downregulatory mechanism for Ras via the recruitment of RasGAP. Inhibition or knockdown of Abl family kinases, Dok1, or RasGAP inhibits this second feedback loop, and likely exacerbates the mislocalization of Ras activation seen in CrkI overexpressing cells. Further experiments will be needed to tease out the effects of this local feedback inhibition on proliferative signaling, and on cell motility and polarity in general.
Our results suggest that CrkI overexpression has both positive and negative effects on cell transformation, through different sets of protein interactions. Inputs that interfere with some of these interactions will shift this equilibrium, pushing the cell toward one extreme or the other (normal vs. tumorigenic). Consistent with this idea, Crk transformation of mammalian cells is relatively weak, presumably at least in part because of the negative feedback provided by phosphorylation of Dok1 by Abl. The ability of Crk to transform cells is likely to depend not only on the level of Crk overexpression, but also on the relative abundance of positive effectors such as Sos, versus potential negative regulators such as Abl, Dok1, and RasGAP. Our results highlight the importance of understanding the role of negative feedback loops when considering therapies that incorporate Abl inhibition for the treatment of human tumors.
Methods and Materials
Cell culture, transfection, and viral infection
NIH3T3 fibroblasts were maintained in DMEM supplemented with 10% super calf serum (SCS) (Gemini Bio-products). Serum starvation was carried out by maintaining the cells in DMEM with 0.1% serum overnight. Transient transfection was performed using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. Retrovirus stocks were produced and used to infect cells as described (11).
DNA constructs
The CrkI MSCVpuro retroviral vector has been described (11). Dok1 and its mutants were cloned into MSCVneo vector (Clontech). The tyrosine-to-phenylalanine Dok1 mutants were generated from WT cDNA (38) using standard site-directed PCR mutagenesis with Platinum Taq polymerase (Life Technologies). The GFP- and Cherry-paxillin constructs were generous gifts of Klaus Hahn (UNC-Chapel Hill).
Construction of Dora-Ras
The Dora-Ras sensor is based on established design principles (58, 64). It contains an N-terminal Ras binding domain (RBD, Cys71-Ser161) derived from Byr2, followed by a fluorescent protein FRET pair (Cerulean3-Venus), and an intact, wild-type H-Ras at the C-terminus. To improve the dynamic range, structural optimization was used to couple the dimerization of fluorescent proteins with the interaction between Byr2 and activated H-Ras (65), hence the name Dimerization Optimized Reporter for Activation (Dora). The detailed characterization of the sensor will be described elsewhere (manuscript in preparation). As a control, a point mutation (R83E) was introduced in the RBD to disrupt Ras binding. The mutant sensor controls for potential alterations of fluorescence in the cell that are independent of Ras activation.
Gene knockdown
The target sequences used for Abl/Arg, Dok1 and RasGAP knockdown were 5′-GAGTACTTGGAGAAGAAGA-3′ (11), 5′-GGTAATGTTCTCCTTTGAA-3′ (modified from (66)), and 5′-AAGATGAAGCCACTACCCTATTT-3′(67) respectively. The 68 bp and 72bp DNA inserts were first hybridized then cloned into pSUPER-hygro retrovirus vector (Oligoengine).
Antibodies
pTyr (P-Tyr-100) was from Cell Signaling; Dok1 (A-3), pDok1 (Tyr362), Actin (I-19) and HA (Y-11) were from Santa Cruz Biotechnology; CrkII (C-18) and Abl (8E9) were from BD Biosciences; RasGAP (B4F8) was from Upstate Biotechnology.
Anchorage independent growth assay
Cells were trypsinized, counted and suspended in 10% FBS Iscove's DMEM with 0.3% Bacto agar (BD Bioscience) on a base agar layer of the same medium with 0.6% agar. 2.5 × 104 cells (Fig. 1) or 5 × 104 cells (Figs. 2, 4) were seeded per 60 mm plate. Plates were fed with 1 ml of fresh medium every week. After 4 weeks, plates were stained with 0.005% crystal violet (Sigma-Aldrich), photographed, and colony numbers were calculated using ImageJ (NIH, Bethesda, MD, USA). For imatinib (LC laboratories) treatment, drug was present at the indicated concentration both in the initial plating medium and in medium used for weekly feeding.
SH2-phosphopeptide binding assay and far-western blotting
Biotinylated synthetic peptides were synthesized in both phosphorylated and unphosphorylated forms (Genescript), carefully spotted onto a gelatin-coated nitrocellulose membrane and fixed with 4% paraformaldehide for 5 min. The membrane was subjected to binding with a panel of GST-tagged pTyr binding domains as described (45). Far-western blotting (Fig. 3D) was carried out as described previously (45) using HRP-conjugated anti-GST antibody for detection.
Protein analysis
Cell lysis, immunoblotting, and immunoprecipitation were performed as previously described (11). For co-IP (Fig. 4a), cells were first treated with 200 μM pervanadate (POV) for 45 min before lysis. Equal amounts of lysate were pre-cleared for 2 h with Protein-A beads, then, supernatant was incubated with anti-HA and Protein-A beads overnight. The next day, beads were washed 4X with a high-salt KLB solution (500 mM NaCl) and once with standard KLB solution before addition of 5X sample buffer.
Live cell imaging and sensor data processing
Cells were seeded onto fibronectin (20 μg/ml) coated coverslips in phenol red- and vitamin-free DMEM imaging medium (US Biological). TIRF (total internal refection fluorescence) imaging was conducted on a customized Ti-E inverted microscope (Nikon, Japan) equipped with a multiline (440/515/594 nm) LMM5 laser merge module (Spectral Applied Research, Canada), a motorized XY stage (Ludl, Hawthorne, NY), and a Stable Z stage heater (Bioptechs, Butler, PA). Images were acquired through a 60x 1.49 NA TIRF objective (Nikon) on an iXon Ultra 897 EMCCD (Andor, Belfast, UK) under the control of MetaMorph software (Molecular Devices, Sunnyvale, CA). Biosensor data were processed using custom routines written in MetaMorph and MATLAB (MathWorks, Natick, MA) for background subtraction, image segmentation, channel registration, and ratiometric arithmetic as described previously (68).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We would like to thank Sofya Borinskaya for technical help in soft agar colony counting, Mari Ogiue-Ikeda for the preparation of the SH2 probes, and Joshua Jadwin for critically reading this manuscript. In addition, we greatly appreciate Pier Paolo Pandolfi from Harvard Medical School for generously providing the human Dok1 cDNA. This study was supported by grant CA82258 from the National Institutes of Health (to BJM).
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Supplementary information: Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Mayer BJ, Hamaguchi M, Hanafusa H. A novel viral oncogene with structural similarity to phospholipase C. Nature. 1988 Mar 17;332(6161):272–5. doi: 10.1038/332272a0. [DOI] [PubMed] [Google Scholar]
- 2.Matsuda M, Tanaka S, Nagata S, Kojima A, Kurata T, Shibuya M. Two species of human CRK cDNA encode proteins with distinct biological activities. Mol Cell Biol. 1992 Aug;12(8):3482–9. doi: 10.1128/mcb.12.8.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.ten Hoeve J, Morris C, Heisterkamp N, Groffen J. Isolation and chromosomal localization of CRKL, a human crk-like gene. Oncogene. 1993 Sep;8(9):2469–74. [PubMed] [Google Scholar]
- 4.Reichman CT, Mayer BJ, Keshav S, Hanafusa H. The product of the cellular crk gene consists primarily of SH2 and SH3 regions. Cell Growth Differ. 1992 Jul;3(7):451–60. [PubMed] [Google Scholar]
- 5.Feller SM, Knudsen B, Hanafusa H. c-Abl kinase regulates the protein binding activity of c-Crk. EMBO J. 1994 May 15;13(10):2341–51. doi: 10.1002/j.1460-2075.1994.tb06518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sriram G, Birge RB. Emerging roles for crk in human cancer. Genes Cancer. 2010 Nov;1(11):1132–9. doi: 10.1177/1947601910397188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller CT, Chen G, Gharib TG, Wang H, Thomas DG, Misek DE, et al. Increased C-CRK proto-oncogene expression is associated with an aggressive phenotype in lung adenocarcinomas. Oncogene. 2003 Sep 11;22(39):7950–7. doi: 10.1038/sj.onc.1206529. [DOI] [PubMed] [Google Scholar]
- 8.Takino T, Nakada M, Miyamori H, Yamashita J, Yamada KM, Sato H. CrkI adapter protein modulates cell migration and invasion in glioblastoma. Cancer Res. 2003 May 1;63(9):2335–7. [PubMed] [Google Scholar]
- 9.Rodrigues SP, Fathers KE, Chan G, Zuo D, Halwani F, Meterissian S, et al. CrkI and CrkII function as key signaling integrators for migration and invasion of cancer cells. Mol Cancer Res. 2005 Apr;3(4):183–94. doi: 10.1158/1541-7786.MCR-04-0211. [DOI] [PubMed] [Google Scholar]
- 10.Linghu H, Tsuda M, Makino Y, Sakai M, Watanabe T, Ichihara S, et al. Involvement of adaptor protein Crk in malignant feature of human ovarian cancer cell line MCAS. Oncogene. 2006 Jun 15;25(25):3547–56. doi: 10.1038/sj.onc.1209398. [DOI] [PubMed] [Google Scholar]
- 11.Zheng J, Machida K, Antoku S, Ng KY, Claffey KP, Mayer BJ. Proteins that bind the Src homology 3 domain of CrkI have distinct roles in Crk transformation. Oncogene. 2010 Dec 2;29(48):6378–89. doi: 10.1038/onc.2010.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bell ES, Park M. Models of crk adaptor proteins in cancer. Genes Cancer. 2012 May;3(5-6):341–52. doi: 10.1177/1947601912459951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayer BJ, Hanafusa H. Mutagenic analysis of the v-crk oncogene: requirement for SH2 and SH3 domains and correlation between increased cellular phosphotyrosine and transformation. J Virol. 1990 Aug;64(8):3581–9. doi: 10.1128/jvi.64.8.3581-3589.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, et al. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993 Mar 12;72(5):767–78. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 15.Birge RB, Fajardo JE, Reichman C, Shoelson SE, Songyang Z, Cantley LC, et al. Identification and characterization of a high-affinity interaction between v-Crk and tyrosine-phosphorylated paxillin in CT10-transformed fibroblasts. Mol Cell Biol. 1993 Aug;13(8):4648–56. doi: 10.1128/mcb.13.8.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakai R, Iwamatsu A, Hirano N, Ogawa S, Tanaka T, Mano H, et al. A novel signaling molecule, p130, forms stable complexes in vivo with v-Crk and v-Src in a tyrosine phosphorylation-dependent manner. EMBO J. 1994 Aug 15;13(16):3748–56. doi: 10.1002/j.1460-2075.1994.tb06684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knudsen BS, Feller SM, Hanafusa H. Four proline-rich sequences of the guanine-nucleotide exchange factor C3G bind with unique specificity to the first Src homology 3 domain of Crk. J Biol Chem. 1994 Dec 30;269(52):32781–7. [PubMed] [Google Scholar]
- 18.Tanaka S, Morishita T, Hashimoto Y, Hattori S, Nakamura S, Shibuya M, et al. C3G, a guanine nucleotide-releasing protein expressed ubiquitously, binds to the Src homology 3 domains of CRK and GRB2/ASH proteins. Proc Natl Acad Sci U S A. 1994 Apr 12;91(8):3443–7. doi: 10.1073/pnas.91.8.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ren R, Ye ZS, Baltimore D. Abl protein-tyrosine kinase selects the Crk adapter as a substrate using SH3-binding sites. Genes Dev. 1994 Apr 1;8(7):783–95. doi: 10.1101/gad.8.7.783. [DOI] [PubMed] [Google Scholar]
- 20.Wang B, Mysliwiec T, Feller SM, Knudsen B, Hanafusa H, Kruh GD. Proline-rich sequences mediate the interaction of the Arg protein tyrosine kinase with Crk. Oncogene. 1996 Oct 3;13(7):1379–85. [PubMed] [Google Scholar]
- 21.Matsuda M, Hashimoto Y, Muroya K, Hasegawa H, Kurata T, Tanaka S, et al. CRK protein binds to two guanine nucleotide-releasing proteins for the Ras family and modulates nerve growth factor-induced activation of Ras in PC12 cells. Mol Cell Biol. 1994 Aug;14(8):5495–500. doi: 10.1128/mcb.14.8.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hasegawa H, Kiyokawa E, Tanaka S, Nagashima K, Gotoh N, Shibuya M, et al. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol Cell Biol. 1996 Apr;16(4):1770–6. doi: 10.1128/mcb.16.4.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abelson HT, Rabstein LS. Lymphosarcoma: virus-induced thymic-independent disease in mice. Cancer Res. 1970 Aug;30(8):2213–22. [PubMed] [Google Scholar]
- 24.Ben-Neriah Y, Daley GQ, Mes-Masson AM, Witte ON, Baltimore D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science. 1986 Jul 11;233(4760):212–4. doi: 10.1126/science.3460176. [DOI] [PubMed] [Google Scholar]
- 25.Apperley JF, Gardembas M, Melo JV, Russell-Jones R, Bain BJ, Baxter EJ, et al. Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor beta. N Engl J Med. 2002 Aug 15;347(7):481–7. doi: 10.1056/NEJMoa020150. [DOI] [PubMed] [Google Scholar]
- 26.Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003 Mar 27;348(13):1201–14. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- 27.Akin C, Fumo G, Yavuz AS, Lipsky PE, Neckers L, Metcalfe DD. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood. 2004 Apr 15;103(8):3222–5. doi: 10.1182/blood-2003-11-3816. [DOI] [PubMed] [Google Scholar]
- 28.Hughes TP, Saglio G, Kantarjian HM, Guilhot F, Niederwieser D, Rosti G, et al. Early molecular response predicts outcomes in patients with chronic myeloid leukemia in chronic phase treated with frontline nilotinib or imatinib. Blood. 2013 Dec 11; doi: 10.1182/blood-2013-06-510396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reichardt P, Reichardt A, Pink D. Molecular targeted therapy of gastrointestinal stromal tumors. Curr Cancer Drug Targets. 2011 Jul;11(6):688–97. doi: 10.2174/156800911796191042. [DOI] [PubMed] [Google Scholar]
- 30.Ashman LK, Griffith R. Therapeutic targeting of c-KIT in cancer. Expert Opin Investig Drugs. 2013 Jan;22(1):103–15. doi: 10.1517/13543784.2013.740010. [DOI] [PubMed] [Google Scholar]
- 31.Yamanashi Y, Baltimore D. Identification of the Abl- and rasGAP-associated 62 kDa protein as a docking protein, Dok. Cell. 1997 Jan 24;88(2):205–11. doi: 10.1016/s0092-8674(00)81841-3. [DOI] [PubMed] [Google Scholar]
- 32.Carpino N, Wisniewski D, Strife A, Marshak D, Kobayashi R, Stillman B, et al. p62(dok): a constitutively tyrosine-phosphorylated, GAP-associated protein in chronic myelogenous leukemia progenitor cells. Cell. 1997 Jan 24;88(2):197–204. doi: 10.1016/s0092-8674(00)81840-1. [DOI] [PubMed] [Google Scholar]
- 33.Mashima R, Hishida Y, Tezuka T, Yamanashi Y. The roles of Dok family adapters in immunoreceptor signaling. Immunol Rev. 2009 Nov;232(1):273–85. doi: 10.1111/j.1600-065X.2009.00844.x. [DOI] [PubMed] [Google Scholar]
- 34.Yamanashi Y, Tamura T, Kanamori T, Yamane H, Nariuchi H, Yamamoto T, et al. Role of the rasGAP-associated docking protein p62(dok) in negative regulation of B cell receptor-mediated signaling. Genes Dev. 2000 Jan 1;14(1):11–6. [PMC free article] [PubMed] [Google Scholar]
- 35.Yasuda T, Bundo K, Hino A, Honda K, Inoue A, Shirakata M, et al. Dok-1 and Dok-2 are negative regulators of T cell receptor signaling. Int Immunol. 2007 Apr;19(4):487–95. doi: 10.1093/intimm/dxm015. [DOI] [PubMed] [Google Scholar]
- 36.Yasuda T, Shirakata M, Iwama A, Ishii A, Ebihara Y, Osawa M, et al. Role of Dok-1 and Dok-2 in myeloid homeostasis and suppression of leukemia. J Exp Med. 2004 Dec 20;200(12):1681–7. doi: 10.1084/jem.20041247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niki M, Di Cristofano A, Zhao M, Honda H, Hirai H, Van Aelst L, et al. Role of Dok-1 and Dok-2 in leukemia suppression. J Exp Med. 2004 Dec 20;200(12):1689–95. doi: 10.1084/jem.20041306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berger AH, Niki M, Morotti A, Taylor BS, Socci ND, Viale A, et al. Identification of DOK genes as lung tumor suppressors. Nat Genet. 2010 Mar;42(3):216–23. doi: 10.1038/ng.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mashima R, Honda K, Yang Y, Morita Y, Inoue A, Arimura S, et al. Mice lacking Dok-1, Dok-2, and Dok-3 succumb to aggressive histiocytic sarcoma. Lab Invest. 2010 Sep;90(9):1357–64. doi: 10.1038/labinvest.2010.121. [DOI] [PubMed] [Google Scholar]
- 40.White D, Saunders V, Lyons AB, Branford S, Grigg A, To LB, et al. In vitro sensitivity to imatinib-induced inhibition of ABL kinase activity is predictive of molecular response in patients with de novo CML. Blood. 2005 Oct 1;106(7):2520–6. doi: 10.1182/blood-2005-03-1103. [DOI] [PubMed] [Google Scholar]
- 41.Shinohara H, Yasuda T, Yamanashi Y. Dok-1 tyrosine residues at 336 and 340 are essential for the negative regulation of Ras-Erk signalling, but dispensable for rasGAP-binding. Genes Cells. 2004 Jun;9(6):601–7. doi: 10.1111/j.1356-9597.2004.00748.x. [DOI] [PubMed] [Google Scholar]
- 42.Greulich H, Hanafusa H. A role for Ras in v-Crk transformation. Cell Growth Differ. 1996 Nov;7(11):1443–51. [PubMed] [Google Scholar]
- 43.Neuzillet C, Tijeras-Raballand A, de Mestier L, Cros J, Faivre S, Raymond E. MEK in cancer and cancer therapy. Pharmacol Ther. 2013 Oct 9; doi: 10.1016/j.pharmthera.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 44.Wagner MJ, Stacey MM, Liu BA, Pawson T. Molecular Mechanisms of SH2- and PTB-Domain-Containing Proteins in Receptor Tyrosine Kinase Signaling. Cold Spring Harb Perspect Biol. 2013;5(12) doi: 10.1101/cshperspect.a008987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Machida K, Thompson CM, Dierck K, Jablonowski K, Karkkainen S, Liu B, et al. High-throughput phosphotyrosine profiling using SH2 domains. Mol Cell. 2007 Jun 22;26(6):899–915. doi: 10.1016/j.molcel.2007.05.031. [DOI] [PubMed] [Google Scholar]
- 46.Chodniewicz D, Klemke RL. Regulation of integrin-mediated cellular responses through assembly of a CAS/Crk scaffold. Biochim Biophys Acta. 2004 Jul 5;1692(2-3):63–76. doi: 10.1016/j.bbamcr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 47.Downey C, Craig DH, Basson MD. Pressure activates colon cancer cell adhesion via paxillin phosphorylation, Crk, Cas, and Rac1. Cell Mol Life Sci. 2008 May;65(9):1446–57. doi: 10.1007/s00018-008-8038-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Watanabe T, Tsuda M, Makino Y, Konstantinou T, Nishihara H, Majima T, et al. Crk adaptor protein-induced phosphorylation of Gab1 on tyrosine 307 via Src is important for organization of focal adhesions and enhanced cell migration. Cell Res. 2009 May;19(5):638–50. doi: 10.1038/cr.2009.40. [DOI] [PubMed] [Google Scholar]
- 49.Gil-Henn H, Patsialou A, Wang Y, Warren MS, Condeelis JS, Koleske AJ. Arg/Abl2 promotes invasion and attenuates proliferation of breast cancer in vivo. Oncogene. 2013 May 23;32(21):2622–30. doi: 10.1038/onc.2012.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Allington TM, Galliher-Beckley AJ, Schiemann WP. Activated Abl kinase inhibits oncogenic transforming growth factor-beta signaling and tumorigenesis in mammary tumors. FASEB J. 2009 Dec;23(12):4231–43. doi: 10.1096/fj.09-138412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Noren NK, Foos G, Hauser CA, Pasquale EB. The EphB4 receptor suppresses breast cancer cell tumorigenicity through an Abl-Crk pathway. Nat Cell Biol. 2006 Aug;8(8):815–25. doi: 10.1038/ncb1438. [DOI] [PubMed] [Google Scholar]
- 52.Nelms K, Snow AJ, Noben-Trauth K. Dok1 encoding p62(dok) maps to mouse chromosome 6 and human chromosome 2 in a region of translocation in chronic lymphocytic leukemia. Genomics. 1998 Oct 15;53(2):243–5. doi: 10.1006/geno.1998.5514. [DOI] [PubMed] [Google Scholar]
- 53.Lee S, Roy F, Galmarini CM, Accardi R, Michelon J, Viller A, et al. Frameshift mutation in the Dok1 gene in chronic lymphocytic leukemia. Oncogene. 2004 Mar 25;23(13):2287–97. doi: 10.1038/sj.onc.1207385. [DOI] [PubMed] [Google Scholar]
- 54.Lambert MP, Paliwal A, Vaissiere T, Chemin I, Zoulim F, Tommasino M, et al. Aberrant DNA methylation distinguishes hepatocellular carcinoma associated with HBV and HCV infection and alcohol intake. J Hepatol. 2011 Apr;54(4):705–15. doi: 10.1016/j.jhep.2010.07.027. [DOI] [PubMed] [Google Scholar]
- 55.Saulnier A, Vaissiere T, Yue J, Siouda M, Malfroy M, Accardi R, et al. Inactivation of the putative suppressor gene DOK1 by promoter hypermethylation in primary human cancers. Int J Cancer. 2012 Jun 1;130(11):2484–94. doi: 10.1002/ijc.26299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Akagi T, Shishido T, Murata K, Hanafusa H. v-Crk activates the phosphoinositide 3-kinase/AKT pathway in transformation. Proc Natl Acad Sci U S A. 2000 Jun 20;97(13):7290–5. doi: 10.1073/pnas.140210297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Songyang Z, Yamanashi Y, Liu D, Baltimore D. Domain-dependent function of the rasGAP-binding protein p62Dok in cell signaling. J Biol Chem. 2001 Jan 26;276(4):2459–65. doi: 10.1074/jbc.M005504200. [DOI] [PubMed] [Google Scholar]
- 58.Mochizuki N, Yamashita S, Kurokawa K, Ohba Y, Nagai T, Miyawaki A, et al. Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature. 2001 Jun 28;411(6841):1065–8. doi: 10.1038/35082594. [DOI] [PubMed] [Google Scholar]
- 59.Yip SC, El-Sibai M, Coniglio SJ, Mouneimne G, Eddy RJ, Drees BE, et al. The distinct roles of Ras and Rac in PI 3-kinase-dependent protrusion during EGF-stimulated cell migration. J Cell Sci. 2007 Sep 1;120(Pt 17):3138–46. doi: 10.1242/jcs.005298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kain KH, Klemke RL. Inhibition of cell migration by Abl family tyrosine kinases through uncoupling of Crk-CAS complexes. J Biol Chem. 2001 May 11;276(19):16185–92. doi: 10.1074/jbc.M100095200. [DOI] [PubMed] [Google Scholar]
- 61.Kain KH, Gooch S, Klemke RL. Cytoplasmic c-Abl provides a molecular ‘Rheostat’ controlling carcinoma cell survival and invasion. Oncogene. 2003 Sep 4;22(38):6071–80. doi: 10.1038/sj.onc.1206930. [DOI] [PubMed] [Google Scholar]
- 62.de Jong R, ten Hoeve J, Heisterkamp N, Groffen J. Tyrosine 207 in CRKL is the BCR/ABL phosphorylation site. Oncogene. 1997 Feb 6;14(5):507–13. doi: 10.1038/sj.onc.1200885. [DOI] [PubMed] [Google Scholar]
- 63.Senechal K, Heaney C, Druker B, Sawyers CL. Structural requirements for function of the Crkl adapter protein in fibroblasts and hematopoietic cells. Mol Cell Biol. 1998 Sep;18(9):5082–90. doi: 10.1128/mcb.18.9.5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pertz O, Hodgson L, Klemke RL, Hahn KM. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature. 2006 Apr 20;440(7087):1069–72. doi: 10.1038/nature04665. [DOI] [PubMed] [Google Scholar]
- 65.Scheffzek K, Grunewald P, Wohlgemuth S, Kabsch W, Tu H, Wigler M, et al. The Ras-Byr2RBD complex: structural basis for Ras effector recognition in yeast. Structure. 2001 Nov;9(11):1043–50. doi: 10.1016/s0969-2126(01)00674-8. [DOI] [PubMed] [Google Scholar]
- 66.Mihrshahi R, Barclay AN, Brown MH. Essential roles for Dok2 and RasGAP in CD200 receptor-mediated regulation of human myeloid cells. J Immunol. 2009 Oct 15;183(8):4879–86. doi: 10.4049/jimmunol.0901531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kunath T, Gish G, Lickert H, Jones N, Pawson T, Rossant J. Transgenic RNA interference in ES cell-derived embryos recapitulates a genetic null phenotype. Nat Biotechnol. 2003 May;21(5):559–61. doi: 10.1038/nbt813. [DOI] [PubMed] [Google Scholar]
- 68.Hodgson L, Shen F, Hahn K. Biosensors for characterizing the dynamics of rho family GTPases in living cells. Curr Protoc Cell Biol. 2010 Mar;Chapter 14:1–26. doi: 10.1002/0471143030.cb1411s46. Unit 1 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.